

Abstract
Major histocompatibility complex class I (MHCI) molecules negatively regulate cortical connections and are implicated in neurodevelopmental disorders, including autism spectrum disorders and schizophrenia. However, the mechanisms that mediate these effects are unknown. Here, we report a novel MHCI signaling pathway that requires the myocyte enhancer factor 2 (MEF2) transcription factors. In young rat cortical neurons, MHCI regulates MEF2 in an activity-dependent manner and requires calcineurin-mediated activation of MEF2 to limit synapse density. Manipulating MEF2 alone alters synaptic strength and GluA1 content, but not synapse density, implicating activity-dependent MEF2 activation as critical for MHCI signaling. The MHCI-MEF2 pathway identified here also mediates the effects of a mouse model of maternal immune activation (MIA) on connectivity in offspring. MHCI and MEF2 levels are higher, and synapse density is lower, on neurons from MIA offspring. Most important, dysregulation of MHCI and MEF2 is required for the MIA-induced reduction in neural connectivity. These results identify a previously unknown MHCI-calcineurin-MEF2 signaling pathway that regulates the establishment of cortical connections and mediates synaptic defects caused by MIA, a risk factor for autism spectrum disorders and schizophrenia.
Introduction
Although major histocompatibility complex class I (MHCI) molecules are most well known for their role in mediating immunity throughout the body, these proteins are also present in the CNS (Corriveau et al., 1998; Needleman et al., 2010; Ribic et al., 2011; Chacon and Boulanger, 2013). During brain development, MHCI plays important roles in activity-dependent refinement of the visual system, synaptic transmission, and long-term plasticity (Elmer and McAllister, 2012). MHCI also negatively regulates the establishment and strength of synapses and controls excitatory/inhibitory balance onto cortical neurons (Glynn et al., 2011). Despite the clear importance of MHCI in brain development, little is known about how these proteins exert their effects on neurons in the CNS.
One approach to identify the molecular mechanisms that mediate MHCI function in the CNS is to test whether other molecules that play similar roles are components of the MHCI signaling pathway. Many molecules promote synapse formation, but relatively few have been identified that limit synapse density early in development. Myocyte enhancer factor 2 (MEF2) transcription factors were one of the first molecular families discovered to play this role (Flavell et al., 2006). Neuronal activity activates MEF2, increasing the expression of many genes, some of which cause synapse elimination (Tsai et al., 2012; Dietrich, 2013). The similarities in the effects of MEF2 and MHCI on synaptic connections suggest the possibility that MHCI activates a signaling pathway that includes MEF2 to restrict synapse density.
In addition to their role in brain development, MHCI genes have also been implicated in neurodevelopmental disorders through both genetic associations and environmental risk factors, especially maternal infection (Brown and Patterson, 2011; Michel et al., 2012; Needleman and McAllister, 2012). Mouse models of maternal immune activation (MIA) strongly support this link. Offspring of pregnant mice injected with the viral mimic poly(I:C) dsRNA display behaviors, gene expression, neuroanatomy, and neurochemistry consistent with both autism spectrum disorder (ASD) and schizophrenia (SZ; Meyer et al., 2009; Patterson, 2011; Malkova et al., 2012; Giovanoli et al., 2013). However, little is known about how MIA affects brain development, including if and when changes in connectivity occur. Because many cytokines are elevated in the brains of newborn MIA offspring (Garay et al., 2013) and regulate MHCI on many cell types (Johnson, 2003), it is possible that MIA-induced cytokines regulate MHCI and its signaling pathways in the brain, thereby causing changes in synapse density and function.
Here, we show that MHCI regulates MEF2 protein levels and transcriptional activity and that this regulation is activity-dependent. MHCI signals through calcineurin (CaN) to activate MEF2 to limit glutamatergic synapse density in cortical neurons. Manipulating MEF2 alone does not regulate synapse density, but rather decreases the strength of excitatory connections by controlling GluA1 synaptic content. Moreover, both MHCI and MEF2 are elevated in neurons from neonatal MIA offspring. Remarkably, neurons cultured from MIA frontal cortex (FC) form half as many synapses as control neurons and this deficit requires increased MHCI-MEF2 signaling. Thus, our results identify a novel MHCI signaling pathway that regulates synapse density and mediates the effects of MIA, a risk factor for neurodevelopmental disorders, on neonatal brain development.
Materials and Methods
Animal care and use.
All studies were conducted with approved protocols from the University of California Davis Animal Care and Use Committee, in compliance with NIH guidelines for the care and use of experimental animals. Timed pregnant Sprague Dawley rats and C56BL/6 mice were purchased from Harlan. For the MIA experiments, C56BL/6 mice were bred in-house.
Reagents.
All reagents were purchased from Sigma-Aldrich unless otherwise specified. Nimodipine (Nim, Tocris Bioscience), TTX, and cyclosporin A (CsA) were used at 1 μm. FK506 (FK, A.G. Scientific) was used at 0.1 μm, APV at 50 μm, and CNQX at 40 μm. Drugs were added 1 h post-transfection. DMSO or ethanol was used as the vehicle control at 0.1% final concentration. (Z)-4-hydroxytamoxifen (4OHT) was made fresh and added to a final concentration of 1 μm in 0.1% ethanol.
MIA.
MIA was performed as previously described (Garay et al., 2013). Pregnant mice at gestational day 12.5 were injected intraperitoneally with fresh poly(I:C) dsRNA at 20 mg/kg, or vehicle control (sterile 0.9% saline). Animals were weighed to confirm sickness behavior. Offspring from the poly(I:C)-injected mothers (MIA offspring) were compared with offspring from saline-injected controls.
Neuronal culture and transfections.
Neurons from male and female P0–P2 rat occipital cortex were dissociated in papain (Worthington) and plated at a density of 15K/cm2 as previously described (Glynn et al., 2011). For “Banker”-style cultures, neurons were grown on poly-l-lysine-coated glass coverslips (Fisher) suspended over a glial-cell feeder layer in media supplemented with N2 (Invitrogen). For “conventional” cultures, neurons were plated directly onto a glial monolayer. Neurons from male and female P0–P2 mouse FC were dissociated with papain and plated at a density of 30K/cm2 on poly-l-lysine-coated glass coverslips and maintained in NS21 media (Chen et al., 2008). Neurons were transfected with Lipofectamine 2000 (Invitrogen) and fixed at the indicated days in vitro (DIV).
Plasmids and RNAi.
MEF2-VP16-ERtm, MEF2ΔDBD-VP16-ERtm, MEF2-Engrailed (MEF2En), 3XMRE-EGFP, MEF2-VP16, MEF2ΔDBD-VP16, pSuper-shMEF2A (shA), pSuper-shMEF2D (shD), and pSuper-shSCRAMBLE control (shSCR) were described previously and were obtained from Drs. M.E. Greenberg (Harvard Medical School, Boston, MA) and C.W. Cowan (McLean Hospital, Harvard University Medical School, Belmont, MA) (Flavell et al., 2006; Pfeiffer et al., 2010). pSuper-shMEF2C (shC) was created from a previously characterized MEF2C siRNA sequence (Pereira et al., 2009) and the manufacturer's protocol (Oligoengine). H2-Kb-YFP and H2-Kb-mCherry were generated by inserting H2-Kb from H2-Kb-CFP (M. Edidin, Johns Hopkins University, Baltimore, MD; Glynn et al., 2011) into pEYFP-N1 or pmCherry-N1. The siβ2m and nontargeting sequence (siNTS) siRNAs were described previously and were purchased from Invitrogen (Glynn et al., 2011). shRNAs targeting mouse β2m and shNTS (same target sequence as siNTS) were generated and inserted into pENTR/U6 using the manufacturers protocol (Invitrogen). Mouse shβ2m targeted the following sequence in β2m (NM_009735.3): 5′-TGATACATACGCCTGCAGAG and this shRNA significantly decreased sMHCI expression (see Fig. 5F). Dominant negative CaN (dnCaN, CaNH151A-YFP), VIVIT-GFP and 3XNFAT-AP1-CFP were obtained from Mark Dell'Acqua (University of Colorado School of Medicine, Aurora, CO).
Figure 5.
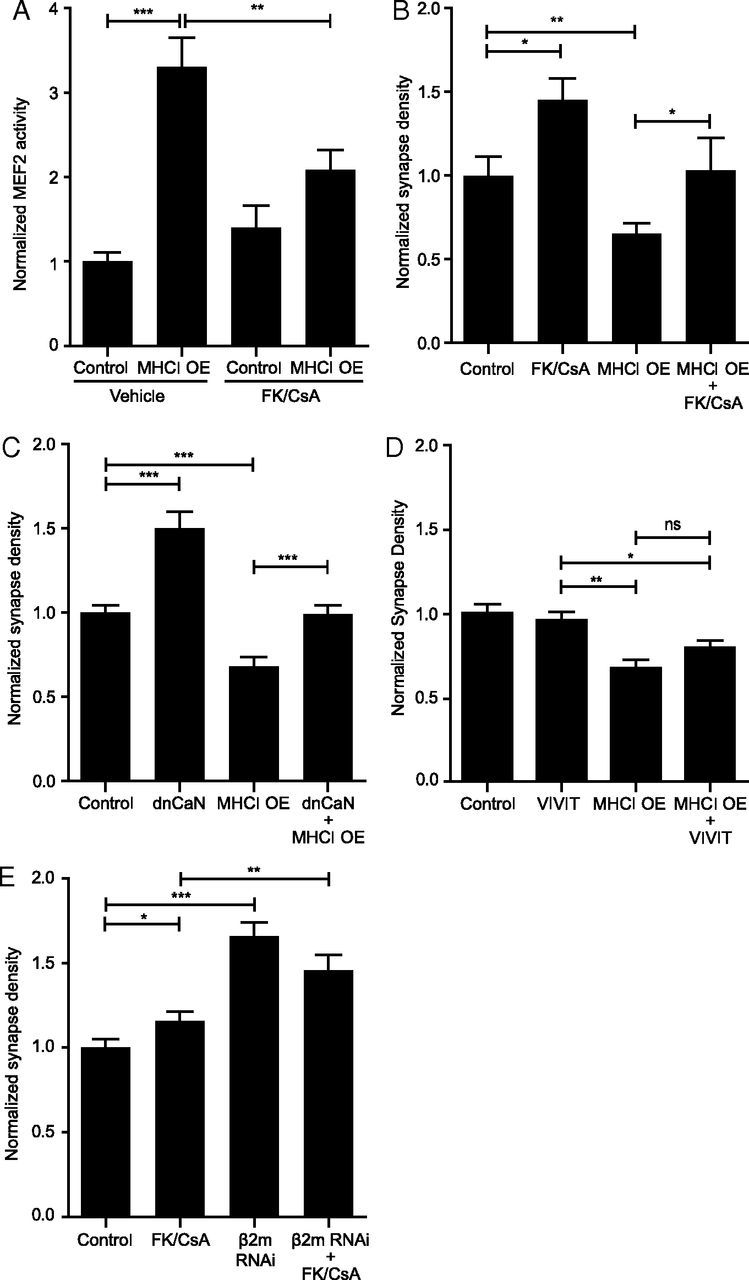
MHCI requires CaN to activate MEF2 and limit synapse density. A, Treatment of neurons with the CaN inhibitors FK506/CsA (FK/CsA) blocks the MHCI-induced increase in MEF2 activity; n ≥ 42 cells per condition, four experiments. B, FK/CsA rescues the MHCI-mediated decrease in synapse density; n ≥ 12 dendrites per condition (4 cells), one experiment. C, Similarly, coexpression of a dominant-negative form of CaN (dnCaN, CaN-H151A-YFP) also rescues the MHCI-mediated decrease in synapse density; n ≥ 51 dendrites (17 cells) per condition, two experiments. D, CaN binding to NFAT does not alter synapse density, nor is it required for the MHCI-induced decrease in synapse density. Neurons were transfected with the indicated constructs at 6 DIV, and synapse density was quantified 48 h later. E, Inhibiting CaN activity does not increase synapse density above that seen by sMHCI KD alone. β2m siRNAs were transfected at 5 DIV with or without FK/CsA, and synapse density was analyzed at 8 DIV; n ≥ 57 dendrites (19 cells) per condition, three experiments. *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant.
Immunoblotting.
Dissociated mouse cortical cultures were lysed at 8 DIV in 2× reducing SDS sample buffer, boiled for 5 min, and resolved by SDS-PAGE. Samples were transferred to PVDF membranes, blocked in Odyssey blocking buffer (LiCor), and probed with the indicated primary antibodies overnight at 4°C. Resolved proteins were visualized in two channels using fluorescent secondary antibodies at 680 and 800 nm on an Odyssey Clx infrared imaging system (LiCor). MEF2 band intensities were quantified using the manufacturer's software, ImageSoft (LiCor), and normalized to a GAPDH loading control. MEF2C protein was quantified after stripping MEF2A and D antibodies and reprobing with a MEF2C antibody. Antibodies used were MEF2A (sc313, 1:200, Santa Cruz Biotechnology), MEF2C (10056-1-AP, 1:500, ProteinTech), MEF2D (M62720, 1:2000, BD/Signal Transduction), MEF2phosphoS408 (1:1000, Dr. M.E. Greenberg, Harvard Medical School, Boston, MA), and GAPDH (G8795, 1:7500, Sigma-Aldrich).
Immunochemistry (ICC).
Cells were fixed in 4% paraformaldehyde, 4% sucrose in PBS at 4°C for 10 min. Neurons were permeabilized with 0.25% Triton X-100 for 5 min, blocked with 10% BSA, and then incubated with primary and secondary antibodies diluted in 3% BSA. Surface MHCI (sMHCI) staining was performed on fixed cells without permeabilization and the sMHCI primary antibody was applied together with a MAP2 primary antibody. Dendritic regions with positive MAP2 staining were excluded from analysis to control for fixation-induced permeabilization (Glynn and McAllister, 2006). Primary antibodies used were as follows: GluA1 (PC246, 1:100; Calbiochem), GluA2 (MAB397, 1:100; Millipore Bioscience Research Reagents), MAP2 (rabbit: AB-5622, 1:2000; Millipore Bioscience Research Reagents, or chicken: Ab5392, 1:2000; Abcam), MEF2A (Ab51153, 1:400; Abcam), MEF2C (10056-1-AP, 1:400; ProteinTech), MEF2D (M62720, 1:400; BD Signal Transduction), MHCI (OX-18, 1:100; AbD Serotec), PSD-95 (K28/43, 1:1000; NeuroMab), VAMP2 (104–202, 1:1000; Synaptic Systems), Synapsin1 (Millipore Bioscience Research Reagents 1:1000), VGluT1 (AB5905, 1:1000; Millipore Bioscience Research Reagents). Secondary antibodies used were AlexaFluor-488, -568, or -647 conjugated anti-rabbit, anti-mouse, anti-chicken and anti-guinea pig [1:400 for all except MAP2 immunocytochemistry (ICC), where a 1:1000 dilution was used, Invitrogen]. For sMHCI ICC, an additional biotin-streptavidin amplification step was performed, whereby biotin-conjugated goat anti-mouse IgG (1:100; Vector Laboratories) was used to label the primary antibody (OX18) followed by AlexaFluor-488 or -546 conjugated streptavidin tertiary label (1:400; Invitrogen). All coverslips were mounted in Fluoromount-G (Southern Biotech).
Flow cytometry.
Brain regions were dissected at P0 from MIA and control offspring and cells were acutely dissociated in papain. Dissociated cells were FcR blocked (Miltenyi Biotec) for 30 min and then stained with primary conjugated antibodies for 60 min at 4°C. Cells were then washed with PBS and resuspended in 4% PFA. Primary antibodies included the differentiated neuronal marker Thy1.2-APC (1:10; Miltenyi Biotec), the MHCI subtype H2-KbDb-PE (1:10, Cederlane) and a viability marker (Violet Dead Cell Stain from Invitrogen). Samples were run on a BD LSRII flow cytometer and analyzed by Flowjo (Treestar). Data were gated on Thy1.2-positive cells and assessed for sMHCI by H2-KbDb-PE fluorescence intensity.
Microscopy and image analysis.
Imaging of neurons was performed using an Olympus Fluoview 300 (software v5.0) laser scanning confocal microscope with a 60× PlanApo oil-immersion objective (1.4 NA). Laser power and photomultiplier tube (PMT) levels were held constant between groups within each experiment. Grayscale images (2048 × 2048, 16 bit) for each channel were acquired sequentially and Kalman-averaged over two scans at 2.5× artificial zoom. Images used for cluster density or colocalization analysis underwent one iteration of deconvolution with the appropriate point spread function for 488, 568, or 647 nm light before image analysis (SVI Huygens Essential).
Pre- and postsynaptic protein colocalization (synapse density) and density were quantified as described previously using custom journals written in MetaMorph software (v7.5, Molecular Devices; Glynn et al., 2011). Briefly, regions of interest (ROI) on dendrites were outlined manually and puncta for each channel within the ROI were detected and measured using a custom journal written to use MetaMorph's adaptive background correction algorithm to label individual clusters of protein in each channel. This type of local background detection enables identification of both intense and the more faint protein clusters found in young, developing neurons that might normally be missed using typical global background subtraction methods. The importance of this approach for accurately detecting synapses in young neurons is illustrated and discussed in detail in (Glynn and McAllister, 2006). To show both the large, bright and small, faint puncta in the images used in the figures, the gain was increased for illustration, which often saturated the brightest puncta but made the faint ones visible. To show all of the puncta that were analyzed, images of representative dendrites are displayed next to their corresponding threshold masks. MEF2 protein levels were measured as the average pixel intensity of staining in the nucleus; specific cytoplasmic staining of MEF2 in neurons was not observed. Transcriptional reporter fluorescence was measured as average reporter protein [EGFP for MEF2 or ECFP for nuclear factor of activated T-cells (NFAT)] pixel intensity in the cell body normalized to pmCherry control. During image acquisition, care was taken to adjust laser PMT levels appropriately to prevent saturation of reporter intensity. All images were prepared in Adobe Photoshop and Illustrator.
Electrophysiology.
Whole-cell patch-clamp recordings were made from 7 DIV rat conventional cultures coexpressing shMEF2A+shMEF2D shRNAs or shSCR for 48 h, together with EGFP for visualization of transfected cells. To isolate mEPSCs, whole-cell patch-clamp recordings were made at −70 mV in the presence of 0.5 μm TTX and 100 μm picrotoxin. The extracellular solution consisted of the following (in mm): 110 NaCl, 3 KCl, 10 HEPES, 10 d-glucose, 10 glycine, 2 CaCl2, and 2 MgCl2, pH 7.3. The osmolarity was adjusted to 295 mOsm using sorbitol. The intracellular solution consisted of the following (in mm):130 potassium gluconate, 10 KCl, 10 HEPES, 10 EGTA, 1 CaCl2, 5 ATP-Mg2+, and 5 GTP-Li2+, pH 7.3. Recordings were filtered at 2 kHz using an Axopatch 200B amplifier and digitized at 5 kHZ using Clampex 8 (Molecular Devices). mEPSCs were detected with MiniAnalysis software (Synaptosoft), at a threshold fourfold larger than the rms noise and confirmed by visual inspection.
Data analysis.
For all experiments, with the exception of the graph in Figure 5B, data were collected from at least two (usually three, and sometimes more) experiments and are presented as the mean ± SEM. Significance was assessed relative to culture prep-matched control by unpaired t test. For all graphs, significance was defined as follows: *p < 0.05, **p < 0.01, ***p < 0.001. When normalized, data are plotted relative to the appropriate, prep-matched control. A dashed line indicates the normalized control values at 1.0 for the cases in which the control values are not plotted. Data were plotted in Graphpad Prism (v5) and exported to Adobe Illustrator for figure preparation.
Results
MHCI negatively regulates glutamatergic synapse density and function; however, the signaling mechanism that mediates these effects is unknown (Elmer and McAllister, 2012). Here, we approached this question by identifying other types of molecules that play similar roles and then testing whether they are components of the MHCI signaling pathway. MEF2 transcription factors were one of the first molecular families discovered to negatively regulate synaptic connectivity in the CNS (Flavell et al., 2006; Shalizi et al., 2006). There are four MEF2 genes, A–D, originally identified for their critical roles in muscle (Potthoff and Olson, 2007). In the brain, MEF2 genes are differentially expressed in distinct but overlapping regions (Lyons et al., 1995, 2012). Previous studies on MEF2 report a wide variety of roles in brain development and function including neuronal survival, migration, hippocampal synaptic plasticity and function, working memory, and motor coordination (Mao et al., 1999; Okamoto et al., 2000; Shalizi et al., 2003, 2006, 2007; Shalizi and Bonni, 2005; Barbosa et al., 2008; Li et al., 2008; Pulipparacharuvil et al., 2008; Fiore et al., 2009; Pazyra-Murphy et al., 2009; Pfeiffer et al., 2010; Akhtar et al., 2012; Chen et al., 2012; Yamada et al., 2013; Zang et al., 2013). Neuronal activity and calcium (Ca2+) signaling activate MEF2, increasing the expression of many genes, some of which cause synapse elimination (Flavell et al., 2006, 2008; Shalizi et al., 2006; Tsai et al., 2012). Based on their similar roles in limiting synapse density, we hypothesized that MHCI and MEF2 interact to limit connectivity in young cortical neurons.
MHCI regulates MEF2 levels and activity
If MHCI and MEF2 are components of the same pathway, then manipulation of one should alter the levels or activation of the other. To test this prediction, MHCI or MEF2 was genetically manipulated in dissociated rat cortical neurons during the initial stages of synapse formation (5–8 DIV) and the effect on the other protein was quantified. MHCI expression on the surface of neurons (sMHCI) and MEF2 levels were visualized using ICC. MEF2 activity was measured using an EGFP-based transcriptional reporter, 3XMRE-EGFP (Pfeiffer et al., 2010; Zang et al., 2013). Data were normalized throughout the paper for accurate comparison to the appropriate culture-matched controls.
To determine whether MHCI regulates MEF2 levels and/or activity in young cortical neurons, sMHCI expression was decreased using RNAi-mediated knockdown of the light chain of MHCI, β2-microglobulin (β2m RNAi or sMHCI knockdown), or increased by transfecting neurons with a fluorescently tagged isoform of MHCI, H2-Kb-YFP or H2-Kb-mCherry (MHCI overexpression; MHCI OE), as previously described (Glynn et al., 2011). Cells were transfected at 5–7 DIV and MEF2 protein levels and transcriptional activity were measured 36–72 h later. MHCI overexpression significantly increased MEF2A, -C, and -D protein levels by 22, 31, and 12%, respectively, whereas knockdown of sMHCI using β2m RNAi decreased them by 62,18, and 25%, respectively, relative to a nontargeting sequence NTS siRNA control (Fig. 1A,B). Similarly, MHCI overexpression increased MEF2 activity twofold, whereas sMHCI knockdown decreased it by almost half (Fig. 1C). These manipulations altered MEF2 family members differentially, with MEF2A and -C showing the largest changes, potentially reflecting differential activation of these isoforms by MHCI. This effect appears to be specific to MEF2, because a transcriptional reporter for another activity-regulated transcription factor, the nuclear factor of activated T-cells (NFAT), did not change upon MHCI overexpression (data not shown). Thus, MHCI specifically regulates MEF2 levels and transcriptional activity in a cell-autonomous manner.
Figure 1.
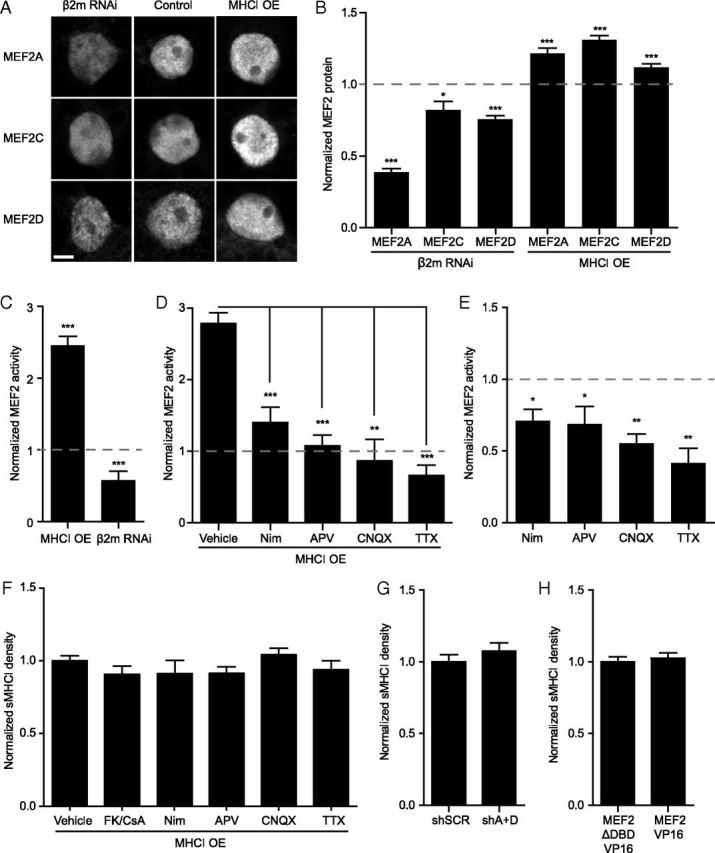
MHCI regulates the levels and activity of MEF2. A, Representative confocal micrographs of MEF2 proteins. Neurons were transfected with H2-Kb-YFP (MHCI OE) or siRNAs for β2m to knockdown sMHCI for 48–72 h (Glynn et al., 2011). Scale bar, 5 μm. B, MHCI regulates MEF2 protein levels. MHCI OE increased levels of nuclear MEF2A, -C, and -D, while decreasing sMHCI (using β2m RNAi) lowered MEF2. MEF2 protein was quantified and plotted normalized relative to the appropriate control group indicated by the dashed line at 1.0 (EGFP for MHCI OE and NTS siRNA for β2m RNAi; n ≥ 25 cells per condition, ≥2 experiments). C, MHCI also regulates MEF2 transcriptional activity. MEF2 activity was quantified after transfecting 7 DIV neurons with H2-Kb-mCherry or β2m siRNAs with the MEF2 transcriptional reporter 3xMRE-EGFP for 36–48 h. EGFP fluorescence intensity was measured and plotted normalized to the appropriate control group indicated by the dashed line at 1.0 (pmCherry for MHCI OE or NTS RNAi + pmCherry for sMHCI KD; n ≥ 31 cells per condition, ≥ 2 experiments). D, The MHCI-induced increase in MEF2 activation is activity-dependent. MEF2 activity following MHCI OE was analyzed as in C), with the indicated pharmacologic manipulations. Drugs or vehicle control were added post-transfection and MEF2 activity was quantified 36–48 h later and normalized to treated controls; n ≥ 21 cells per condition, ≥2 experiments. E, MEF2 is activated by synaptic activity and Ca2+ influx through L-VSCCs. Neurons were transfected with pmCherry and 3XMRE-EGFP at 6 DIV, and cells were fixed and analyzed at 8 DIV. Drugs were added as in D, and MEF2 activity was quantified 36–48 h later; n ≥ 8 cells, ≥2 experiments. F, sMHCI overexpression is not dependent on synaptic activity or Ca2+ influx through L-VSCCs or CaN activation. Neurons were transfected with H2-Kb-YFP at 6 DIV. Inhibitors of CaN (FK506/CsA), L-VSCCs (Nim), NMDARs (APV), AMPARs (CNQX), and Na-dependent action potentials (TTX) were added 1 h post-transfection as in D and sMHCI on transfected cells was quantified 48 h later; n ≥ 8 cells, two experiments. G, MEF2 knockdown does not alter sMHCI density. sMHCI density was quantified 48 h after transfection of MEF2A+MEF2D shRNAs (shA+D) or a scrambled shRNA control (shSCR); n ≥ 22 cells per condition, ≥2 experiments. H, MEF2 activation also does not alter sMHCI density. MEF2-VP16 was expressed for 48 h and sMHCI was measured relative to cells expressing a mutant form of MEF2-VP16 unable to bind to DNA, MEF2-ΔDBD-VP16. *p < 0.05, **p < 0.01, ***p < 0.001.
MEF2 transcription factors are activated by Ca2+ influx through L-type voltage sensitive Ca2+ channels (L-VSCCs) and ionotropic glutamate receptors (Flavell et al., 2006; Shalizi et al., 2006). To determine whether MHCI requires these pathways to increase MEF2 activity, MHCI was overexpressed while blocking synaptic activity or Ca2+ influx through L-VSCCs and MEF2 reporter expression was measured as in Figure 1C. Blockade of L-VSCCs with nimodipine prevented the MHCI-induced increase in MEF2 activity (Fig. 1D). Similarly, addition of the NMDAR antagonist APV, the AMPAR and kainate receptor antagonist CNQX, as well as blocking sodium-dependent action potentials with TTX, all prevented the MHCI-induced increase in MEF2 reporter activity (Fig. 1D). Importantly, addition of these drugs decreased MEF2 transcriptional activity in cortical neurons as expected (Fig. 1E; Flavell et al., 2006), but did not prevent sMHCI overexpression (Fig. 1F). These results show that MHCI requires synaptic activity and Ca2+ influx through L-VSCCs to activate MEF2. Thus, MHCI positively regulates MEF2 protein levels and activity, and increases MEF2 transcriptional activity in an activity-dependent manner.
To determine whether the reciprocal interaction between MHCI and MEF2 might also occur, cultured neurons were transfected with MEF2A and MEF2D shRNAs (shA+D), or a scrambled shRNA control (shSCR; Flavell et al., 2006), and sMHCI expression was quantified on the dendrites of transfected cells 48 h later. MEF2 knockdown did not change sMHCI density relative to control (Fig. 1G). In addition, expression of a constitutively active form of MEF2, which has its transactivation domain replaced with the viral transactivator VP16 (MEF2-VP16; Black et al., 1996; Pulipparacharuvil et al., 2008), did not alter sMHCI density compared with that found in cells transfected with a form of MEF2VP16 that lacks the DNA-binding domain (MEF2-ΔDBD-VP16; Fig. 1H). Thus, increasing (with MEF2-VP16) or decreasing (with shA+D) MEF2 activity does not regulate sMHCI density, indicating that MEF2 does not act upstream of MHCI. Together, results from this section suggest that MHCI acts upstream of MEF2 to activate signaling cascades to increase MEF2 protein levels and transcriptional activity in cortical neurons.
MHCI requires MEF2 to limit synapse density
Because MHCI and MEF2 negatively regulate neural connectivity and MHCI regulates MEF2 levels and activity (Flavell et al., 2006; Shalizi et al., 2006; Glynn et al., 2011; Yamada et al., 2013), we next tested whether MHCI requires MEF2 to limit synapse density. If so, then MEF2 knockdown should prevent the MHCI-dependent reduction in glutamatergic synapse density. To test this prediction, MHCI was overexpressed in cortical neurons alone or with shRNAs to MEF2A, MEF2C, and MEF2D or combinations of shRNAs to knockdown MEF2A+MEF2D, MEF2A+MEF2C, or MEF2C+MEF2D (shA+D, shA+C, and shC+D). Neurons were transfected at 6 DIV, the start of synaptogenesis in our cultures, and fixed at 8 DIV for ICC using antibodies against the presynaptic protein vesicular glutamate transporter-1 (VGluT1) and the postsynaptic density protein PSD-95. Synapses were defined as sites of colocalization between these two proteins. VGluT1 is expressed at the majority of glutamatergic presynaptic terminals and is commonly used as a reliable marker for excitatory synapses (Micheva et al., 2010). The low transfection efficiency (1–5%) of our experimental system enabled visualization of synapses made onto transfected neurons by untransfected neurons.
MHCI overexpression significantly decreased synapse density by almost half, consistent with previous work (Fig. 2; Glynn et al., 2011). Although knockdown of single isoforms of MEF2 did not rescue the MHCI-induced reduction in synapse density (data not shown), knockdown of any two isoforms of MEF2 was sufficient to prevent the MHCI-induced decrease in synapse density (Fig. 2A–D). Repeating this rescue experiment with MEF2En, a dominant negative form of MEF2 that has its transactivation domain replaced with the engrailed transcriptional repressor (Arnold et al., 2007), gave similar results (Fig. 2E), although shRNAs produced a more robust rescue. Together, these data demonstrate that MHCI requires MEF2 to limit excitatory synapse density and that reduction of MEF2 activity by MEF2En, or by knockdown of at least two MEF2 isoforms, is sufficient to block MHCI signaling.
Figure 2.
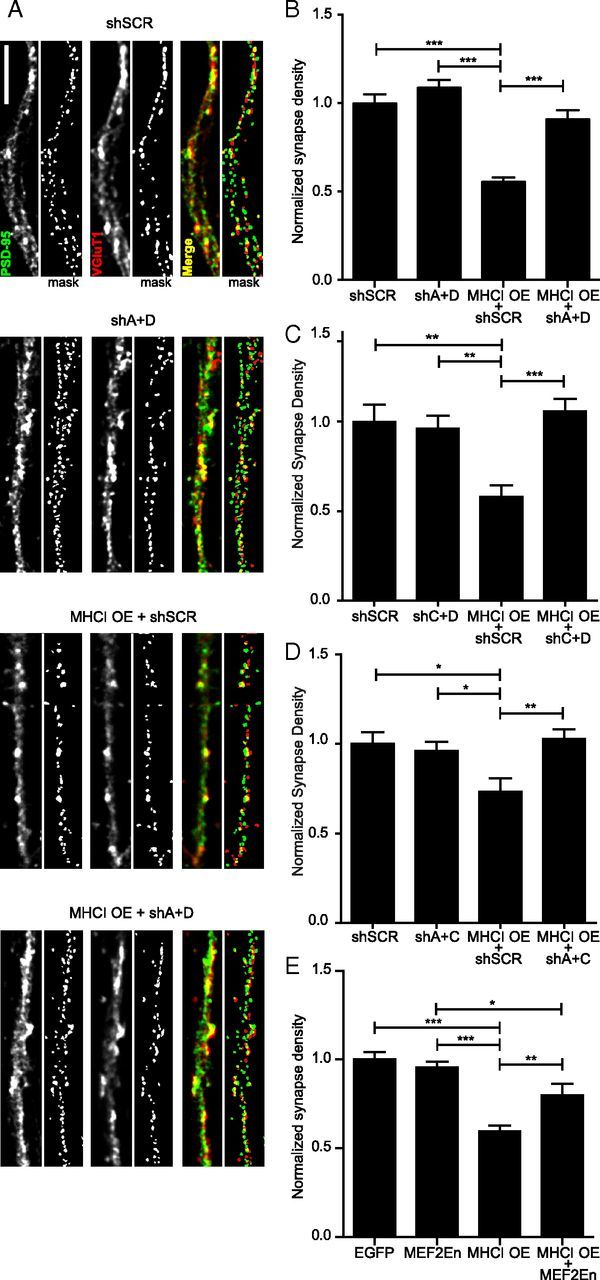
MHCI requires MEF2 to limit synapse density. A, Representative images of dendrites from 8 DIV cortical neurons transfected with the indicated constructs for 48 h. Excitatory glutamatergic synapses were quantified as the density of postsynaptic PSD-95 puncta (green) colocalized (yellow) with presynaptic VGluT1 puncta (red). Scale bar, 5 μm. Each representative image shown includes an adjacent “mask” panel displaying the regions obtained following thresholding that were used for quantification. The intensity of the images shown was increased for publication to enable visualization of the faint puncta, in addition to the bright ones, because both are detected by our image analysis, as shown in the mask panels. The brighter puncta are saturated in those images by this increase in intensity. B, MEF2A+MEF2D knockdown alone does not alter synapse density, but completely prevents the MHCI-mediated decrease in synapse density. Expression of shRNAs against MEF2A and D (shA+D) together with MHCI OE rescued the decrease in synapse density observed in neurons expressing a scrambled shRNA (shSCR) with MHCI OE. Results are normalized to shSCR control; n ≥ 61 dendrites, 21 cells per condition; four experiments. C, MEF2C+MEF2D knockdown and MEF2A+MEF2C knockdown (D) had similar effects as MEF2A+MEF2D knockdown shown in B. E, Similarly, expression of MEF2-En, a dominant negative form of MEF2, does not alter synapse density alone, but partially rescues the MHCI-mediated decrease in synapse density; n ≥ 51 dendrites (17 cells) per condition, two experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Manipulating MEF2 alone alters synaptic strength, but not synapse density, in young cortical neurons
Although MHCI clearly requires MEF2 to limit synapse density, MEF2 knockdown alone did not increase synapse density (Fig. 2A–D). This result was surprising given previous reports that MEF2A+MEF2D knockdown increases synapse density in cultured hippocampal neurons (Flavell et al., 2006). Although MEF2A and MEF2D are expressed at similar levels in cortex and hippocampus, MEF2C is expressed at relatively higher levels in cortex (Lyons et al., 1995, 2012). To investigate the possibility that different isoforms or combinations of heterodimers of MEF2 might alter synapse density, synapses were quantified following individual knockdown of MEF2A, MEF2C, or MEF2D or after knockdown of combinations of MEF2 isoforms (shA+D, shA+C, and shC+D). No combination of MEF2 shRNAs, or the dominant-negative construct MEF2En, altered synapse density in young cortical neurons (Fig. 3A). Synapse density was also unaltered after increasing MEF2 activity using MEF2-VP16 (Fig. 3B) or a more potent 4OHT-inducible form of this mutant (MEF2-VP16-ER; Fig. 3C). These results were obtained by defining a synapse as colocalization of VGluT1 + PSD-95 clusters (Fig. 3A–C), and were also reproduced using VAMP2 + PSD-95 and synapsin1 + PSD-95 colocalization (the combination used previously in hippocampal neurons; Flavell et al., 2006; Fig. 3D). Densities of the presynaptic markers synapsin1 and VAMP2 were unchanged following knockdown of MEF2A+MEF2D (Fig. 3E), whereas the density of VGluT1 was slightly decreased by both MEF2En and MEF2-VP16 (Fig. 3F). Although a recent paper demonstrated that MEF2 decreases PSD-95 cluster area in mature hippocampal cultures (Tsai et al., 2012), the density (Fig. 3F) and area (Fig. 3G) of PSD-95 clusters were not regulated by MEF2 in our cortical neurons. All of these manipulations were confirmed to alter MEF2 transcriptional activity in the expected manner (Fig. 3H), despite their inability to alter synapse density. Together, these results definitively show that manipulating MEF2 alone does not alter the density of PSD-95-containing synapses in young cortical neurons.
Figure 3.
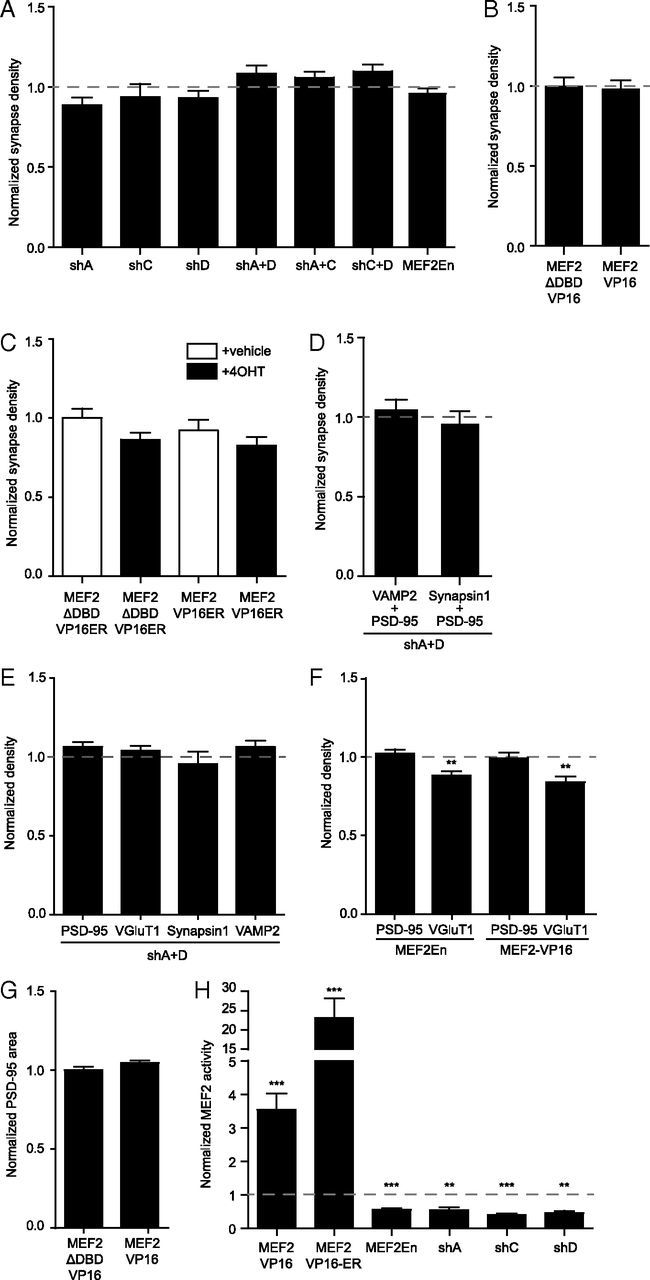
MEF2 does not alter synapse density or the density of synaptic proteins. A, Decreasing MEF2 activity does not change synapse density. No combination of shRNAs targeting MEF2 isoforms changes synapse density; n ≥ 63 dendrites (21 cells) per condition, two experiments. Values are normalized to shSCR control. Similarly, MEF2-En does not change synapse density from control (EGFP) levels. Cells were transfected at 6 DIV and fixed at 8 DIV. Normalized, control levels are indicated by the dashed gray line (shSCR for RNAi and EGFP for MEF2En); n ≥ 152 dendrites (51 cells) per condition, four experiments. B, Expression of MEF2-VP16 also does not alter synapse density. Cells were transfected at 6 DIV and fixed at 8 DIV; n ≥ 71 dendrites (23 cells) per condition, four experiments. C, A tamoxifen inducible form of MEF2-VP16 (MEF2-VP16-ER) does not alter synapse density. Cells were transfected with the indicated forms of MEF2 at 5 DIV. 4OHT or vehicle control (ethanol to 0.1%) was added 24 h later and cells were fixed at 8 DIV; n ≥ 21 dendrites (7 cells), two experiments. D, MEF2A+D knockdown in young cortical neurons does not alter synapse density defined by the colocalization of PSD-95 and VAMP2 (data are the same as the control shown in Fig. 7C) or by the colocalization of PSD-95 and synapsin1. Neurons were transfected at 6 DIV and fixed at 8 DIV. Normalized control (shSCR) levels are indicated by the dashed gray line; n ≥ 21 dendrites (7 cells), one experiment. E, MEF2A+MEF2D knockdown does not alter the density of PSD-95, VGluT1, synapsin1 or VAMP2. Densities of the indicated proteins were quantified from experiments shown in D, Figure 3A, and 7C. Normalized control (shSCR) levels are indicated by the dashed gray line. F, Changing MEF2 activity by dominant negative MEF2En or constitutively active MEF2-VP16 does not alter PSD95 density, but slightly decreases VGluT1. Cells were transfected at 6 DIV and fixed at 8 DIV. Normalized control (EGFP for MEF2En or MEF2-DBD-VP16 for MEF2-VP16) levels are indicated by the dashed gray line. MEF2En; n ≥ 131 dendrites (43 cells), four experiments. MEF2-VP16; n ≥ 71 dendrites (23 cells), four experiments. G, MEF2VP16 expression does not alter PSD-95 cluster area as compared with control. Cells were transfected at 6 DIV, and fixed at 8 DIV; n ≥ 71 dendrites (23 cells), four experiments. H, MEF2 shRNAs, MEF2En, and MEF2-VP16 all alter MEF2 activity as expected. shRNAs targeting MEF2A, C, or D and MEF2En decrease MEF2 activity. MEF2-VP16 increases MEF2 activity ∼3.5-fold, and a 4OH-tamoxifen inducible form of MEF2, MEF2-VP16-ER, increases MEF2 activity ∼23-fold relative to mutant versions that cannot bind DNA (MEF2-DBD-VP16 or MEF2-DBD-VP16-ER) or to MEF2-VP16-ER without 4OHT. Respective, normalized control levels are indicated by the dashed gray line; n ≥ 8 cells per condition. *p < 0.05, **p < 0.01, ***p < 0.001.
Because synapses on young cortical neurons are often silent (AMPAR-lacking) (Malinow and Malenka, 2002), it is possible that defining synapses by PSD-95 staining might not reflect changes in the proportion of AMPAR-containing synapses. To test this hypothesis, the density of synapses that contained either GluA1 or GluA2 apposed to presynaptic VGluT1 was quantified (Fig. 4A–C). Remarkably, the density of GluA1-containing synapses was increased by 27% following MEF2A+MEF2D knockdown (Fig. 4A,B), whereas the density of GluA2-containing synapses was unchanged (Fig. 4C). Thus, MEF2A+MEF2D knockdown increases the proportion of synapses that contain GluA2-lacking, Ca2+-permeable AMPARs, while leaving the total number of PSD-95-containing synapses unchanged.
Figure 4.

MEF2 limits synaptic strength and synaptic GluA1 content. A, Representative images of synapse density defined by colocalized GluA1 and VGluT1 in 8 DIV neurons transfected with shSCR or shA+D at 6 DIV. B, MEF2 A+D knockdown increases the density of GluA1-containing synapses compared with shSCR-expressing cells; n ≥ 44 dendrites (15 cells) per condition, two experiments. C, GluA2 synapse density was unchanged following MEF2A+D knockdown; n ≥ 38 dendrites (13 cells) per condition, two experiments. D, Representative traces of mEPSCs from cells expressing shSCR or shMEF2A+D shRNAs. E, F, MEF2 A+D knockdown significantly increases mEPSC amplitude (E), but not frequency (F); n ≥ 16 cells per condition, five experiments. *p < 0.05, **p < 0.01.
The increase in GluA1-, but not GluA2-, containing synapses in neurons expressing lower levels of MEF2 would be expected to enhance synaptic strength (Swanson et al., 1997; Liu and Zukin, 2007). To test this prediction mEPSCs were obtained using whole-cell patch-clamp recording of cultured cortical neurons transfected for 48 h with shMEF2A+D or shSCR control (Fig. 4D). Consistent with our prediction, MEF2A+MEF2D knockdown increased the amplitude of mEPSCs by 23% (Fig. 4E). Although there was a trend toward increased mEPSC frequency (Fig. 4F), this effect remained nonsignificant even after increasing our sample size well above that typically used for mEPSC analysis (shSCR: n = 16 cells, shA+D: n = 19 cells). The increase in mEPSC amplitude is consistent with the idea that synapses with GluA1-containing Ca2+-permeable AMPARs have enhanced conductance (Swanson et al., 1997; Liu and Zukin, 2007). Therefore, our results show that in young neurons, MEF2 does not regulate the total number of PSD-95-containing synapses, but does limit the proportion of GluA1-containing synapses.
CaN is required for the MHCI-induced decrease in synapse density
The Ca2+ and calmodulin-regulated phosphatase, CaN, dephosphorylates and activates MEF2 in response to synaptic activity, leading to an increase in MEF2 transcriptional activity (Flavell et al., 2006; Shalizi et al., 2006). Based on the central role that CaN plays in activating MEF2, we hypothesized that MHCI might signal through CaN to activate MEF2. If so, then inhibiting CaN should prevent MHCI overexpression from increasing MEF2 activity. To test this hypothesis, the CaN inhibitors FK and CsA were added to 6 DIV neurons transfected with H2-Kb-mCherry and the MEF2 reporter, 3XMRE-EGFP. MEF2 activity was quantified 36–48 h later. CaN inhibition largely prevented the MHCI-induced increase in MEF2 transcriptional activity, indicating that MHCI does signal through CaN to activate MEF2 (Fig. 5A).
Because CaN mediates MHCI-induced MEF2 activation, and MHCI requires MEF2 to limit synapse density, we next predicted that CaN inhibition would prevent the MHCI-induced decrease in connectivity. Indeed, blocking CaN activation using FK/CsA prevented the decrease in synapse density caused by MHCI overexpression (Fig. 5B). A dominant negative mutant of CaN (dnCaN) produced similar results, and further indicated that the effects of CaN on synapse density are cell autonomous (Fig. 5C). Interestingly, these effects were not dependent on NFAT, which is a cofactor for MEF2 and is activated by CaN in response to synaptic activity (Blaeser et al., 2000; Groth et al., 2003). Expression of a peptide inhibitor that prevents CaN from activating NFAT (VIVIT-GFP; Aramburu et al., 1999) did not increase synapse density or prevent the MHCI-induced decrease in synapse density (Fig. 5D). Thus, CaN does not limit synapse density through NFAT, and MHCI regulates connectivity through CaN in a manner that is not dependent on proteins, such as NFAT, that contain the “PxIxIT” CaN docking motif.
Although these results suggest that MHCI requires CaN to activate MEF2 and limit synapse density, it is also possible that MHCI and CaN regulate synapse density through parallel, independent pathways because blocking CaN alone increased synapse density. To test this possibility, genetic epistasis/occlusion analysis was used to determine whether reducing both MHCI and CaN in the same neuron increases synapse density in an additive manner (indicating that they work through independent pathways) or in a nonadditive manner (indicating a shared pathway). To that end, CaN inhibitors were added to neurons transfected with β2m or NTS siRNAs from 5 to 8 DIV. Both CaN inhibition and β2m RNAi increased excitatory synapse density, consistent with previous work (Flavell et al., 2006; Glynn et al., 2011), but blocking CaN did not increase synapse density over that observed with sMHCI knockdown alone (Fig. 5E). Thus, MHCI requires CaN to activate MEF2 transcription and limit synapse density.
MIA increases MHCI on cortical neurons and decreases their ability to form synapses
Although MHCI clearly limits synapse density through CaN and MEF2-dependent signaling when overexpressed, the physiological relevance of this phenotype was unclear. Therefore, we next asked whether MHCI levels are elevated on neurons by more physiologically relevant mechanisms, and if so, whether this increase limits connectivity in a MEF2-dependent manner. Given that cytokines are potent regulators of MHCI in the immune system, we addressed this issue using a mouse model of MIA in which cytokines are elevated in the brains of offspring (Meyer et al., 2006, 2008b; Fatemi et al., 2008; Arrode-Brusés and Brusés, 2012; Garay et al., 2013). Pregnant mice were injected intraperitoneally with poly(I:C) (MIA) or saline (control) at gestational day 12.5 (Garay et al., 2013) and two approaches were used to measure neuronal sMHCI on neurons from the FC of newborn offspring. First, neurons were acutely dissociated and sMHCI levels were measured using flow cytometry. Neuronal sMHCI levels from MIA offspring were increased 1.9-fold over control (Fig. 6A). Second, ICC was used to detect sMHCI clusters on dendrites of neurons cultured from newborn MIA and control offspring (Fig. 6B). The density of sMHCI was 1.8-fold higher on neurons from MIA offspring compared with those from control offspring (Fig. 6C), similar to the flow cytometry data. Thus, an acute immune insult during mid-gestation causes a dramatic elevation in MHCI levels on the surface of cortical neurons from newborn offspring.
Figure 6.
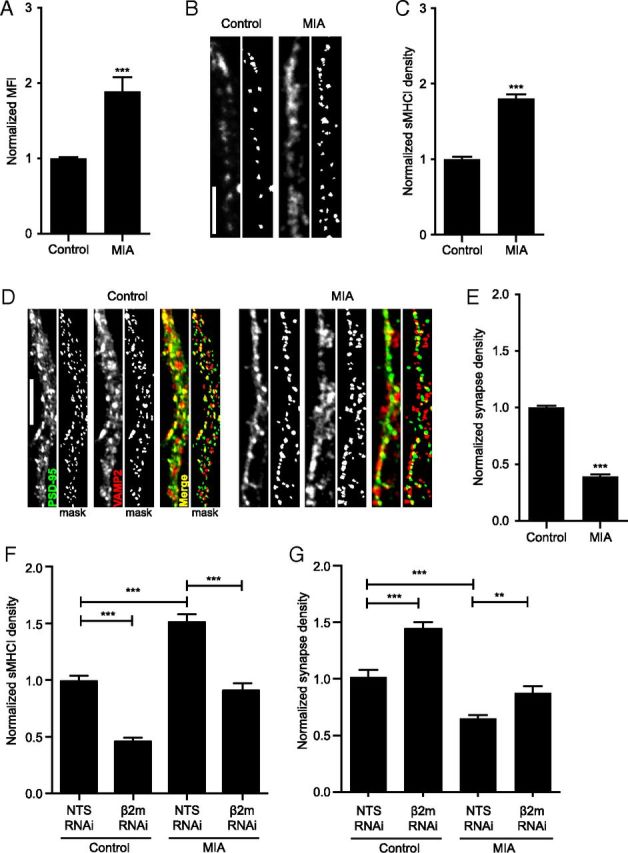
Maternal immune activation increases sMHCI and decreases glutamatergic synapse density on cortical neurons from newborn offspring. A, Surface MHCI (H2-Kb/H2-Db) is almost doubled on acutely dissociated neurons from P0 FC from newborn offspring of poly(I:C)-injected (MIA) mothers compared to those from saline-injected (control) offspring, as assessed using flow cytometry; n ≥ 5 experiments. B, Representative images of dendritic sMHCI on 8 DIV mouse cortical neurons cultured from FC of MIA or control offspring. Scale bar, 5 μm. C, The density of sMHCI puncta is almost doubled in neurons from MIA offspring; n ≥ 89 cells per condition, ≥7 experiments. D, Representative images of synapses on dendrites from cultured MIA and control neurons immunostained for excitatory synapse density using antibodies against PSD-95 (green) and VAMP2 (red). Scale bar, 5 μm. E, Excitatory synapse density is decreased over twofold on neurons cultured from MIA offspring compared with control offspring; n ≥ 88 cells (264 dendrites) per condition, six experiments. F, The increase in sMHCI on MIA neurons returns to control levels following β2m RNAi. MIA or saline neurons were transfected with EGFP + β2m or NTS shRNAs for 48 h before quantification of sMHCI; n ≥ 24 dendrites (8 cells) per condition, three experiments. G, The MIA-induced decrease in synapse density is rescued by preventing the MIA-induced increase in sMHCI. Synapse density is graphed normalized to control (saline neurons transfected with NTS RNAi); n ≥ 21 dendrites (7 cells), two experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
An increase in sMHCI on neurons from MIA offspring would be expected to cause a decrease in glutamatergic synapse density compared with control cultures, similar to that observed following MHCI overexpression (Fig. 2; Glynn et al., 2011). To test this hypothesis, synapse density was measured on 8 DIV neurons cultured from MIA and control offspring. Indeed, neurons from MIA offspring formed less than half the number of synapses that neurons from control offspring formed (Fig. 6D,E). To determine whether the decrease in synapse density between MIA neurons was caused by elevated sMHCI, we transfected MIA neurons with a β2m shRNA to reduce sMHCI to control (control + shNTS) levels (Fig. 6F) and then measured synapse density. Remarkably, synapse density returned to control levels in neurons from MIA offspring in which sMHCI was returned to normal (Fig. 6G). These results indicate that the MIA-induced deficiency in synapse formation is mediated by an increase in neuronal MHCI.
The MHCI-MEF2 signaling pathway is required for the MIA-induced deficiency in synapse formation
Next, we tested the hypothesis that MHCI signals through MEF2 to limit synapse density between neurons from MIA offspring. If the MIA-induced increase in sMHCI activates the same pathway as MHCI overexpression, then levels of MEF2 protein would be expected to increase in MIA neurons. Western blotting was used to measure MEF2 protein levels from lysates of cultured MIA or control neurons. Neurons from MIA offspring contained significantly more MEF2 protein than did controls (Fig. 7A), consistent with our hypothesis. MEF2A and MEF2C were both increased twofold, whereas MEF2D was increased threefold, in MIA cultures compared with controls (Fig. 7B). If the elevated level of sMHCI on MIA neurons activates the same pathway as MHCI overexpression, then preventing the MIA-induced increase in MEF2 should rescue the MIA-induced decrease in synapse density. Indeed, knocking down MEF2A+D in neurons from MIA offspring was sufficient to return synapse density to control levels (Fig. 7C), mimicking the results from the rescue experiments shown in Figure 2. Together, our results demonstrate that the MHCI-MEF2 signaling pathway identified here mediates the effects of MIA in limiting cortical synapse density.
Figure 7.

MEF2 mediates the MIA-induced reduction in synapse density. A, Representative Western blots showing increased MEF2 protein from 8 DIV cultured neurons from MIA or saline control offspring. B, Densitometry shows increased levels of MEF2A, -C, and –D protein relative to GAPDH loading controls. Data were averaged from four different cultures of each group. C, Preventing the MIA-induced increase in MEF2 rescues the MIA-induced reduction in synapse density. MIA or saline control neurons were transfected with shSCR or MEF2A+D shRNAs from 6 to 8 DIV. Synapse density was quantified as in Figure 5; n ≥ 65 dendrites (22 cells) per condition, three experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
Despite the many roles that MHCI proteins play in CNS development, plasticity, and repair (Huh et al., 2000; Goddard et al., 2007; Datwani et al., 2009; McConnell et al., 2009; Fourgeaud et al., 2010; Washburn et al., 2011; Wu et al., 2011; Adelson et al., 2012; Bilousova et al., 2012), almost nothing is known about the signaling mechanisms underlying these functions. Here, we report a novel MHCI signaling pathway that converges on MEF transcription factors. Activity-dependent, CaN-mediated activation of MEF2 transcription factors is an important downstream signaling pathway that mediates the effects of MHCI in limiting synaptic connectivity. This signaling pathway is active not only following MHCI overexpression, but also in neurons from offspring following MIA. Neurons from MIA offspring form less than half as many synapses compared with controls because of elevated MHCI-MEF2 signaling. Thus, we have identified a novel signaling pathway that mediates both the effect of MHCI in limiting synapse density during brain development and the deficiency in synapse formation caused by MIA, a risk factor for ASD and SZ.
MEF2 limits glutamatergic synapse density between hippocampal neurons (Dietrich, 2013). It was therefore surprising that acute MEF2 knockdown in young cortical neurons did not alter synapse density, defined by colocalization of PSD-95 with three different presynaptic markers. Moreover, mEPSC frequency was not significantly different in neurons with lower MEF2 levels. Although these results are inconsistent with previous reports that MEF2 limits synapse density (Flavell et al., 2006; Barbosa et al., 2008; Pulipparacharuvil et al., 2008; Pfeiffer et al., 2010; Tsai et al., 2012; Zang et al., 2013), they are consistent with more recent studies showing that MEF2 alone does not alter synapse density, but rather limits active remodeling of existing connections (Tian et al., 2010; Vetere et al., 2011; Akhtar et al., 2012; Cole et al., 2012; Zang et al., 2013). Thus, the effects of MEF2 on neural connectivity are dependent on brain region, age, and activity level.
Despite a lack of effect on synapse density, MEF2 does regulate synaptic strength in young cortical neurons. mEPSC amplitude was increased in MEF2 knockdown neurons, suggesting that AMPARs are either more abundant or more effective at their synapses. Consistent with the latter interpretation, MEF2 knockdown for 48 h increased the density of GluA1-, but not GluA-2-, containing synapses, suggesting that newly inserted, Ca2+-permeable AMPARs mediate the increase in mEPSC amplitude through their enhanced conductance (Swanson et al., 1997; Liu and Zukin, 2007). The lack of a significant increase in mEPSC frequency may reflect an increase in the insertion of GluA1 homomers into preexisting, AMPAR-containing synapses. The ability of MEF2 to alter synaptic strength in young cortical neurons is different from the initial report of no such effect in more mature hippocampal neurons (Flavell et al., 2006). Although recent papers have shown that MEF2 can actually limit synaptic strength in hippocampal neurons (Pfeiffer et al., 2010; Zang et al., 2013), the mechanism underlying this effect —destabilization of PSD-95 (Tsai et al., 2012)—is distinct from that in young cortical neurons, because up- or downregulation of MEF2 in these cells did not alter PSD-95. Thus, MEF2 uses distinct mechanisms to regulate the composition of synapses in a region- and age-specific manner.
The necessity, but not sufficiency, of MEF2 in MHCI signaling suggests that MHCI activates additional factors that work with MEF2 to decrease synapse density. These factors are likely downstream of both MHCI and CaN, because CaN is both necessary and sufficient to mediate the effects of MHCI in negatively regulating synapse density. In nonneuronal cells, MEF2 interacts with a large number of cofactors, including NFAT, Cabin1/Cain, GSK3β, Cdk5, MASH-1, p300, and HDACs (Sun et al., 1998; Dietrich, 2013). Because NFAT is one of the primary cofactors for MEF2 and is a major mediator of CaN signaling (Blaeser et al., 2000; Groth et al., 2003), we tested its role in mediating the effects of MHCI on synapse density. However, MHCI overexpression did not alter NFAT transcriptional activity and specific inhibition of CaN:NFAT signaling did not rescue the MHCI-mediated decrease in synapse density (Fig. 5D). Thus, other cofactors may be necessary for MHCI to limit synapse density.
How MHCI signaling leads to changes in synaptic activity, Ca2+ influx, and CaN activation in a cell-autonomous manner is unknown. Most research into MHCI signaling in the immune system has focused on activation of immune receptors. Indeed, some of the effects of MHCI in regulating activity-dependent plasticity are mimicked in mice lacking the paired Ig-like receptor B (Syken et al., 2006; Datwani et al., 2009). Fewer studies have addressed the possibility that MHCI might signal in a cell-autonomous manner (Pedersen et al., 1999). This type of “reverse” signaling has been investigated in non-neuronal cells, where antibody-mediated MHCI clustering causes Ca2+ influx (Gilliland et al., 1989; Dissing et al., 1990; Skov et al., 1995, 1997; Jin et al., 2004). On cortical neurons, a subset of sMHCI exists as clustered heavy chains and this clustering is required for its synapse-limiting activity (Glynn et al., 2011), supporting the idea that MHCI clustering could mediate the MHCI-induced activation of MEF2.
MHCI molecules could also modulate Ca2+ influx (and CaN signaling) by altering the function of other transmembrane proteins, including ion channels. NMDAR activation is required for MHCI to activate MEF2 and moreover, MHCI limits NMDAR currents and AMPAR trafficking following NMDAR stimulation (Fourgeaud et al., 2010). A reduction in NMDAR conductance by MHCI could result in stimulus-induced Ca2+ levels more conducive to CaN activation and LTD induction (Groth et al., 2003), consistent with the lack of LTD in mice deficient in sMHCI (Huh et al., 2000). Alternatively, MHCI might interact with as yet unknown proteins to regulate MEF2 activation. Many cell-surface proteins unrelated to the immune response interact with, and are regulated by, MHCI (Arosa et al., 2007). Thus, identifying novel MHCI effectors in neurons is an important goal for the future.
One of the most exciting implications of the role for MHCI in regulating neural development and plasticity is its potential involvement in mediating the effects of immune dysregulation on brain development in disease. MIA is a risk factor for ASD and SZ in humans (Atladóttir et al., 2010, 2012; Meyer et al., 2011; Brown, 2012). Moreover, mouse models of this risk factor show both ASD- and SZ-like behaviors in offspring (Shi et al., 2003, 2005; Meyer et al., 2008a, 2009; Ito et al., 2010; Vuillermot et al., 2010; Patterson, 2011; Malkova et al., 2012; Giovanoli et al., 2013) and long lasting changes in cytokine levels in the brains of postnatal MIA offspring (Garay et al., 2013). Cytokines regulate MHCI expression in most cell types, but whether neuronal MHCI levels in MIA offspring are altered was unexplored. Using flow cytometry and ICC, we show here that MHCI levels on neurons from newborn MIA offspring are doubled relative to controls, providing evidence for the first time that sMHCI levels on young cortical neurons are highly responsive to a peripheral immune response.
Although MIA leads to ASD- and SZ-like neuropathology and behaviors as well as decreased mEPSC frequency in hippocampal slices from young adult offspring (Ito et al., 2010), whether it also alters connections in the brains of offspring is unknown. Our results indicate that MIA does indeed alter connectivity during development, causing a profound deficit in the ability of cortical neurons to form synapses. Most important, this MIA-induced deficit in synapse formation requires elevated neuronal sMHCI and MEF2 levels. Together, these results imply that MIA requires the MHCI-MEF2 signaling pathway to limit the ability of cortical neurons to form synapses. Future experiments aimed at discovering how this pathway is altered by MIA, the developmental progression of those changes, and whether this signaling pathway mediates disease-linked behaviors in offspring will be critical for determining the relevance of this MHCI-MEF2 signaling pathway to disease.
Because MIA is a risk factor for both ASD and SZ (Atladóttir et al., 2010, 2012; Meyer et al., 2011; Brown, 2012), the MHCI-MEF2 signaling pathway identified here may be critical for the pathogenesis of these disorders. Increasing evidence supports a central role for immune dysregulation in ASD and SZ. Both disorders are linked to aberrations on chromosome 6, which is densely packed with immune genes, including MHCI (Needleman and McAllister, 2012; Torres et al., 2012a). Specific MHCI haplotypes are associated with ASD (Needleman and McAllister, 2012; Torres et al., 2012b; Mostafa et al., 2013) and the MHC region is one of the few replicable sites associated with SZ in large-scale genome-wide association studies (Purcell et al., 2009; Shi et al., 2009; Stefansson et al., 2009; Li et al., 2010; Kano et al., 2011; Jia et al., 2012; Irish Schizophrenia Genomics Consortium and the Wellcome Trust Case Control Consortium 2, 2012; Andreassen et al., 2013). MEF2C is also linked to ASD and regulates several risk factors, including protocadherin-10 and Deleted-In-Autism 1 (Morrow et al., 2008; Novara et al., 2010; Mikhail et al., 2011; Neale et al., 2012; Tsai et al., 2012). Moreover, the gene that causes Rett syndrome, MeCP2, represses MEF2 expression (Chahrour et al., 2008) and MEF2 requires the fragile-X mental retardation protein to eliminate synapses on mature hippocampal neurons (Pfeiffer et al., 2010; Tsai et al., 2012; Zang et al., 2013). Together, these observations suggest the intriguing hypothesis that the MHCI-MEF2 signaling pathway identified here not only regulates synapse density in the developing cortex, but may also represent a common molecular pathway downstream of both genetic mutations and environmental factors that contribute to ASDs and SZ.
Footnotes
This work was funded by NINDS R01 NS060125 (A.K.M.), a grant from the UC Davis Research Investments in Science and Engineering Program (A.K.M.), and Dennis Weatherstone Predoctoral Fellowships (B.M.E. No. 6339 and M.L.E. No. 7825), and was supported by the National Center for Advancing Translational Sciences, NIH, through Grant UL1 TR 000002 and linked award TL1 TR 000133 (B.M.E.). We thank Drs. Stephanie Barrow, Samantha Spangler, Leigh Needleman, and Vlasta Lyles for generating neuronal cultures, and Drs. Needleman and Spangler for creating H2-Kb-YFP and H2-Kb-mCherry, respectively. Faten El-Sabeawy contributed valuable technical support. We would particularly like to thank Dr. Johannes Hell's lab at UC Davis for providing NS21 supplement. We would also like to thank the Flow Cytometry Shared Resource facility at UC Davis for their expertise and instrumentation. Drs. Chris Cowan, Mike Greenberg, and Mark Dell'Acqua provided valuable advice throughout the project.
The authors declare no competing financial interests.
References
- Adelson JD, Barreto GE, Xu L, Kim T, Brott BK, Ouyang YB, Naserke T, Djurisic M, Xiong X, Shatz CJ, Giffard RG. Neuroprotection from stroke in the absence of MHCI or PirB. Neuron. 2012;73:1100–1107. doi: 10.1016/j.neuron.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar MW, Kim MS, Adachi M, Morris MJ, Qi X, Richardson JA, Bassel-Duby R, Olson EN, Kavalali ET, Monteggia LM. In vivo analysis of MEF2 transcription factors in synapse regulation and neuronal survival. PLoS One. 2012;7:e34863. doi: 10.1371/journal.pone.0034863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen OA, Thompson WK, Schork AJ, Ripke S, Mattingsdal M, Kelsoe JR, Kendler KS, O'Donovan MC, Rujescu D, Werge T, Sklar P, Roddey JC, Chen CH, McEvoy L, Desikan RS, Djurovic S, Dale AM. Improved detection of common variants associated with schizophrenia and bipolar disorder using pleiotropy-informed conditional false discovery rate. PLoS Genet. 2013;9:e1003455. doi: 10.1371/journal.pgen.1003455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramburu J, Yaffe MB, López-Rodriguez C, Cantley LC, Hogan PG, Rao A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science. 1999;285:2129–2133. doi: 10.1126/science.285.5436.2129. [DOI] [PubMed] [Google Scholar]
- Arnold MA, Kim Y, Czubryt MP, Phan D, McAnally J, Qi X, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell. 2007;12:377–389. doi: 10.1016/j.devcel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Arosa FA, Santos SG, Powis SJ. Open conformers: the hidden face of MHC-I molecules. Trends Immunol. 2007;28:115–123. doi: 10.1016/j.it.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Arrode-Brusés G, Brusés JL. Maternal immune activation by poly(I:C) induces expression of cytokines IL-1beta and IL-13, chemokine MCP-1 and colony stimulating factor VEGF in fetal mouse brain. J Neuroinflammation. 2012;9:83. doi: 10.1186/1742-2094-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atladóttir HO, Thorsen P, Østergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord. 2010;40:1423–1430. doi: 10.1007/s10803-010-1006-y. [DOI] [PubMed] [Google Scholar]
- Atladóttir HÓ, Henriksen TB, Schendel DE, Parner ET. Autism after infection, febrile episodes, and antibiotic use during pregnancy: an exploratory study. Pediatrics. 2012;130:e1447–1454. doi: 10.1542/peds.2012-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa AC, Olson EN, Kim MS, Ertunc M, Adachi M, Nelson ED, McAnally J, Richardson JA, Kavalali ET, Monteggia LM, Bassel-Duby R. MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc Natl Acad Sci U S A. 2008;105:9391–9396. doi: 10.1073/pnas.0802679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilousova T, Dang H, Xu W, Gustafson S, Jin Y, Wickramasinghe L, Won T, Bobarnac G, Middleton B, Tian J, Kaufman DL. Major histocompatibility complex class I molecules modulate embryonic neuritogenesis and neuronal polarization. J Neuroimmunol. 2012;247:1–8. doi: 10.1016/j.jneuroim.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black BL, Ligon KL, Zhang Y, Olson EN. Cooperative transcriptional activation by the neurogenic basic helix-loop-helix protein MASH1 and members of the myocyte enhancer factor-2 (MEF2) family. J Biol Chem. 1996;271:26659–26663. doi: 10.1074/jbc.271.43.26659. [DOI] [PubMed] [Google Scholar]
- Blaeser F, Ho N, Prywes R, Chatila TA. Ca(2+)-dependent gene expression mediated by MEF2 transcription factors. J Biol Chem. 2000;275:197–209. doi: 10.1074/jbc.275.1.197. [DOI] [PubMed] [Google Scholar]
- Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol. 2012;72:1272–1276. doi: 10.1002/dneu.22024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Patterson PH. Maternal infection and schizophrenia: implications for prevention. Schizophr Bulle. 2011;37:284–290. doi: 10.1093/schbul/sbq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon MA, Boulanger LM. MHC class I protein is expressed by neurons and neural progenitors in mid-gestation mouse brain. Mol Cell Neurosci. 2013;52:117–127. doi: 10.1016/j.mcn.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SX, Cherry A, Tari PK, Podgorski K, Kwong YK, Haas K. The transcription factor MEF2 directs developmental visually driven functional and structural metaplasticity. Cell. 2012;151:41–55. doi: 10.1016/j.cell.2012.08.028. [DOI] [PubMed] [Google Scholar]
- Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW. NS21: redefined and modified supplement B27 for neuronal cultures. J Neurosci Methods. 2008;171:239–247. doi: 10.1016/j.jneumeth.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole CJ, Mercaldo V, Restivo L, Yiu AP, Sekeres MJ, Han JH, Vetere G, Pekar T, Ross PJ, Neve RL, Frankland PW, Josselyn SA. MEF2 negatively regulates learning-induced structural plasticity and memory formation. Nat Neurosci. 2012;15:1255–1264. doi: 10.1038/nn.3189. [DOI] [PubMed] [Google Scholar]
- Corriveau R, Huh GS, Shatz CJ. Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron. 1998;21:505–520. doi: 10.1016/S0896-6273(00)80562-0. [DOI] [PubMed] [Google Scholar]
- Datwani A, McConnell MJ, Kanold PO, Micheva KD, Busse B, Shamloo M, Smith SJ, Shatz CJ. Classical MHCI molecules regulate retinogeniculate refinement and limit ocular dominance plasticity. Neuron. 2009;64:463–470. doi: 10.1016/j.neuron.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich JB. The MEF2 family and the brain: from molecules to memory. Cell Tissue Res. 2013;352:179–190. doi: 10.1007/s00441-013-1565-2. [DOI] [PubMed] [Google Scholar]
- Dissing S, Geisler C, Rubin B, Plesner T, Claesson MH. T cell activation: II. Activation of human T lymphoma cells by cross-linking of their MHC class I antigens. Cell Immunol. 1990;126:196–210. doi: 10.1016/0008-8749(90)90312-F. [DOI] [PubMed] [Google Scholar]
- Elmer BM, McAllister AK. Major histocompatibility complex class I proteins in brain development and plasticity. Trends Neurosci. 2012;35:660–670. doi: 10.1016/j.tins.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Reutiman TJ, Folsom TD, Huang H, Oishi K, Mori S, Smee DF, Pearce DA, Winter C, Sohr R, Juckel G. Maternal infection leads to abnormal gene regulation and brain atrophy in mouse offspring: implications for genesis of neurodevelopmental disorders. Schizophr Res. 2008;99:56–70. doi: 10.1016/j.schres.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore R, Khudayberdiev S, Christensen M, Siegel G, Flavell SW, Kim TK, Greenberg ME, Schratt G. Mef2-mediated transcription of the miR379–410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J. 2009;28:697–710. doi: 10.1038/emboj.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S, Griffith EC, Hu LS, Chen C, Greenberg ME. Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science. 2006;311:1008–1012. doi: 10.1126/science.1122511. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, Greenberg ME. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourgeaud L, Davenport CM, Tyler CM, Cheng TT, Spencer MB, Boulanger LM. MHC class I modulates NMDA receptor function and AMPA receptor trafficking. Proc Natl Acad Sci U S A. 2010;107:22278–22283. doi: 10.1073/pnas.0914064107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garay PA, Hsiao EY, Patterson PH, McAllister AK. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun. 2013;31:54–68. doi: 10.1016/j.bbi.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland LK, Norris NA, Grosmaire LS, Ferrone S, Gladstone P, Ledbetter JA. Signal transduction in lymphocyte activation through crosslinking of HLA class I molecules. Hum Immunol. 1989;25:269–289. doi: 10.1016/0198-8859(89)90089-X. [DOI] [PubMed] [Google Scholar]
- Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, Winter C, Riva MA, Mortensen PB, Schedlowski M, Meyer U. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science. 2013;339:1095–1099. doi: 10.1126/science.1228261. [DOI] [PubMed] [Google Scholar]
- Glynn MW, McAllister AK. Immunocytochemistry and quantification of protein colocalization in cultured neurons. Nat Protoc. 2006;1:1287–1296. doi: 10.1038/nprot.2006.220. [DOI] [PubMed] [Google Scholar]
- Glynn MW, Elmer BM, Garay PA, Liu XB, Needleman LA, El-Sabeawy F, McAllister AK. MHCI negatively regulates synapse density during the establishment of cortical connections. Nat Neurosci. 2011;14:442–451. doi: 10.1038/nn.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard CA, Butts DA, Shatz CJ. Regulation of CNS synapses by neuronal MHC class I. Proc Natl Acad Sci U S A. 2007;104:6828–6833. doi: 10.1073/pnas.0702023104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth RD, Dunbar RL, Mermelstein PG. Calcineurin regulation of neuronal plasticity. Biochem Biophys Res Commun. 2003;311:1159–1171. doi: 10.1016/j.bbrc.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ. Functional requirement for class I MHC in CNS development and plasticity. Science. 2000;290:2155–2159. doi: 10.1126/science.290.5499.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irish Schizophrenia Genomics Consortium and the Wellcome Trust Case Control Consortium 2. Genome-wide association study implicates HLA-C*01:02 as a risk factor at the major histocompatibility complex locus in schizophrenia. Biol Psychiatry. 2012;72:620–628. doi: 10.1016/j.biopsych.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito HT, Smith SE, Hsiao E, Patterson PH. Maternal immune activation alters nonspatial information processing in the hippocampus of the adult offspring. Brain Behav Immun. 2010;24:930–941. doi: 10.1016/j.bbi.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia P, Wang L, Fanous AH, Chen X, Kendler KS, Zhao Z. A bias-reducing pathway enrichment analysis of genome-wide association data confirmed association of the MHC region with schizophrenia. J Med Genet. 2012;49:96–103. doi: 10.1136/jmedgenet-2011-100397. [DOI] [PubMed] [Google Scholar]
- Jin YP, Fishbein MC, Said JW, Jindra PT, Rajalingam R, Rozengurt E, Reed EF. Anti-HLA class I antibody-mediated activation of the PI3K/Akt signaling pathway and induction of Bcl-2 and Bcl-xL expression in endothelial cells. Hum Immunol. 2004;65:291–302. doi: 10.1016/j.humimm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Johnson DR. Locus-specific constitutive and cytokine-induced HLA class I gene expression. J Immunol. 2003;170:1894–1902. doi: 10.4049/jimmunol.170.4.1894. [DOI] [PubMed] [Google Scholar]
- Kano S, Nwulia E, Niwa M, Chen Y, Sawa A, Cascella N. Altered MHC class I expression in dorsolateral prefrontal cortex of nonsmoker patients with schizophrenia. Neurosci Res. 2011;71:289–293. doi: 10.1016/j.neures.2011.07.1818. [DOI] [PubMed] [Google Scholar]
- Li H, Radford JC, Ragusa MJ, Shea KL, McKercher SR, Zaremba JD, Soussou W, Nie Z, Kang YJ, Nakanishi N, Okamoto S, Roberts AJ, Schwarz JJ, Lipton SA. Transcription factor MEF2C influences neural stem/progenitor cell differentiation and maturation in vivo. Proc Natl Acad Sci U S A. 2008;105:9397–9402. doi: 10.1073/pnas.0802876105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Li Z, Chen P, Zhao Q, Wang T, Huang K, Li J, Li Y, Liu J, Zeng Z, Feng G, He L, Shi Y. Common variants in major histocompatibility complex region and TCF4 gene are significantly associated with schizophrenia in Han Chinese. Biol Psychiatry. 2010;68:671–673. doi: 10.1016/j.biopsych.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Liu SJ, Zukin RS. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 2007;30:126–134. doi: 10.1016/j.tins.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Lyons GE, Micales BK, Schwarz J, Martin JF, Olson EN. Expression of mef2 genes in the mouse central nervous system suggests a role in neuronal maturation. J Neurosci. 1995;15:5727–5738. doi: 10.1523/JNEUROSCI.15-08-05727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons MR, Schwarz CM, West AE. Members of the myocyte enhancer factor 2 transcription factor family differentially regulate Bdnf transcription in response to neuronal depolarization. J Neurosci. 2012;32:12780–12785. doi: 10.1523/JNEUROSCI.0534-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. 2012;26:607–616. doi: 10.1016/j.bbi.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- McConnell MJ, Huang YH, Datwani A, Shatz CJ. H2-K(b) and H2-D(b) regulate cerebellar long-term depression and limit motor learning. Proc Natl Acad Sci U S A. 2009;106:6784–6789. doi: 10.1073/pnas.0902018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008a;22:469–486. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Schwendener S, Knuesel I, Yee BK, Feldon J. Relative prenatal and postnatal maternal contributions to schizophrenia-related neurochemical dysfunction after in utero immune challenge. Neuropsychopharmacology. 2008b;33:441–456. doi: 10.1038/sj.npp.1301413. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Fatemi SH. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev. 2009;33:1061–1079. doi: 10.1016/j.neubiorev.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Meyer U, Feldon J, Dammann O. Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr Res. 2011;69:26R–33R. doi: 10.1203/PDR.0b013e318212c196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel M, Schmidt MJ, Mirnics K. Immune system gene dysregulation in autism and schizophrenia. Dev Neurobiol. 2012;72:1277–1287. doi: 10.1002/dneu.22044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Busse B, Weiler NC, O'Rourke N, Smith SJ. Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron. 2010;68:639–653. doi: 10.1016/j.neuron.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhail FM, Lose EJ, Robin NH, Descartes MD, Rutledge KD, Rutledge SL, Korf BR, Carroll AJ. Clinically relevant single gene or intragenic deletions encompassing critical neurodevelopmental genes in patients with developmental delay, mental retardation, and/or autism spectrum disorders. Am J Med Genet A. 2011;155A:2386–2396. doi: 10.1002/ajmg.a.34177. [DOI] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, Mukaddes NM, Balkhy S, Gascon G, Hashmi A, Al-Saad S, Ware J, Joseph RM, Greenblatt R, Gleason D, Ertelt JA, Apse KA, Bodell A, Partlow JN, Barry B, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostafa GA, Shehab AA, Al-Ayadhi LY. The link between some alleles on human leukocyte antigen system and autism in children. J Neuroimmunol. 2013;255:70–74. doi: 10.1016/j.jneuroim.2012.10.002. [DOI] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman LA, McAllister AK. The major histocompatibility complex and autism spectrum disorder. Dev Neurobiol. 2012;72:1288–1301. doi: 10.1002/dneu.22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman LA, Liu XB, El-Sabeawy F, Jones EG, McAllister AK. MHC class I molecules are present both pre- and postsynaptically in the visual cortex during postnatal development and in adulthood. Proc Natl Acad Sci U S A. 2010;107:16999–17004. doi: 10.1073/pnas.1006087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novara F, Beri S, Giorda R, Ortibus E, Nageshappa S, Darra F, Dalla Bernardina B, Zuffardi O, Van Esch H. Refining the phenotype associated with MEF2C haploinsufficiency. Clin Genet. 2010;78:471–477. doi: 10.1111/j.1399-0004.2010.01413.x. [DOI] [PubMed] [Google Scholar]
- Okamoto S, Krainc D, Sherman K, Lipton SA. Antiapoptotic role of the p38 mitogen-activated protein kinase-myocyte enhancer factor 2 transcription factor pathway during neuronal differentiation. Proc Natl Acad Sci U S A. 2000;97:7561–7566. doi: 10.1073/pnas.130502697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. 2011;17:389–394. doi: 10.1016/j.molmed.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazyra-Murphy MF, Hans A, Courchesne SL, Karch C, Cosker KE, Heerssen HM, Watson FL, Kim T, Greenberg ME, Segal RA. A retrograde neuronal survival response: target-derived neurotrophins regulate MEF2D and bcl-w. J Neurosci. 2009;29:6700–6709. doi: 10.1523/JNEUROSCI.0233-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen AE, Skov S, Bregenholt S, Ruhwald M, Claesson MH. Signal transduction by the major histocompatibility complex class I molecule. APMIS. 1999;107:887–895. doi: 10.1111/j.1699-0463.1999.tb01488.x. [DOI] [PubMed] [Google Scholar]
- Pereira AH, Clemente CF, Cardoso AC, Theizen TH, Rocco SA, Judice CC, Guido MC, Pascoal VD, Lopes-Cendes I, Souza JR, Franchini KG. MEF2C silencing attenuates load-induced left ventricular hypertrophy by modulating mTOR/S6K pathway in mice. PLoS One. 2009;4:e8472. doi: 10.1371/journal.pone.0008472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, Zang T, Wilkerson JR, Taniguchi M, Maksimova MA, Smith LN, Cowan CW, Huber KM. Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron. 2010;66:191–197. doi: 10.1016/j.neuron.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development. 2007;134:4131–4140. doi: 10.1242/dev.008367. [DOI] [PubMed] [Google Scholar]
- Pulipparacharuvil S, Renthal W, Hale CF, Taniguchi M, Xiao G, Kumar A, Russo SJ, Sikder D, Dewey CM, Davis MM, Greengard P, Nairn AC, Nestler EJ, Cowan CW. Cocaine regulates MEF2 to control synaptic and behavioral plasticity. Neuron. 2008;59:621–633. doi: 10.1016/j.neuron.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribic A, Flügge G, Schlumbohm C, Mätz-Rensing K, Walter L, Fuchs E. Activity-dependent regulation of MHC class I expression in the developing primary visual cortex of the common marmoset monkey. Behav Brain Funct. 2011;7:1. doi: 10.1186/1744-9081-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalizi AK, Bonni A. brawn for brains: the role of MEF2 proteins in the developing nervous system. Curr Top Dev Biol. 2005;69:239–266. doi: 10.1016/S0070-2153(05)69009-6. [DOI] [PubMed] [Google Scholar]
- Shalizi A, Lehtinen M, Gaudilliere B, Donovan N, Han J, Konishi Y, Bonni A. Characterization of a neurotrophin signaling mechanism that mediates neuron survival in a temporally specific pattern. J Neurosci. 2003;23:7326–7336. doi: 10.1523/JNEUROSCI.23-19-07326.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalizi A, Gaudillière B, Yuan Z, Stegmüller J, Shirogane T, Ge Q, Tan Y, Schulman B, Harper JW, Bonni A. A calcium-regulated MEF2 sumoylation switch controls postsynaptic differentiation. Science. 2006;311:1012–1017. doi: 10.1126/science.1122513. [DOI] [PubMed] [Google Scholar]
- Shalizi A, Bilimoria PM, Stegmüller J, Gaudillière B, Yang Y, Shuai K, Bonni A. PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. J Neurosci. 2007;27:10037–10046. doi: 10.1523/JNEUROSCI.0361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Levinson DF, Duan J, Sanders AR, Zhang Y, Pe'er I, Dudbridge F, Holmans PA, Whittemore AS, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Bocola NG, Byerley WF, Black DW, Crowe RR, Oksenberg JR, Mirel DB, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Tu N, Patterson PH. Maternal influenza infection is likely to alter fetal brain development indirectly: the virus is not detected in the fetus. Int J Dev Neurosci. 2005;23:299–305. doi: 10.1016/j.ijdevneu.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Skov S, Odum N, Claesson MH. MHC class I signaling in T cells leads to tyrosine kinase activity and PLC-gamma 1 phosphorylation. J Immunol. 1995;154:1167–1176. [PubMed] [Google Scholar]
- Skov S, Bregenholt S, Claesson MH. MHC class I ligation of human T cells activates the ZAP70 and p56lck tyrosine kinases, leads to an alternative phenotype of the TCR/CD3 zeta-chain, and induces apoptosis. J Immunol. 1997;158:3189–3196. [PubMed] [Google Scholar]
- Stefansson H, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Youn HD, Loh C, Stolow M, He W, Liu JO. Cabin 1, a negative regulator for calcineurin signaling in T lymphocytes. Immunity. 1998;8:703–711. doi: 10.1016/S1074-7613(00)80575-0. [DOI] [PubMed] [Google Scholar]
- Swanson GT, Kamboj SK, Cull-Candy SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syken J, Grandpre T, Kanold PO, Shatz CJ. PirB restricts ocular-dominance plasticity in visual cortex. Science. 2006;313:1795–1800. doi: 10.1126/science.1128232. [DOI] [PubMed] [Google Scholar]
- Tian X, Kai L, Hockberger PE, Wokosin DL, Surmeier DJ. MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Mol Cell Neurosci. 2010;44:94–108. doi: 10.1016/j.mcn.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres AR, Westover JB, Rosenspire AJ. HLA immune function genes in autism. Autism Res Treat. 2012a;2012:959073. doi: 10.1155/2012/959073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres AR, Westover JB, Gibbons C, Johnson RC, Ward DC. Activating killer-cell immunoglobulin-like receptors (KIR) and their cognate HLA ligands are significantly increased in autism. Brain Behav Immun. 2012b;26:1122–1127. doi: 10.1016/j.bbi.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai NP, Wilkerson JR, Guo W, Maksimova MA, DeMartino GN, Cowan CW, Huber KM. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell. 2012;151:1581–1594. doi: 10.1016/j.cell.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetere G, Restivo L, Cole CJ, Ross PJ, Ammassari-Teule M, Josselyn SA, Frankland PW. Spine growth in the anterior cingulate cortex is necessary for the consolidation of contextual fear memory. Proc Natl Acad Sci U S A. 2011;108:8456–8460. doi: 10.1073/pnas.1016275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillermot S, Weber L, Feldon J, Meyer U. A longitudinal examination of the neurodevelopmental impact of prenatal immune activation in mice reveals primary defects in dopaminergic development relevant to schizophrenia. J Neurosci. 2010;30:1270–1287. doi: 10.1523/JNEUROSCI.5408-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn LR, Zekzer D, Eitan S, Lu Y, Dang H, Middleton B, Evans CJ, Tian J, Kaufman DL. A potential role for shed soluble major histocompatibility class I molecules as modulators of neurite outgrowth. PLoS One. 2011;6:e18439. doi: 10.1371/journal.pone.0018439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZP, Washburn L, Bilousova TV, Boudzinskaia M, Escande-Beillard N, Querubin J, Dang H, Xie CW, Tian J, Kaufman DL. Enhanced neuronal expression of major histocompatibility complex class I leads to aberrations in neurodevelopment and neurorepair. J Neuroimmunol. 2011;232:8–16. doi: 10.1016/j.jneuroim.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Huang J, Coppola G, Geschwind DH, Bonni A. Sumoylated MEF2A coordinately eliminates orphan presynaptic sites and promotes maturation of presynaptic boutons. J Neurosci. 2013;33:4726–4740. doi: 10.1523/JNEUROSCI.4191-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang T, Maksimova MA, Cowan CW, Bassel-Duby R, Olson EN, Huber KM. Postsynaptic FMRP bidirectionally regulates excitatory synapses as a function of developmental age and MEF2 activity. Mol Cell Neurosci. 2013;56C:39–49. doi: 10.1016/j.mcn.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]