

Abstract
Objective
Activation of metabotropic glutamate receptor 5 (mGluR5) has neuroprotective properties in vitro and has been reported to limit postischemic lesion volume in vivo. Previously, mGluR5 has been identified on microglia in vitro, but the effects of mGluR5 activation on inflammation in vivo or on recovery after spinal cord injury is unknown.
Methods
Rats received intrathecal infusion of the selective mGluR5 agonist (RS)-2-chloro-5-hydroxyphenylglycine (CHPG) for 7 days after moderate impact spinal cord injury at T9. Complementary studies examined CHPG effects on activated spinal microglia cultures.
Results
Functional motor recovery was significantly increased by CHPG treatment up to 28 days after injury, with improvements in weight bearing, step taking, and coordination of stepping behavior. CHPG treatment significantly reduced lesion volume and increased white matter sparing at 28 days after injury. Administration of CHPG attenuated microglial-associated inflammatory responses in a dose-dependent fashion, including expression of ED1, Iba-1, Galectin-3, NADPH oxidase components, tumor necrosis factor-α, and inducible nitric oxide synthase. Because mGluR5 is expressed by microglial cells in the rat spinal cord, such effects may be mediated by direct action on microglial cells. mGluR5 stimulation also reduced microglial activation and decreased microglial-induced neurotoxicity in spinal cord microglia cultures; the latter effects were blocked by the selective mGluR5 antagonist MTEP.
Interpretation
These data demonstrate that mGluR5 activation can reduce microglial-associated inflammation, suggesting that the protective effects of mGluR5 agonists may reflect this action.
Spinal cord injury (SCI) results in a prolonged pathological response involving chronic inflammation, microglial activation, and astroglial scar formation that ultimately results in the development of a large cavity at the site of the lesion, the loss of gray matter/neurons, and persistent functional deficits.1,2 Inflammation, including the activation of resident microglial cells, plays an important role in this “secondary injury.”3–5
Microglia may have either neurotoxic or neurorestorative properties, although the preponderance of evidence suggests that they contribute to secondary injury and neuronal cell death.6 – 8 Products known to be produced by microglia, such as nitric oxide (NO), reactive oxygen species, and various proinflammatory cytokines (eg, tumor necrosis factor-α [TNF-α]) have been shown to directly induce neuronal death in vitro and in vivo.9 –16 Inhibition of microglial activation has been shown to have neuroprotective properties after SCI.17,18
Metabotropic glutamate receptors (mGluRs) are G-protein–coupled receptors that modulate glutamatergic activity. Within the central nervous system (CNS), these receptors have been studied largely in neurons, although they are also found in glia.19,20 mGluRs have been identified on microglia,21–23 but their roles in neuroinflammation have received limited attention. Taylor and colleagues22,23 have reported modulatory effects by group II and III receptors in opposite directions in vitro, but such data are hard to reconcile with in vivo studies showing virtually identical neuroprotective profiles after activation of these receptors.24 –29 Recently, we reported that microglia express functional mGluR5 receptors.30 Activation of these receptors results in significant suppression of microglial activity in vitro, including reductions in production of proinflammatory mediators and microglial-induced neurotoxicity. These functions are dependent on the mGluR5 receptor, because knock-out of the receptor or pharmacological inhibition reverses the effects of agonist treatment.
In this article, we show that treatment with the selective mGluR5 agonist (RS)-2-chloro-5-hydroxyphenyl-glycine (CHPG) reduces lesion size and improves neurological recovery after SCI in adult rats. These effects may be mediated, in part, through modulation of microglial-associated inflammation. We find that microglia in the rat spinal cord express mGluR5, and that mGluR5 activation negatively regulates the release of microglial-associated inflammatory factors and related neurotoxicity.
Materials and Methods
Spinal Cord Injury
Contusion SCI was performed in adult male Sprague–Dawley rats as described previously.31 In brief, rats (275–325gm) were anesthetized with sodium pentobarbital (67mg/kg intraperitoneally), and moderate injury was induced by dropping a 10gm weight from 25mm onto an impounder positioned on the exposed spinal cord at vertebral level T9. Shaminjured rats underwent laminectomy without weight drop. All experiments complied fully with the principles set forth in the Guide for the Care and Use of Laboratory Animals prepared by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Resources, National Research Council (DHEW pub. No. [NIH] 85-23, 2985) and were approved by the Georgetown University Institutional Animal Care and Use Committee.
CHPG Administration
Thirty minutes after contusion injury, an intrathecal catheter (P-100, 1.52 outside diameter) was inserted two segments below the injury site (spinal T11). The catheter was attached to an Alzet miniosmotic pump (Model 2001; Alzet, Cupertino, CA), loaded with either CHPG (41.6mM in 1% dimethylsulfoxide in saline; Tocris Biosciences, Ellisville, MI) or vehicle (1% dimethylsulfoxide in 0.9% saline). The administration rate was 1μl drug or vehicle per hour for 7 days (2.9pmol/day CHPG), for a final dosage similar to what has been previously used in the literature.32 A dose–response study for inflammatory responses compared a 7-day infusion of CHPG at 10mM (in 1% dimethylsulfoxide in saline) with 41.6mM CHPG and vehicle. Pumps were weighed before insertion and after removal to ensure function during implantation.
Functional Assessment
The Basso–Beattie–Bresnahan scale was used to assess neurological function (n = 15/group; Table), as detailed previously.33–35 All functional scores were obtained at days 1, 7, 14, 21, and 28 by two individuals blinded to treatment. Data are presented as an average of raw score per group, as well as slope of the recovery curve from 1 to 28 days after injury.
Table 1.
Table Definition of Groups
| Survival Time | Number per Group | Outcome Measures |
|---|---|---|
| 24 hours | 5 CHPG 41.6mM | Tumor necrosis factor-α enzyme-linked immunosorbent assay |
| 5 vehicle | ||
| 3 sham | ||
| 72 hours | 4 CHPG 41.6mM | Immunohistochemistry |
| 4 vehicle | ||
| 2 sham | ||
| 7 days | 4 CHPG 41.6mM | Immunohistochemistry |
| 4 CHPG 10mM | ||
| 4 vehicle | ||
| 7 days | 4 CHPG 4.16mM | Western blot |
| 4 vehicle | ||
| 2 sham | ||
| 28 days | 15 CHPG 41.6mM |
Functional assessment |
| 15 vehicle | ||
| Subgroups (randomly selected): | ||
| 5/group | Magnetic resonance imaging | |
| 4/group | Immunohistochemistry | |
| 4/group | Western blot |
CHPG = (RS)-2-chloro-5-hydroxyphenylglycine.
Magnetic Resonance Imaging Analysis
At 28 days after injury, a random sample of rats (n = 5/group) was chosen by an investigator blinded to functional outcome and underwent magnetic resonance imaging (MRI) using a 7-Tesla 20cm bore MRI (Bruker Biospin, Billerica, MA) with a two-dimensional T2-weighted imaging protocol: TR = 3,640 milliseconds, TE = 121 milliseconds, matrix size = 256 × 256. Hyperintense areas, which appeared as white regions in the normally gray spinal cord, on MRI images were assessed using Image J analysis software by a blinded investigator. In brief, regions of interest were outlined and Image J analysis software was utilized to measure the area of the region. Areas were then used to extrapolate lesion volume throughout the injured cord.
Histology
The day after T2-weighted MRI, a random sample of rats (n = 4/group) chosen by an investigator blinded to functional outcome was anesthetized (100mg/Kg sodium pentobarbital, intraperitoneally) and intracardially perfused with 10% buffered formalin. To determine volume of cavitation and white matter sparing, we processed with a standard Eriochrome cyanine R (Sigma, St. Louis, MO) staining protocol every 20th section (20μm) of the 1cm spinal cord block centered at the lesion epicenter, with a random starting section. Cavitation and spared white matter volume were assessed using unbiased stereology and the Stereologer Program (Systems Planning and Analysis, Alexandria, VA).
Immunohistochemistry
Standard fluorescent immunohistochemistry was performed on sections from animals obtained at 72 hours, 7 days, or 28 days after injury (n = 4/group; 28-day rats were randomly chosen by an investigator blinded to functional outcome), obtained as described earlier. Anti-ED1 (Serotec, Raleigh, NC), OX42 (Serotec), Iba-1 (Wako, Richmond, VA), galectin-3 (Abcam, Cambridge, MA), gp91phox (BD Transduction Laboratories, San Jose, CA), and mGluR5 (Chemicon, Billerica, MA) were used as primary antibodies. Immunofluorescence was detected and quantified in twelve 20μm sections, selected with a random start and consistent interslice distance, using confocal microscopy as described previously.36 In brief, the proportional area of tissue occupied by immunohistochemically stained cellular profiles within a defined target area (the lesion site and surrounding tissue) was measured using the Scion Image Analysis system using a method modified from Popovich and colleagues37 description.
Western Blot
At 7 or 28 days after injury, the 1cm section of the spinal cord encompassing the lesion site was dissected from 4 rats/group. Rats from the 28-day group were randomly chosen by an investigator blinded to functional outcome. Tissue was homogenized and underwent Western blotting as described previously36 for p22phox (Santa Cruz Biotechnologies, Santa Cruz, CA), ED1 (Serotec), galectin-3 (Abcam), and inducible nitric oxide synthase (iNOS; BD Transduction Laboratories).
Tumor Necrosis Factor-α Enzyme-Linked Immunosorbent Assay
At 24 hours after injury, rats (n = 5/group + 3 sham) were perfused with cooled saline, and the 1cm section of spinal cord encompassing the lesion site was carefully dissected and frozen on dry ice. The concentration of TNF-α was measured and enzyme-linked immunosorbent assay was performed as per the manufacturer’s instructions (BD Bioscience, San Jose, CA). Absorbance was read at 450nm, and values were corrected for protein concentration and expressed as picograms per milligram protein.
Microglial Cultures
Microglial cells were obtained from postnatal day 2 Sprague–Dawley rat pup spinal cords and cultured as described previously.38 After the initial incubation, the cells were shaken for 1 hour at 100 rpm and 37°C. Detached microglia were collected and replated at 2 × 105 cells/ml into 96-well plates for proliferation and NO assays, or at 5 × 105 cells/ml into Transwell inserts (Fischer Scientific, Fair Lawn, NJ) for coculture assays.
Microglial Proliferation
At 24 hours after application of the mGluR agonist CHPG and stimulation with lipopolysaccharide (LPS), proliferation of microglia in 96-well plates was assessed using the MTS assay (MTS tetrazolium compound; Cell Titer 96 Aqueous One Solution; Promega, Madison, WI), performed according to the manufacturer’s protocols. Each treatment was performed in triplicate and the experiment was repeated three times; data shown are a representative outcome.
Nitric Oxide Production
NO production was assayed at 24 hours after administration of LPS using the Griess Reagent Assay (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. Each treatment was performed in triplicate and the experiment was repeated three times; data shown are a representative outcome.
Neurotoxicity
Rat primary cortical neuronal cultures were derived from embryonic day 18 (E18) rat cortices (Taconic, Germantown, NY) as described previously.39 Microglia were stimulated with LPS for 24 hours, with or without pretreatments, and washed in warmed media before insertion into 24-well plates containing neurons (neurons were at day 5 in vitro). Twenty-four hours later, microglia were removed, and the neurons were fixed with 4% paraformaldehyde and stained with neuronal nuclei (NeuN). The number of NeuN+ cells were counted in at least 10 areas around the 24-well plate. Each treatment was performed in triplicate and the experiment was repeated three times; data shown are a representative outcome.
Statistical Analysis
Quantitative data are presented as mean ± standard error of the mean. Lesion volume, functional data, Western blot, and immunohistochemical data were obtained by an investigator blinded to treatment group. Basso–Beattie–Bresnahan scores were analyzed with two-way analysis of variance (ANOVA) and repeated measures. Remaining data were analyzed using Student’s t test or one-way ANOVA, where appropriate. All statistical tests were performed using the GraphPad Prism Program, Version 3.02 for Windows (GraphPad Software, San Diego, CA). A p value < 0.05 was considered statistically significant.
Results
CHPG Treatment Improves Recovery after Spinal Cord Injury
To test the neuroprotective effects of mGluR5 activation in vivo, we subjected adult male Sprague–Dawley rats to moderate SCI and treated them with the selective mGluR5 agonist CHPG or vehicle (7-day intrathecal infusion beginning at 30 minutes after injury of 41.6mM CHPG or vehicle; 1μl/hr infusion rate; n = 15/group). Function was assessed weekly, beginning at day 1 after injury, to determine recovery. By day 1, all animals had a score of 0 or 1, indicating complete or nearly complete loss of motor function (Fig 1A). By day 14 after injury, rats that received CHPG infusion had significantly improved Basso–Beattie–Bresnahan scores (12.3 ± 1.3; p < 0.05 with repeated-measures ANOVA) compared with vehicle-treated rats (8.50 ± 0.9). This improvement remained through 28 days after injury (12.4 ± 1.3 for CHPG vs 9.5 ± 0.7 for vehicle; p < 0.05 with repeated-measures ANOVA). For function, CHPG-treated rats showed coordinated walking steps, whereas vehicle-treated rats showed an ability to bear weight on their hind limbs but were unable to take walking steps.
Fig 1.

Functional effects of metabotropic glutamate receptor 5 (mGluR5) activation after traumatic spinal cord injury. Hind-limb locomotor function was assessed using the Basso–Beattie–Bresnahan (BBB) score at days 1, 7, 14, 21, and 28 after injury (A). CHPG treatment resulted in a significant improvement in BBB score by day 14 after injury, which continued through day 28. Squares = vehicle; triangles = CHPG. The slope of the line was also assessed (B). CHPG treatment resulted in a significantly greater rate of recovery than vehicle. Bars represent mean ± standard error of the mean. *p < 0.05.
The slope of the recovery curve was also assessed (see Fig 1B). CHPG treatment resulted in a significant increase in slope in comparison with vehicle treatment, suggesting an increased rate of recovery.
Metabotropic Glutamate Receptor 5 Activation Reduces Lesion Volume after Spinal Cord Injury
Rat spinal cords were imaged at day 28 after injury using a T2-weighted MRI protocol. Analysis of hyper-intense areas on resultant images demonstrated longitudinal lesions centered at T9 (Figs 2A, B). Quantitation of hyperintense areas demonstrated a significant reduction in lesion volume with CHPG infusion (p < 0.05, Student’s t test; see Fig 2C).
Fig 2.

Magnetic resonance imaging (MRI)–based lesion volume measurements after traumatic spinal cord injury. At 28 days after injury, rats underwent T2-weighted MRI. Hyperin-tense regions (arrows) indicate lesion sites in vehicle-treated (A) and CHPG-treated (B) animals. The MRI-based lesion volume of these hyperintense regions was assessed and demonstrated a significant reduction with CHPG treatment (C). Bars represent mean ± standard error of the mean. *p < 0.05.
To determine whether MRI-based lesion volumes reflected tissue cavitation, we quantified cavity volume in Eriochrome-stained tissue sections (Fig 3A). CHPG infusion resulted in a marked reduction in tissue cavity volume (p < 0.05, Student’s t test; see Fig 3B). This reduction in tissue cavity was accompanied by a significant increase in the amount of spared white matter (p < 0.05, Student’s t test; see Fig 3C).
Fig 3.
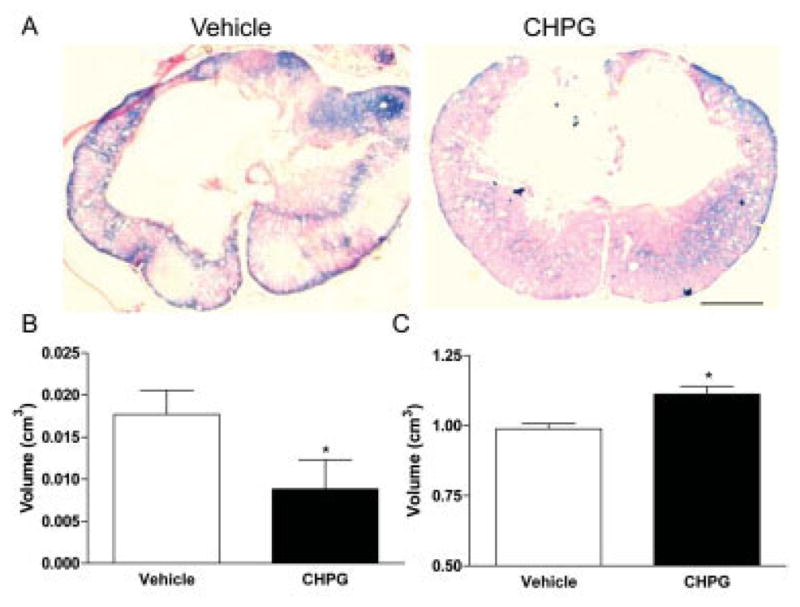
Histological effects of metabotropic glutamate receptor 5 (mGluR5) activation after traumatic spinal cord injury. Using Eriochrome-stained tissue slides (A), we calculated cavity (B) and spared white matter volume (C). CHPG infusion resulted in a significant reduction in cavitation, supporting magnetic resonance imaging (MRI) findings. CHPG treatment also increased remaining white matter around the lesion area, which stains blue with the Eriochrome dye. Representative images obtained from the lesion epicenter. Bars represent mean ± standard error of the mean. *p < 0.05. Scale bar = 500μm.
Analysis of neuronal responses to injury and CHPG treatment demonstrated little evidence of neuronal apoptosis, as measured by double labeling of NeuN and cleaved caspase-3 at the lesion site, rostral or caudal to the lesion epicenter. No significant difference in NeuN/cleaved-caspase-3 double immunolabeling was observed between vehicle and CHPG-treated tissue (data not shown).
Metabotropic Glutamate Receptor 5 Activation Reduces Inflammation after Spinal Cord Injury
Previously, we have demonstrated that microglia express mGluR5 in vitro and respond to mGluR5 activation by reducing microglial reactivity.30 To test the hypothesis that the effects of mGluR5 stimulation in vivo are mediated, in part, by suppression of inflammation, we measured the expression of the activated microglia/macrophage marker Iba-1 in the spinal cord 7 days after injury after vehicle or CHPG administration. Vehicle-treated tissue demonstrated strong Iba-1 staining throughout the spinal cord (Fig 4A). In contrast, administration of CHPG markedly reduced SCI-induced Iba-1 staining (see Figs 4B, C). The reduction in Iba-1 staining was dependent on the dosage of CHPG applied, as increasing CHPG concentrations dose-dependently reduced Iba-1 immunolabeling (see Fig 4D), with a significant reduction in Iba-1 staining with 41.6mM CHPG treatment (p < 0.05, one-way ANOVA).
Fig 4.
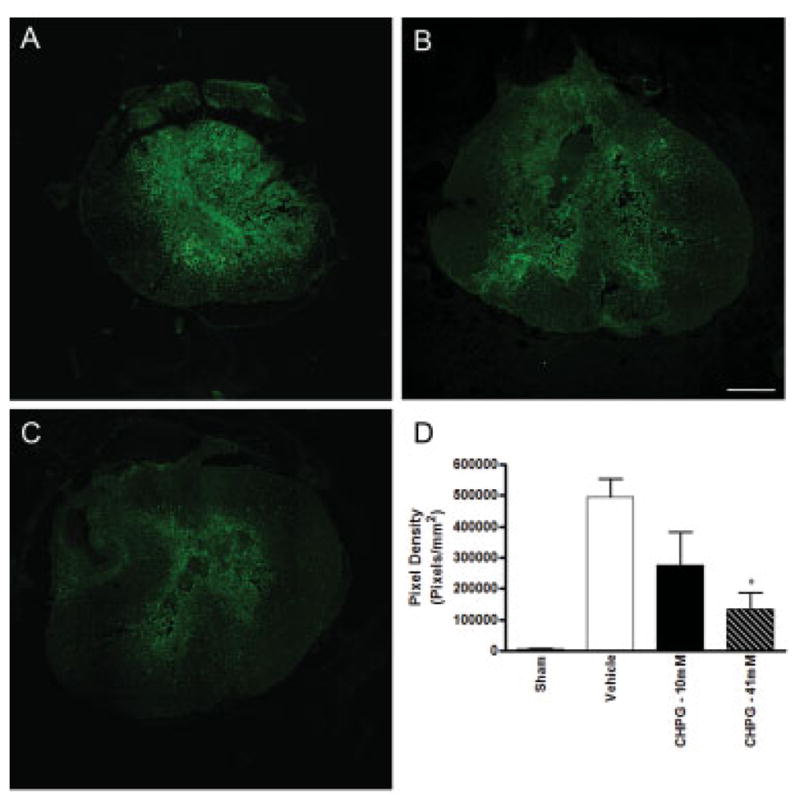
Metabotropic glutamate receptor 5 (mGluR5) activation reduces Iba-1 staining after spinal cord injury in a dose-dependent manner. At 7 days after injury, spinal cord tissue at the lesion site was immunolabeled with Iba-1, a marker for activated microglia/macrophages. Immunolabeling was greatest in tissue that received infusion of vehicle (A). Infusion of 10mM CHPG reduced Iba-1 immunostaining (B). Immuno-staining was further reduced after treatment with 41mM CHPG (C). Quantitation of immunolabeling demonstrated a dose-dependent trend with CHPG treatment, and 41mM CHPG significantly reduced immunopositive pixel density in comparison with vehicle-treated tissue. Representative images obtained at 1mm caudal to lesion epicenter. Scale bar = 500μm. Bars represent mean ± standard error of the mean. *p < 0.05.
To further investigate the role of mGluR5 on inflammation, we assessed after SCI the presence of other inflammatory proteins and markers of microglial activation such as p22phox and gp91phox, components of the NADPH oxidase enzyme expressed in microglial cells,38 ED1, the common lysosomal marker for activated microglia and macrophages, Galectin-3 (MAC-2), a cell-surface marker reportedly found only in microglia,40 iNOS, the enzyme responsible for NO, and the proinflammatory cytokine, TNF-α. Spinal cord tissue homogenate was obtained at 24 hours after injury, and TNF-α protein expression was measured. Vehicle-treated tissue demonstrated a significant increase in TNF-α protein content, in comparison with sham-injured tissue (Fig 5). Treatment with CHPG significantly reduced the TNF-α expression (p < 0.05, one-way ANOVA).
Fig 5.
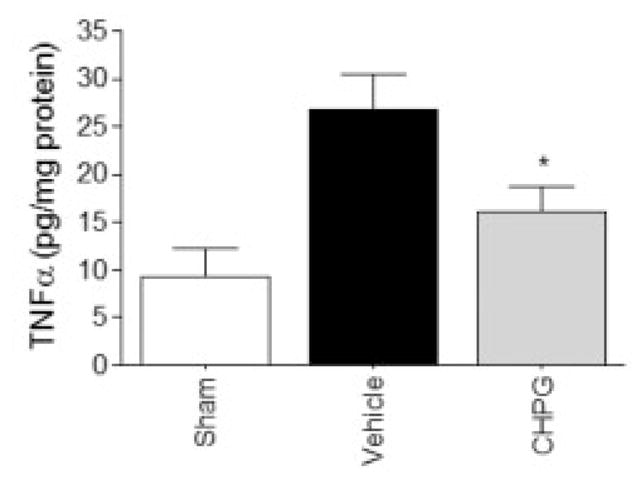
Metabotropic glutamate receptor 5 (mGluR5) activation reduces tumor necrosis factor-α (TNF-α) secretion at 24 hours after spinal cord injury. TNF-α was measured by enzyme-linked immunosorbent assay in the spinal cord tissue 24 hours after contusion injury. TNF-α was significantly increased in animals receiving contusion injury and vehicle treatment in comparison with sham-injured rats. Infusion of CHPG into the spinal cord at 30 minutes after injury resulted in a significant reduction in TNF-α protein detection. Bars represent mean ± standard error of the mean. *p < 0.05.
Tissue was obtained for Western blot at 7 and 28 days after injury. At 7 and 28 days after injury, CHPG treatment significantly reduced the expression of p22phox, a membrane-bound component of NADPH oxidase (Fig 6A), when compared with vehicle-treated rats (p < 0.05, one-way ANOVA). Although there was a nonsignificant trend toward reduction in ED1 protein expression at 7 days, a marked reduction was observed at 28 days after injury (p < 0.05, one-way ANOVA; see Fig 6B). Also at 28 days, CHPG infusion significantly reduced expression of Galectin-3/MAC2, a marker of microglial activation38 (p < 0.05, Student’s t test; see Fig 6C), and iNOS, the enzyme responsible for production of NO (p < 0.05, Student’s t test; see Fig 6D).
Fig 6.

Metabotropic glutamate receptor 5 (mGluR5) activation alters the inflammatory response after spinal cord injury (SCI). Microglial-related inflammatory products p22 phox (A), ED1 (B), Galectin-3 (C), and inducible nitric oxide synthase (iNOS) (D) were found to be significantly suppressed 7 or 28 days after SCI in animals treated with CHPG in comparison with control, as measured by Western blotting. Representative Western blots for 7- or 28-day samples are shown. Bars represent mean ± standard error of the mean. *p < 0.05.
Furthermore, quantitation of immunohistochemical labeling for ED1 expression at 28 days demonstrated a significant reduction with CHPG treatment in comparison with vehicle treatment (Student’s t test; Figs 7A–C). Analysis of immunolabeling for gp91phox, the membrane-bound catalytic subunit of NADPH oxidase, shows that there was no significant difference between vehicle and treated tissue at 72 hours. However, a significant reduction in gp91phox staining was observed at 28 days with CHPG treatment (p < 0.05, one-way ANOVA; see Fig 7).
Fig 7.
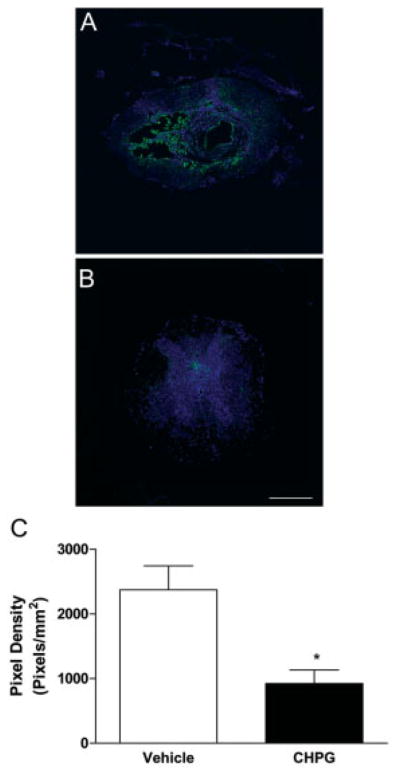
Metabotropic glutamate receptor 5 (mGluR5) activation reduces ED1 expression at 28 days after spinal cord injury (SCI). ED1 expression was measured in spinal cord tissue samples using fluorescence immunohistochemistry. Vehicle-treated tissue (A) demonstrated a large amount of staining for ED1 (green) around a large cavity in the center of the tissue, which was decreased in CHPG-treated tissue (B). TO-PRO-3–stained nuclei (blue) provide contrast. Images are taken from 2mm rostral to the lesion epicenter. Quantitation of ED1-positive pixel density showed that this reduction is statistically significant (C). Bars represent mean ± standard error of the mean. *p < 0.05. Scale bar = 500μm.
Metabotropic Glutamate Receptor 5 Is Expressed on Microglia after Spinal Cord Injury
To investigate whether microglia express mGluR5 in vivo, we performed double immunohistochemical labeling with mGluR5 and microglial markers. In injured spinal cord tissue, mGluR5 colabeled with markers specific for microglia in the CNS, galectin-341 (Fig 8A) and OX42 (CD11b; see Fig 8B). This double labeling was observed in the lesioned area at both the lesion epicenter and in the periphery. Negative controls, in which the primary antibodies were not included, failed to show the same labeling pattern (see Fig 8C).
Fig 8.

Metabotropic glutamate receptor 5 (mGluR5) activation reduces gp91phox expression at 28 days after spinal cord injury (SCI). Immunolabeling for gp91phox (green) was performed at 72 hours (A, B) and 28 days (C, D) after injury in vehicle- (A, C) and CHPG-treated (B, D) tissue. TO-PRO-3–stained nuclei (blue) provide contrast. Quantitation of gp91phox-positive pixel density demonstrated a significant reduction in CHPG-treated tissue at 28 days after injury (E). Images are taken from 2mm caudal to the lesion epicenter. Bars represent mean ± standard error of the mean. *p < 0.05. Scale bar = 500μm.
Metabotropic Glutamate Receptor 5 Stimulation Reduces Spinal Cord Microglial Activation
Because microglia demonstrate regional heterogeneity within the CNS,42 microglia were cultured from rat spinal cord to determine whether spinal cord microglia are responsive to mGluR5 activation. These cells were treated with LPS (100ng/ml) with and without 1-hour CHPG (100μM) pretreatment. LPS stimulation of spinal cord microglia for 24 hours resulted in a marked increase in NO production and proliferation (p < 0.001, one-way ANOVA; Figs 9A, B). These increases were significantly reduced by CHPG treatment (p < 0.05, one-way ANOVA; see Figs 9A, B), demonstrating that activation of mGluR5 on spinal cord microglia attenuates microglial reactivity. All experiments were performed in triplicate and repeated at least twice.
Fig 9.

Microglia express metabotropic glutamate receptor 5 (mGluR5). Double labeling for mGluR5 and various markers of microglia was performed to determine whether microglia express mGluR5 in the spinal cord. mGluR5 (green) is expressed on Galectin-3–(red; A, B) or OX42 (red, C)-positive microglia (arrows) in injured spinal cord. Double labeling is shown magnified in (B). Areas of double labeling are indicated by yellow. Negative controls, in which the primary antibody for mGluR5 is OX42, are shown (D), and lack cellular labeling observed in (A–C).
Metabotropic Glutamate Receptor 5 Stimulation Reduces Microglial-Induced Neurotoxicity
To determine whether mGluR5 plays a role in microglial-induced neurotoxicity, we cultured spinal cord microglia in transwell plates and stimulated them with LPS (100ng/ml) with or without CHPG (100μM) pretreatment. Twenty-four hours after stimulation with LPS, transwell plates were transferred into wells containing neurons. Stimulation of microglia with LPS before coculture significantly decreased the number of NeuN+ neurons at 24 hours after coculture (p < 0.05, one-way ANOVA; Fig 10). Pretreatment of microglia with CHPG resulted in a significant increase in NeuN+ cells (p < 0.05, one-way ANOVA). Microglia were washed before addition to neurons to eliminate the possibility of a direct effect of CHPG on neurons in this study. Neither LPS nor CHPG had any direct effect on neurons when added without microglia (data not shown). Addition of the selective mGluR5 antagonist MTEP (3-[(2-methyl-1,3 thiazol-4-yl) ethynyl]-pyridine) (100μM) to microglia before CHPG pretreatment reversed the protective effect of CHPG, reducing neuronal viability (p < 0.05; see Fig 10), suggesting that CHPG acts through the mGluR5 receptor.
Fig 10.
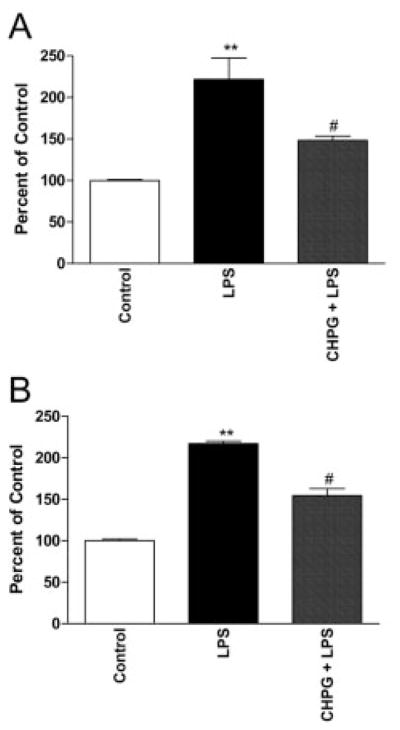
CHPG inhibits lipopolysaccharide (LPS)-induced activation in spinal cord–derived microglia. Spinal cord microglial activation was measured by proliferation (MTS assay, A) and nitric oxide production (B) at 24 hours after stimulation. Both measurements were significantly inhibited by pretreatment with the metabotropic glutamate receptor 5 (mGluR5) agonist, CHPG (100μM). Bars represent mean ± standard error of the mean. **p < 0.01 vs control; #p < 0.05 vs LPS.
Discussion
Our results provide the first evidence that mGluR5 agonists can significantly improve behavioral outcome and reduce histopathological changes after SCI. Administration of the mGluR5 agonist CHPG for 7 days after injury markedly improved motor function, beginning as early as 14 days after injury. In addition, CHPG treatment significantly reduced lesion volume and spinal cord tissue loss. Previously, CHPG treatment was reported to have neuroprotective effects, as measured by improvements in neurological function and reduced infarct volume, in a rat model of focal brain ischemia32; however, the mechanism of how CHPG mediated these effects was not explored.
A number of cells in the CNS express mGluRs, with expression profiles dependent on region. In the spinal cord, mGluR5 is expressed on neurons19,20 and astrocytes.20 In vitro, microglia have been shown to be mGluR5-positive.30 To our knowledge, this is the first demonstration that microglia in vivo express mGluR5 (see Fig 7).
mGluR5 activation inhibits caspase-dependent neuronal apoptosis in multiple cell culture models; administration of CHPG or the less selective group I agonist DHPG (s)-3,5-dihydroxyphenylglycine to rat cortical neuronal cultures significantly reduces cell death when neurons are challenged with apoptotic inducers staurosporine and etoposide.43 However, the effect of mGluR5 agonists on apoptosis in vivo has not been examined. In our SCI model, little neuronal apoptosis was found rostral or caudal to the lesion site, and CHPG treatment did not significantly reduce cleaved caspase-3 staining in neurons at 72 hours after injury (data not shown). These data suggest that inhibition of neuronal apoptosis is not a mechanism of functional recovery in this model. Furthermore, neuronal sparing in the lesion site would not be expected to have significant functional consequences in the spinal cord, because T9 level neurons do not innervate hind-limb muscles. Increased white matter sparing resulting from CHPG treatment, as shown in Figure 2, does, however, correlate with recovered hind-limb function.
Activation of mGluR5 on spinal cord–derived microglia with CHPG significantly suppresses markers of microglial activation, such as NO production and proliferation, as well as associated microglial-induced neuronal toxicity (Figs 10 and 11). Our previous work has shown similar effects in brain-derived microglia: presence of functional mGluR5 receptors that reduce NO and reactive oxygen species production, as well as neurotoxicity, in a Gαq-protein–mediated fashion that involves phospholipase C phosphorylation, hydrolysis of phosphatidyl inositol, protein kinase C activation, and calcium release.30 In vivo, our data demonstrate a significant reduction in markers of inflammation and microglial activation, including expression of ED1, galectin-3, p22phox, gp91phox, and iNOS, and the production of TNF-α. Further, galectin-3 appears to be expressed on activated microglia but not macrophages after CNS injury,40 supporting a microglial-specific influence. These in vitro and in vivo data suggest that activation of microglial mGluR5 may contribute to the protective effects of CHPG after SCI via an antiinflammatory mechanism. Although the effects on galectin-3 indicate involvement of microglia specifically, we cannot rule out possible CHPG effects on circulating macrophages. It is unclear whether macrophages express mGluR5, a question that should be addressed in the future. This report shows that spinal cord microglia express mGluR5, and that microglial-related inflammation is reduced by mGluR5 activation. Multiple studies have reported neuroprotection in vitro41,44 or an improvement in histological and functional measurements of recovery after SCI17,36,45,46 after other treatments that reduce microglial activity or inflammation.
Fig 11.
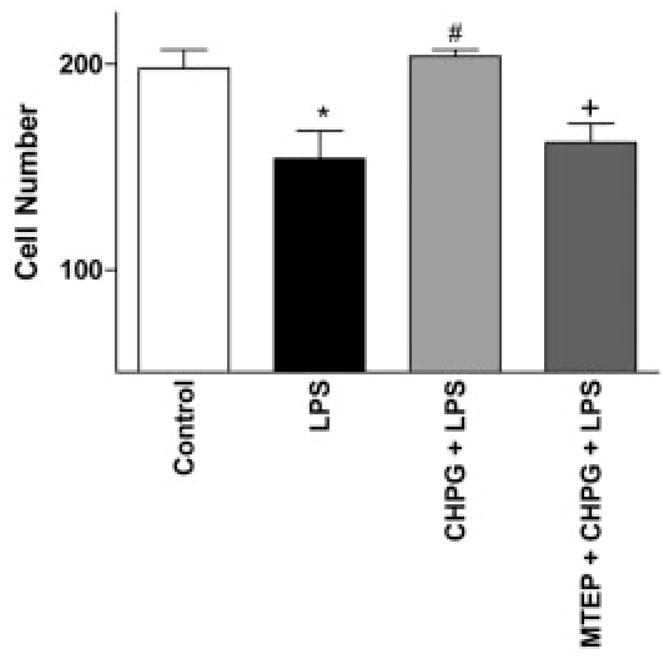
CHPG inhibits microglial-induced neurotoxicity. Neuronal number, as measured by counting neuronal nuclei–positive(NeuN+) cells 24 hours after microglia/neuron coculture. The number of NeuN+ cells was reduced by the coincubation of lipopolysaccharide (LPS)-stimulated microglia with neurons; this was reversed by the pretreatment of microglia with CHPG. Addition of the metabotropic glutamate receptor 5 (mGluR5) antagonist, MTEP (100μM), inhibited the effect of CHPG on microglial-induced neurotoxicity, demonstrating an mGluR5-mediated effect by CHPG. Bars represent mean ± standard error of the mean. *p < 0.05 vs control; #p < 0.05 vs LPS; +p < 0.05 vs CHPG + LPS.
It is important to note that the influence of microglia and macrophages after SCI is a controversial topic. For example, acute inhibition of TNF-α, as well as other markers of inflammation, is associated with functional recovery after SCI.47 Furthermore, this report, as well as others,48,49 demonstrates a restoration of function after reduction of microglial/macrophage activity. However, several groups have reported a neuroprotective effect of both microglia and macrophages.50,51 It is possible that the timing of treatment (acute vs chronic) or length of treatment determine neuroprotective and neurotoxic effects of activated microglia/macrophages. For example, during the time of axonal regeneration through the lesion site, activated microglia/macrophages have been shown to induce retraction of axons.52 Moreover, differential activation of microglia/macrophages at different time points may result in tissue loss or preservation.53 Consistent with the concept that neuroprotective effects of microglial modulation after SCI are time dependent, it has been shown that, although early inhibition of TNF improves recovery, knocking out the gene serves to impair recovery.54,55 The antiinflammatory effects demonstrated here indicate that mGluR5 activation may have multipotential neuroprotective actions. Multipotential drugs provide an attractive therapeutic option because they modulate multiple pathways involved in secondary injury. Because mGluR5 receptors are expressed on microglia, neurons, oligodendrocytes, and astrocytes, mGluR5 agonists may have multiple modulatory effects in vivo (for review, see Byrnes and colleagues56). Because both neuroinflammation and caspase-dependent neuronal apoptosis have been implicated in many acute and chronic neurodegenerative disorders,7,57,58 mGluR5 agonist therapy may have broad therapeutic relevance.
Acknowledgments
This work was supported by the NIH (National Institute of Neurological Disorders and Stroke, R01 5R01NS037313-08). (A.I.F.)
Footnotes
Potential conflict of interest: Drs. Faden and Byrnes are co-inventors on a patent application that has been filed by Georgetown University related to the technology that is described in this paper.
References
- 1.Dumont RJ, Okonkwo DO, Verma S, et al. Acute spinal cord injury, part I: pathophysiologic mechanisms. Clin Neuropharmacol. 2001;24:254–264. doi: 10.1097/00002826-200109000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Tator CH. Experimental and clinical studies of the pathophysiology and management of acute spinal cord injury. J Spinal Cord Med. 1996;19:206–214. doi: 10.1080/10790268.1996.11719436. [DOI] [PubMed] [Google Scholar]
- 3.Popovich PG, Guan Z, McGaughy V, et al. The neuropathological and behavioral consequences of intraspinal microglial/macrophage activation. J Neuropathol Exp Neurol. 2002;61:623– 633. doi: 10.1093/jnen/61.7.623. [DOI] [PubMed] [Google Scholar]
- 4.Keane RW, Davis AR, Dietrich WD. Inflammatory and apoptotic signaling after spinal cord injury. J Neurotrauma. 2006;23:335–344. doi: 10.1089/neu.2006.23.335. [DOI] [PubMed] [Google Scholar]
- 5.Hausmann ON. Post-traumatic inflammation following spinal cord injury. Spinal Cord. 2003;41:369–378. doi: 10.1038/sj.sc.3101483. [DOI] [PubMed] [Google Scholar]
- 6.Bethea JR, Castro M, Keane RW, et al. Traumatic spinal cord injury induces nuclear factor-kappaB activation. J Neurosci. 1998;18:3251–3260. doi: 10.1523/JNEUROSCI.18-09-03251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Chavarria A, Alcocer-Varela J. Is damage in central nervous system due to inflammation? Autoimmun Rev. 2004;3:251–260. doi: 10.1016/j.autrev.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 9.He Y, Imam SZ, Dong Z, et al. Role of nitric oxide in rotenone-induced nigrostriatal injury. J Neurochem. 2003;86:1338–1345. doi: 10.1046/j.1471-4159.2003.01938.x. [DOI] [PubMed] [Google Scholar]
- 10.Iravani MM, Kashefi K, Mander P, et al. Involvement of inducible nitric oxide synthase in inflammation-induced dopaminergic neurodegeneration. Neuroscience. 2002;110:49–58. doi: 10.1016/s0306-4522(01)00562-0. [DOI] [PubMed] [Google Scholar]
- 11.Jeohn GH, Kim WG, Hong JS. Time dependency of the action of nitric oxide in lipopolysaccharide-interferon-gamma-induced neuronal cell death in murine primary neuronglia co-cultures. Brain Res. 2000;880:173–177. doi: 10.1016/s0006-8993(00)02737-2. [DOI] [PubMed] [Google Scholar]
- 12.Munch G, Gasic-Milenkovic J, Dukic-Stefanovic S, et al. Microglial activation induces cell death, inhibits neurite outgrowth and causes neurite retraction of differentiated neuroblastoma cells. Exp Brain Res. 2003;150:1– 8. doi: 10.1007/s00221-003-1389-5. [DOI] [PubMed] [Google Scholar]
- 13.Zou JY, Crews FT. TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res. 2005;1034:11–24. doi: 10.1016/j.brainres.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Taylor DL, Jones F, Kubota ES, Pocock JM. Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor alpha-induced neurotoxicity in concert with microglial-derived Fas ligand. J Neurosci. 2005;25:2952–2964. doi: 10.1523/JNEUROSCI.4456-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juurlink BH, Paterson PG. Review of oxidative stress in brain and spinal cord injury: suggestions for pharmacological and nutritional management strategies. J Spinal Cord Med. 1998;21:309–334. doi: 10.1080/10790268.1998.11719540. [DOI] [PubMed] [Google Scholar]
- 16.Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma. 2000;17:871– 890. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- 17.Tian DS, Xie MJ, Yu ZY, et al. Cell cycle inhibition attenuates microglia induced inflammatory response and alleviates neuronal cell death after spinal cord injury in rats. Brain Res. 2007;1135:177–185. doi: 10.1016/j.brainres.2006.11.085. [DOI] [PubMed] [Google Scholar]
- 18.Dijkstra S, Duis S, Pans IM, et al. Intraspinal administration of an antibody against CD81 enhances functional recovery and tissue sparing after experimental spinal cord injury. Exp Neurol. 2006;202:57– 66. doi: 10.1016/j.expneurol.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 19.Walker K, Reeve A, Bowes M, et al. mGlu5 receptors and nociceptive function II. mGlu5 receptors functionally expressed on peripheral sensory neurones mediate inflammatory hyperalgesia. Neuropharmacology. 2001;40:10–19. doi: 10.1016/s0028-3908(00)00114-3. [DOI] [PubMed] [Google Scholar]
- 20.Gwak YS, Hulsebosch CE. Upregulation of Group I metabotropic glutamate receptors in neurons and astrocytes in the dorsal horn following spinal cord injury. Exp Neurol. 2005;195:236–243. doi: 10.1016/j.expneurol.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 21.Biber K, Laurie DJ, Berthele A, et al. Expression and signaling of group I metabotropic glutamate receptors in astrocytes and microglia. J Neurochem. 1999;72:1671–1680. doi: 10.1046/j.1471-4159.1999.721671.x. [DOI] [PubMed] [Google Scholar]
- 22.Taylor DL, Diemel LT, Cuzner ML, Pocock JM. Activation of group II metabotropic glutamate receptors underlies microglial reactivity and neurotoxicity following stimulation with chromogranin A, a peptide up-regulated in Alzheimer’s disease. J Neurochem. 2002;82:1179–1191. doi: 10.1046/j.1471-4159.2002.01062.x. [DOI] [PubMed] [Google Scholar]
- 23.Taylor DL, Diemel LT, Pocock JM. Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. J Neurosci. 2003;23:2150–2160. doi: 10.1523/JNEUROSCI.23-06-02150.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Movsesyan VA, Faden AI. Neuroprotective effects of selective group II mGluR activation in brain trauma and traumatic neuronal injury. J Neurotrauma. 2006;23:117–127. doi: 10.1089/neu.2006.23.117. [DOI] [PubMed] [Google Scholar]
- 25.Allen JW, Ivanova SA, Fan L, et al. Group II metabotropic glutamate receptor activation attenuates traumatic neuronal injury and improves neurological recovery after traumatic brain injury. J Pharmacol Exp Ther. 1999;290:112–120. [PubMed] [Google Scholar]
- 26.Faden AI, Ivanova SA, Yakovlev AG, Mukhin AG. Neuroprotective effects of group III mGluR in traumatic neuronal injury. J Neurotrauma. 1997;14:885– 895. doi: 10.1089/neu.1997.14.885. [DOI] [PubMed] [Google Scholar]
- 27.Vernon AC, Zbarsky V, Datla KP, et al. Selective activation of group III metabotropic glutamate receptors by L-(+)-2-amino-4-phosphonobutryic acid protects the nigrostriatal system against 6-hydroxydopamine toxicity in vivo. J Pharmacol Exp Ther. 2007;320:397– 409. doi: 10.1124/jpet.106.108159. [DOI] [PubMed] [Google Scholar]
- 28.Folbergrova J, Druga R, Otahal J, et al. Seizures induced in immature rats by homocysteic acid and the associated brain damage are prevented by group II metabotropic glutamate receptor agonist (2R,4R)-4-aminopyrrolidine-2,4-dicarboxylate. Exp Neurol. 2005;192:420– 436. doi: 10.1016/j.expneurol.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 29.Bond A, Ragumoorthy N, Monn JA, et al. LY379268, a potent and selective Group II metabotropic glutamate receptor agonist, is neuroprotective in gerbil global, but not focal, cerebral ischaemia. Neurosci Lett. 1999;273:191–194. doi: 10.1016/s0304-3940(99)00663-1. [DOI] [PubMed] [Google Scholar]
- 30.Byrnes KR, Stoica B, Loane DJ, et al. Metabotropic glutamate receptor 5 activation inhibits microglial associated inflammation and neurotoxicity. Glia. 2009;57:550–560. doi: 10.1002/glia.20783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yakovlev AG, Faden AI. Sequential expression of c-fos protoon-cogene, TNF-alpha, and dynorphin genes in spinal cord following experimental traumatic injury. Mol Chem Neuropathol. 1994;23:179–190. doi: 10.1007/BF02815410. [DOI] [PubMed] [Google Scholar]
- 32.Bao WL, Williams AJ, Faden AI, Tortella FC. Selective mGluR5 receptor antagonist or agonist provides neuroprotection in a rat model of focal cerebral ischemia. Brain Res. 2001;922:173–179. doi: 10.1016/s0006-8993(01)03062-1. [DOI] [PubMed] [Google Scholar]
- 33.Basso DM, Beattie MS, Bresnahan JC, et al. MASCIS evaluation of open field locomotor scores: effects of experience and teamwork on reliability. Multicenter Animal Spinal Cord Injury Study. J Neurotrauma. 1996;13:343–359. doi: 10.1089/neu.1996.13.343. [DOI] [PubMed] [Google Scholar]
- 34.Faden AI, Halt P. Platelet-activating factor reduces spinal cord blood flow and causes behavioral deficits after intrathecal administration in rats through a specific receptor mechanism. J Pharmacol Exp Ther. 1992;261:1064–1070. [PubMed] [Google Scholar]
- 35.Kerasidis H, Wrathall JR, Gale K. Behavioral assessment of functional deficit in rats with contusive spinal cord injury. J Neurosci Methods. 1987;20:167–179. doi: 10.1016/0165-0270(87)90048-3. [DOI] [PubMed] [Google Scholar]
- 36.Byrnes KR, Stoica BA, Fricke S, et al. Cell cycle activation contributes to post-mitotic cell death and secondary damage after spinal cord injury. Brain. 2007;130:2977–2992. doi: 10.1093/brain/awm179. [DOI] [PubMed] [Google Scholar]
- 37.Popovich PG, Wei P, Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol. 1997;377:443– 464. doi: 10.1002/(sici)1096-9861(19970120)377:3<443::aid-cne10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 38.Byrnes KR, Garay J, Di Giovanni S, et al. Expression of two temporally distinct microglia-related gene clusters after spinal cord injury. Glia. 2006;53:420– 433. doi: 10.1002/glia.20295. [DOI] [PubMed] [Google Scholar]
- 39.Mukhin AG, Ivanova SA, Allen JW, Faden AI. Mechanical injury to neuronal/glial cultures in microplates: role of NMDA receptors and pH in secondary neuronal cell death. J Neurosci Res. 1998;51:748–758. doi: 10.1002/(SICI)1097-4547(19980315)51:6<748::AID-JNR8>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 40.Lalancette-Hebert M, Gowing G, Simard A, et al. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chao CC, Molitor TW, Hu S. Neuroprotective role of IL-4 against activated microglia. J Immunol. 1993;151:1473–1481. [PubMed] [Google Scholar]
- 42.Kim WG, Mohney RP, Wilson B, et al. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci. 2000;20:6309– 6316. doi: 10.1523/JNEUROSCI.20-16-06309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allen JW, Knoblach SM, Faden AI. Activation of group I metabotropic glutamate receptors reduces neuronal apoptosis but increases necrotic cell death in vitro. Cell Death Differ. 2000;7:470– 476. doi: 10.1038/sj.cdd.4400678. [DOI] [PubMed] [Google Scholar]
- 44.Liu B, Du L, Kong LY, et al. Reduction by naloxone of lipopolysaccharide-induced neurotoxicity in mouse cortical neuron-glia co-cultures. Neuroscience. 2000;97:749–756. doi: 10.1016/s0306-4522(00)00057-9. [DOI] [PubMed] [Google Scholar]
- 45.Stirling DP, Khodarahmi K, Liu J, et al. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal die-back, and improves functional outcome after spinal cord injury. J Neurosci. 2004;24:2182–2190. doi: 10.1523/JNEUROSCI.5275-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sribnick EA, Wingrave JM, Matzelle DD, et al. Estrogen attenuated markers of inflammation and decreased lesion volume in acute spinal cord injury in rats. J Neurosci Res. 2005;82:283–293. doi: 10.1002/jnr.20622. [DOI] [PubMed] [Google Scholar]
- 47.Genovese T, Esposito E, Mazzon E, et al. Effect of cyclopentanone prostaglandin 15-deoxy-delta12,14PGJ2 on early functional recovery from experimental spinal cord injury. Shock. 2008;30:142–152. doi: 10.1097/SHK.0b013e31815dd381. [DOI] [PubMed] [Google Scholar]
- 48.Cronin M, Anderson PN, Cook JE, et al. Blocking connexin43 expression reduces inflammation and improves functional recovery after spinal cord injury. Mol Cell Neurosci. 2008;39:152–160. doi: 10.1016/j.mcn.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 49.Yune TY, Lee JY, Jung GY, et al. Minocycline alleviates death of oligodendrocytes by inhibiting pro-nerve growth factor production in microglia after spinal cord injury. J Neurosci. 2007;27:7751–7761. doi: 10.1523/JNEUROSCI.1661-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwartz M, Yoles E. Immune-based therapy for spinal cord repair: autologous macrophages and beyond. J Neurotrauma. 2006;23:360–370. doi: 10.1089/neu.2006.23.360. [DOI] [PubMed] [Google Scholar]
- 51.Yaguchi M, Ohta S, Toyama Y, et al. Functional recovery after spinal cord injury in mice through activation of microglia and dendritic cells after IL-12 administration. J Neurosci Res. 2008;86:1972–1980. doi: 10.1002/jnr.21658. [DOI] [PubMed] [Google Scholar]
- 52.Horn KP, Busch SA, Hawthorne AL, et al. Another barrier to regeneration in the CNS: activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J Neurosci. 2008;28:9330–9341. doi: 10.1523/JNEUROSCI.2488-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rolls A, Shechter R, London A, et al. Two faces of chondroitin sulfate proteoglycan in spinal cord repair: a role in microglia/macrophage activation. PLoS Med. 2008;5:e171. doi: 10.1371/journal.pmed.0050171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Farooque M, Isaksson J, Olsson Y. Improved recovery after spinal cord injury in neuronal nitric oxide synthase-deficient mice but not in TNF-alpha-deficient mice. J Neurotrauma. 2001;18:105–114. doi: 10.1089/089771501750055811. [DOI] [PubMed] [Google Scholar]
- 55.Kim GM, Xu J, Song SK, et al. Tumor necrosis factor receptor deletion reduces nuclear factor-kappaB activation, cellular inhibitor of apoptosis protein 2 expression, and functional recovery after traumatic spinal cord injury. J Neurosci. 2001;21:6617– 6625. doi: 10.1523/JNEUROSCI.21-17-06617.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Byrnes KR, Loane DJ, Faden AI. Metabotropic glutamate receptors as targets for multipotential treatment of neurological disorders. Neurotherapeutics. 2009;6:94–107. doi: 10.1016/j.nurt.2008.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tansey MG, McCoy MK, Frank-Cannon TC. Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol. 2007;208:1–25. doi: 10.1016/j.expneurol.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yakovlev AG, Faden AI. Caspase-dependent apoptotic pathways in CNS injury. Mol Neurobiol. 2001;24:131–144. doi: 10.1385/MN:24:1-3:131. [DOI] [PubMed] [Google Scholar]