

Abstract
CD8 engagement is believed to be a critical event in the activation of naive T cells. In this communication, we address the effects of peptide-MHC (pMHC)/TCR affinity on the necessity of CD8 engagement in T cell activation of primary naive cells. Using two peptides with different measured avidities for the same pMHC-TCR complex, we compared biochemical affinity of pMHC/TCR and the cell surface binding avidity of pMHC/TCR with and without CD8 engagement. We compared early signaling events and later functional activity of naive T cells in the same manner. Although early signaling events are altered, we find that high-affinity pMHC/TCR interactions can overcome the need for CD8 engagement for proliferation and CTL function. An integrated signal over time allows T cell activation with a high-affinity ligand in the absence of CD8 engagement.
During T cell interactions with APCs, CD8 and CD4 co-receptors bind to MHC class I and II, respectively (1). Although the role of CD4 coreceptor in stabilization of peptide-MHC and TCR complexes (pMHC3/TCR) and activation of naive CD4+ T cells has been well characterized (2–10), less work has been done on CD8 coreceptor requirements. Studies using surface plasmon resonance (SPR) have yielded contradictory results (11, 12). Some TCRs bind class I MHC despite the presence of anti-CD8 Abs or mutations in the CD8 binding site of MHC (13–17). However, these studies do not examine the effects of CD8 engagement on downstream T cell activation. Work done in human T cell lines suggests that although CD8 engagement is not necessary for pMHC binding to TCR, full activation of the T cell does not occur in the absence of CD8 ligation (18). Other studies suggest that CD8 may be necessary for T cell activation events, such as phosphorylation of TCR signaling molecules, cytokine production, and CTL activity (19–22), as well as positive selection in the thymus (23).
CD8 coreceptors may promote T cell activation in two distinct ways: extracellular stabilization of pMHC-TCR interactions and activation of intracellular signaling cascades (21, 24, 25). The co-receptors might stabilize pMHC/TCR binding to allow sufficient time for pMHC-TCR interaction (21, 24). Alternatively, CD8 may promote T cell activation through association with signaling molecules such as p56Lck, thus bringing them into proximity of TCR-associated signaling molecules (21, 25). Recent studies suggest that CD8 may be preassociated with TCR in T cell lines and that TCR may contain a CD8-binding motif (26, 27). It has been proposed that affinity of the pMHC for TCR dictates the amount of a T cell response (28), although others have argued for an optimum affinity (29, 30). However, little work has been reported to directly address the effect of pMHC/TCR binding affinity along with the requirement of CD8 engagement in functional T cell activation events. Recent work by our laboratory and others using T cell clones indicate that affinity may determine the necessity of CD8 engagement in T cell activation (31, 32). Unlike these articles, the data in this communication describe the requirements for CD8 engagement throughout early signaling and later functional stages of T cell activation in primary, naive T cells.
MHC tetramer studies provide a system to examine specific pMHC-TCR interactions in the absence of costimulatory molecules. Tetramers can be used to stain T cells (6, 13, 33) and elicit T cell responses similar to those elicited by Ag-containing APCs (34–36). We have used this system to study the requirement of CD8 engagement in pMHC/TCR stabilization and CD8+ T cell activation using pMHC-TCR interactions of known affinities.
To uncouple CD8 binding to MHC, we produced pMHC tetramers containing the Db D227K mutation, which has been shown to abrogate CD8 interaction with MHC (18, 37–40). These mutated pMHC tetramers were compared with wild-type Db tetramers to study pMHC/TCR binding, early signaling events, T cell proliferation, cytokine production, and CTL activity in the lymphocytic choriomeningitis virus (LCMV) TCR-transgenic (P14) mouse specific for the wild-type KAVYNFATC epitope of LCMV gp33 (41). This mouse has been used extensively to study CD8+ T cell activation (28, 41–44). Experiments were conducted using the gp33-altered peptide ligand KAVYNFATM (C9M), which we show here has higher affinity binding for TCR. In this study, we show that CD8 greatly stabilizes pMHC-TCR interactions on the cell surface of both moderate (gp33) and high (C9M)-affinity ligands. CD8 coreceptor engagement is required during moderate affinity pMHC-TCR interactions to provide optimal T cell activation. However, higher pMHC/TCR affinity ligands do not require CD8 engagement for development of CD8+ T cell effector functions. These high-affinity interactions allow a level of signaling that accumulates over time, thus allowing T cell activation without CD8 engagement.
Materials and Methods
Mice and cell preparation
P14 (B6; D2-TgN (TCR-Lcmv) 327 Sdz) mice expressing the H2Db-restricted P14 TCR specific for the gp33 epitope (aa 33–41) of LCMV were purchased from The Jackson Laboratory (Bar Harbor, ME), backcrossed an additional six times to C57BL/6 mice (purchased from The Jackson Laboratory or Charles River (Raleigh, NC)) and then intercrossed. Mice were kept under specific pathogen-free conditions at the University of North Carolina at Chapel Hill Department of Laboratory Animal Medicine facilities. We purified CD8+ P14 T cells from single-cell splenocyte preparations by negative selection using MACS anti-CD4 and anti-MHC class II microbeads (Miltenyi Biotec, Auburn, CA) as described previously (35). Anti-NK microbeads were used in addition to deplete NK cells in signaling experiments. Experiments were conducted following the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee guidelines.
Tetramer mutation
H2Db cDNA (modified to include a BirA site (35)) was altered using a Quick Change site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer’s protocol. Mutagenesis primers (forward, GGAGGAGCTGACCCAGAAAATGGAGCTTGTGGAGACC and reverse, GGTCTCCACAAGCTCCATTTTCTGGGTCAGCTCCTCC) introduced two nucleotide changes resulting in a single amino acid change of residue 227 in the α3 domain of the monomer. The resulting clones were sequenced at the University of North Carolina Automated DNA Sequencing Facility and analyzed by GCG Wisconsin Package software (Accelrys, San Diego, CA) to confirm the proper mutation and the absence of other unintended changes.
Peptides and tetramer preparation
gp33 (KAVYNFATC), C9M (KAVYNFATM), HY (KCSRNRQYL), and influenza (ASNENMETM) peptides were synthesized at the University of North Carolina Peptide Synthesis Facility. Peptides were purified using HPLC, and purity was confirmed by mass spectroscopy. Tetramers were produced as described elsewhere (35). Streptavidin-PE (Leinco, St. Louis, MO) was used to label tetramers unless otherwise indicated. To ensure optimal tetramer formation, streptavidin was added to pMHC monomer at a 1:6 ratio. Quality control of tetramers was assured by gel shift analysis and staining of cells by flow cytometry.
CD8 binding assay
CD8α and/or β chains were expressed in COS-7 cells, and tetramer binding was analyzed as described previously (45).
Surface plasmon resonance (SPR)
P14 TCR was produced as previously described (46). Approximately 5000 response units of H57-597 (monoclonal TCR Ab against Cβ) were covalently bound to SPR sensor chip CM5 (Biacore, Uppsala, Sweden) using standard amine coupling. Soluble P14 TCR (ligand) was then added to the Ab at a concentration of 0.67 μM (equal to 300–500 response units). Soluble class I pMHC (analyte) was injected onto the surface at a flow rate of 100 μl/min in a 30-s pulse. TCR and pMHC were removed from the surface with 0.1 M glycine and 0.5 M NaCl, and the procedure was repeated until at least two curves were obtained for the different concentrations of analyte. Curves obtained at each concentration were subtracted from a reference surface of Ab alone. After background subtraction, curves were smoothed by deletion of aberrant data points (injection spikes) and imported into BIAevaluation 3.0 (Biacore). Following normalization along the x- and y-axes, the curves were fit globally for affinity and kinetics. All data were used in fitting except for 1 s before the start of dissociation. Scatchard plots were calculated (see below) from the average of equilibrium binding (Req) for at least two curves of each concentration.
Analysis of tetramer binding to living T cells
Splenocytes were stained with anti-CD8α mAb (clone 53.6.7; BD Phar-Mingen, San Diego, CA) and increasing concentrations of PE-conjugated tetramer for 1 h at 4°C, washed twice with FACS buffer, and staining was measured by FACS. Median fluorescence intensity of tetramer staining was calculated using Summit software (Cytomation, Fort Collins, CO), and background fluorescence (from tetramer containing irrelevant influenza peptide) was subtracted to determine mean fluorescence intensity values of specific tetramer staining. Data were normalized to the saturation concentration of the Db/gp33 tetramer. Mean channel shift is expressed as fluorescence units (FU). Bound (FU) vs bound/free (FU/nM) data were plotted in a standard Scatchard analysis (47). As in Ref. 47, we assumed that tetramer is in excess, so that the amount of free tetramer is equal to the amount of tetramer added. We estimate that at the lowest tetramer concentration tested the amount of bound tetramer is <1 in 105 molecules, thus this simplification is reasonable. The apparent dissociation constant (KD) was calculated from slope of line (KD = −1/slope) of the Scatchard plot. The median KD value from multiple Scatchard analyses is shown with the SEM. The CD8α mAb (53.6.7) has been reported to enhance tetramer binding (20, 21). We confirm this, but note an increase of <10% in mean channel fluorescence, which has no effect on subsequent analysis.
Detection of signaling events
For tyrosine phosphorylation (p-tyr) immunoblots, CD8+ cells were purified from LCMV TCR-transgenic (tg) mice as described above. Five × 106 cells/sample were prewarmed in a 37°C water bath for 10 min before addition of tetramer. After stimulation with 10 μg of tetramer for 3 min at 37°C, the cells were pelleted and lysed in 10 mM Tris buffer (pH 7.5) with 1% Nonidet P-40, 150 mM NaCl, 2 mM sodium o-vanadate, 1 mM PMSF, 0.4 mM EDTA, 10 mM NaF, and protease inhibitors (1 mg/ml aprotinin, leupeptin, and α1-anti-trypsin). After incubation at 4°C for 10 min, nuclear and cytoskeletal components were removed by centrifugation. Supernatants were run on an SDS-PAGE gel, transferred to polyvinylidene difluoride membrane, and probed with AB-2 anti-phosphotyrosine Ab (Oncogene Research Products, San Diego, CA). Anti-mouse IgG conjugated to HRP (Sigma-Aldrich, St. Louis, MO) and ECL Plus reagent (Amersham International, Buckinghamshire, U.K.) were used for detection. ECL was visualized and bands were quantitated on a Storm 860 phosphor imager (Amersham Biosciences, Piscataway, NJ), as recommended by the manufacturer.
Ca2+ mobilization was measured in purified CD8+ LCMV TCR tg splenocytes as described previously (48). Cells were loaded with Indo1-AM and resuspended at 2 × 105/ml. After a baseline level of calcium was recorded, tetramer was added to each sample and measured for calcium mobilization based on the ratio of fluorescence at 405 and 485 nm for a minimum of 8 min. Data analysis was performed using FlowJo software (Tree Star, San Carlos, CA). Ca2+ levels were estimated using a standard curve based on fluorescence of known calcium concentrations.
Measurement of p56Lck kinase activity was performed by immunopre-cipitating p56Lck and assaying kinase activity on a peptide substrate as previously described (31).
Confocal microscopy
Two × 105 purified CD8+ P14 splenocytes in 100 μl of 1× PBS were allowed to adhere to electrostatically charged slides (Fisher Scientific, Pittsburgh, PA) and then blocked with 100 μl of 10% FCS in PBS for 30 min on ice. Cells were then washed three times with 250 μl of wash buffer (PBS with 0.5% FCS (w/v)). The third wash remained on the cells until stimulation with tetramer and/or Abs. Alexa 568-conjugated tetramer (Molecular Probes, Eugene, OR) was centrifuged for 10 min in an airfuge (Beckman, Fullerton, CA) to remove aggregates. Wash solution was aspirated from each sample before 10 μg of tetramer and/or 1.5 μg of Ab (as indicated) in a 100-μl total volume were added to the cells for 5 min. Samples remained on ice and were protected from light throughout staining. Cells were washed three times with 250 μl of wash buffer and then fixed (1× PBS with 3% sucrose, 0.02% azide, and 3% paraformaldehyde). After 10 min at room temperature, the fixation buffer was aspirated off the slide and SlowFade mounting medium with glycerol (Molecular Probes) was added to the samples and secured with a 1.5 glass coverslip (Fisher Scientific). Coverslips were sealed with clear nail polish to prevent drying, and the prepared slides were stored at 4°C for up to 48 h until examination with a digital deconvolusion microscope (Intellegent Imaging Innovations (3I), Denver, CO). Images were collected and analyzed using Slidebook software (3I). Multiple photographs of cells through the z-plane were collected (z-stack). Images were deconvolved under the most stringent conditions (constrained iterative) for all analyses. Colocalization calculations were performed by the creation of a two-dimensional projection image of the z-stack, with subsequent analysis of pixels containing tetramer (Cy3) and/or CD8 (FITC). Percent colocalization was calculated as follows: (the number of pixels containing FITC and Cy3/number of pixels containing either FITC or Cy3) × 100. Calculations were based on multiple images from independent experiments for each tetramer.
Fluorescence resonance energy transfer (FRET)
Splenocytes from P14 mice were harvested, made into a single-cell suspension, and RBCs were lysed as described elsewhere (35). Cells were processed in FRET buffer (1× PBS containing 2% FCS) and kept at 4°C until just before analysis. H57-597 Ab (anti-Cβ TCR) was purified from hybridoma supernatant and conjugated to tetramethylrhodamine isothiocyanate (TRITC) fluorophore. Cells were stained with FITC-labeled anti-CD8α and/or TRITC-labeled anti-TCR Ab as indicated. FITC-labeled anti-CD2 or anti-CD62 ligand (BD PharMingen) were used as negative controls. After staining 3 × 106 cells on ice for 20 min, cells were washed once with FRET buffer and kept on ice until analysis on a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). Each sample was shielded from light and prewarmed to room temperature 10 min before analysis. Twenty nanomolar unlabeled tetramer or TRITC-H57 Ab was added after a 1-min baseline was established. FRET was detected by excitation using a single 488-nm laser and monitoring TRITC signal at 575 nm. All samples were analyzed over a 6-min time period. In all samples where FRET was apparent in the TRITC channel, a corresponding decrease in FITC channel fluorescence was observed, indicating energy transfer. Data are plotted as TRITC FU over time. Data were collected using CellQuest software (BD Biosciences) and analyzed using FloJo software (Tree Star). Negative controls and acceptor or donor fluorophore-stained cells were used to establish a baseline, above which was considered a positive FRET signal (reviewed in Ref. 49). Statistics were calculated by determining the mean fluores-cence of 20 regions (17 s each) for each sample, then correcting for sample photobleaching. Photobleaching was determined by measurement of decreased fluorescence in CD8 FITC alone or, for samples with tetramer, CD8 FITC plus TCR TRITC with added Db/influenza (flu). Statistics were calculated by comparison of the first and last five regions in a sample through Student’s t test. Levels of FITC-conjugated anti-CD2 and anti-CD62 ligand, which were used a negative controls, were sufficient to induce FRET if in proximity to TCR. In samples where tetramer was used, both fluorophores were already bound to the cell before addition of unlabeled tetramer, thus any change seen is a function of tetramer engagement.
Proliferation assays
Purified CD8+ P14 T cells at 1 × 105/well were incubated in medium (RPMI 1640 with 10% FCS and antibiotics) with varying tetramer concentrations at a total volume of 200 μl. Cells were cultured at 37°C with 5% CO2 for 24 – 48 h, then pulsed with [3H]thymidine (1 μCi/well) for 6–18 h. In some assays, the tetramer was removed at the indicated time points by centrifugation of the cells and removal of the supernatants. Fresh medium was added and the cells were pelleted again for a total of two washes before further incubation for the remainder of the assay. We estimated that this resulted in a 1000-fold dilution of unbound tetramer. Samples were harvested using a multiple sample harvester (Otto Hiller, Madison WI) and [3H]thymidine incorporation was measured using a Beckman LS5000 counter. All measurements were performed in triplicate and the mean [3H]thymidine incorporation was determined.
For CFSE staining, P14 splenocytes were harvested and labeled with CFSE as previously described (50). Approximately 2 × 107 CFSE-labeled P14 cells were injected i.p. into recipient C57BL/6J mice. After 24 h, 20 μg of Db/gp33, Db D227K/gp33, Db/C9M, or Db D227K/C9M tetramer in 1× PBS was injected i.p. into each mouse. Control mice received Db/ irrelevant tetramer or none at all. Mice were sacrificed at 48 h following tetramer injection, and splenocytes were harvested and stained with anti-P14 TCR (anti-Vβ2 PE clone B20.1; BD PharMingen). Data were collected using Summit software and analyzed using Modfit (BD Biosciences). Statistical analyses for all data were conducted using Student’s t test.
Measurement of cytokines
Supernatants from tetramer-stimulated purified CD8+ P14 cells (as described above) were assayed using the Mouse Th1/Th2 Cytokine Cytometric Bead Array kit (BD PharMingen) according to the manufacturer’s protocol and analyzed with a FACSCalibur flow cytometer (BD PharMingen). The mean value of multiple sample dilutions was calculated, and data were normalized to cytokine production by Db/gp33-stimulated cells.
CTL assays
Purified CD8+ P14 T cells at 1 × 105/well were stimulated with a concentration of tetramer which was predetermined to result in maximum proliferation. After 48 h, the cells were pooled, washed, and used as effector cells. EL4 cells were labeled with 51Cr, then pulsed with 10 μM of either gp33 or irrelevant (HY) peptide and used as targets in a standard CTL assay (previously described in Ref. 35). Samples were assayed in triplicate and the mean 51Cr release was plotted.
Results
Db D227K mutation of the MHC α3 domain abrogates binding of MHC to CD8
pMHC tetramers allow analysis of T cell responses without additional costimulatory molecules (35). However, class I pMHC binds to CD8 coreceptor as well as TCR (21). To determine the relative contribution of CD8 in naive T cell activation, we produced pMHC tetramers containing a charge reversal mutation of aspartic acid to lysine at position 227 of the α3 domain (Db D227K), which has been shown to abrogate CD8 interaction with MHC (18, 37–39). To confirm uncoupling of CD8 engagement, we stained CD8-expressing COS-7 cells with Db/C9M or Db D227K/C9M (Fig. 1A). Since COS-7 cells do not express TCR, binding is mediated only through CD8. Cells expressing CD8αα, or predominantly CD8αβ dimers, bound Db/C9M but not Db D227K/C9M, indicating that CD8 binding is indeed abrogated by the mutation.
FIGURE 1.

D227K mutation abrogates CD8 binding, but does not abrogate binding of MHC and TCR. A, Db/C9M and Db D227K/C9M tetramers were used to stain untransfected COS-7 cells or COS-7 cells transfected with CD8 α- and/or β-chains. Data are representative of two independent experiments. B, Monomers containing either gp33 or C9M peptides were measured for their ability to bind P14 TCR by SPR. Data are the average of two independent experiments. C and D, The ability of tetramers to bind live cells was measured by Scatchard analysis. The binding data were fitted to a standard Scatchard plot (C) and the dissociation constant (Kd) was calculated by the slope of the line (D). Kd values for Db/C9M and Db D227K/C9M differ significantly (p ≤ 0.05), as do Db/gp33 and Db D227K/gp33 (p ≤ 0.005).
C9M peptide confers a higher affinity MHC-TCR interaction than gp33
To confirm previous functional data that suggested the C9M altered peptide ligand caused a higher affinity MHC-TCR interaction than the wild-type gp33 peptide (51), SPR was performed using monomers containing both of these peptides. Fig. 1B shows the results of a Scatchard analysis of monomer binding to soluble P14 TCR. As illustrated, Db/C9M binds tighter to TCR then Db/gp33, with KD values of 17 ± 1.3 μM and 45 ± 21 μM, respectively. Therefore, although subsequent experiments use tetrameric MHC complexes, we will refer to Db/C9M as having higher binding affinity for the remainder of the communication.
Db D227K mutation does not abrogate TCR binding of pMHC
It has been previously shown that other mutations in the CD8 binding site of MHC do not affect the ability of pMHC to bind TCR (52). However, to ensure that this was true specifically for the D227K mutation, monomeric Db/C9M or Db D227K/C9M were used in SPR experiments to assay binding to P14 TCR (Fig. 1B). Both Db/C9M and Db D227K/C9M monomer bind similarly to P14 TCR, with KD values of 17 ± 1.3 μM and 19 ± 5 μM, respectively. The KD measurements for Db/C9M are in reasonable agreement with the SPR value of 6 μM previously reported (46). This experiment clearly indicates that the Db D227K mutation does not alter the interaction of pMHC and TCR.
CD8 binding to MHC strengthens the avidity of pMHC/TCR on live cells
Although SPR measures the affinity of pMHC monomer to TCR, it does not account for multiple or serial engagements of pMHC/ TCR or the effects of membrane redistribution that bring these molecules into closer proximity, both of which contribute to pMHC/TCR avidity on live cells. Also, SPR cannot easily estimate the multivalent binding of pMHC/TCR with the CD8 coreceptor. Binding to the cell surface reflects more realistically the effects of pMHC/TCR binding. Therefore, we measured tetramer binding to live CD8+ P14 T cells by flow cytometry (FACS) (47). Plots and apparent KD values from this Scatchard analysis are shown in Fig. 1, C and D, respectively. The apparent avidities are ~4- to 14-fold lower without CD8 engagement. This analysis allows us to rank the avidities of these pMHC complexes: Db/C9M > Db/gp33 > Db D227K/C9M ≈ Db D227K/gp33. These data confirm previous results from our laboratory that ~5- to 10-fold more gp33 peptide is required for equivalent lysis of Db targets by P14 CTL than C9M (51). The 1000-fold difference between the SPR affinity measurements of Db/C9M (micromolar) (Fig. 1B) and the avidity on live cells (nanomolar) is accounted for by the differences in avidity between monomeric and tetrameric ligands, as well as potential binding cooperativity of multiple TCR that cannot occur with SPR. Thus, while the D227K mutation has no effect on biochemical affinity, it produces a profound effect on avidity when binding is measured on CD8+ T cells (Fig. 1D).
CD8 engagement increases the ability of CD8+ T cells to proliferate, secrete cytokines, and kill target cells
We wanted to determine the contribution of CD8 engagement to naive CD8+ T cell activation. Upon initial activation, naive CD8+ T cells divide rapidly, thus clonally expanding the reactive T cell population. The ability of Db and Db D227K tetramers to induce proliferation was measured in vitro. Purified P14 CD8+ T cells were stimulated with increasing concentrations of Db/gp33, Db D227K/gp33, or irrelevant Db/influenza peptide, and [3H]thymidine incorporation was measured. Fig. 2A shows that Db/gp33-stimulated cells induce a greater in vitro proliferative response than Db D227K/gp33-stimulated cells. Since in vitro assays cannot replicate the microenvironment of cytokines and other factors that naive T cells experience in vivo, we devised an in vivo proliferation assay using CFSE labeling (50). CFSE-labeled P14 splenocytes were transferred into B6 mice. After 24 h, Db or Db D227K tetramers were injected and splenocytes were stained at 48 h postinjection with anti-TCR and anti-CD8 Abs. CFSE-labeled P14 cells proliferate more in response to Db/gp33 tetramer than Db D227K/gp33 tetramer (Fig. 2B), thus, both in vivo and in vitro, CD8 engagement results in increased T cell proliferation. In both assays, Db D227K/gp33 is more effective than the tetramer containing irrelevant peptide, suggesting only a change in the efficacy of the proliferative response.
FIGURE 2.

CD8 engagement increases CD8+ T cell proliferation, IFN-γ and TNF-α production, and CTL activity. A, CD8+ P14 splenocytes were stimulated with Db/gp33 (●) and Db D227K/gp33 (○) or control (x) tetramers, then measured for their ability to proliferate in vitro. Each concentration was assayed in triplicate. All error bars are ±SEM. Data are representative of five independent experiments. B, In vivo stimulation of splenocytes and proliferation based upon CFSE staining. The percentage of divided cells was calculated using Modfit software. Proliferation by Db D227K/gp33-stimulated CD8+ T cells is significantly decreased in vivo (*, p ≤ 0.05) compared with cells stimulated with Db/gp33 tetramers. Each group contained four mice and the data are representative of two independent experiments. C, Production of IFN-γ and TNF-α by in vitro-stimulated CD8+ T cells. IFN-γ and TNF-α production were significantly decreased (***, p ≤ 0.001 and **, p ≤ 0.01) without CD8 engagement compared with stimulation with wild-type tetramer. Each sample was assayed in triplicate by a cytometric bead array and normalized to the amount of cytokine induced by Db/gp33. Data are representative of two independent experiments. D, CTL activity of tetramer-stimulated P14 cells. Target cells were pulsed with relevant (●, ○) and irrelevant peptide (▲,△). Db D227K/gp33-stimulated cells are shown with open symbols, while Db/gp33-stimulated cells are shown with filled symbols. Triplicate wells at each E:T ratio were averaged. Data are representative of two independent experiments.
Although proliferation is a good measurement of naive CD8+ T cell activation, it does not clearly indicate downstream effector function. Cells may proliferate in response to both agonist and antagonist ligands, yet only gain full effector cell function when exposed to agonist peptide (53, 54). Naive T cells can also undergo homeostatic proliferation without becoming effector cells (55, 56). Cytokine production is an important effector function of CD8+ T cells. After 2 days of culture, supernatants from Db/gp33 or Db D227K/gp33-stimulated purified CD8+ T cells were assayed for IL-2, IL-4, IL-5, TNF-α, and IFN-γ. Both IFN-γ and TNF-α production were significantly decreased in Db D227K/gp33-stimulated cells as compared with Db/gp33 (Fig. 2C; p < 0.001 and p < 0.01, respectively). No significant differences were seen in IL-2, IL-4, or IL-5 production (data not shown), although IL-2 production was low and IL-4 and IL-5 were undetectable. These data indicate that CD8 engagement is required for optimum production of IFN-γ and TNF-α in activated CD8+ T cells.
A major effector function of CD8+ T cells is cell-mediated cytotoxicity. To determine whether CD8 engagement is necessary for generation of CTL activity, purified P14 CD8+ splenocytes were stimulated with Db/gp33 or Db D227K/gp33 and measured for their ability to lyse specific target cells (Fig. 2D). Db/gp33 tetramer-stimulated cells elicit markedly greater specific lysis than cells stimulated with Db D227K/gp33. Effector cells do not lyse targets pulsed with irrelevant peptide. Thus, CD8 engagement provides a much more effective signal than TCR alone for production of CTL activity.
Higher affinity pMHC-TCR interaction largely compensates for lack of CD8 engagement
We wanted to next determine whether higher affinity of pMHC/ TCR altered the need for CD8 engagement. We used Db/C9M and Db D227K/C9M tetramers in functional T cell activation experiments to address this question. Db/C9M binds CD8+ P14 T cells with ~4-fold higher apparent avidity than Db/gp33 (Fig. 1D). Indeed, the titration of Db/C9M tetramer concentrations to optimally stimulate cells is 10-fold less than concentrations needed for Db/ gp33-stimulated cells (Figs. 2A and 3A). This is approximately the same ratio as peptide sensitization for P14 CTL lysis (51). Db/C9M and Db D227K/C9M were used to stimulate proliferation, cytokine production, and CTL activity of naive purified P14 CD8+ T cells. All experiments were performed alongside Db/gp33 and Db D227K/gp33 tetramers to allow for subsequent comparison. Cells stimulated with Db/C9M or Db D227K/C9M tetramers proliferate equally, both in vitro and in vivo (Fig. 3, A and B). In contrast, cells cultured with Db D227K/gp33 proliferate poorly compared with Db/gp33 (Fig. 2, A and B). Despite equal proliferative activity, IFN-γ levels remained significantly decreased in Db D227K/ C9M-stimulated cells as compared with Db/C9M ( p < 0.001; Fig. 3C). Unlike Db/gp33 and Db D227K/gp33 stimulation, no significant differences in TNF-α production were measured in Db D227K/C9M-stimulated cells. Similar to Db/gp33 and Db D227K/ gp33, IL-2 production was low and IL-4 and IL-5 were undetectable (data not shown). Finally, CTL function was equal in Db/C9M and Db D227K/C9M-stimulated cells (Fig. 3D). This was in contrast to Db D227K/gp33-cultured cells, which have reduced killing ability compared with Db/gp33-stimulated cells (Fig. 2D). Collectively, these data indicate that higher affinity of pMHC/TCR can compensate for the absence of CD8 in proliferation and CTL activity, but not IFN-γ production.
FIGURE 3.
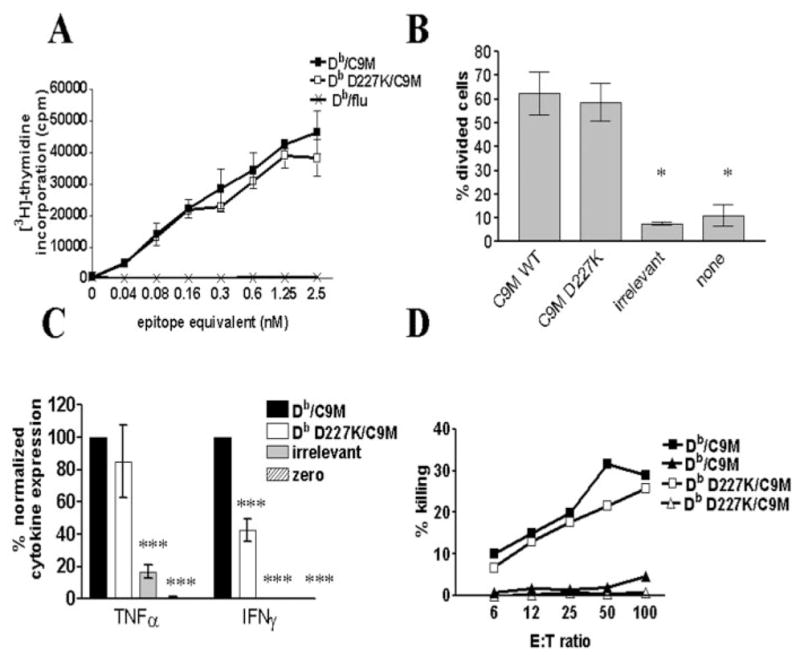
Higher affinity MHC-TCR interaction overcomes need for CD8 engagement. A, CD8+ P14 splenocytes were stimulated with Db/C9M (■), Db D227K/C9M (□), or control (×) tetramers, then measured for their ability to proliferate in vitro. Samples were assayed in triplicate, and the average was plotted with the error bars indicating ±SEM. Data are representative of five independent experiments. B, Measurement of in vivo proliferative ability of cells stimulated with Db/C9M or Db D227K/C9M. Db D227K/C9M-stimulated CD8+ T cells illustrate similar proliferative ability, as compared with cells stimulated with Db/C9M tetramers (*, p ≤ 0.05). In vivo proliferation was performed using four mice per group, the percentage of divided cells was calculated as in Fig. 2, and the data are representative of two independent experiments. C, IFN-γ production by Db D227K/C9M-stimulated cells is significantly lower as compared with Db/C9M stimulation (***, p ≤ 0.001). Each sample was assayed in triplicate and the amount of cytokines produced was normalized to cytokine levels produced by Db/gp33 stimulation. Data are representative of two independent experiments. D, CTL activity of tetramer-stimulated P14 cells. Target cells were pulsed with relevant (■,□) and irrelevant peptide (▲,△). Db D227K/C9M-stimulated cells are shown with open symbols, while Db/C9M-stimulated cells are shown with filled symbols. The average of three replicates per ratio is plotted, and data are representative of three independent experiments.
CD8 engagement induces optimal TCR signaling
The functional data above showed the impact of CD8 engagement in naive CD8+ T cell activation based upon the affinity of the pMHC-TCR interaction. These differences were apparent at the earliest functional activity measurement of proliferation. Therefore, we decided to investigate earlier signaling events to determine the contribution of CD8 ligation in naive CD8+ T cell activation. We examined the p-tyr patterns of naive T cells following activation with either Db or Db D227K/gp33 and C9M tetramers for 3 min at 37°C. As shown in Fig. 4A, stimulation with Db D227K tetramers containing either gp33 or C9M peptides (lanes 2 and 4, respectively) produces an altered p-tyr pattern as compared with cells induced with Db/gp33 or C9M tetramer (lanes 1 and 3, respectively). Indeed, at least two p-tyr bands were almost completely missing in Db D227K-stimulated cell lysates, which was consistently observed in three independent experiments. This agrees with a previous report using a human T cell clone (18). New bands are not apparent. Using a phosphor imager, we quantitated the apparent decrease in band intensity (Fig. 4B). Cells stimulated with Db D227K tetramers illustrate a greater than 100-fold decrease in bands #1 and #2 as compared with cells stimulated with wild-type Db tetramers. Quantitation of band 3 illustrates similar protein loading of all samples. Decreased bands in Db D227K/ C9M-stimulated cells suggest a disconnect between early signaling events and downstream functional activity, as CTL activity in Db D227K/C9M-stimulated cells is comparable to Db/C9M-stimulated cells.
FIGURE 4.

CD8 engagement with MHC/ TCR induces altered signaling patterns, as compared with MHC/TCR alone. A, CD8 P14 splenocytes were stimulated for 3 min with tetramers at 37°C, and phosphotyrosine activity was detected by Western blot. Cells were unstimulated (US) or stimulated with Db/gp33 (lane 1), Db D227K/gp33 (lane 2), Db/C9M (lane 3), or Db D227K/C9M (lane 4). Data are representative of three independent experiments. B, Quantitation of band intensity of tetramer-stimulated cells. To measure the intensity of the bands, pixel volume of identical areas of the gel were determined for each lane and corrected for local area background. Two bands missing in the Db D227K stimulated and one control band are plotted. C, Calcium mobilization of cells treated with indicated tetramers. Data are representative of two independent experiments.
We measured intracellular calcium flux as another indicator of early signaling events. As shown in Fig. 4C, Db/gp33 and C9M tetramers were able to quickly induce Ca2+ mobilization. Neither Db D227K/gp33 nor Db D227K/C9M tetramers were able to induce Ca2+ mobilization in naive CD8+ P14 T cells. This lack of response is equivalent to that of tetramer containing irrelevant peptide and correlates with the lack of p-tyr signals. These experiments indicate that both pMHC-TCR interaction along with CD8 ligation are necessary to induce optimal early signaling events.
CD8 is present in lipid rafts without engagement by MHC
CD8 is associated with p56Lck, which is an early signaling molecule critical for CD8+ T cell activation (24, 25). Upon pMHC/ TCR binding, p56Lck phosphorylates ZAP-70, then both of these molecules phosphorylate immunoreceptor tyrosine-based activation motifs on the TCR, leading to an activation signaling cascade. We hypothesized that CD8 may be in close proximity to TCR, even in the absence of CD8 ligation by MHC. This might explain the T cell effector function that we observed in the absence of CD8 ligation. Indeed, a recent article by Cawthon and Alexander-Miller (26) showed that CD8 could be preassociated with TCR in lipid rafts. To examine this possibility, we performed confocal microscopy to examine colocalization of CD8 and Db or Db D227K tetramer on the cell surface. As shown in Fig. 5A, CD8 and TCR colocalize in foci, irrespective of whether the MHC binds directly to CD8. Indeed, the average calculated percent colocalization was >50% and was similar in both Db/C9M and Db D227K/C9M-stained cells ( p = 0.25; Fig. 5B). Colocalization between TCR and CD8 with Db/C9M staining is expected, since Db/C9M can bind both TCR and CD8. Thus, the colocalization observed cannot be attributed to random circumstance. Lack of significant differences in colocalization between cells stained with Db/C9M or Db D227K/C9M tetramer indicates that CD8 is indeed in close proximity to TCR, even when CD8 is not bound by MHC. Staining is specific, as Db/irrelevant tetramer does not stain the cells (Fig. 5A). Similar staining was seen using Db/gp33 and Db D227K/gp33 (data not shown). We believe these foci to be signaling synapses, because they contain GM1, a component of lipid rafts (our unpublished data and Refs. 57 and 58).
FIGURE 5.

CD8 is present in mi-crodomains without engagement by MHC. A, CD8+ P14 splenocytes were stained with anti-CD8α and Db/C9M (top), Db D227K/C9M (middle), or Db tetramer containing an irrelevant peptide (Db/flu, bottom). Tetramer staining is shown in red, CD8 staining in green, and colocalization of tetramer and CD8 in yellow. CD8 colocalizes with tetramer in discrete patches (foci, arrows) with and without tetramer engagement of CD8. Data are representative of at least three independent experiments. B, Calculated colocalization of CD8 and tetramer.
To confirm proximity of CD8 to pMHC/TCR without direct engagement by MHC, we performed experiments using FRET, as detected by flow cytometry. FRET flow cytometry has been often used to detect distances between molecules and has been used successfully to determine CD4 interactions with TCR (59–62). Fig. 6A illustrates that the rhodamine (TRITC) signal is not apparent by FITC excitation with the 488-nm laser (red line), and that the presence of anti-CD2 FITC and anti-TCR TRITC together (two molecules that should not be in proximity on the cell surface) does not cause bleed-through emission in the TRITC channel (green line). Thus, this establishes a baseline above which TRITC emission is considered a positive FRET signal. Indeed, statistical analysis revealed no significant FRET in the TRITC channel of these negative controls. Simultaneous staining with anti-CD8 FITC and anti-TCR TRITC illustrates significant FRET (Fig. 6A, black line; p ≤ 0.01). To demonstrate this more strikingly, we stained cells with anti-TCR TRITC and added anti-CD8 FITC to measure FRET in real time (Fig. 6A, blue line). Indeed, addition of anti-CD8 FITC results in an immediate, significant increase in FRET signal, which plateaus within ~2 min ( p ≤ 0.001). All samples that illustrated TRITC FRET also had a corresponding significant decrease in FITC fluorescence. These results clearly show that CD8 and TCR are within close proximity before ligation of pMHC.
FIGURE 6.
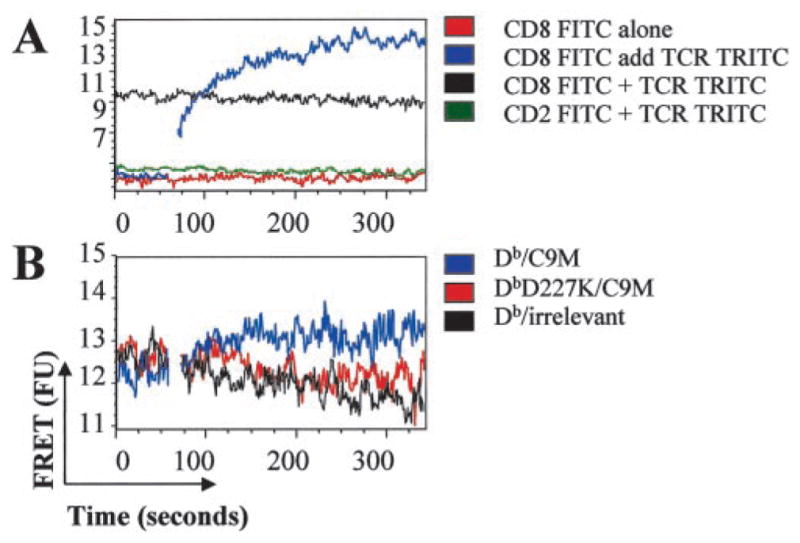
CD8 is in close proximity to TCR on the cell surface. P14 splenocytes were labeled with the indicated Abs and analyzed by FACS for FRET signal. A, The addition TCR-TRITC to CD8-FITC stained cells produced significantly increased FRET, thus indicating that CD8 and TCR are closely associated (p ≤ 0.01). B, Addition of Db/C9M (blue), but not Db D227K/C9M (red) or irrelevant tetramer (black) significantly increases FRET of cells prelabeled with both TCR-TRITC and CD8-FITC (p ≤ 0.001). Data are representative of three independent experiments.
To determine whether ligation of pMHC/TCR causes additional movement of the CD8 coreceptor, we compared the FRET signal between anti-CD8 FITC and anti-TCR TRITC in the presence of either Db or Db D227K tetramers (Fig. 6B). In this experiment, the baseline FRET signal was determined on cells prestained with anti-CD8 FITC and anti-TCR TRITC, then unlabeled tetramer was added in real time, as indicated. As shown in Fig. 6B, addition of Db/C9M tetramer resulted in significantly increased FRET (blue line; p ≤ 0.001). No increase in FRET was seen with the addition of either Db D227K/C9M or Db/irrelevant (red and black lines, respectively). Therefore, binding of specific pMHC to TCR causes CD8 coreceptor to move closer to TCR. Tetramer that cannot bind CD8 or tetramer containing irrelevant peptide do not cause the movement of CD8 closer to TCR. Taken together, the confocal and FRET data suggest that CD8 and TCR are within close proximity to each other, even without pMHC binding. However, binding of cognate pMHC does result in increased proximity of CD8 to TCR.
Activation of p56Lck can occur without CD8 engagement
Since p56Lck is associated with CD8 coreceptor, we investigated the efficacy of p56Lck activation with and without CD8 engagement. To determine the relative activity of p56Lck upon stimulation of T cells by Db and Db D227K tetramers, we measured p56Lck kinase activity. As shown in Fig. 7A, Db/gp33 induced significantly higher p56Lck activity than Db D227K/gp33 (*, p < 0.005). Interestingly, Db D227K/gp33 stimulated greater p56Lck kinase activity than irrelevant or no tetramer (**, p < 0.005). Thus, although engagement of CD8 may provide an optimal level of p56Lck activation, it can still be activated without CD8 ligation. It is especially striking that p56Lck activity can be measured in Db D227K/gp33-stimulated cells, since this decreased activity correlates with decreased T cell activation compared with Db/gp33-stimulated cells.
FIGURE 7.

Higher affinity MHC-TCR interactions produce an additive activation signal over time. A, Purified P14 CD8+ cells were stimulated with Db/gp33 or Db D227K/gp33 tetramers and activation of p56Lck kinase was measured. Db D227K/gp33 stimulated significantly higher kinase activity than irrelevant or no tetramer (**, p < 0.005). However, Db/gp33 induces significantly greater p56Lck activity than Db D227K/gp33 (*, p < 0.005). Data are representative of three independent experiments. B, Purified P14 CD8+ cells were stimulated with Db/C9M or Db D227K/C9M tetramers for 15 min or 2 h and washed away. Control cells received tetramer stimulation for the entire course of the assay (36 h). Data are representative of two independent experiments.
Despite the absence of CD8 engagement, higher affinity pMHC-TCR interaction produces an additive T cell activation signal over time
Thus far, the data show a discrepancy between the absolute requirement to engage CD8 for optimal early signaling events and later functional data, which shows that increased pMHC/TCR affinity can overcome the need for CD8 engagement to achieve proliferation and CTL activity. However, it is clear that CD8 and TCR are in close proximity and that p56Lck activation can occur without CD8 engagement. Therefore, we hypothesized that a low level of signaling produced by Db D227K/C9M was integrated over time, allowing Db D227K/C9M-stimulated cells to have equivalent function to Db/C9M-stimulated cells. To investigate this possibility, we determined the length of time tetramer needed to be present in culture for proliferation to occur. Control samples were incubated with tetramer for the entire time course of the assay (36 h) or, alternatively, tetramer was washed out at the indicated times. When exposed to tetramer for <2 h, Db D227K/C9M-stimulated cells proliferated less than those stimulated by Db/C9M (Fig. 7B). Indeed, these data are comparable to the proliferative differences between Db/gp33- and Db D227K/gp33-stimulated cells (Fig. 2A). However, by 2 h of tetramer exposure, the Db D227K/C9M-stimulated cells had equal proliferative ability as compared with Db/ C9M-stimulated cells. Identical responses to Db/C9M and Db D227K/C9M were seen in cultures with Db/C9M or Db D227K/ C9M tetramers present for the entire time (Fig. 7B). This indicates that over time at higher pMHC/TCR affinity, activation signals may overcome lack of CD8 engagement, thus explaining the differences between early signaling events (3 min) and later functional activity (36 h).
Discussion
The experiments described above are a comprehensive study on the role of the CD8 coreceptor in primary naive P14 CD8+ T cell activation of two different pMHC/TCR affinities. Our results show that while CD8 coreceptor does stabilize pMHC-TCR interactions, its engagement by MHC is not absolutely required for T cell activation, as previously reported (18, 21, 22). The requirement for CD8 ligation is dependent upon the affinity of MHC for TCR. This agrees with recent data published by our group using xenoreactive T cell clones and by Holler and Kranz using T cell hybridomas (31, 32). We have shown that the absence of CD8 engagement during moderate affinity pMHC-TCR interactions can affect all stages of T cell activation and effector function, from early signaling events to proliferation, cytokine production, and the ability to become effector CTL. However, higher affinity pMHC-TCR interactions allow an accumulation of activation signals over time that compensates for lack of CD8 engagement and allows T cell proliferation and CTL effector function. Indeed, accumulation of signals over time leading to full T cell activation has been previously reported (63).
The nature of the altered signaling by the p-tyr blots is intriguing. Although the extensive study of altered signaling is beyond the scope of this report, preliminary analysis of lysates immuno-precipitated with anti- CD3ζ and antiextracellular signal-regulated kinase from Db/C9M and Db D227K/C9M-stimulated CD8+ P14 T cells indicates that phosphorylation of CD3ζ and extracellular signal-regulated kinase is decreased in cells stimulated with Db D227K/C9M (data not shown).
It has been proposed that the stabilization and signaling properties of the CD8 coreceptor are separate entities (21). Our data support this hypothesis. Our findings show that CD8 engagement greatly stabilizes pMHC-TCR interaction on the cell surface. Although early signaling patterns are altered without CD8 engagement, CTL activity remains intact. Studies investigating the role of CD8 in the high-affinity 2C TCR-Ld interaction (64) have yielded similar results. Allospecific CD8−/− 2C-transgenic TCR cells can still mount an effective CTL response and, at large doses of antigenic stimulus, CD8+ cells can proliferate in the presence of anti-CD8 Abs (65, 66). Using T cell hybridomas, Holler and Kranz (32) have recently shown that high-affinity pMHC-TCR interactions can stimulate T cells without CD8 engagement. Daniels and Jameson (21) showed that cell staining with OVA/Kb multimers, coupled with lower affinity altered peptide ligands, can be blocked or enhanced by using various anti-CD8 Abs. They also show that blocking anti-CD8β Abs prevent calcium mobilization in cells stimulated with these multimers. This mirrors our own data using blocking and nonblocking anti-CD8 Abs with Db/C9M and Db/ gp33 tetramers (data not shown) and our calcium mobilization data presented here. Experiments previously conducted in our laboratory also show that addition of anti-CD8 Abs decreases the ability of Db/gp33 tetramers to stimulate CD8+ T cell proliferation and CTL activity (35).
Recent work indicates that a TCR binding motif, anti-CPM, may also play a role in CD8 colocalization with pMHC/TCR (27). This is consistent with the FRET signal seen in our experiments even before addition of MHC tetramer (Fig. 6A). Although CD8 may still be associated with TCR through anti-C9M in our experiments, we still observed decreased signaling when CD8 cannot bind MHC. Thus, though possibly still bound by TCR via anti-CPM, binding of CD8 by MHC is critical for T cell activation. This suggests that at least the same monomer binds both TCR and CD8. As positive selection in the thymus is impaired in cells lacking anti-CPM (27), perhaps this interaction of TCR and CD8 results in some signaling through CD8 when MHC is engaged. This would explain the p56Lck activity seen in Db D227K/gp33-stimulated cells and the additive signal that we see in Db D227K/C9M-stimulated cells (Fig. 7). This is further bolstered by the increased FRET seen after addition of unlabeled tetramer (Fig. 6B).
We cannot formally eliminate the possibility that T cells stimulated with Db D227K tetramers could be receiving necessary signaling by engaging MHC on neighboring T cells in our experiments. However, in early signaling events (<3 min), we see drastic differences with and without CD8 engagement in both Ca2+ flux and p-tyr patterns. In this time frame, CD8 engagement by class I molecules on other T cells in suspension is unlikely. The differences seen in functional activity at later time points may reflect CD8 engagement by neighboring cells, which rescues T cell functionality. However, this is possible only in Db D227K/C9M-stimulated cells, as Db D227K/gp33 stimulation results in decreased T cell activation, suggesting again that this is unlikely. Experiments mixing Db/irrelevant with Db D227K/gp33 tetramers do not result in restoration of activity, providing further data consistent with the requirement for the same pMHC molecule to engage both TCR and CD8 (data not shown). This is further supported by structural studies from our group on a CD8-independent TCR (31).
Recent articles indicate that peptide may fall off of soluble pMHC multimers and may be represented on cell surface MHC of neighboring T cells (67, 68). Although the data presented in these articles show that peptide representation can occur, we do not believe that this is a major factor in our experiments. Work done by Schott et al. (67) indicates that the dose-response curve of OT-1 T cells is identical for peptide and tetramer, when normalized for peptide molarity (67). However, P14 T cells, as well as HY TCR tg T cells, require 50- to 100-fold more free peptide than tetramer to respond equally; thus, it is unlikely that transferred peptide is responsible (35, 36). In addition, we reported that single T cells cultured with tetramer divide, thus no other T cells are present to function as APCs (35). More importantly, if peptide transfer occurred in our experiments, differences between Db and Db D227K tetramers would not be seen. Preferential transfer of peptide by Db tetramers is unlikely, as there is no biochemical evidence that Db or Db D227K molecules differ in their peptide-binding stability. Lastly, experiments with tetramer performed at 37°C indicate that Db tetramers with gp33 and C9M peptides are very stable, with half-lives in excess of 24 h in vivo (35).
We propose a model where a high-affinity interaction between pMHC and TCR obviates the requirement for binding to CD8. However, at moderate affinity, CD8 engagement along with the presenting MHC molecule is necessary to stabilize and promote signaling. We addressed this possibility with a [3H]thymidine incorporation assay that stimulated cells with Db/gp33 or Db D227K/ gp33 in the presence of wild-type Db tetramer containing irrelevant peptide. Addition of Db/irrelevant tetramer should provide a source of CD8 engagement to Db D227K/gp33-stimulated cells, which can only be used if the cells do not require simultaneous engagement of MHC/TCR/CD8 in the same complex. This experiment showed no increase in Db D227K/gp33-stimulated cell proliferation in the presence of Db/irrelevant tetramer (data not shown). Indeed, proliferation of cells in the presence of both Db/gp33 or Db D227K/gp33 and Db/irrelevant tetramers was similar to cells stimulated with Db/gp33 or Db D227K/gp33 alone. This indicates that, at moderate affinities, CD8 engagement from neighboring T cells cannot compensate for the absence of direct CD8 engagement with pMHC/TCR.
Thus, we have demonstrated that the requirement for CD8 engagement in naive CD8+ T cells rests on the affinity of the pMHC-TCR interaction. This creates a complex model for full activation of naive CD8+ T cells that depends upon both the intrinsic affinity of TCR and the engagement of CD8.
Acknowledgments
We thank Katherine Midkiff, Carie Barnes, Linda Rogozinski, and Brian Miller for excellent technical assistance and Larry Arnold for assistance with flow cytometry.
Footnotes
This research was funded by National Institutes of Health Grants GM67143, T32A1007273 (to S.E.K.) and T32A1007001 (to L.A.C.).
Abbreviations used in this paper: pMHC, peptide MHC; SPR, surface plasmon resonance; LCMV, lymphocytic choriomeningitis virus; MFI, mean fluorescence intensity; FU, fluorescence unit; tg, transgenic; p-tyr, tyrosine phosphorylation; FRET, fluorescence resonance energy transfer; TRITC, tetramethylrhodamine isothiocyanate.
References
- 1.Zamoyska R. CD4 and CD8: modulators of T-cell receptor recognition of antigen and of immune responses? Curr Opin Immunol. 1998;10:82. doi: 10.1016/s0952-7915(98)80036-8. [DOI] [PubMed] [Google Scholar]
- 2.Janeway CA., Jr The T cell receptor as a multicomponent signalling machine: CD4/CD8 coreceptors and CD45 in T cell activation. Annu Rev Immunol. 1992;10:645. doi: 10.1146/annurev.iy.10.040192.003241. [DOI] [PubMed] [Google Scholar]
- 3.Madrenas J, Chau LA, Smith J, Bluestone JA, Germain RN. The efficiency of CD4 recruitment to ligand-engaged TCR controls the agonist/ partial agonist properties of peptide-MHC molecule ligands. J Exp Med. 1997;185:219. doi: 10.1084/jem.185.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hampl J, Chien YH, Davis MM. CD4 augments the response of a T cell to agonist but not to antagonist ligands. Immunity. 1997;7:379. doi: 10.1016/s1074-7613(00)80359-3. [DOI] [PubMed] [Google Scholar]
- 5.Hamad AR, O’Herrin SM, Lebowitz MS, Srikrishnan A, Bieler J, Schneck J, Pardoll D. Potent T cell activation with dimeric peptide-major histocompatibility complex class II ligand: the role of CD4 coreceptor. J Exp Med. 1998;188:1633. doi: 10.1084/jem.188.9.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crawford F, Kozono H, White J, Marrack P, Kappler J. Detection of antigen-specific T cells with multivalent soluble class II MHC covalent peptide complexes. Immunity. 1998;8:675. doi: 10.1016/s1074-7613(00)80572-5. [DOI] [PubMed] [Google Scholar]
- 7.Boniface JJ, Rabinowitz JD, Wulfing C, Hampl J, Reich Z, Altman JD, Kantor RM, Beeson C, McConnell HM, Davis MM. Initiation of signal transduction through the T cell receptor requires the multivalent engagement of peptide/MHC ligands [corrected] Immunity. 1998;9:459. doi: 10.1016/s1074-7613(00)80629-9. [DOI] [PubMed] [Google Scholar]
- 8.Chirmule N, Avots A, LakshmiTamma SM, Pahwa S, Serfling E. CD4-mediated signals induce T cell dysfunction in vivo. J Immunol. 1999;163:644. [PubMed] [Google Scholar]
- 9.Xiong Y, Kern P, Chang H, Reinherz E. T cell receptor binding to a pMHCII ligand is kinetically distinct from and independent of CD4. J Biol Chem. 2001;276:5659. doi: 10.1074/jbc.M009580200. [DOI] [PubMed] [Google Scholar]
- 10.Krummel MF, Sjaastad MD, Wulfing C, Davis MM. Differential clustering of CD4 and CD3ζ during T cell recognition. Science. 2000;289:1349. doi: 10.1126/science.289.5483.1349. [DOI] [PubMed] [Google Scholar]
- 11.Wyer JR, Willcox BE, Gao GF, Gerth UC, Davis SJ, Bell JI, van der Merwe PA, Jakobsen BK. T cell receptor and coreceptor CD8αα bind peptide-MHC independently and with distinct kinetics. Immunity. 1999;10:219. doi: 10.1016/s1074-7613(00)80022-9. [DOI] [PubMed] [Google Scholar]
- 12.Garcia KC, Scott CA, Brunmark A, Carbone FR, Peterson PA, Wilson IA, Teyton L. CD8 enhances formation of stable T-cell receptor/MHC class I molecule complexes. Nature. 1996;384:577. doi: 10.1038/384577a0. [DOI] [PubMed] [Google Scholar]
- 13.Altman JD, Moss PA, Goulder PJ, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94. doi: 10.1126/science.274.5284.94. [DOI] [PubMed] [Google Scholar]
- 14.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 15.Busch DH, I, Pilip M, Vijh S, Pamer EG. Coordinate regulation of complex T cell populations responding to bacterial infection. Immunity. 1998;8:353. doi: 10.1016/s1074-7613(00)80540-3. [DOI] [PubMed] [Google Scholar]
- 16.Flynn KJ, Belz GT, Altman JD, Ahmed R, Woodland DL, Doherty PC. Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity. 1998;8:683. doi: 10.1016/s1074-7613(00)80573-7. [DOI] [PubMed] [Google Scholar]
- 17.Muller D, Pederson K, Murray R, Frelinger JA. A single amino acid substitution in an MHC class I molecule allows heteroclitic recognition by lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes. J Immunol. 1991;147:1392. [PubMed] [Google Scholar]
- 18.Purbhoo MA, Boulter JM, Price DA, Vuidepot AL, Hourigan CS, Dunbar PR, Olson K, Dawson SJ, Phillips RE, Jakobsen BK, et al. The human CD8 coreceptor effects cytotoxic T cell activation and antigen sensitivity primarily by mediating complete phosphorylation of the T cell receptor ζ chain. J Biol Chem. 2001;276:32786. doi: 10.1074/jbc.M102498200. [DOI] [PubMed] [Google Scholar]
- 19.Connolly JM, Hansen TH, Ingold AL, Potter TA. Recognition by CD8 on cytotoxic T lymphocytes is ablated by several substitutions in the class I α3 domain: CD8 and the T-cell receptor recognize the same class I molecule. Proc Natl Acad Sci USA. 1990;87:2137. doi: 10.1073/pnas.87.6.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luescher IF, Vivier E, Layer A, Mahiou J, Godeau F, Malissen B, Romero P. CD8 modulation of T-cell antigen receptor-ligand interactions on living cytotoxic T lymphocytes. Nature. 1995;373:353. doi: 10.1038/373353a0. [DOI] [PubMed] [Google Scholar]
- 21.Daniels MA, Jameson SC. Critical role for CD8 in T cell receptor binding and activation by peptide/major histocompatibility complex multimers. J Exp Med. 2000;191:335. doi: 10.1084/jem.191.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denkberg G, Cohen CJ, Reiter Y. Critical role for CD8 in binding of MHC tetramers to TCR: CD8 antibodies block specific binding of human tumor-specific MHC-peptide tetramers to TCR. J Immunol. 2001;167:270. doi: 10.4049/jimmunol.167.1.270. [DOI] [PubMed] [Google Scholar]
- 23.Bosselut R, Kubo S, Guinter T, Kopacz JL, Altman JD, Feigenbaum L, Singer A. Role of CD8β domains in CD8 coreceptor function: importance for MHC I binding, signaling, and positive selection of CD8+ T cells in the thymus. Immunity. 2000;12:409. doi: 10.1016/s1074-7613(00)80193-4. [DOI] [PubMed] [Google Scholar]
- 24.Xu H, Littman DR. A kinase-independent function of Lck in potentiating antigen-specific T cell activation. Cell. 1993;74:633. doi: 10.1016/0092-8674(93)90511-n. [DOI] [PubMed] [Google Scholar]
- 25.Thome M, Germain V, DiSanto JP, Acuto O. The p56lck SH2 domain mediates recruitment of CD8/p56lck to the activated T cell receptor/ CD3/ζ complex. Eur J Immunol. 1996;26:2093. doi: 10.1002/eji.1830260920. [DOI] [PubMed] [Google Scholar]
- 26.Cawthon AG, Alexander-Miller MA. Optimal colocalization of TCR and CD8 as a novel mechanism for the control of functional avidity. J Immunol. 2002;169:3492. doi: 10.4049/jimmunol.169.7.3492. [DOI] [PubMed] [Google Scholar]
- 27.Naeher D, I, Luescher F, Palmer E. A role for the α-chain connecting peptide motif in mediating TCR-CD8 cooperation. J Immunol. 2002;169:2964. doi: 10.4049/jimmunol.169.6.2964. [DOI] [PubMed] [Google Scholar]
- 28.Bachmann MF, Oxenius A, Speiser DE, Mariathasan S, Hengartner H, Zinkernagel RM, Ohashi PS. Peptide-induced T cell receptor down-regulation on naive T cells predicts agonist/partial agonist properties and strictly correlates with T cell activation. Eur J Immunol. 1997;27:2195. doi: 10.1002/eji.1830270912. [DOI] [PubMed] [Google Scholar]
- 29.Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375:148. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- 30.Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- 31.Buslepp J, Kerry SE, Loftus D, Frelinger JA, Appella E, Collins EJ. High affinity xenoreactive TCR:MHC interaction recruits CD8 in absence of binding to MHC. J Immunol. 2003;170:373. doi: 10.4049/jimmunol.170.1.373. [DOI] [PubMed] [Google Scholar]
- 32.Holler PD, Kranz DM. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity. 2003;18:255. doi: 10.1016/s1074-7613(03)00019-0. [DOI] [PubMed] [Google Scholar]
- 33.Burrows SR, Kienzle N, Winterhalter A, Bharadwaj M, Altman JD, Brooks A. Peptide-MHC class I tetrameric complexes display exquisite ligand specificity. J Immunol. 2000;165:6229. doi: 10.4049/jimmunol.165.11.6229. [DOI] [PubMed] [Google Scholar]
- 34.Delon J, Gregoire C, Malissen B, Darche S, Lemaitre F, Kourilsky P, Abastado JP, Trautmann A. CD8 expression allows T cell signaling by monomeric peptide-MHC complexes. Immunity. 1998;9:467. doi: 10.1016/s1074-7613(00)80630-5. [DOI] [PubMed] [Google Scholar]
- 35.Wang B, Maile R, Greenwood R, Collins EJ, Frelinger JA. Naive CD8+ T cells do not require costimulation for proliferation and differentiation into cytotoxic effector cells. J Immunol. 2000;164:1216. doi: 10.4049/jimmunol.164.3.1216. [DOI] [PubMed] [Google Scholar]
- 36.Maile R, Wang B, Schooler W, Meyer A, Collins EJ, Frelinger JA. Antigen-specific modulation of an immune response by in vivo administration of soluble MHC class I tetramers. J Immunol. 2001;167:3708. doi: 10.4049/jimmunol.167.7.3708. [DOI] [PubMed] [Google Scholar]
- 37.Potter TA, Bluestone JA, Rajan TV. A single amino acid substitution in the α3 domain of an H-2 class I molecule abrogates reactivity with CTL. J Exp Med. 1987;166:956. doi: 10.1084/jem.166.4.956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Potter TA, Rajan TV, Dick RF, 2nd, Bluestone JA. Substitution at residue 227 of H-2 class I molecules abrogates recognition by CD8-dependent, but not CD8-independent, cytotoxic T lymphocytes. Nature. 1989;337:73. doi: 10.1038/337073a0. [DOI] [PubMed] [Google Scholar]
- 39.Salter RD, Benjamin RJ, Wesley PK, Buxton SE, Garrett TP, Clayberger C, Krensky AM, Norment AM, Littman DR, Parham P. A binding site for the T-cell co-receptor CD8 on the α3 domain of HLA-A2. Nature. 1990;345:41. doi: 10.1038/345041a0. [DOI] [PubMed] [Google Scholar]
- 40.Killeen N, Moriarty A, Teh HS, Littman DR. Requirement for CD8-major histocompatibility complex class I interaction in positive and negative selection of developing T cells. J Exp Med. 1992;176:89. doi: 10.1084/jem.176.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 42.Brandle D, Burki K, Wallace VA, Rohrer UH, Mak TW, Malissen B, Hengartner H, Pircher H. Involvement of both T cell receptor Vα and Vβ variable region domains and α chain junctional region in viral antigen recognition. Eur J Immunol. 1991;21:2195. doi: 10.1002/eji.1830210930. [DOI] [PubMed] [Google Scholar]
- 43.Ashton-Rickardt PG, Bandeira A, Delaney JR, Van Kaer L, Pircher HP, Zinkernagel RM, Tonegawa S. Evidence for a differential avidity model of T cell selection in the thymus. Cell. 1994;76:651. doi: 10.1016/0092-8674(94)90505-3. [DOI] [PubMed] [Google Scholar]
- 44.Bachmann MF, Sebzda E, Kundig TM, Shahinian A, Speiser DE, Mak TW, Ohashi PS. T cell responses are governed by avidity and co-stimulatory thresholds. Eur J Immunol. 1996;26:2017. doi: 10.1002/eji.1830260908. [DOI] [PubMed] [Google Scholar]
- 45.Devine L, Rogozinski L, Naidenko OV, Cheroutre H, Kavathas PB. The complementarity-determining region-like loops of CD8α interact differently with β2-microglobulin of the class I molecules H-2Kb and thymic leu- kemia antigen, while similarly with their α3 domains. J Immunol. 2002;168:3881. doi: 10.4049/jimmunol.168.8.3881. [DOI] [PubMed] [Google Scholar]
- 46.Tissot AC, Pecorari F, Pluckthun A. Characterizing the functionality of recombinant T-cell receptors in vitro: a pMHC tetramer based approach. J Immunol Methods. 2000;236:147. doi: 10.1016/s0022-1759(99)00226-4. [DOI] [PubMed] [Google Scholar]
- 47.Savage PA, Boniface JJ, Davis MM. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity. 1999;10:485. doi: 10.1016/s1074-7613(00)80048-5. [DOI] [PubMed] [Google Scholar]
- 48.Cambier J, Chen ZZ, Pasternak J, Ransom J, Sandoval V, Pickles H. Ligand-induced desensitization of B-cell membrane immunoglobulin-mediated Ca2+ mobilization and protein kinase C translocation. Proc Natl Acad Sci USA. 1988;85:6493. doi: 10.1073/pnas.85.17.6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sekar RB, Periasamy A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J Cell Biol. 2003;160:629. doi: 10.1083/jcb.200210140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Methods. 1994;171:131. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 51.Wang B, Sharma A, Maile R, Saad M, Collins EJ, Frelinger JA. Peptidic termini play a significant role in TCR recognition. J Immunol. 2002;169:3137. doi: 10.4049/jimmunol.169.6.3137. [DOI] [PubMed] [Google Scholar]
- 52.Bodinier M, Peyrat MA, Tournay C, Davodeau F, Romagne F, Bonneville M, Lang F. Efficient detection and immunomagnetic sorting of specific T cells using multimers of MHC class I and peptide with reduced CD8 binding. Nat Med. 2000;6:707. doi: 10.1038/76292. [DOI] [PubMed] [Google Scholar]
- 53.Bachmann MF, Speiser DE, Zakarian A, Ohashi PS. Inhibition of TCR triggering by a spectrum of altered peptide ligands suggests the mechanism for TCR antagonism. Eur J Immunol. 1998;28:3110. doi: 10.1002/(SICI)1521-4141(199810)28:10<3110::AID-IMMU3110>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 54.Sloan-Lancaster J, Allen PM. Altered peptide ligand-induced partial T cell activation: molecular mechanisms and role in T cell biology. Annu Rev Immunol. 1996;14:1. doi: 10.1146/annurev.immunol.14.1.1. [DOI] [PubMed] [Google Scholar]
- 55.Goldrath AW, Bogatzki LY, Bevan MJ. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med. 2000;192:557. doi: 10.1084/jem.192.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murali-Krishna K, Ahmed R. Cutting edge: naive T cells masquerading as memory cells. J Immunol. 2000;165:1733. doi: 10.4049/jimmunol.165.4.1733. [DOI] [PubMed] [Google Scholar]
- 57.Tuosto L, Parolini I, Schroder S, Sargiacomo M, Lanzavecchia A, Viola A. Organization of plasma membrane functional rafts upon T cell activation. Eur J Immunol. 2001;31:345. doi: 10.1002/1521-4141(200102)31:2<345::aid-immu345>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 58.Arcaro A, Gregoire C, Bakker TR, Baldi L, Jordan M, Goffin L, Boucheron N, Wurm F, van der Merwe PA, Malissen B, Luescher IF. CD8β endows CD8 with efficient coreceptor function by coupling T cell receptor/CD3 to raft-associated CD8/p56lck complexes. J Exp Med. 2001;194:1485. doi: 10.1084/jem.194.10.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vignali DA, Carson RT, Chang B, Mittler RS, Strominger JL. The two membrane proximal domains of CD4 interact with the T cell receptor. J Exp Med. 1996;183:2097. doi: 10.1084/jem.183.5.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szollosi J, Damjanovich S, Matyus L. Application of fluorescence resonance energy transfer in the clinical laboratory: routine and research. Cytometry. 1998;34:159. [PubMed] [Google Scholar]
- 61.Chan SS, Arndt-Jovin DJ, Jovin TM. Proximity of lectin receptors on the cell surface measured by fluorescence energy transfer in a flow system. J Histochem Cytochem. 1979;27:56. doi: 10.1177/27.1.374620. [DOI] [PubMed] [Google Scholar]
- 62.Damjanovich S, Tron L, Szollosi J, Zidovetzki R, Vaz WL, Regateiro F, Arndt-Jovin DJ, Jovin TM. Distribution and mobility of murine histocompatibility H-2Kk antigen in the cytoplasmic membrane. Proc Natl Acad Sci USA. 1983;80:5985. doi: 10.1073/pnas.80.19.5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosette C, Werlen G, Daniels MA, Holman PO, Alam SM, Travers PJ, Gascoigne NR, Palmer E, Jameson SC. The impact of duration versus extent of TCR occupancy on T cell activation: a revision of the kinetic proofreading model. Immunity. 2001;15:59. doi: 10.1016/s1074-7613(01)00173-x. [DOI] [PubMed] [Google Scholar]
- 64.Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, Chien Y. Ligand recognition by αβ T cell receptors. Annu Rev Immunol. 1998;16:523. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 65.Cai Z, Sprent J. Influence of antigen dose and costimulation on the primary response of CD8+ T cells in vitro. J Exp Med. 1996;183:2247. doi: 10.1084/jem.183.5.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cai Z, Sprent J. Resting and activated T cells display different requirements for CD8 molecules. J Exp Med. 1994;179:2005. doi: 10.1084/jem.179.6.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schott E, Bertho N, Ge Q, Maurice MM, Ploegh HL. Class I negative CD8 T cells reveal the confounding role of peptide-transfer onto CD8 T cells stimulated with soluble H2-Kb molecules. Proc Natl Acad Sci USA. 2002;99:13735. doi: 10.1073/pnas.212515399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ge Q, Stone JD, Thompson MT, Cochran JR, Rushe M, Eisen HN, Chen J, Stern LJ. Soluble peptide-MHC monomers cause activation of CD8+ T cells through transfer of the peptide to T cell MHC molecules. Proc Natl Acad Sci USA. 2002;99:13729. doi: 10.1073/pnas.212515299. [DOI] [PMC free article] [PubMed] [Google Scholar]