

Abstract
De novo mutations in the X-linked gene encoding the transcription factor methyl-CpG binding protein 2 (MECP2) are the most frequent cause of the neurological disorder Rett syndrome (RTT). Hemizygous males usually die of neonatal encephalopathy. Heterozygous females survive into adulthood but exhibit severe symptoms including microcephaly, loss of purposeful hand motions and speech, and motor abnormalities, which appear after a period of apparently normal development. Most studies have focused on male mouse models because of the shorter latency to and severity in symptoms, yet how well these mice mimic the disease in affected females is not clear. Very few therapeutic treatments have been proposed for females, the more gender-appropriate model. Here, we show that self-complementary AAV9, bearing MeCP2 cDNA under control of a fragment of its own promoter (scAAV9/MeCP2), is capable of significantly stabilizing or reversing symptoms when administered systemically into female RTT mice. To our knowledge, this is the first potential gene therapy for females afflicted with RTT.
Introduction
In 1999, mutations in the epigenetic factor methyl-CpG binding protein 2 (MECP2) were identified as an underlying cause of Rett syndrome (RTT) in girls (Amir et al., 1999). Since that time, studies from many different investigators have been directed toward understanding the in vitro and in vivo mechanisms of MeCP2 action and the consequences of MeCP2 loss in different mouse models (Guy et al., 2011). Experiments provide support for MeCP2's role as transcriptional repressor, activator, and RNA-binding protein, among other functions, but, to date, no unifying principle to explain MeCP2 function has emerged. Further, RNA profiling of MeCP2-deficient mouse and human brains shows hundreds of changed transcripts (Colantuoni et al., 2001; Tudor et al., 2002; Chahrour et al., 2008). Possibly for these reasons, no single therapeutic has been shown to reverse or ameliorate RTT-like symptoms in female mouse models. Therefore, conceptually, gene replacement is well suited as a strategy for this disease. In particular, widespread neuronal cell death is not observed in mice or upon human autopsy (Reiss et al., 1993), suggesting that the cellular damage due to loss of MeCP2 is reversible. Indeed, global replacement of Mecp2 in otherwise Mecp2-null mice results in a reversal of key RTT-like symptoms and restoration of normal neuronal morphology (Guy et al., 2007; Robinson et al., 2012).
Previous studies have pointed to scAAV9 as an optimal virus for delivering genes in a widespread manner in the CNS (Foust et al., 2009). Two strong advantages are that it crosses the blood–brain barrier and infects both neuronal and non-neuronal cells in the brain (Foust et al., 2009; Gray et al., 2011). Expression in non-neuronal cells is particularly relevant for RTT because restoration of MeCP2 just in glia results in significant improvement in RTT-like symptoms in mice (Lioy et al., 2011; Derecki et al., 2012). Further, scAAV9 virus persists as a stable episome in nondividing cells both inside and outside of the nervous system. This attribute is desirable due to the potential involvement of peripheral tissues in RTT (O'connor et al., 2009; De Felice et al., 2010; Hofstaetter et al., 2010; Motil et al., 2012) and for the continued need for MeCP2 throughout adult life (McGraw et al., 2011; Cheval et al., 2012; Nguyen et al., 2012).
An important question is whether viral delivery of MeCP2 cDNA can be engineered to deliver physiological levels of MeCP2 in female RTT mouse models. To test this idea, we first used recombinant scAAV9 virus containing cre recombinase to rescue RTT-like symptoms in mice containing a floxed stop sequence within the endogenous Mecp2 gene. In this proof of principle, after systemic delivery of the virus, MeCP2 is expressed in the context of all of the normal regulatory elements, providing a benchmark for the extent of possible recovery. We then cloned MeCP2 cDNA, under control of a fragment of its own promoter and lacking other regulatory elements into scAAV9 and delivered it systemically into MeCP2-deficient mice. Both strategies resulted in widespread delivery of MeCP2 and dramatic improvements or reversal of RTT-like symptoms in the gender-appropriate female RTT mouse models. Moreover, the MeCP2 protein that was made, like the native protein, bound to heterochromatin and also restored the somal size of neurons to normal, indicating functionality. Our work provides the new findings that scAAV9 can deliver physiological levels of a tightly regulated transcription factor throughout the brain and that not all cells need to be corrected to significantly ameliorate disease progression. This work is important clinically in suggesting that gene replacement strategies are viable for reversible neurological diseases such as RTT.
Materials and Methods
All animal procedures were approved by Oregon Health and Science University Institutional Animal Care and Use Committee and UK Home Office regulations and licenses.
Virus preparation
AAV9 was produced by transient transfection procedures using a double-stranded AAV2-ITR–based CAG-Cre or MeCP2 minimal promoter–MeCP2 (E1) vector, with a plasmid encoding Rep2Cap9 sequence as described previously, along with the adenoviral helper plasmid pHelper (Stratagene) in 293 cells (Fu et al., 2003; Gao et al., 2004; Ayuso et al., 2010). Virus was purified by cesium chloride density gradient purification steps as described previously, dialyzed against PBS formulated with 0.001% Pluronic-F68 to prevent virus aggregation, and stored at 4°C. All vector preparations were titered by quantitative PCR using TaqMan technology. Purity of vectors was assessed by 4–12% SDS-acrylamide gel electrophoresis and silver staining (Invitrogen).
Maintenance, breeding, and genotyping
Mice were group housed with littermates in standard housing on a 12:12 h light/dark cycle. Mecp2stop (catalog #006849; Guy et al., 2007) and Mecp2Bird.knockout (Mecp2Bnull, catalog #003890; Guy et al., 2001) mice were obtained from The Jackson Laboratory and were on a C57BL/6 background. Wild-type (WT) male mice were crossed to female Mecp2+/stop and Mecp2+/Bnull mice to yield the male and female Mecp2stop and Mecp2Bnull genotypes. The floxed Stop sequence was identified from tail biopsies using the following primers: common 5′-AACAGTGCCAGCTGCTCTTC-3′, WT 5′-CTGTATCCTTGGGTCAAGCTG-3′, and mutant 5′-GCCAGAGGCCACTTGTGTAG-3′. For Bird-null, the following primers were used: 5′-CCACCCTCCAGTTTGGTTTA-3′ and 5′-GACCCCTTGGGACTGAAGTT-3′ (Lioy et al., 2011).
scAAV9 delivery
Vascular delivery.
Mice were placed in a restraint that positioned the tail in a lighted, heated groove. The tail was swabbed with alcohol and then injected intravenously with a 300 μl of viral solution containing 3 × 1012 DNase-resistant particles of scAAV9 in PBS. After the injection, mice were returned to their cages.
Cranial delivery.
Five- to 6-week-old male and 33- to 49-week-old female C57BL/6 mice were anesthetized using isoflurane and received analgesia (Vetergesic, 0.05 mg/kg) at the start of the surgical procedure and 24 h postsurgery. Using a glass capillary (diameter of 50 μm), mice were injected bilaterally at three sites (protocol modified from Sondhi et al., 2008): striatum (A/P 0.60 mm, L −7 mm, D −3.0 mm), thalamus (A/P −2.0 mm, L −1.0 mm, D −3.0 mm) and deep cerebellar nuclei (DCN, A/P 5.7 mm, L 1.8 mm, D −2.6 mm). As described previously (Sondhi et al., 2008), the capillary was introduced slowly into the parenchyma and, once at the right depth, was left for 2 min before and after the administration of the vector and then slowly withdrawn. The following vectors were administered at an infusion rate of 0.5 μl/min with a final volume of 3 μl: scAAV9/cre (1.3 × 1012 vg/ml) and scAAV9/Control (1 × 1012 vg/ml).
Tissue preparation, immunolabeling, and cell counts
Immunostaining (vascular delivery).
Mice were anesthetized by intraperitoneal injection of Avertin (2,2,2-tribromoethanol) and killed by transcardial perfusion of 4% PFA in PBS. Brains were equilibrated in 30% sucrose overnight at 4°C. Sagittal sections (40 μm) were cut at −20°C using a cryostat (Leica) and stored at −20°C. Sections were immunolabeled overnight at 4°C using the following primary antibodies: rabbit-MeCP2 (1:500; Covance), mouse-GFAP (1:500; Millipore), chicken-GFAP (1:200; Abcam), and mouse-NeuN (1:200; Millipore). Appropriate Alexa Fluor/Dylight Fluor secondary antibodies (1:500; Invitrogen) were used for 1 h at room temperature. DAPI was present in the ProLong Gold Antifade (Invitrogen) mounting reagent. Nissl staining (at either 594 or 640 nm) was performed as instructed by the manufacturer (NeuroTrace; Invitrogen). All images were collected on a Zeiss confocal laser scanning LSM 510 microscope.
MeCP2 expressing cells were identified as described previously (Lioy et al., 2011) with some modifications: nuclei of astrocytes (GFAP+ at 555 or 640 nm; NeuN− at 555 or 640 nm) and neurons (NeuN+ at 555 or 640 nm) were first identified by DAPI staining. Cells with clearly identified nuclei were then assessed for MeCP2 expression by analyzing 505 nm signal (excitation: 488 nm) in the nucleus.
Immunostaining (direct brain delivery).
Once removed, brains were snap-frozen and stored at −80°C until use. Fourteen-micrometer longitudinal sections were fixed in 4% PFA, followed by permeabilization in 0.1% Triton X-100. Sections were stained with a rabbit anti-MeCP2 primary antibody (1:200; Millipore) and a mouse anti-NeuN primary antibody conjugated with Cy3 (1:250; Millipore). An Alexa Fluor 488 goat anti-rabbit antibody (1:500; Invitrogen) was used as a secondary antibody for the MeCP2 staining. After being stained in DAPI (Sigma), sections were mounted in Prolong Gold (Invitrogen) and analyzed using a TCS SP1 confocal laser microscope (Leica). Colocalization of MeCP2, NeuN, and DAPI in the nucleus was evaluated in serial sections spaced throughout the entire brain.
Estimation of neuronal soma size.
Somal diameters of Nissl-stained neurons were determined by averaging the lengths of the long and short perpendicular axes across the cell body. Only cells with a clearly visible DAPI-stained nucleus were considered.
Fluorescent intensity measurements.
Brainstem sections were processed for immunolabeling as above. The fluorescent signal was generated on an inverted microscope equipped with an X-cite 120 fluorescence supply and a Fluar 20× objective (Zeiss) with a 0.75 numerical aperture. Images were captured using an AxioCam HRc camera (Zeiss) in grayscale mode (1388 × 1040 pixels). Under these conditions, an individual cell soma corresponded to ∼250 pixels. Digital images obtained in AxioVision Software (Zeiss) were exported to ImageJ. The fluorescence intensity associated with each pixel was determined for each of 10 randomly chosen fields corresponding to 750 × 550 μm. Excitation and acquisition parameters were adjusted to fully eliminate pixel saturation and all images were collected using the identical settings.
Phenotype scoring
Mice were removed from their home cages and placed onto a metal laminar flow hood for observation. Scoring was as follows. For mobility: 0 = WT; 1 = reduced movement compared with WT, with extended freezing periods or extended delay to movement when first placed on the surface; and 2 = complete loss of movement when placed on the surface. For gait: 0 = WT; 1 = hindlimbs spread wider than WT when ambulating and/or a lowered pelvis when ambulating; and 2 = lack of full strides by hindlimbs resulting in a dragging of hindquarters. For hindlimb clasping: 0 = WT; hindlimbs splay outward when suspended by the tail; 1 = one hindlimb is pulled into the body or forelimbs are stiff and splayed outward without motion; and 2 = one hindlimb is pulled into the body and forelimbs are stiff and splayed outward without motion and might form a widened bowl shape or both hindlimbs are pulled into the body with or without abnormal forelimb posture. For tremor: 0 = no tremor; 1 = intermittent mild tremor; and 2 = continuous tremor or intermittent violent tremor. For general condition: 0 = shiny coat, clear and opened eyes, normal body stance; 1 = dull or squinty eyes, dull or ungroomed coat, somewhat hunched stance; and 2 = piloerection, hunched stance.
Behavioral testing
All tests were performed at the same time of day (12:00–18:00) and in the same dedicated observation room. Mice were never subjected to multiple tasks on the same day.
Open field activity.
Mice were placed singly into the center of an open field arena (14 × 14 inches) equipped to record live images from the top. Activity was recorded for 20 min using StereoScan Software (Clever Systems) on a Dell computer fitted with a Microsoft Windows operating system. Software calculated the total distance traveled and average velocity of the movements from recorded movies. The mice could not see the experimenter during recordings.
Rotarod.
Mice were placed on an elevated rotating rod (diameter: 7 cm, elevated: 45 cm, Economex; Columbus Instruments) initially rotating at 5.0 rpm. The rod accelerated 5.0 rpm/s. The latency to fall (in seconds) was recorded manually using individual mouse-specific stopwatches. Each mouse received three trials per day, with no delay between trials, on three consecutive days.
Platform test.
The platform test was performed as described previously (Grady et al., 2006) with some modifications. Each mouse was timed for how long it remained on an elevated, circular platform (3.0 cm in diameter) with rounded edges. A maximum score of 60 s was assigned if the mouse remained on the platform for the entire test trial without falling. Two trials were administered for each test with 4 h intervening between trials and means were calculated across the trials for each mouse.
Inverted screen test.
The inverted screen test was performed as described previously (Grady et al., 2006) with some modifications. Each mouse was placed in the middle of wire grid (parallel metal wires 0.5 cm apart) that was inverted to 180°. A mouse was timed for how long it remained upside down on the screen, with a maximum score of 60 s being given if the animal did not fall. Two trials were administered for each test, with 4 h intervening between trials, and means were calculated across the trials for each mouse.
Nesting ability.
Mice were placed in individual cages and provided with a nest-building material (5 cm × 5 cm × 0.5 cm). The material was placed in top left corner of cage and nesting ability was scored overnight based on the interaction of individual mouse with nesting material. The scores of 0, 1, 2, or 3 were assigned. The score 0 was assigned to a mouse that did not interact at all with the material and the score 3 was assigned to a mouse that completely used the material to build a nest.
Novel object recognition test.
The novel object recognition test was conducted in the open field arena used to evaluate motor activity. The two objects (a sphere and a box) were selected based on similar volume and unbiased interaction of WT mice. During habituation, the mice were allowed to explore an empty arena for 5 min. Twenty-four hours after habituation, the mice were exposed to the familiar arena with two identical objects (spheres) placed at an equal distance for 5 min. The next day, same exercise was repeated. On third day of the test, the mice were allowed to explore the open field in the presence of the familiar and a novel object (a box) for 5 min to test cognition. The time spent exploring each object on the second and final day of the test was recorded to estimate the extent of novel object recognition by calculating the discrimination index (DI) as (Tn − Tf)/(Tn + Tf) where Tn is time with the novel object and Tf is time with the familiar object. The DI value can vary between +1 and −1, where a positive score indicates more time spent with the novel object, a negative score indicates more time spent with the familiar object, and a zero score indicates a null preference.
Respiration
Respiratory parameters were determined in a body plethysmograph. Individual nonanesthetized animals were placed in a 65 ml chamber with their head exposed through a close-fitting hole in Parafilm. A pneumotachograph was connected to the chamber and a differential pressure transducer (Model PT5A; Grass Instrument). The pressure signal was integrated to give tidal volume. Volume changes were calibrated by injecting known amounts of air into the chamber. The analog signal from the transducer was amplified, converted to digital, displayed on a monitor, and stored to disk by computer for later analysis. Apnea was defined as an expiratory time of 1.0 s or greater. Irregularity score was determined from: absolute (TTOTn − TTOTn + 1)/(TTOTn + 1).
Statistics
The following measurements were analyzed using one-way ANOVA followed, when appropriate (p < 0.05), by Newman–Keuls post hoc test: anatomical and cell-type expression patterns of transduced MeCP2, whole body and brain weights, respiratory parameters, open field activity, time on rotarod, and somal diameters. The following measurements were analyzed using Kruskal–Wallis test followed, when appropriate (p < 0.05), by Dunn's multiple-comparisons test: phenotype severity scores, nesting scores, time on an inverted grid, time on a platform, and novel object recognition. Survival curves were compared using the log-rank method. All statistics were performed using GraphPad version 5.0 software (Prism).
Results
Systemic delivery of AAV9 bearing Cre recombinase results in stabilization and reversal of symptoms in male and female Mecp2Stop mice
To provide a benchmark for the extent of recovery possible with recombinant scAAV9, we first sought to repeat the genetic study in which RTT-like symptoms in adult Mecp2stop/y mice were reversed by mating to mice containing a conditional cre transgene under control of the ubiquitous CAG promoter (Guy et al., 2007). To this end, we generated scAAV9 virus encoding cre recombinase under this promoter (Fig. 1A), injected it into the tail vein of adult 4- to 8-week-old Mecp2stop/y mice, and determined the cellular distribution of MeCP2 expression 3 weeks later. Immunolabeling and Western blotting at 3 weeks after injection showed MeCP2 expression throughout the brain (Fig. 1B–D). MeCP2 protein was detected in a high percentage (15–50%) of neurons (NeuN+), GFAP+ astrocytes (30–75%), and cell types that were not positive for either marker (Fig. 1E,F). In peripheral tissues, MeCP2 protein was present at ∼20–100% of WT levels (Fig. 1G).
Figure 1.
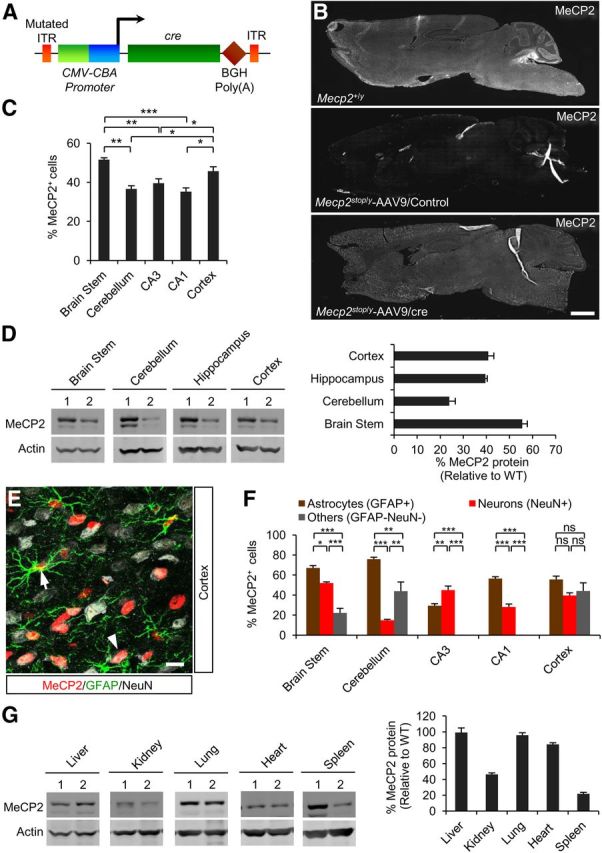
MeCP2 expression is restored throughout the brain after systemic delivery of scAAV9/cre into Mecp2stop/y mice. A, Schematic of the scAAV9/cre viral construct. ITR, inverted terminal repeat; CMV, cytomegalovirus enhancer; CBA, chicken-β-actin promoter; CRE, cre recombinase; BGH, bovine growth hormone. B, Representative sagittal brain sections from 10-week-old WT or virally injected mice. Scale bar, 1 mm. C, Efficiency of virally mediated MeCP2 expression in specified brain regions relative to total DAPI-positive cells (18 sections, n = 3 mice). D, Left: Western blot of MeCP2 protein levels in injected mice. Lane 1, Mecp2+/y; Lane 2, Mecp2stop/y-AAV9/cre. Right: Quantified data of Western blot (n = 3). E, Immunostaining of representative cortical section showing MeCP2 expression in neurons (NeuN+) and astrocytes (GFAP+). Arrow and arrowhead indicate a MeCP2+ astrocyte and neuron, respectively. Scale bar, 10 μm. F, Immunostaining for MeCP2 restoration in Mecp2stop/y mice after scAAV9/cre injection. Cell counts are relative to indicated cell type-specific markers (18 sections, n = 3 mice). G, Western blot (left) of MeCP2 protein levels in different peripheral tissues. Lane 1, Mecp2+/y; Lane 2, Mecp2stop/y-AAV9/cre. Right: Quantified data of Western blot (n = 3). Data are means ± SEM. In C and F, *p < 0.05, **p < 0.01, ***p < 0.001, and NS by one-way ANOVA (Newman–Keuls multiple-comparisons test). Data are means ± SEM.
The virally transduced MeCP2 expression resulted in robust behavioral recovery. We found that four of seven Mecp2stop/y mice injected with scAAV9/cre survived past 40 weeks of age before three were processed for immunocytochemistry (Fig. 2A,B). The remaining mouse survived for over 1 year before being killed for analysis. In contrast, the mice injected with control virus showed severe symptoms and a median survival time of 18–19 weeks (Fig. 2B). The prolonged longevity of scAAV9/cre–injected males was accompanied by overall more robust health, as measured by an observational scoring system based on hindlimb clasping, tremors, locomotion, gait, and general condition (Guy et al., 2007; see also Materials and Methods). The scoring revealed stabilization at a moderately symptomatic score of ∼4 (where 10 = lethality) over a 23-week monitoring period (Fig. 2C). In addition, open field activity was improved significantly and respiration was reversed to the WT level (Fig. 2D–G).
Figure 2.
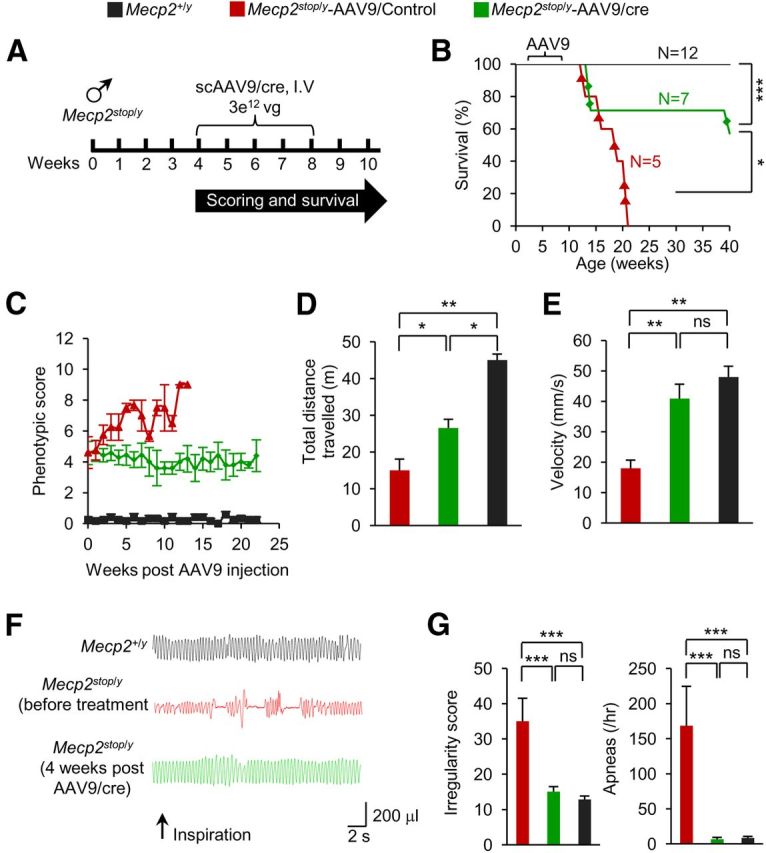
Systemic delivery of scAAV9/cre into Mecp2stop/y mice rescues abnormal respiration and open field motor activities. A, Experimental paradigm. Mice were analyzed 4–5 weeks after injection. B, Kaplan-Meier survival curve shows significant extension in longevity in scAAV9/cre mice. ***p < 0.001, *p < 0.05, log-rank test. C, Observational scores show stabilization in Mecp2stop/y mice injected with scAAV9/cre (n = 7), scAAV9/Control (n = 5), and noninjected Mecp2+/y mice (n = 12). D, E, Open field activity in scAAV9/cre (n = 3), scAAV9/Control (n = 3), and Mecp2+/y (n = 4) mice. F, Representative plethysmographic recordings from an age-matched Mecp2+/y mouse (top), Mecp2Stop/y mouse before (middle), and 5 weeks post scAAV9/cre injection (bottom). Scale shows time and tidal volume. G, Respiratory pattern returns to normal in scAAV9/cre (n = 6), scAAV9/Control (n = 3), and Mecp2+/y (n = 12) mice. In D, E, and G, *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA (Newman–Keuls multiple-comparison test). Data are means ± SEM.
To test for effects of cre-mediated restoration of MeCP2 in females, we injected 7- to 10-month-old symptomatic Mecp2Stop/+ mice with a similar load of scAAV9/cre or control scAAV9 virus lacking a cDNA insert (Fig. 3A). We performed weekly observational scoring over a 35-week period. We found that, unlike control injected mice, symptomatic Mecp2Stop/+-scAAV9/cre mice did not progress in severity of symptoms over this time period (Fig. 3B). Further, they lacked the overweight phenotype associated normally with RTT female mice. Three of 10 control-injected mice exhibited seizures, another prominent RTT phenotype, compared with 0/10 of the Mecp2Stop/+-scAAV9/cre–injected females, which performed as well as WT age-matched controls in the rotarod, platform, and inverted grid tests and nest building (Fig. 3C–F). We also examined cognition using a novel object recognition test. The Mecp2Stop/+-scAAV9/cre mice performed at WT level in this test (Fig. 3G) and had increased brain weights compared with controls (Fig. 3H).
Figure 3.

Systemic delivery of scAAV9/cre virus into Mecp2stop/+ mice prevents progression of behavioral abnormalities. A, Experimental paradigm for injection and scoring. B, Observational scores of mice (n = 10) injected with indicated virus or control, noninjected Mecp2+/+ mice (n = 10). Arrow indicates time of behavioral analysis. C, Rotarod analysis on third day of test. D, Platform test for balance. E, Inverted grid test for strength (n = 9). F, Nesting ability (n = 9) with scoring guide at right. G, Novel object recognition analysis showing discrimination index on last day of test (n = 9). H, Brain weights analyzed 40 weeks after injection or in age-matched noninjected mice (n = 4). *p < 0.05, **p < 0.01, ***p < 0.001, NS by one-way ANOVA (Newman–Keuls multiple-comparison test for C and H) and one-way ANOVA (Dunn's multiple-comparison test for D–G). Data are means ± SEM.
Direct brain injection of AAV9 bearing cre recombinase results in predominantly neuronal-specific MeCP2 expression and modest behavioral improvement
Direct cranial injection of AAV reduces certain brain pathologies (Sondhi et al., 2007). To compare the efficacy of direct and systemic injection regimes, we injected scAAV9/cre or scAAV9/control virus into three sites in the brains of symptomatic male and female Mecp2Stop mice (Fig. 4A,B). In the injected males, immunocytochemistry showed that the percentage of MeCP2+DAPI+ cells varied between 20 and 50% in the different brain regions but, unlike the case for systemic delivery, most of the expressing cells were neurons (90–95%; Fig. 4C–G). This neurotropism may be a consequence of circumventing the cellular anatomy of the blood–brain barrier that is likely a factor in systemic delivery. With cranial delivery into males, there was a 6 week extension of median survival in cre-injected compared with control-injected mice and some improvement in observational scores (data not shown). In females, observational scoring indicated modest but significant improvement of symptoms by 20 weeks (Fig. 4H,I). However, interpretation was confounded by the appearance of a parkinsonian-like phenotype and cre toxicity that was evident at autopsy in female mice injected specifically with scAAV9/cre virus. Therefore, it is likely that in the cranial injection paradigm, local overexpression of cre (Ciesielska et al., 2013) combined with restricted distribution of MeCP2-expressing cells compromised rescue. Therefore, we focused on systemic AAV9 injections in all further studies.
Figure 4.

Direct brain injections of scAAV9/cre virus into Mecp2stop mice result in predominantly neuronal-specific MeCP2 expression and a mild improvement of behavioral phenotypes. A, Schematic showing site of injections (*). B, Experimental paradigm for males. C, Immunostaining of representative striatal section from a Mecp2stop/y mouse injected with scAAV9/cre shows MeCP2 expression in neurons (NeuN+). Scale bar, 200 μm. D–G, Quantification of immunostaining data (n = 3). Data are means ± SEM. Cell counts are relative to total DAPI-positive cells (D), total NeuN-positive cells (E), total NeuN-negative cells (F), and total MeCP2-positive cells (G). H, Experimental paradigm for females. I, Observational scores for female Mecp2Stop/+ mice injected with scAAV9/cre (n = 5), scAAV9/Control (n = 5), and noninjected Mecp2+/+ mice (n = 12).
Systemic delivery of AAV9 bearing Mecp2 results in physiological levels of MeCP2 protein in the brain, localization of MeCP2 to heterochromatin, and rescued neuronal soma size
The cre experiments provided proof of principle that gene therapy is well suited to RTT by showing that the number of cells transduced by AAV9 is sufficient to rescue RTT-like phenotypes. However, gene therapy for RTT requires AAV9 bearing MeCP2 and the amount of MeCP2 expressed/cell may need to be regulated tightly because MeCP2 gene duplication gives rise to a neurological disease (Friez et al., 2006; del Gaudio et al., 2006). To counteract possible overexpression and to better mimic the expression pattern of virally mediated MeCP2, we generated a new vector containing MeCP2 (E1) cDNA under control of an ∼700 bp fragment of its own promoter (Rastegar et al., 2009; scAAV9/MeCP2; Fig. 5A). Because epitopes can alter protein localization and other functions (Jarvik and Telmer, 1998), we did not use a tagged MeCP2 protein in this study. Primary cultures of both neurons and glia prepared from Mecp2-null mice expressed MeCP2 after transfection of the scAAV9/MeCP2 construct (data not shown and Rastegar et al., 2009). In vivo, MeCP2 expressed from scAAV9/MeCP2 was detected by immunolabeling throughout the brain (Fig. 5B,C). However, with the exception of cerebellum, MeCP2 expression, unlike the case for the scAAV9/cre–injected mice, was not overrepresented in astrocytes (Fig. 5D). This may reflect the cell-specific regulatory elements in the cloned promoter fragment because MeCP2 is expressed generally at lower levels in astrocytes than in neurons (Ballas et al., 2009; Skene et al., 2010). Ectopically expressed MeCP2 migrated to the correct size, as determined by Western blot analysis (Fig. 5E).
Figure 5.

Systemic injection of Mecp2Bnull/y mice with scAAV9/MeCP2 virus results in MeCP2 expression in different cell types in brain. A, Schematic of scAAV9/MeCP2 vector. Mouse Mecp2-e1 is cloned downstream of 730 bp fragment of Mecp2 promoter. Other abbreviations are as noted in the legend to Figure 1. B, Experimental paradigm. C, Immunostaining of MeCP2 expression in different brain regions. Cell counts are relative to total DAPI-positive cells (36 sections, n = 2 mice). D, Expression of MeCP2 in neurons and non-neuronal cells varies with brain region. Cell counts are relative to indicated cell-specific marker (12 sections, n = 2 mice). *p < 0.05, **p < 0.01, ***p < 0.001, NS by one-way ANOVA (Newman–Keuls multiple-comparison test). Data are means ± SEM. E, Western blot of MeCP2 protein in different brain regions. Lane 1, Mecp2+/y; Lane 2, Mecp2Bnull/y; and Lane 3, Mecp2Bnull/y-AAV9/MeCP2.
In Mecp2Bnull/y mice, virally expressed MeCP2 was detected immunochemically in heterochromatic puncta of both cell types, indicating WT DNA-binding function (Fig. 6A). Further, the brains in human RTT girls and male and female mouse models are microcephalic and associated with smaller neuronal somas (Chen et al., 2001; Armstrong, 2005; Chahrour and Zoghbi, 2007). MeCP2-positive neurons in the CA3 region and mitral cell layer of the olfactory bulb of scAAV9/MeCP2–injected and WT males had significantly larger somal sizes than MeCP2-negative neurons (Fig. 6B). Consistent with all of these metrics, in the present study, the injected male mice had prolonged lifespans and better observational scores compared with control injected mice (Fig. 6C, D).
Figure 6.

MeCP2 expressed from virus binds to DNA, restores normal neuronal somal size, and improves survival. A, Ectopic MeCP2 localizes to DAPI+ heterochromatin puncta in Mecp2Bnull/y-scAAV9/MeCP2–injected mice. Shown is colocalization of DAPI and MeCP2 in olfactory neuron (top), CA3 pyramidal neuron (middle), and dentate gyrus astrocyte (bottom). Pearson's correlation coefficient = 0.943, 0.932, and 0.985, respectively. Scale bar, 5 μm. B, Image (left) shows representative MeCP2-positive (arrow) and MeCP2-negative (arrowhead) CA3 pyramidal neurons. Scale bar, 10 μm. Average somal diameters (right) of MeCP2-positive (purple) and MeCP2-negative (red) CA3 pyramidal neurons and olfactory bulb mitral cells from Mecp2Bnull/y-scAAV9/MeCP2–injected mice (n = 2). Also shown are measurements from WT mice (black bars; n = 2). The number of cells analyzed is indicated above each bar. ***p < 0.001 by one-way ANOVA (Newman–Keuls multiple-comparison test). Data are means ± SEM. C, Kaplan-Meier survival curve. D, Observational scores. Mecp2Bnull/y-scAAV9/MeCP2 (n = 5), Mecp2Bnull/y-AAV9/Control (n = 6), and Mecp2+/y (n = 6). Data are means ± SEM. E, Field pixel intensities of MeCP2-Cy3 immunofluorescence measured from brainstem sections of Mecp2+/y and Mecp2Bnull/y-scAAV9/MeCP2 mice. Traces represent pixel intensities from individual fields and each field is indicated by a differently colored trace, n = 2 mice per genotype, 5 fields per mouse.
To determine further whether MeCP2 was expressed from AAV9 close to physiological levels, we estimated MeCP2 protein expression at the cellular level using immunofluorescence. For this purpose, we measured the pixel intensities of 10 fields selected randomly from the brainstem of Mecp2+/y and scAAV9/MeCP2-transduced Mecp2Bnull/y mice (Fig. 6E). Fields from WT mice contained ∼400 cell bodies each and each cell body comprised ∼250 pixels. The fluorescence intensity and acquisition parameters were adjusted to eliminate saturation. The same settings were used for all of the analyses and the raw data from each image are plotted (Fig. 6E). Measurements from Mecp2+/y brains resulted in three peaks of fluorescence (Fig. 6E, left). The first peak occurred between 0 and 100 arbitrary units (AU) of fluorescence intensity and corresponded to background fluorescence. Two additional peaks, corresponding to nuclear MeCP2 signals, occurred at 171–174 AU and 202–206 AU. Both of these peaks were recapitulated in AAV9/MeCP2–transduced Mecp2Bnull/y mice (172–174 AU and 202–206 AU; Fig. 6E, right). Our analysis did not show peaks at higher intensity values in AAV9/MeCP2–transduced Mecp2Bnull/y mice relative to Mecp2+/y mice, which would indicate overexpression. Therefore, we conclude that scAAV9/MeCP2 injection resulted in MeCP2 protein levels similar to endogenous levels.
Robust improvement of symptoms in Mecp2Bnull/+ mice after systemic Mecp2 delivery
Having established that scAAV9/MeCP2 programmed MeCP2 expression to approximately physiological levels in multiple cell types in brain, we next analyzed rescue parameters in 15- to 17-month-old symptomatic Mecp2Bnull/+ mice that were systemically injected with scAAV9/MeCP2 or control virus at 10 to 12 months old (Fig. 7A). Like the males, there was no evidence for overexpression of MeCP2 (Fig. 7B) and viral therapy did not compromise survival. The observational scores increased initially from 2 to 3. Strikingly, by 12 weeks, scAAV9/MeCP2–injected females stabilized at an improved score of 1 until the end of scoring at 24 weeks, whereas females injected with control virus progressed to a score of nearly 6 (Fig. 7C). The scAAV9/MeCP2–injected Mecp2Bnull/+ mice also performed significantly better than scAAV9/control females in the rotarod, inverted grid, and platform tests and nesting ability (Fig. 7D–G). None of the injected females exhibited seizures, unlike the females injected with control virus (2/5 with seizures). We also examined respiration. Unlike the other motor phenotypes tested, the effect on respiration was inconclusive in this cohort of mice. Of 5 Mecp2Bnull/+ mice injected with scAAV9/MeCP2, 2 showed improvement, with 1.3- and 3-fold decreases in apnea rate at 6 months after injection. However, 2 other mice progressed to 1.6- and 10-fold increases in apnea rates and another mouse, which was WT in respiration initially, showed no change at 6 months after injection. All 3 control injected mice progressed over a 4- to 48-fold range in apnea rates over their initial values during this time interval. The lack of consistent effect on respiration by injection with AAV9/MeCP2 virus may reflect the need for higher numbers of cells to correct this phenotype.
Figure 7.

Systemic delivery of scAAV9/MeCP2 virus into Mecp2Bnull/+ mice prevents progression of aberrant behaviors. A, Experimental paradigm. Mice were analyzed 5 months after injection. B, Field pixel intensities of MeCP2-Cy3 immunofluorescence measured from brainstem sections of WT and scAAV9/MeCP2–injected females. Traces represent pixel intensities from individual fields and each field is indicated by a differently colored trace. n = 2 mice per genotype, 5 fields per mouse. C, Observational scores of Mecp2Bnull/+ mice injected with scAAV9/MeCP2 (n = 8), scAAV9/Control (n = 5), and noninjected (Mecp2+/+) mice (n = 8). Arrow indicates time of behavioral analysis. D, Rotarod activity on third day of test. E, Inverted grid test. F, Platform test for scAAV9/MeCP2 (n = 8), scAAV9/Control (n = 5), and Mecp2+/+ (n = 8) mice. G, Nesting ability of scAAV9/MeCP2 (n = 8), scAAV9/Control (n = 5), and Mecp2+/+ (n = 8) mice. *p < 0.05, **p < 0.01, ***p < 0.001, NS by one-way ANOVA (Newman–Keuls multiple-comparison test for D and one-way ANOVA with Dunn's multiple-comparison test for E–G). Data are means ± SEM.
Discussion
Our results are important because they suggest for the first time that symptoms in human RTT female patients may be reversible by ectopic expression of MeCP2 in virus that infects peripheral tissue and multiple cell types within the CNS. Further, the widespread expression of MECP2 programmed from scAAV9/MeCP2 in different brain regions was likely an important factor in the rescue, because regional MeCP2 expression levels in mosaic MeCP2-deficient mice correlates with different behavioral outcomes (Wither et al., 2013). A previous study showed some improvement of symptoms in male mouse models of RTT injected with recombinant scAAV9 virus, but was discouraging in that only a very small percentage of cells gained MeCP2 expression in brain and recovery was correspondingly very modest (Gadalla et al., 2013). More importantly, the disease initiates and progresses differently in females and males due to the mosaic nature of MeCP2 loss of function in females. Therefore, therapeutics designed especially for affected females are required, as we tested here.
MeCP2 was first identified as a protein that binds to methylated DNA and this property is reflected in its association with heterochromatin (Nan et al., 1996). The MeCP2 programmed from our virus binds to heterochromatin and, further, was sufficient to restore somal size to normal. It will be important in the future to determine the extent to which any molecular hallmarks, as they are identified, are also restored by expression of ectopically supplied MeCP2. Perhaps most surprisingly, our experiments indicate that not all cells need to be repaired with MeCP2 to stabilize or reverse some key phenotypes in the female mice. This may reflect redundancy of cells in behavioral circuits or compensation by other mechanisms to supplant circuits weakened by insufficient neuronal/glial MeCP2.
The use of AAV is gaining momentum for treating a variety of human genetic disorders (Nathwani et al., 2011; McClements and Maclaren, 2013), but CNS disorders represent a particular challenge. Our study is an important first step in highlighting the potential for AAV9 in treating the neurological symptoms in RTT. Future studies optimizing AAV9 vector size, mode of delivery, and promoter efficiency should improve our promising gene therapy results for the neurological disorder RTT.
Footnotes
The work was supported by the Rett Syndrome Research Trust, the National Institutes of Health (to G.M.), the Wellcome Trust (Grants #077224 and #091580), Medical Research Council, UK (Grant #G0800401), Action Medical Research in association with the Henry Smith Charity, and the R.S. MacDonald Charitable Trust (Grant #SP4443 to A.B.). We thank Travis Polston and Andrea Ansari (Oregon Health and Science University) and Kyla Brown and Jim Selfridge (University of Edinburgh) for help with phenotypic analysis and mouse care.
The authors declare no competing financial interests.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005;20:747–753. doi: 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- Ayuso E, Mingozzi F, Montane J, Leon X, Anguela XM, Haurigot V, Edmonson SA, Africa L, Zhou S, High KA, Bosch F, Wright JF. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010;17:503–510. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
- Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Cheval H, Guy J, Merusi C, De Sousa D, Selfridge J, Bird A. Postnatal inactivation reveals enhanced requirement for MeCP2 at distinct age windows. Hum Mol Genet. 2012;21:3806–3814. doi: 10.1093/hmg/dds208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciesielska A, Hadaczek P, Mittermeyer G, Zhou S, Wright JF, Bankiewicz KS, Forsayeth J. Cerebral infusion of AAV9 vector-encoding non-self proteins can elicit cell-mediated immune responses. Mol Ther. 2013;21:158–166. doi: 10.1038/mt.2012.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C, Jeon OH, Hyder K, Chenchik A, Khimani AH, Narayanan V, Hoffman EP, Kaufmann WE, Naidu S, Pevsner J. Gene expression profiling in postmortem Rett Syndrome brain: differential gene expression and patient classification. Neurobiol Dis. 2001;8:847–865. doi: 10.1006/nbdi.2001.0428. [DOI] [PubMed] [Google Scholar]
- De Felice C, Guazzi G, Rossi M, Ciccoli L, Signorini C, Leoncini S, Tonni G, Latini G, Valacchi G, Hayek J. Unrecognized lung disease in classic Rett syndrome: a physiologic and high-resolution CT imaging study. Chest. 2010;138:386–392. doi: 10.1378/chest.09-3021. [DOI] [PubMed] [Google Scholar]
- del Gaudio D, Fang P, Scaglia F, Ward PA, Craigen WJ, Glaze DG, Neul JL, Patel A, Lee JA, Irons M, Berry SA, Pursley AA, Grebe TA, Freedenberg D, Martin RA, Hsich GE, Khera JR, Friedman NR, Zoghbi HY, Eng CM, Lupski JR, Beaudet AL, Cheung SW, Roa BB. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med. 2006;8:784–792. doi: 10.1097/01.gim.0000250502.28516.3c. [DOI] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, Kipnis J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friez MJ, Jones JR, Clarkson K, Lubs H, Abuelo D, Bier JA, Pai S, Simensen R, Williams C, Giampietro PF, Schwartz CE, Stevenson RE. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. 2006;118:e1687–1695. doi: 10.1542/peds.2006-0395. [DOI] [PubMed] [Google Scholar]
- Fu H, Muenzer J, Samulski RJ, Breese G, Sifford J, Zeng X, McCarty DM. Self-complementary adeno-associated virus serotype 2 vector: global distribution and broad dispersion of AAV-mediated transgene expression in mouse brain. Mol Ther. 2003;8:911–917. doi: 10.1016/j.ymthe.2003.08.021. [DOI] [PubMed] [Google Scholar]
- Gadalla KK, Bailey ME, Spike RC, Ross PD, Woodard KT, Kalburgi SN, Bachaboina L, Deng JV, West AE, Samulski RJ, Gray SJ, Cobb SR. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther. 2013;21:18–30. doi: 10.1038/mt.2012.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady RM, Wozniak DF, Ohlemiller KK, Sanes JR. Cerebellar synaptic defects and abnormal motor behavior in mice lacking alpha- and beta-dystrobrevin. J Neurosci. 2006;26:2841–2851. doi: 10.1523/JNEUROSCI.4823-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther. 2011;19:1058–1069. doi: 10.1038/mt.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- Hofstaetter JG, Roetzer KM, Krepler P, Nawrot-Wawrzyniak K, Schwarzbraun T, Klaushofer K, Roschger P. Altered bone matrix mineralization in a patient with Rett syndrome. Bone. 2010;47:701–705. doi: 10.1016/j.bone.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Jarvik JW, Telmer CA. Epitope tagging. Annu Rev Genet. 1998;32:601–618. doi: 10.1146/annurev.genet.32.1.601. [DOI] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, Mandel G. A role for glia in the progression of Rett's syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClements ME, MacLaren RE. Gene therapy for retinal disease. Transl Res. 2013;161:241–254. doi: 10.1016/j.trsl.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science. 2011;333:186. doi: 10.1126/science.1206593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motil KJ, Caeg E, Barrish JO, Geerts S, Lane JB, Percy AK, Annese F, McNair L, Skinner SA, Lee HS, Neul JL, Glaze DG. Gastrointestinal and nutritional problems occur frequently throughout life in girls and women with Rett syndrome. J Pediatr Gastroenterol Nutr. 2012;55:292–298. doi: 10.1097/MPG.0b013e31824b6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Tate P, Li E, Bird A. DNA methylation specifies chromosomal localization of MeCP2. Mol Cell Biol. 1996;16:414–421. doi: 10.1128/mcb.16.1.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, Chowdary P, Riddell A, Pie AJ, Harrington C, O'Beirne J, Smith K, Pasi J, Glader B, Rustagi P, Ng CY, Kay MA, Zhou J, Spence Y, Morton CL, Allay J, Coleman J, Sleep S, Cunningham JM, Srivastava D, Basner-Tschakarjan E, Mingozzi F, High KA, Gray JT, Reiss UM, Nienhuis AW, Davidoff AM. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MV, Du F, Felice CA, Shan X, Nigam A, Mandel G, Robinson JK, Ballas N. MeCP2 is critical for maintaining mature neuronal networks and global brain anatomy during late stages of postnatal brain development and in the mature adult brain. J Neurosci. 2012;32:10021–10034. doi: 10.1523/JNEUROSCI.1316-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor RD, Zayzafoon M, Farach-Carson MC, Schanen NC. Mecp2 deficiency decreases bone formation and reduces bone volume in a rodent model of Rett syndrome. Bone. 2009;45:346–356. doi: 10.1016/j.bone.2009.04.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastegar M, Hotta A, Pasceri P, Makarem M, Cheung AY, Elliott S, Park KJ, Adachi M, Jones FS, Clarke ID, Dirks P, Ellis J. MECP2 isoform-specific vectors with regulated expression for Rett syndrome gene therapy. PloS one. 2009;4:e6810. doi: 10.1371/journal.pone.0006810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss AL, Faruque F, Naidu S, Abrams M, Beaty T, Bryan RN, Moser H. Neuroanatomy of Rett syndrome: a volumetric imaging study. Annal Neurol. 1993;34:227–234. doi: 10.1002/ana.410340220. [DOI] [PubMed] [Google Scholar]
- Robinson L, Guy J, McKay L, Brockett E, Spike RC, Selfridge J, De Sousa D, Merusi C, Riedel G, Bird A, Cobb SR. Morphological and functional reversal of phenotypes in a mouse model of Rett syndrome. Brain. 2012;135:2699–2710. doi: 10.1093/brain/aws096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondhi D, Hackett NR, Peterson DA, Stratton J, Baad M, Travis KM, Wilson JM, Crystal RG. Enhanced survival of the LINCL mouse following CLN2 gene transfer using the rh. 10 rhesus macaque-derived adeno-associated virus vector. Mol Ther. 2007;15:481–491. doi: 10.1038/sj.mt.6300049. [DOI] [PubMed] [Google Scholar]
- Sondhi D, Peterson DA, Edelstein AM, del Fierro K, Hackett NR, Crystal RG. Survival advantage of neonatal CNS gene transfer for late infantile neuronal ceroid lipofuscinosis. Exp Neurol. 2008;213:18–27. doi: 10.1016/j.expneurol.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002;99:15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wither RG, Lang M, Zhang L, Eubanks JH. Regional MeCP2 expression levels in the female MeCP2-deficient mouse brain correlate with specific behavioral impairments. Exp Neurol. 2013;239:49–59. doi: 10.1016/j.expneurol.2012.09.005. [DOI] [PubMed] [Google Scholar]