

Abstract
PKNs form a subfamily of the AGC serine/threonine protein kinases, and have a catalytic domain homologous with that of PKC (protein kinase C) in the C-terminal region and three characteristic ACC (antiparallel coiled-coil) domain repeats in the N-terminal region. The preferred peptide phosphorylation motif for PKNs determined by a combinatorial peptide library method was highly similar to that of PKCs within a 10-amino-acid stretch. Previously reported PKN inhibitory compounds also inhibit PKCs to a similar extent, and no PKN selective inhibitors have been commercially available. We have identified a 15-amino-acid peptide inhibitor of PKNs based on amino acids 485–499 of the C-terminal region of the C2-like domain of PKN1. This peptide, designated as PRL, selectively inhibits the kinase activity of all isoforms of PKN (Ki = 0.7 μM) towards a peptide substrate, as well as autophosphorylation activity of PKN in vitro, in contrast with PKC. Reversible conjugation by a disulfide bond of a carrier peptide bearing a penetration accelerating sequence to PRL, facilitated the cellular uptake of this peptide and significantly inhibited phosphorylation of tau by PKN1 at the PKN1-specific phosphorylation site in vivo. This peptide may serve as a valuable tool for investigating PKN activation and PKN-mediated responses.
Keywords: cell-penetrating peptide (CPP), penetration accelerating sequence octa-arginine (LPasR8), PRL, protein kinase C (PKC), PKN inhibitor
INTRODUCTION
PKN (protein kinase novel) is a serine/threonine protein kinase with a catalytic domain highly homologous with that of the PKC (protein kinase C) family at its C-terminal region and three repeats of an ACC (antiparallel coiled-coil) domain and a C2-like domain at its N-terminal region [1]. PKN can bind to and be activated by the small GTPase Rho, and it can also be activated by fatty acids, such as arachidonic acid, in vitro [1]. The PKN pathway has been so far analysed primarily using expression of PKN constructs or RNAi (RNA interference) techniques in cells. These studies have implicated PKNs in a range of physiological processes such as cytoskeletal regulation, vesicle transport and cell-cycle regulation (reviewed in [1], [2–5]), as well as in some pathological conditions including cancer development and metastasis [6–8], Alzheimer’s disease [9], retinitis pigmentosa [10] and amyotrophic lateral sclerosis [11].
Mammalian PKN has three isoforms derived from different genes: PKN1 (PKNα/PRK1/PAK1), PKN2 (PRK2/PAK2/PKNγ) and PKN3 (PKNβ). It is also recognized that at least two isoforms result from the alternative splicing of the PKN1 gene (H. Mukai, unpublished work). These PKN isoforms have well-conserved ACC, C2-like and catalytic domains, but show distinct enzymatic properties and tissue distributions. PKN isoform-selective targeted knockout or knockdown experiments would therefore appear to be beneficial for understanding the in vivo function of PKN. However, interpretation of knockout phenotypes can be confounded by possible functional redundancy between isoforms, and by the potential for alteration in signalling fidelity accompanying prolonged change in the levels of signalling molecules. PKN has been suggested to bind to various proteins (reviewed in [1], [12–14]) and also to have a scaffolding function in cells [15], suggesting that simple knockout or knockdown may disrupt protein complexes or impair functional interactions among proteins irrespective of the protein kinase activity of PKN. Accordingly, inhibitors of the PKN pathway would be useful tools that can be rapidly applied and would not alter the expression of PKNs to achieve direct and specific inhibition.
As speculated from the structural resemblance among catalytic domains of PKNs and PKCs, PKNs have been reported to efficiently phosphorylate established substrates for PKCs in vitro [1]. For example, synthetic oligopeptides based on the pseudo-substrate sites of PKCs are good substrates for PKNs [16], and PKN1 efficiently phosphorylates identical sites on MARCKS (myristoylated alanine-rich C-kinase substrate) [17] and vimentin [18] to PKCs in vitro. In Drosophila the kinase domain of PKC53E, a PKC family kinase, could functionally substitute for the kinase domain of Pkn during development, although the rescue efficiency was low [19]. This observation suggests that these two kinases can overlap in their spectrum of potential phosphorylation substrates in vivo in Drosophila. The well known kinase inhibitor reagents for PKC such as bisindolylmaleimide I and H-7 also potently inhibit kinase activity of PKN [20], and commercially available peptide inhibitors sold as being specific for PKC also affect the kinase activity of PKN with similar Ki values as PKC (K. Shiga and H. Mukai, unpublished work). In light of these observations, progress in evaluating the function of the PKC subfamily that has been facilitated by these so-called PKC inhibitors, should be re-evaluated. Although high sequence similarity suggests that the PKN isoform selectivity may be difficult to achieve with small molecule or peptide inhibitors, reagents that can selectively inhibit PKNs, but not PKCs, would be an important tool to focus on and evaluate the physiological function of PKNs as distinct from PKCs.
In the present study we describe a peptide inhibitor that is selective for PKN family members over PKCs. This peptide could be efficiently delivered into cells by conjugation to a CPP (cell-penetrating peptide) bearing a novel penetration accelerating sequence (PasR8: FFLIPKGRRRRRRRR; where Pas is penetration accelerating sequence) in the presence of pyrenebutyrate, and selectively inhibited PKN1-induced phosphorylation of tau in vivo.
MATERIALS AND METHODS
Peptide synthesis
Peptides for library screening were synthesized as described previously [21]. Table 1 lists the range of the peptide sequences used in the present study. δpeptide, S6, TAT-PRL, NER, AEV, PVI, PRL and QKK peptides were commercially synthesized by BEX. For the preparation of PRL-SS-PasR8, the PRL and PasR8 segments bearing a cysteamine and cysteine at the C-termini (PRL-SH and FFLIPKGRRRRRRRRGC-amide) were prepared by Fmoc (9-fluorenylmethoxycarbonyl)-solid-phase peptide synthesis using the cysteamine 4-methoxytrityl (Novabiochem) and TGS-RAM (Shimadzu) resins respectively, followed by deprotection using trifluoroacetic acid/ethanedithiol (95:5) and purification by HPLC [22,23]. The thiol group of the PRL-SH peptide thus obtained was activated by treatment with 2,2′-dithiodipyridine [23], and the selective disulfide cross-linkage was formed with FFLIPKGRRRRRRRRGC-amide to yield PRL-SS-PasR8. Other disulfide-linked peptides were similarly prepared. The structure of the products was confirmed by MALDI–TOF-MS (matrix-assisted laser-desorption ionization–time-of-flight MS)
Table 1.
Amino acid sequences of the peptides used in the present study
| Peptide name | Peptide sequence | Parent protein |
|---|---|---|
| δpeptide | AMFPTMNRRGSIKQAKI | 137–153 of PKCδ |
| S6 | AKRRRLSSLRA | 229–239 of bovine ribosomal protein S6 |
| Kemptide | LRRASLG | [60] |
| tau320 | LSKVTSKCGS | 315–324 of human tau |
| NER | NERHEVQLDMEPQGCLV | 455–471 of human PKN1 |
| AEV | AEVTFRNPVIERIPRLRR | 472–489 of human PKN1 |
| PVI | PVIERIPRLRRQKKI | 479–493 of human PKN1 |
| PRL | PRLRRQKKIFSKQQG | 485–499 of human PKN1 |
| QKK | QKKIFSKQQGKAFQAARQMN | 490–509 of human PKN1 |
| PRL-PasR8 | PRLRRQKKIFSKQQGFFLIPKGRRRRRRRRG | 485–499 of human PKN1 |
| PasR8-PRL | FFLIPKGRRRRRRRRGPRLRRQKKIFSKQQG | 485–499 of human PKN1 |
| TAT-PRL | GYGRKKRRQRRRGPRLRRQKKIFSKQQG | 485–499 of human PKN1 |
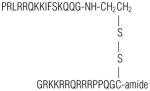
| PRL-SH | PRLRRQKKIFSKQQG-NH-CH2CH2-SH | 485–499 of human PKN1 |
| PRL-SS-PasR8 |

|
485–499 of human PKN1 |
| PRL-SS-FHV |

|
485–499 of human PKN1 |
| PRL-SS-TAT |
|
485–499 of human PKN1 |
| DNER-SS-PasR8 |

|
454–468 of human PKN1 |
Recombinant proteins
pDESTvectors were used for GST (glutathione transferase) fused to the catalytic domain of rat PKCα (corresponding to amino acids 332–end), rat PKCβII (corresponding to amino acids 329–end), rat PKCγ(corresponding to amino acids 344–end), rat PKCδ (corresponding to amino acids 328–end), mouse PKCλ(corresponding to amino acids 235–end; full length PKCλcDNA was kindly provided by Dr Ohno, Yokohama City University, Yokohama, Japan), rat PKCζ(corresponding to amino acids 850–end), human PKN1 (corresponding to amino acids 543–end), human PKN3 (corresponding to amino acids 517–end) and mouse PKN3 (corresponding to amino acids 521–end). Each plasmid was transformed into DH10Bac cells (Bac-to-Bac baculovirus expression system; Invitrogen) to generate recombinant baculovirus bacmid DNA. Bacmid DNA was transfected into Sf9 insect cells using Cellfectin reagent (Invitrogen) to produce recombinant baculovirus. Production and affinity purification of GST-fusion proteins was carried out as previously described [24]. GST-fused human PKN2 and rat PKCε were prepared as described previously [24,25]. GST-tagged oligopeptides were constructed by subcloning DNA encoding each peptide into pGEX-5X-1, as listed in Table 2.
Table 2.
The amino acid sequences of peptides analysed
| Peptide name | Position
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| − 5 | − 4 | − 3 | − 2 | − 1 | 0 | + 1 | + 2 | + 3 | + 4 | |
| NS1 | K | R | R | K | P | S | F | R | N | P |
| NS2 | K | R | R | K | P | S | F | R | E | P |
| NS3 | K | R | R | K | D | S | F | R | N | P |
| NS4 | K | R | R | K | P | S | F | P | N | P |
| NS5 | K | R | R | K | P | S | F | C | N | P |
| NS6 | K | R | R | K | P | S | D | R | N | P |
| NS7 | K | R | R | K | P | S | N | R | N | P |
| NS8 | K | R | R | K | D | S | F | P | N | P |
| NS9 | K | R | Q | K | P | S | F | C | N | P |
| NS10 | K | R | G | K | P | S | F | C | N | P |
The letters in bold indicate the substituted peptide from NS1.
Peptide library screening
Phosphorylation-site-specificity was determined using a positional scanning peptide library as described previously [26].
In vitro kinase assay
In order to assess the peptide kinase activity of PKN and PKC, 10 ng of purified kinase was incubated for 5 min at 30 °C in a reaction mixture (final volume of 25 μl) containing 20 mM Tris/HCl (pH 7.5), 4 mM MgCl2, 40 μM ATP, 18.5 kBq of [γ-32P]ATP, 100 μM ‘δpeptide’ based on amino acids 137–153 of PKCδ, substituting a serine residue for Ala147 (see Table 1) as the phosphate acceptor, 0.1 mg/ml BSA or recombinant GST as a stabilizer, and in the presence or absence of the synthetic oligopeptides. The assays were conducted in a linear range. Reactions were terminated by spotting the mixture on to P81 phosphocellulose paper (Whatman) and submerging and washing in 75 mM phosphate buffer three times for 10 min each. The incorporation of [32P]phosphate into δpeptide was assessed by Cherenkov counting. In order to assess the autophosphorylation activity of PKN and PKCs, 10 ng of purified kinase was incubated for 5 min at 30 °C in a reaction mixture (final volume of 25 μl) containing 20 mM Tris/HCl (pH 7.5), 4 mM MgCl2, 40 μM ATP, 18.5 kBq of [γ-32P]ATP, 0.1 mg/ml BSA or recombinant GST as a stabilizer, and in the presence or absence of inhibitors. Reactions were terminated by the addition of 2 × Laemmli SDS/PAGE sample buffer and boiling, and then a 10 μl aliquot was subjected to SDS/PAGE (10 % gel) followed by detection of radioactivity using a BAS2500 image analyser (Fujifilm). In order to assay phosphorylation of GST-tagged oligopeptides by PKN1 and PKCs, 10 ng of purified kinase was incubated for 5 min at 30 °C in a reaction mixture (final volume of 25 μl) containing 20 mM Tris/HCl (pH 7.5), 4 mM MgCl2, 40 μM ATP, 18.5 kBq of [γ-32P]ATP, 0.1 mg/ml BSA as a stabilizer, and in the presence or absence of the synthetic oligopeptide. Reactions were terminated by the addition of 2 × Laemmli SDS/PAGE sample buffer and boiling, and then a 10 μl aliquot was subjected to SDS/PAGE (15 % gel) followed by detection of radioactivity using a BAS2500 image analyser.
Mammalian expression constructs
pSG5/tau vector for the expression of tau was generated by insertion of cDNA encoding the open reading frames of human brain tau (383 residues) into the mammalian expression vector pSG5 (Stratagene) as described previously [27]. pTB701/ PKN1/AF3 vector expressing a catalytically active form of PKN1 (aPKN) and pTB701/PKN1/AF3(K644E) vector expressing a catalytically inactive mutant of PKN1 (iPKN) have been described previously [27].
Cell culture, transfection, peptide treatment and Western blot analysis
HeLa cells were cultured at 37 °C in 5 % CO2 in DMEM (Dulbecco’s modified Eagle’s medium) supplemented with 10 % FBS (fetal bovine serum) and antibiotics (50 units/ml penicillin and 50 μg/ml streptomycin). HeLa cells seeded at 70 % confluency in 24-well plates were transfected with 0.5 μg of pSG5/tau vector in combination with 0.2 μg of pTB701 vector, or 0.2 μg of pTB701/PKN1/AF3 vector, or 0.2 μg of pTB701/PKN1/AF3(K644E) vector using 2 μl of Hilymax (Dojindo) for 20 h. After removing the medium, the cells were washed twice with PBS. The cells were incubated with or without the synthetic oligopeptides and pyrenebutyrate as described previously [28] for 20 min. Cell lysates were prepared and subjected to immunoblot analysis using appropriate antibodies.
Antibodies
A polyclonal antibody, HIA3, was raised in rabbit against a synthetic phosphopeptide, CLSKVTS(P)KCGSL, where S(P) represents phosphoserine, as described in [27]. A monoclonal antibody, HIHT1, was raised in mouse against a recombinant N-terminal protein corresponding to residues 1–190 without residues 45–102 of tau, as described in [27]. Animal experimentation was performed in accordance with the regulations of the Hyogo Institute for Aging Brain and Cognitive Disorders.
RESULTS
Determination of PKN substrate specificity
Many protein kinases identify their target substrates via recognition of a specific sequence of amino acids surrounding a phosphorylation site. Peptides that correspond to this motif have the potential to be substrate competitive inhibitors [29]. We used a previously developed positional scanning peptide library assay to identify the optimal phosphorylation motifs for two PKN family members, PKN1 and PKN3 [26,30]. This assay utilizes 198 distinct biotinylated peptide libraries. Each library has a 1:1 mixture of serine and threonine fixed at the central position and one additional position fixed to one of the 20 natural amino acids. Other positions surrounding the central serine/threonine mixture contain a degenerate mixture of amino acids. Phosphothreonine and phosphotyrosine were included at the fixed positions to identify potential requirements for priming phosphorylation events. A GST-tagged catalytic fragment of PKN1 or PKN3 was purified from Sf9 cells and a kinase assay was performed in solution on all peptide libraries simultaneously using radiolabelled ATP. The biotinylated peptides were then captured with a streptavidin-coated membrane and the extent of incorporation of radiolabelled phosphate into each of the peptides was visualized as shown in Figure 1. PKN1 and PKN3 both demonstrated strong selections for arginine at the − 3 position and also preferred hydrophobic amino acids, such as phenylalanine, at the + 1 position and basic residues at several positions downstream of the phosphorylation site (Figure 1). In addition, we observed that this phosphorylation motif is very similar to the phosphorylation motifs for a number of PKC family members [30–32], supporting the conclusion that PKNs and PKCs are likely to share overlapping substrate pools.
Figure 1. Determination of phosphorylation motifs for PKN1 and PKN3.

The top panel shows a schematic representation of the 198 peptide substrates that comprise the positional scanning peptide library (Z, fixed positions; X, degenerate positions). The remaining panels show the degree of phosphorylation of each component of the library by human PKN1 and mouse PKN3.
Quantification of selectivity values for each amino acid at each position relative to the phosphorylation site suggested the sequence ‘KRRKPSFRNP’ as the preferred PKN peptide substrate, which we designate ‘NS1’. NS1 peptide fused to GST was purified from bacteria and subjected to an in vitro PKN1 kinase assay. Whereas PKN1 did not phosphorylate GST alone [20], GST–NS1 was efficiently phosphorylated by PKN1. As NS1 was also efficiently phosphorylated by PKCα and PKCδ, we attempted to design alternative peptide substrates based on the sequence of NS1 which could be phosphorylated by PKN and not PKCs (Table 2). However, we were unable to identify a 10-aminoacid peptide that could be preferentially phosphorylated by PKN (Figure 2).
Figure 2. Favoured substrate motifs for PKN.

Each peptide from Table 2 was fused to GST and subjected to an in vitro kinase assay using the same amount of the catalytic domain of human PKN1, rat PKCα, rat PKCδ and rat PKCζ respectively.
Ser320 of tau, a microtubule-associated protein can be phosphorylated in vitro by PKN, but not PKC [27]. We therefore also prepared GST fused to a 10-amino-acid peptide corresponding to the sequence surrounding Ser320 of tau (tau320 in Table 1). We subjected this fusion protein to an in vitro kinase assay, and found that this peptide was efficiently phosphorylated by both PKN1 and PKCδ (see Supplementary Figure S1 at http://www.BiochemJ.org/bj/425/bj4250445add.htm). Taken together, these results suggest that although PKN substrate specificity is largely determined by the sequence context of phosphorylation sites, other factors, such as alternative protein–protein interactions between PKN and its substrates, are also likely to play an important role in fine-tuning PKN substrate specificity.
Design of the specific inhibitory peptide for PKN based on the Iα region
Competitive inhibitors for some protein kinases have been derived from autoinhibitory pseudosubstrate regions located outside the catalytic domain [33,34]. The region comprising amino acid residues 455–511 of PKN1 (designated as Iα), corresponding to the C-terminal part of the C2-like region of PKN1, was reported to function as an autoinhibitory domain of this enzyme [24]. Arachidonic acid, a potential regulator of PKN1, was shown to directly interfere with the interaction between Iα and the catalytic domain of PKN1, and to relieve the catalytic activity of wild-type PKN1 from autoinhibition by Iα [24]. His6-tagged Iα inhibited the kinase activity of the catalytic domain of all isoforms of PKNs, but had no inhibitory effect on PKA and PKCδ [24]. To determine the minimal region of Iα that can retain PKN1-selective inhibitory properties, we synthesized three non-overlapping 17–20 amino acid peptides that collectively cover the entire Iα region (designated as NER, AEV and QKK in Figure 3A). We also prepared two additional 15-amino-acid peptides (designated as PVI and PRL in Figures 3A and 3B), each of which carries conserved arginine and hydrophobic amino acids separated by three amino acid residues, which we suspected could constitute pseudosubstrate regions based on the results from the peptide library screen (Figure 1). The kinase activity of the catalytic domain of PKN1 was assayed in the presence of various concentrations of each synthetic peptide, using a peptide substrate based on the pseudosubstrate region of PKCδ (see δpeptide in Table 1). As shown in Figure 3(C), the PRL peptide potently inhibited the peptide kinase activity of PKN1 in a concentration-dependent manner. The AEV, QKK and PVI peptides, which share a portion of the PRL sequence, partially inhibited the activity of PKN1, whereas the NER peptide did not inhibit the activity of PKN1, even at concentrations up to 50 μM. Although the PRL peptide contains a serine residue, the inhibition of PKN1 was not due to PRL being a competitive substrate because it was not detectably phosphorylated by PKN1 (results not shown). To assess whether the PRL peptide inhibits PKN specifically, we next examined its ability to inhibit the activity of each PKN family member (human PKN1 and PKN2, and mouse PKN3), as well as various PKC isoforms. As shown in Figure 3(D), the PRL peptide potently inhibited the activities of the catalytic domain of all of the PKN family members, whereas it partially inhibited PKCζ and PKCλ (members of the atypical PKC group) and PKCε (a member of the novel PKC group) and did not inhibit PKCα, PKCβII and PKCγ (members of the conventional PKC group). The PRL peptide rather slightly enhanced the kinase activity of PKCδ (a member of the novel PKC group). The PRL peptide did not inhibit the catalytic domain of PKA and Rho-kinase (ROCK2/ROKα) either, both of which are also classified as AGC kinases (see Supplementary Figure S2 at http://www.BiochemJ.org/bj/425/bj4250445add.htm). Next we examined the effect of PRL on the kinase activity of the enzyme using GST-fused NS1 as a substrate. As shown in Figure 3(E), PRL potently inhibited the kinase activity of PKN1 against GST–NS1, as well as GST–δpeptide, compared with that of PKCβII. Kinetic analyses were carried out to characterize the inhibition of PKN1 kinase activity by PRL. The data were fitted to the Michaelis–Menten equation, which revealed that increasing concentrations of the PRL peptide increased the apparent Km for the substrate δpeptide, without affecting Vmax (Figure 3F). A re-plot of the apparent K m/Vmax values against inhibitor concentration demonstrated that the PRL peptide inhibited PKN1 activity in a manner that was competitive with the substrate δpeptide, with a Ki of 0.7 μM (Figure 3G). These observations suggest that the PRL peptide is a promising candidate for a selective substrate competitive inhibitor of PKN.
Figure 3. Design of the selective inhibitory peptide for PKN.

(A) Iα region and peptide fragments. Synthetic oligopeptides (NER, AEV, PVI, PRL and QKK) were designed based on the Iα region of human PKN1. (B) Alignment of the autoinhibitory regions of PKN family members. Shaded amino acids indicate conservation. hPKN1, human PKN1; rPKN1, rat PKN1; mPKN1, mouse PKN1; xPKN1, frog PKN; sPKN2, starfish PKN2; con, conservation. (C) In vitro kinase assay of PKN in the presence of various synthetic oligopeptides. The kinase activity of the catalytic domain of PKN1 was assayed for δpeptide in the presence of various concentrations of PRL (●), PVI (○), QKK (▲), AEV (■) and NER peptide (×). The δpeptide kinase activity of PKN in the absence of additional peptides was taken as 100 % activity. The result shown is a representative of three independent experiments. (D) In vitro kinase assay of PKNs and PKCs in the presence of PRL. The activity of the catalytic domain of PKN isoforms [human PKN1 (◆), human PKN2 (●) and mouse PKN3 (▲)] and PKC isoforms [rat PKCδ (△), rat PKCβιII (○), rat PKCγ(◇), rat PKCα (+), rat PKCε (□), rat PKCζ(×) and mouse PKCλ(−)] was assayed for δpeptide in the presence of various concentrations of the PRL peptide. The δpeptide kinase activity of the enzyme in the absence of additional peptides was taken as 100 % activity. The result shown is a representative of three independent experiments. (E) Phoshorylation of GST-tagged oligopeptides in the presence of PRL. Each δpeptide and the NS1 peptide was fused to GST and subjected to an in vitro kinase assay by PKN1 and PKCβII in the presence of various concentrations of the PRL peptide. 32P, autoradiography; CBB, Coomassie Brilliant Blue staining. (F) Double-reciprocal plots. The PRL peptide was evaluated for the ability to inhibit human PKN1 as a function of substrate (δpeptide) concentration. The activation of the catalytic domain of human PKN1 was assayed in the absence (●) or the presence of the PRL peptide at a final concentration of 2 μM (○), 4 μM (▲) or 8 μM (×). Concentration of the δpeptide was 12.5, 25, 50 and 100 μM respectively. Results were expressed as double-reciprocal plots of substrate concentration (μM) against enzymatic activity expressed as nanomoles of 32P/min per mg of protein. (G) Secondary plot of apparent Km /Vmax against PRL inhibitor concentration.
PRL inhibits the autophosphorylation of PKN
We have previously reported that the autophosphorylation of PKN1 is involved in the autoactivation of this enzyme [24,35]. We investigated whether the PRL peptide affects the autophosphorylation activity of PKN1. As shown in Figure 4, the PRL peptide potently inhibited the autophosphorylation of PKN1 in a concentration-dependent manner, but did not significantly affect the autophosphorylation activity of conventional PKC isoforms even at the 20 μM level.
Figure 4. The effect of PRL on the autophosphorylation of PKN and PKCs.

Autophosphorylation activity of human PKN1 or PKC was detected by autoradiography. The concentration of the PRL peptide was 0, 0.5, 1, 5 and 20 μM respectively. The results were plotted as a percentage of kinase activity observed in the absence of the PRL peptide (lower panel). The kinase activity in the absence of the PRL peptide was taken as 100 % activity. The results shown are representative of three independent experiments. Human PKN1, ●; rat PKCβII, ○; rat PKCα, +; rat PKCγ, ◇; rat PKCε, □; mouse PKCλ, −; rat PKCζ, ×.
Delivery of PRL into cells and inhibition of PKN in vivo
Next, we examined whether the PRL peptide could work as a cell-permeable PKN inhibitor. We previously reported that Ser320 of tau protein is specifically phosphorylated by PKN, and that the phosphorylated Ser320 is dephosphorylated by the calcium/calmodulin-dependent protein phosphatase calcineurin in cultured cells [27]. In order to monitor the kinase activity of PKN1 and to examine the effect of the PRL peptide in vivo, we co-expressed tau protein with either active kinase or inactive mutant forms of PKN1 in cultured cells, and subjected cell extracts to immunoblotting with a phosphospecific antibody (HIA3) directed against the phosphorylated Ser320 of tau [27]. As expected from the previous experiments, the HIA3 antibody did not react with lysates from mock-transfected HeLa cells or from HeLa cells transfected with kinase-inactive PKN1. However, this antibody clearly showed reactivity with lysates from cells transfected with active PKN1 (Figure 5A), indicating that the immunoreactivity monitors in vivo PKN activity. Although the PRL peptide inhibited PKN activation in vitro, when HeLa cells were treated with 100 μM PRL for 20 min before preparation of the extract, no inhibition of PKN activation was observed in vivo, presumably because of the inability of the peptide to cross cellular membranes (see Supplementary Figure S3A at http://www.BiochemJ.org/bj/425/bj4250445add.htm). To allow for efficient entry of peptide into cells in culture, we modified the peptide with membrane-translocating moieties. We first tried myristoylation of the inhibitor peptide in order to increase its hydrophobicity and hence its cellular uptake [36]. Myristoylation of PRL efficiently inhibited the activity of PKN, but lost its selectivity for PKN and also significantly inhibited PKC in vitro (see Supplementary Figure S4A at http://www.BiochemJ.org/bj/425/bj4250445add.htm). This may be related to a previous report that myristoylation itself confers inhibitory activity to peptides against PKC [37]. We then tried a second modification, linking a CPP to facilitate the intracellular delivery of PRL [38,39]. Several CPPs, including HIV-1 Tat (48–60) (TAT) [40], flock house virus coat (35–49) (FHV) [22] and octaarginine (R8) [22], capable of transporting peptides have been described previously. To satisfactorily increase the intracellular concentration of the PRL peptide, we employed a novel extension sequence to improve the translocation of arginine-rich CPPs (Pas: FFLIPKG) (K. Takayama, unpublished work) together with the R8 sequence (PasR8). The TAT and FHV peptides were also employed for comparison of the efficiency in the delivery of PasR8.
Figure 5. The effect of PRL on the kinase activity of PKN in vivo.

(A) Immunoblotting using a phosphospecific antibody against Ser320 of tau protein (HIA3). HeLa cells were transfected with pSG5/tau in combination with pTB701/PKN1/AF3 (aPKN), pTB701/PKN1/AF3 K644E (iPKN) or pTB701 (mock) respectively. The total amount of tau protein irrespective of its phosphorylation state was visualized using antibody HIHT1. (B) The effect of various PRL-SS-CPPs on Ser320 phosphorylation. HeLa cells, expressing recombinant tau and aPKN, were treated with or without 10 μM PRL-SS-PasR8, PRL-SS-FHV, PRL-SS-TAT and control DNER-SS-PasR8 peptide for 20 min.
We initially synthesized the PRL peptide tandemly fused directly to these CPPs (sequences) and subjected them to an in vitro kinase assay, but this resulted in the loss of selectivity for PKN and in potent inhibition of both PKN and PKC (see Supplementary Figure S4B). We therefore modified our approach by conjugating the PRL peptide to the CPP sequence through a disulfide bond [41,42]. Owing to the reducing environment inside cells, the disulfide bond is cleaved, releasing the peptide cargo upon intracellular delivery [43]. Cysteamine was employed as a linker to introduce a thiol to the PRL segment to minimize steric hindrance to PRL (PRL-SH). PRL-SH, which should be liberated in cytosol from PRL-SS-CPPs, showed selective inhibition of PKN in vitro, as for the original free PRL (see Supplementary Figure S4C). Application of PRL-SS-CPPs to cells is therefore expected to selectively inhibit PKN in vivo and is unlikely to be hampered by CPP-induced changes in efficacy and localization.
Tau- and PKN-transfected HeLa cells were pre-incubated with pyrenebutyrate for 10 min, followed by incubation with PRL-SS-CPPs for 20 min. Pyrenebutyrate is a counteranion bearing an aromatic hydrophobic moiety which has been reported to improve the ability of CPP-fused peptides to translocate across cellular membranes [28]. As shown in Figure 5(B), PRL-SS-PasR8 strongly diminished immunoreactivity of tau with the HIA3 antibody, whereas PRL-SS-FHV mildly and PRL-SS-TAT only slightly diminished the immunoreactivity. DNER is a control 15-amino-acid peptide derived from the NER peptide sequence that did not inhibit in vitro PKN kinase activity (results not shown). As shown in Figure 5(B), DNER-SS-PasR8 did not significantly change immunoreactivity of tau with HIA3. Similar results were obtained when we used CHO (Chinese-hamster ovary)-K1 cells instead of HeLa cells (see Supplementary Figure S5 at http://www.BiochemJ.org/bj/425/bj4250445add.htm). These results suggest that PRL-SS-PasR8 efficiently translocates the cellular membrane and the liberated PRL peptide works as an inhibitor of PKN in cultured cells.
DISCUSSION
Several lines of evidence indicate that Rho family GTPases mediate the activation of PKN [44–47]. Rho GTPases function as molecular switches and have been implicated in numerous cellular functions (recent review in [48]). These proteins function by interacting with and stimulating various downstream targets, including not only protein kinases such as PKN and Rho-kinase, but actin nucleators and phospholipases. Previous experiments using the expression of kinase-negative forms of PKN as dominant-negative constructs, may be complicated by blockage of other Rho-mediated signalling events through the simple sequestration of Rho GTPases in cells. Knocking down of PKN by RNAi methods also potentially changes the balance between Rho GTPases and other target proteins. Thus, in assessing the specific roles of PKN in signal transduction, it is helpful to obtain a peptide fragment that acts as a specific dominant inhibitor of PKN by competing with PKN substrate utilization and not by interfering with the interaction of PKN with binding proteins, such as Rho GTPases. We previously reported that the Iα region of PKN, corresponding to the C-terminal part of the C2-like domain, functions as an autoinhibitory domain, and His-tagged Iα potently inhibited PKN kinase activity with marked specificity through direct effects on the catalytic region of the enzyme [24]. We have attempted to transfer His-tagged Iα into cells by conjugation with the HIV1-TAT-(48–60) peptide. However, application of 10 μM TAT-conjugated His-tagged Iα to cultured cells for 20 min did not inhibit in vivo phosphorylation of tau protein by PKN1, probably due to insufficient transfer of this protein into cells (see Supplementary Figure S3B). In the present study we have shown that the PRL-SS-CPP peptide, which is much smaller than the whole Iα fragment, could be transferred into cells and that this peptide worked as an in vivo inhibitor of PKN. This shortened peptide sequence can be readily synthesized by standard peptide chemistry for further use in evaluating the biological roles of PKNs.
The PRL peptide fused to PasR8 most efficiently inhibited in vivo phosphorylation of tau by PKN compared with peptides fused to other CPPs we tried. The addition of the Pas sequence of argininerich CPPs often results in improvement of the efficiency in intracellular delivery, and the detailed properties of this extension sequence will be reported elsewhere.
Whereas ATP-competitive inhibitor compounds of a kinase are expected to uniformly inhibit phosphorylation of all protein substrates of a kinase, pseudosubstrate peptide inhibitors might act with various potencies against different protein substrates. However, the PRL peptide not only inhibited peptide substrate phosphorylation by PKN, but also significantly inhibited PKN autophosphorylation, which is regarded to be involved in the activation process of this enzyme [24,35]. This result suggests that this peptide inhibitor potentially broadly inhibits PKN. PRL displays a modest affinity (Ki = 0.7 μM) for PKN in vitro. However, one should consider that in vitro potency does not always reflect the in vivo potential, mainly because in vitro assays use large excesses of substrates, whereas in cellular conditions the concentration of the substrate is far below the micromolar range. In our experiment shown in Figure 5(B), 10 μM of the PRL peptide could efficiently inhibit phosphorylation of tau overexpressed in cultured cells. The PRL peptide may show better efficacy in cells for native physiological substrates of PKN. It should be noted that PRL partially, but relatively significantly, inhibited some isoforms of PKC such as PKCλ, PKCε and PKCζ in a dose-dependent manner. In the future, more potent and specific inhibitors of PKN or each isoform of PKN could be identified via addition of some amino acids or stepwise combinatorial modification of the features of the PRL peptide [49]. In the present study, we checked only the short-term effect (less than 20 min) of the PRL peptide treatment and have not examined the protease resistance of this peptide in vivo. It is also noted that modification of this peptide might be necessary to generate a long-acting, stable inhibitor.
PKN has been suggested to be implicated in some pathological conditions [6–11]. Metzger et al. [7] reported that PKN1 is pivotal to androgen receptor function and that microRNA-mediated knockdown of PKN1 or inhibition of PKN1 by Ro318220 (generally used as a PKC inhibitor) blocks proliferation of androgen receptor-induced tumour cell proliferation, suggesting specific inhibition of PKN1 as a potential therapeutic strategy in the treatment of prostate cancer. Leenders et al. [6] reported that inducible knockdown of PKN3 expression strongly inhibited the formation of lymph node metastasis in an orthotopic mouse prostate tumour model, indicating that the development of inhibitors against PKN3 holds promise for therapeutic intervention in cancers regulated by activated PI3K (phosphoinositide 3-kinase). Loh et al. [10] reported that PKN1 is essential for neurite retraction in a human neuroblastoma cell line, and that haploinsufficiency of Drosophila PKN1 suppresses light-induced retinal degeneration in a fly model of class III ADRP (autosomal dominant retinitis pigmentosa), indicating that PKN1 is a potential therapeutic target for neurodegenerative disease as well. The use of the PRL peptide in animal models for these diseases could provide further information as to whether PKN inhibition is an effective therapeutic strategy. Traditionally, peptides are considered less attractive compared with small molecule drugs due to limited delivery into tissues and susceptibility to degradation. However, a great deal of progress has been made recently to overcome these problems [29]. For example, CPP-fused peptide inhibitors against JNKs (c-Jun N-terminal kinases), which are non-selective with respect to JNK isoforms, have been recently regarded as a potential therapeutic agent for the treatment of various diseases such as cerebral ischaemia/stroke [50–52], viral encephalitis [53], pancreatic β-cell death [54,55], hearing disorder induced by trauma and neomycin toxicity [56,57], neuropathic pain [58] and myocardial ischaemia [59]. Delivery of a PKN inhibitory peptide such as PRL in vivo may also provide new therapeutic approaches to the treatment of cancer and neurodegenerative disease.
Supplementary Material
Acknowledgments
We thank Benjamin E. Turk for providing helpful comments on the manuscript. We also thank Dr Takuya Maeda for encouragement during this work.
FUNDING
This work was supported in part by a Grant-in-Aid for Scientific Research from the Global Center of Excellence (COE) programme of the Ministry of Education, Culture, Sports, Science and Technology, Japan; by a Grant-in-Aid for Scientific Research on Priority Areas “Life Surveyor”, from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to S.F.); and by a grant from the Mitsubishi Foundation. K.T. is grateful for a JSPS Research Fellowship for Young Scientists.
Abbreviations used
- ACC
antiparallel coiled-coil
- CPP
cell-penetrating peptide
- FHV
flock house virus
- GST
glutathione transferase
- JNK
c-Jun N-terminal kinase
- Pas
penetration accelerating sequence
- PKC
protein kinase C
- RNAi
RNA interference
Footnotes
AUTHOR CONTRIBUTION
Kazuhiro Shiga was involved in the overall kinase assays. Kentaro Takayama and Shiroh Futaki were involved in the preparation of the peptides. Jessica Hutti and Lewis Cantley were involved in the positional scanning peptide library assay. Katsuko Ueki was involved in the preparation of the purified protein substrates. Yoshitaka Ono edited the manuscript prior to submission. Hideyuki Mukai was involved in the design of the experiments and the overall kinase assays, and wrote the paper.
References
- 1.Mukai H. The structure and function of PKN, a protein kinase having a catalytic domain homologous to that of PKC. J Biochem. 2003;133:17–27. doi: 10.1093/jb/mvg019. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt A, Durgan J, Magalhaes A, Hall A. Rho GTPases regulate PRK2/PKN2 to control entry into mitosis and exit from cytokinesis. EMBO J. 2007;26:1624–1636. doi: 10.1038/sj.emboj.7601637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lim MA, Yang L, Zheng Y, Wu H, Dong LQ, Liu F. Roles of PDK-1 and PKN in regulating cell migration and cortical actin formation of PTEN-knockout cells. Oncogene. 2004;23:9348–9358. doi: 10.1038/sj.onc.1208147. [DOI] [PubMed] [Google Scholar]
- 4.Fischer A, Stuckas H, Gluth M, Russell TD, Rudolph MC, Beeman NE, Bachmann S, Umemura S, Ohashi Y, Neville MC, Theuring F. Impaired tight junction sealing and precocious involution in mammary glands of PKN1 transgenic mice. J Cell Sci. 2007;120:2272–2283. doi: 10.1242/jcs.03467. [DOI] [PubMed] [Google Scholar]
- 5.Su C, Deaton RA, Iglewsky MA, Valencia TG, Grant SR. PKN activation via transforming growth factor-β1 (TGF-β1) receptor signaling delays G2/M phase transition in vascular smooth muscle cells. Cell Cycle. 2007;6:739–749. doi: 10.4161/cc.6.6.3985. [DOI] [PubMed] [Google Scholar]
- 6.Leenders F, Mopert K, Schmiedeknecht A, Santel A, Czauderna F, Aleku M, Penschuck S, Dames S, Sternberger M, Rohl T, et al. PKN3 is required for malignant prostate cell growth downstream of activated PI 3-kinase. EMBO J. 2004;23:3303–3313. doi: 10.1038/sj.emboj.7600345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Metzger E, Yin N, Wissmann M, Kunowska N, Fischer K, Friedrichs N, Patnaik D, Higgins JM, Potier N, Scheidtmann KH, et al. Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat Cell Biol. 2008;10:53–60. doi: 10.1038/ncb1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng PY, Rane N, Du W, Chintapalli J, Prendergast GC. Role for RhoB and PRK in the suppression of epithelial cell transformation by farnesyltransferase inhibitors. Oncogene. 2003;22:1124–1134. doi: 10.1038/sj.onc.1206181. [DOI] [PubMed] [Google Scholar]
- 9.Kawamata T, Taniguchi T, Mukai H, Kitagawa M, Hashimoto T, Maeda K, Ono Y, Tanaka C. A protein kinase, PKN, accumulates in Alzheimer neurofibrillary tangles and associated endoplasmic reticulum-derived vesicles and phosphorylates tau protein. J Neurosci. 1998;18:7402–7410. doi: 10.1523/JNEUROSCI.18-18-07402.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loh SH, Francescut L, Lingor P, Bahr M, Nicotera P. Identification of new kinase clusters required for neurite outgrowth and retraction by a loss-of-function RNA interference screen. Cell Death Differ. 2008;15:283–298. doi: 10.1038/sj.cdd.4402258. [DOI] [PubMed] [Google Scholar]
- 11.Manser C, Stevenson A, Banner S, Davies J, Tudor EL, Ono Y, Leigh PN, McLoughlin DM, Shaw CE, Miller CC. Deregulation of PKN1 activity disrupts neurofilament organisation and axonal transport. FEBS Lett. 2008;582:2303–2308. doi: 10.1016/j.febslet.2008.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Metzger E, Muller JM, Ferrari S, Buettner R, Schule R. A novel inducible transactivation domain in the androgen receptor: implications for PRK in prostate cancer. EMBO J. 2003;22:270–280. doi: 10.1093/emboj/cdg023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gotoh Y, Oishi K, Shibata H, Yamagiwa A, Isagawa T, Nishimura T, Goyama E, Takahashi M, Mukai H, Ono Y. Protein kinase PKN1 associates with TRAF2 and is involved in TRAF2-NF-κB signaling pathway. Biochem Biophys Res Commun. 2004;314:688–694. doi: 10.1016/j.bbrc.2003.12.148. [DOI] [PubMed] [Google Scholar]
- 14.Bourguignon LY, Gilad E, Peyrollier K, Brightman A, Swanson RA. Hyaluronan-CD44 interaction stimulates Rac1 signaling and PKNγ kinase activation leading to cytoskeleton function and cell migration in astrocytes. J Neurochem. 2007;101:1002–1017. doi: 10.1111/j.1471-4159.2007.04485.x. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi M, Gotoh Y, Isagawa T, Nishimura T, Goyama E, Kim HS, Mukai H, Ono Y. Regulation of a mitogen-activated protein kinase kinase kinase, MLTK by PKN. J Biochem. 2003;133:181–187. doi: 10.1093/jb/mvg022. [DOI] [PubMed] [Google Scholar]
- 16.Mukai H, Kitagawa M, Shibata H, Takanaga H, Mori K, Shimakawa M, Miyahara M, Hirao K, Ono Y. Activation of PKN, a novel 120-kDa protein kinase with leucine zipper-like sequences, by unsaturated fatty acids and by limited proteolysis. Biochem Biophys Res Commun. 1994;204:348–356. doi: 10.1006/bbrc.1994.2466. [DOI] [PubMed] [Google Scholar]
- 17.Palmer RH, Schonwasser DC, Rahman D, Pappin DJ, Herget T, Parker PJ. PRK1 phosphorylates MARCKS at the PKC sites: serine 152, serine 156 and serine 163. FEBS Lett. 1996;378:281–285. doi: 10.1016/0014-5793(95)01454-3. [DOI] [PubMed] [Google Scholar]
- 18.Matsuzawa K, Kosako H, Azuma I, Inagaki N, Inagaki M. Possible regulation of intermediate filament proteins by Rho-binding kinases. Subcell Biochem. 1998;31:423–435. [PubMed] [Google Scholar]
- 19.Betson M, Settleman J. A rho-binding protein kinase C-like activity is required for the function of protein kinase N in Drosophila development. Genetics. 2007;176:2201–2212. doi: 10.1534/genetics.107.072967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukai H, Ono Y. Purification and kinase assay of PKN. Methods Enzymol. 2006;406:234–250. doi: 10.1016/S0076-6879(06)06017-4. [DOI] [PubMed] [Google Scholar]
- 21.Sheridan DL, Kong Y, Parker SA, Dalby KN, Turk BE. Substrate discrimination among mitogen-activated protein kinases through distinct docking sequence motifs. J Biol Chem. 2008;283:19511–19520. doi: 10.1074/jbc.M801074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Futaki S, Suzuki T, Ohashi W, Yagami T, Tanaka S, Ueda K, Sugiura Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem. 2001;276:5836–5840. doi: 10.1074/jbc.M007540200. [DOI] [PubMed] [Google Scholar]
- 23.Futaki S, Kitagawa K. Peptide-unit assembling via disulfide cross-linking: a versatile approach which enables the creation of artificial. Tetrahedron. 1997;53:7479–7492. [Google Scholar]
- 24.Yoshinaga C, Mukai H, Toshimori M, Miyamoto M, Ono Y. Mutational analysis of the regulatory mechanism of PKN: the regulatory region of PKN contains an arachidonic acid-sensitive autoinhibitory domain. J Biochem. 1999;126:475–484. doi: 10.1093/oxfordjournals.jbchem.a022476. [DOI] [PubMed] [Google Scholar]
- 25.Misaki K, Mukai H, Yoshinaga C, Oishi K, Isagawa T, Takahashi M, Ohsumi K, Kishimoto T, Ono Y. PKN delays mitotic timing by inhibition of Cdc25C: possible involvement of PKN in the regulation of cell division. Proc Natl Acad Sci USA. 2001;98:125–129. doi: 10.1073/pnas.021541498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turk BE, Hutti JE, Cantley LC. Determining protein kinase substrate specificity by parallel solution-phase assay of large numbers of peptide substrates. Nat Protoc. 2006;1:375–379. doi: 10.1038/nprot.2006.57. [DOI] [PubMed] [Google Scholar]
- 27.Taniguchi T, Kawamata T, Mukai H, Hasegawa H, Isagawa T, Yasuda M, Hashimoto T, Terashima A, Nakai M, Mori H, et al. Phosphorylation of tau is regulated by PKN. J Biol Chem. 2001;276:10025–10031. doi: 10.1074/jbc.M007427200. [DOI] [PubMed] [Google Scholar]
- 28.Takeuchi T, Kosuge M, Tadokoro A, Sugiura Y, Nishi M, Kawata M, Sakai N, Matile S, Futaki S. Direct and rapid cytosolic delivery using cell-penetrating peptides mediated by pyrenebutyrate. ACS Chem Biol. 2006;1:299–303. doi: 10.1021/cb600127m. [DOI] [PubMed] [Google Scholar]
- 29.Kaidanovich-Beilin O, Eldar-Finkelman H. Peptides targeting protein kinases: strategies and implications. Physiology (Bethesda) 2006;21:411–418. doi: 10.1152/physiol.00022.2006. [DOI] [PubMed] [Google Scholar]
- 30.Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, Turk BE. A rapid method for determining protein kinase phosphorylation specificity. Nat Methods. 2004;1:27–29. doi: 10.1038/nmeth708. [DOI] [PubMed] [Google Scholar]
- 31.Fujii K, Zhu G, Liu Y, Hallam J, Chen L, Herrero J, Shaw S. Kinase peptide specificity: improved determination and relevance to protein phosphorylation. Proc Natl Acad Sci USA. 2004;101:13744–13749. doi: 10.1073/pnas.0401881101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishikawa K, Toker A, Johannes FJ, Songyang Z, Cantley LC. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–960. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- 33.Kemp BE, Pearson RB. Protein kinase recognition sequence motifs. Trends Biochem Sci. 1990;15:342–346. doi: 10.1016/0968-0004(90)90073-k. [DOI] [PubMed] [Google Scholar]
- 34.Soderling TR. Protein kinases. Regulation by autoinhibitory domains. J Biol Chem. 1990;265:1823–1826. [PubMed] [Google Scholar]
- 35.Peng B, Morrice NA, Groenen LC, Wettenhall RE. Phosphorylation events associated with different states of activation of a hepatic cardiolipin/protease-activated protein kinase. Structural identity to the protein kinase N-type protein kinases. J Biol Chem. 1996;271:32233–32240. doi: 10.1074/jbc.271.50.32233. [DOI] [PubMed] [Google Scholar]
- 36.Eichholtz T, de Bont DB, de Widt J, Liskamp RM, Ploegh HL. A myristoylated pseudosubstrate peptide, a novel protein kinase C inhibitor. J Biol Chem. 1993;268:1982–1986. [PubMed] [Google Scholar]
- 37.O’Brian CA, Ward NE, Liskamp RM, de Bont DB, van Boom JH. N-myristyl-Lys-Arg-Thr-Leu-Arg: a novel protein kinase C inhibitor. Biochem Pharmacol. 1990;39:49–57. doi: 10.1016/0006-2952(90)90647-4. [DOI] [PubMed] [Google Scholar]
- 38.Futaki S. Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv Drug Delivery Rev. 2005;57:547–558. doi: 10.1016/j.addr.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 39.Futaki S. Oligoarginine vectors for intracellular delivery: design and cellular-uptake mechanisms. Biopolymers. 2006;84:241–249. doi: 10.1002/bip.20421. [DOI] [PubMed] [Google Scholar]
- 40.Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- 41.Chen L, Wright LR, Chen CH, Oliver SF, Wender PA, Mochly-Rosen D. Molecular transporters for peptides: delivery of a cardioprotective epsilonPKC agonist peptide into cells and intact ischemic heart using a transport system, R(7) Chem Biol. 2001;8:1123–1129. doi: 10.1016/s1074-5521(01)00076-x. [DOI] [PubMed] [Google Scholar]
- 42.Nakase I, Niwa M, Takeuchi T, Sonomura K, Kawabata N, Koike Y, Takehashi M, Tanaka S, Ueda K, Simpson JC, et al. Cellular uptake of arginine-rich peptides: roles for macropinocytosis and actin rearrangement. Mol Ther. 2004;10:1011–1022. doi: 10.1016/j.ymthe.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 43.Inomata K, Ohno A, Tochio H, Isogai S, Tenno T, Nakase I, Takeuchi T, Futaki S, Ito Y, Hiroaki H, Shirakawa M. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature. 2009;458:106–109. doi: 10.1038/nature07839. [DOI] [PubMed] [Google Scholar]
- 44.Lu Y, Settleman J. The Drosophila Pkn protein kinase is a Rho/Rac effector target required for dorsal closure during embryogenesis. Genes Dev. 1999;13:1168–1180. doi: 10.1101/gad.13.9.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amano M, Mukai H, Ono Y, Chihara K, Matsui T, Hamajima Y, Okawa K, Iwamatsu A, Kaibuchi K. Identification of a putative target for Rho as the serine-threonine kinase protein kinase N. Science. 1996;271:648–650. doi: 10.1126/science.271.5249.648. [DOI] [PubMed] [Google Scholar]
- 46.Watanabe G, Saito Y, Madaule P, Ishizaki T, Fujisawa K, Morii N, Mukai H, Ono Y, Kakizuka A, Narumiya S. Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase Rho. Science. 1996;271:645–648. doi: 10.1126/science.271.5249.645. [DOI] [PubMed] [Google Scholar]
- 47.Calautti E, Grossi M, Mammucari C, Aoyama Y, Pirro M, Ono Y, Li J, Dotto GP. Fyn tyrosine kinase is a downstream mediator of Rho/PRK2 function in keratinocyte cell-cell adhesion. J Cell Biol. 2002;156:137–148. doi: 10.1083/jcb.200105140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 49.Lee JH, Nandy SK, Lawrence DS. A highly potent and selective PKCα inhibitor generated via combinatorial modification of a peptide scaffold. J Am Chem Soc. 2004;126:3394–3395. doi: 10.1021/ja037300b. [DOI] [PubMed] [Google Scholar]
- 50.Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, Bogousslavsky J, Bonny C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 51.Esneault E, Castagne V, Moser P, Bonny C, Bernaudin M. D-JNKi, a peptide inhibitor of c-Jun N-terminal kinase, promotes functional recovery after transient focal cerebral ischemia in rats. Neuroscience. 2008;152:308–320. doi: 10.1016/j.neuroscience.2007.12.036. [DOI] [PubMed] [Google Scholar]
- 52.Hirt L, Badaut J, Thevenet J, Granziera C, Regli L, Maurer F, Bonny C, Bogousslavsky J. D-JNKI1, a cell-penetrating c-Jun-N-terminal kinase inhibitor, protects against cell death in severe cerebral ischemia. Stroke. 2004;35:1738–1743. doi: 10.1161/01.STR.0000131480.03994.b1. [DOI] [PubMed] [Google Scholar]
- 53.Beckham JD, Goody RJ, Clarke P, Bonny C, Tyler KL. Novel strategy for treatment of viral central nervous system infection by using a cell-permeating inhibitor of c-Jun N-terminal kinase. J Virol. 2007;81:6984–6992. doi: 10.1128/JVI.00467-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. Cell-permeable peptide inhibitors of JNK: novel blockers of β-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- 55.Noguchi H, Matsumoto S, Onaca N, Naziruddin B, Jackson A, Ikemoto T, Shimoda M, Fujita Y, Chujo D, Iwanaga Y, et al. Ductal injection of JNK inhibitors before pancreas preservation prevents islet apoptosis and improves islet graft function. Hum, Gene Ther. 2009;20:73–85. doi: 10.1089/hum.2008.125. [DOI] [PubMed] [Google Scholar]
- 56.Eshraghi AA, Wang J, Adil E, He J, Zine A, Bublik M, Bonny C, Puel JL, Balkany TJ, Van De Water TR. Blocking c-Jun-N-terminal kinase signaling can prevent hearing loss induced by both electrode insertion trauma and neomycin ototoxicity. Hearing Res. 2007;226:168–177. doi: 10.1016/j.heares.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Van De Water TR, Bonny C, de Ribaupierre F, Puel JL, Zine A. A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J Neurosci. 2003;23:8596–8607. doi: 10.1523/JNEUROSCI.23-24-08596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milano G, Morel S, Bonny C, Samaja M, von Segesser LK, Nicod P, Vassalli G. A peptide inhibitor of c-Jun NH2-terminal kinase reduces myocardial ischemia-reperfusion injury and infarct size in vivo. Am J Physiol Heart Circ Physiol. 2007;292:H1828–H1835. doi: 10.1152/ajpheart.01117.2006. [DOI] [PubMed] [Google Scholar]
- 60.Kemp BE, Pearson RB, House C. Role of basic residues in the phosphorylation of synthetic peptides by myosin light chain kinase. Proc Natl Acad Sci USA. 1983;80:7471–7475. doi: 10.1073/pnas.80.24.7471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.