

Abstract
Monoclonal antibodies against GD2 ganglioside, such as ch14.18, the human–mouse chimeric antibody, have been shown to be effective for the treatment of neuroblastoma. However, treatment is associated with generalized, relatively opiate-resistant pain. We investigated if a point mutation in ch14.18 antibody (hu14.18K332A) to limit complement-dependent cytotoxicity (CDC) would ameliorate the pain behavior, while preserving antibody-dependent cellular cytotoxicity (ADCC). In vitro, CDC and ADCC were measured using europium-TDA assay. In vivo, allodynia was evaluated by measuring thresholds to von Frey filaments applied to the hindpaws after injection of either ch14.18 or hu14.18K332 into wild type rats or rats with deficient complement factor 6. Other rats were pretreated with complement factor C5a receptor antagonist and tested following ch14.18 injection. The mutation reduces the antibody’s ability to activate complement, while maintaining its ADCC capabilities. Injection of hu14.18K322 (1 or 3 mg/kg) produced faster resolving allodynia than that engendered by ch14.18 (1 mg/kg). Injection of ch14.18 (1 mg/kg) into rats with C6 complement deficiency further reduced antibody-induced allodynia, while pre-treatment with complement factor C5a receptor antagonist completely abolished ch14.18-induced allodynia. These findings showed that mutant hu14.18 K322 elicited less allodynia than ch14.18 and that ch14.18-elicited allodynia is due to activation of the complement cascade: in part, to formation of membrane attack complex, but more importantly to release of complement factor C5a. Development of immunotherapeutic agents with decreased complement-dependent lysis while maintaining cellular cytotoxicity may offer treatment options with reduced adverse side effects, thereby allowing dose escalation of therapeutic antibodies.
Keywords: Anti-GD2, Pain, C5a complement factor, Membrane attack complex, Neuroblastoma, Neuroblastoma therapy, Complement-dependent cytotoxicity, Antibody-dependent cellular cytotoxicity
1. Introduction
The GD2 ganglioside is enriched in plasma membranes of tumor cells of neuroectodermal origin [29] and to a lesser extent, in peripheral nerves [33]. Anti-GD2 antibodies mediate lysis of neuroblastoma and melanoma cells via complement-dependent cytotoxicity (CDC) and by activation of Fc receptors on granulocytes and mononuclear cells. This latter effect leads to antibody-dependent cellular cytotoxicity (ADCC) [2]. Thus, a chimeric anti-GD2 antibody (ch14.18) demonstrated therapeutic efficacy for patients with refractory neuroblastoma in early clinical trials [38] and significantly improved event-free survival and overall survival in a phase III randomized clinical trial in patients with high risk neuroblastoma [37]. However, systemic administration of GD2 antibodies elicits spontaneous intense visceral pain and perceived pain in response to light touch (allodynia) in patients [7,35] and increased mechanical sensitivity in rats [31,32]. The pain is relatively morphine resistant [35], occurs with the same time course in both humans and rats and, in our experimental model, is associated with ectopic activity in afferent C fibers [36]. Although several mechanisms have been postulated, activation of the complement cascade, which then elicits local neuronal activity, has long been suspected. In clinical trials, anti-GD2 administration is associated with initial increases of complement component C3a and concurrent decreases of C3c along with a maintained decrease in C4 [17]. Local application of complement cascade activators such as zymosan, to a sensory nerve causes pain behavior with characteristics similar to that seen for anti-GD2, i.e. sensitization to touch, but not to temperature [5,32]. Activation of the complement cascade is well established as an initiator of pain and hyperalgesia [23] and blockade of the alternative complement pathway prevents zymosan-induced pain [34]. To ameliorate the undesirable side effect of pain, a new version of the anti-GD2 antibody (hu14.18K322A) has been made which contains a point mutation that is expected to keep it from fully activating complement. Importantly, a reduction in ADCC resulting from this mutation was overcome by expression of the mutant in a cell line that enhances binding to FcRIII and thus, ADCC should be minimally affected by this mutation. To facilitate clinical development of hu14.18K322A, we aimed first to demonstrate that this mutation indeed reduces complement activation with minimal reduction of ADCC. The second aim was to determine to what extent this mutation and decrement of complement activation altered evoked pain behavior in comparison with pain behavior elicited by the original antibody. Last, the ability of intravenous treatment with ch14.18, the antibody form that is currently under clinical trials, was tested in animals pretreated with a C5a complement receptor antagonist and separately in a rat strain with deficient C6 complement to demonstrate by alternative strategies that an intact complement pathway is, in fact, necessary for the anti-GD2- associated allodynia.
2. Materials and methods
2.1. Anti-GD2 monoclonal antibodies
The human-mouse chimeric anti-GD2 antibody (ch14.18) is composed of the variable regions of the murine anti-GD2 monoclonal antibody (mAb), 14.18 and the constant regions of human IgG1-k [14]. Stock solutions were kept at 4 mg/ml at 4 °C and diluted with sterile isotonic saline to 1 mg/ml at the time of the experiment. The GD2 antibody variant hu14.18K322A is humanized and shares identical C regions of IgG1-k as ch14.18 with the exception of a mutation to alanine at lysine 322 that limits its ability to fix complement. The molecule was expressed in YB2/0 rat myeloma cells following electroporation and subsequent selection in culture medium containing methotrexate (100 nM). Surviving clones were tested for human antibody secretion and ADCC activity, relative to the non-mutated antibody (hu14.18). Following purification with protein A Sepharose, this agent was kept as a stock solution at 2.8 mg/ml at 4 °C before saline dilution to 1 mg/ml for delivery at 1 mg/kg. In order to achieve doses of 3 mg/ml the stock solution was not diluted and 1.07 ml/kg was injected. Hu14.18K322A has similar affinity for the GD2 ganglioside as ch14.18 (unpublished data) and is expected to have a similar pharmacokinetic profile.
2.2. Isolation of natural killer (NK) cells
Peripheral blood mononuclear cells from healthy donors were collected at St. Jude Children’s Research Hospital (Memphis, TN) using a protocol approved by the St. Jude Hospital Institutional Review Board. Natural killer cells (CD56+ CD3−) were harvested from peripheral blood mononuclear cells using an indirect magnetic labeling system for the isolation of untouched NK cells with subsequent enrichment on the AutoMACS device (Miltenyi Biotech, Auburn, CA). Labeling and enrichment were performed according to manufacturer’s instructions.
2.3. Flow-cytometric analysis
Purity of the isolated NK cells was determined by flow cytometry. Expression of the natural killer cell markers CD16 and CD56 was determined using anti-CD16 labeled with fluorescein isothiocyanate (FITC), (BD Bioscience, San Diego, CA), and anti-CD56 labeled with allophycocyanin (APC) (Beckman-Coulter, Miami, FL). Cells were stained with a saturating amount of antibody according to the manufacturer’s protocol. Flow-cytometric data acquisition and analysis was performed on a BD LSR flow cytometer (Beckton–Dickinson, Mountain View, CA) using Cell Quest software. The overall purity of the isolated NK cells was >85% (Fig. 1A).
Fig. 1.

(A) Flow-cytometric analysis of one isolated natural killer cell population as defined by cells expressing both CD56 (y axis) and CD16 (x axis), indicating greater than 85% purity. (B) ADCC was measured by Europium-TDA release assay. NK cells from 10 healthy donors were incubated with 5 × 103 NB-1691 cells prelabeled with BATDA at an effector: target ratio of 10:1. Antibody-dependent cellular cytotoxicity is shown for each sample alone or following 2 h incubation with ch14.18 or hu14.18K322A. Results indicate that hu14.18K322A incubation results in only a modest decrease in ADCC compared to ch14.18.
2.4. In Vitro studies: comparison of ch14.18 with hu14.18K322A
2.4.1. Antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent lysis (CDC)
Antibody-dependent cellular cytotoxicity and CDC were measured in a conventional 2-h europium-TDA assay (Perkin–Elmer Wallac, Turku, Finland) as described previously [3]. Briefly, NB-1691 neuroblastoma cells (kindly provided by Children’s Oncology Group, Arcadia, CA) were labeled with the hydrophobic fluorescence-enhancing ligand (BATDA) according to the manufacturer’s protocol. Under basal conditions, intracellular hydrolysis of ester bonds renders the ligand hydrophilic and BATDA is unable to pass through the cell membrane. Cytolysis results in ligand release from the cytosol into the supernatant where it reacts with europium to form a stable, fluorescent chelate. Labeled NB-1691 neuroblastoma cells were incubated for 2 h in a 96-well plate, 5 × 103/well at 37 °C in a 5% CO2 atmosphere and 95% humidity with NK cells in an effector-to-target cell ratio of 10:1 in triplicate, with or without 1 μg/ml anti-GD2 (ch14.18) or mutated anti-GD2 (hu14.18K322A). Fluorescence was measured using a Perkin-Elmer Wallac Victor 2 device.
For CDC, target cells were incubated with ch14.18 or hu14.18K322A (0.1–10 μg/ml) in the presence of 0.5% human complement serum (Sigma–Aldrich, St. Louis, MO). Following incubation, supernatant was allowed to react with europium solution and the time-resolved fluorescence was measured using a Perkin-Elmer Wallac Victor 2 device. To calculate spontaneous and specific cytotoxicity, the following formulae were used:
Spontaneous release was determined by incubation of target cells in culture medium without effector cells. Maximal release was determined by incubating target cells in 0.2% SDS before harvesting supernatant.
2.5. In vivo studies
Experiments using adult (200–300 g) male Sprague–Dawley, PVG (C+) (Harlan Industries, Indianapolis, IN), and PVG (C−) rats, were approved by the Institutional Animal Care and Use Committee of the University of California, San Diego. PVG (C−) rats have a profound deficiency in C6 complement; they were bred at Johns Hopkins University [4] by WMB. Absence of C6 complement does not effect generation of upstream complement components C3a or C5a, but eliminates the ability of the activated complement cascade to generate membrane attack complex (MAC). All rats were allowed to recover from shipping for at least 72 h prior to experiments. Animals were acclimated to the test facility for a minimum of 1 h before being placed in individual plastic compartments (26 × 11 × 20 cm) with wire mesh floors for 30 min prior to the start of the experiment. After acclimation, basal 50% probability mechanical withdrawal threshold for each rat was determined. Animals were then lightly anesthetized with isoflorane (about 2%) and injected with antibody or vehicle through the tail vein, using a 30 g needle. Anesthesia was discontinued after injection and the animal placed back in the testing compartment. Rats were awake within 3 min and in most experiments, mechanical withdrawal thresholds were re-measured every 15 min for the first 2 h after injection of agent and every 30 min for an additional 2.5 h. Experiments began between 9 and 10 a.m. Animals used in the study that directly compared ch14.18 and hu14.18K322A were returned to their home cages for the night and brought back to the testing room, re-acclimated and tested at 24 h and again at 48 h post-injection. Animals were kept two to a cage under a 12/12 h day/night cycle with food and water available ad libitum.
2.5.1. Comparison of ch14.18 with hu14.18K322A
Sprague–Dawley rats were injected with chimeric anti-GD2 (1 mg/kg; N = 8) (ch14.18), an equal amount of the mutated hu14.18K322A, (N = 8) (hu-1), an increased dose (3 mg/kg; N = 7) of the mutated antibody (hu-3) or saline (N = 8). One mg/kg of ch14.18 anti-GD2 produces maximum allodynia in the rat [31]. The increased dose of hu14.18K322A was used to determine if the reduction in complement activation resulted in a right shift of the dose-allodynia curve, i.e. more hu14.18K322A was required to observe the maximal behavioral effect. The person performing the behavioral testing was blinded to the contents of the syringe. Mechanical withdrawal thresholds following the various treatments were measured and compared.
2.5.2. Allodynic actions of ch14.18 in rats with compromised complement systems
2.5.2.1. Blockade of C5a complement receptor
One h before the start of the experiment Sprague–Dawley rats were subcutaneously injected mid-trunk with either saline (N = 5) or the C5a complement receptor antagonist (3 mg/kg; N = 5). The C5a receptor antagonist, a synthetic cyclic AcF-[OPdChaWR] peptide was produced in the peptide core of the Genomics Research Center, Academia Sinica, Taiwan [12]. Following determination of baseline withdrawal thresholds, animals were lightly anesthetized and injected with 1 mg/ml of ch14.18 through the tail vein. The experimenter was blinded as to the pre-treatment. Animals were examined at 30 min intervals for 5.0 h after the tail vein injection of GD2 antibody.
2.5.3. Elimination of membrane attack complex via loss of C6 complement factor
Lightly anesthetized PVG (C+; N = 7) and PVG (C−; N = 9) rats were injected with 1 mg/ml of ch14.18 through the tail vein. Animals were examined only for the first 4.5 h and not at the later time points. Due to low availability of PVG (C−) rats, both strains of PVG rat were re-used at minimal intervals of 1 week. Results did not change between first and second usage. A second set of PVG (C−) rats were injected with 1 or 3 mg/kg of hu14.18K322A or saline through the tail vein (N = 4–9/group). In all cases, the experimenter was blinded as to the genetic make-up of the animals. All testing was as described above.
2.6. Behavioral testing
Mechanical withdrawal threshold was measured with a set of von Frey filaments (Stoelting) with exponentially incremental bending forces ranging from 0.41 to 15.1 g. When the animal was quiet and resting on all four paws, a filament was presented perpendicular to the plantar surface of the hindpaw with sufficient force to elicit a slight bend. Filaments, beginning with the 2.0 g filament, were presented in ascending order of stiffness until an abrupt paw withdrawal (escape) or the stiffest (15.10 g) filament in the set was applied. Stimuli were maintained for 6 s. Successive stimuli were separated by several seconds or until the animal was again calm with hindpaws placed flat on the mesh flooring. Testing followed an up–down paradigm [6], i.e., when a response was made, filaments of decreasing strength were applied until the animal no longer responded, at which point filaments were again presented in ascending order. This pattern was repeated for four stimulus presentations after the first withdrawal response. The 50% probability withdrawal threshold was calculated [11]. Animals that did not respond to the stiffest filament, were considered to have thresholds at cut-off (15.1 g). This process was repeated for both the left and right hindpaws at each timepoint, the average value of the two hindpaws was considered to be the animal’s response. Area over the curve was calculated for 0–2 h, heuristically defined as induction and early allodynia; and 2–4.5 h, which we defined as maintenance of allodynia.
2.7. Statistics
Group results are illustrated as means ± standard error of the mean (S.E.M.). Analysis of variance (ANOVA) and post hoc Dunnetts tests were used to compare ADCC data. Percent allodynia area over the curve (AOC) was calculated for each animal based on their individual baseline responses using the trapezoidal method. This was then normalized such that 0 = no change from baseline and 100 = maximal measurable allodynia. Statistics were performed on the 50% probability withdrawal threshold using ANOVA for repeated measures and Bonferroni’s post hoc test. Unpaired t-tests or one way ANOVAs were used to compare percent allodynia, area under the curve, for different treatment (strain) groups. p ≥ 0.05 was considered to be significant.
3. Results
3.1. In vitro experiments
3.1.1. Comparison of ADCC mediated by ch14.18 and hu14.18K322A
The capacity of ch14.18 and hu14.18 to mediate ADCC was determined using NK cells isolated from 10 healthy donors. As shown in Fig. 1B, the natural cytotoxicities for these individuals ranged from 8% to 32% specific lysis of target cells; with a mean of 18.4% ± 2.5. In the presence of 1.0 μg/ml anti-GD2, specific lysis of target cells rose to 66–98% (mean 81.2% ± 3.6) for ch14.18 and 26–96% (mean 64.4% ± 7.1) for hu14.18K322A. As expected, for all individuals, the ADCC activities mediated by both ch14.18 and hu14.18K322A were substantially higher than seen in the untreated NK cells (p ≥ 0.0001; analysis of variance (ANOVA)). Importantly, cytotoxicity elicited by hu14.18K322A was almost comparable to that seen following incubation with ch14.18 (p = 0.053, unpaired t test). In samples from 4/10 donors, specific lysis elicited by hu14.18K322A was equivalent to that induced by ch14.18, higher than ch14.18 in one sample, and lower than ch14.18 in the remaining individuals. Overall, hu14.18K322A clearly retains significant ADCC activity.
3.2. Comparison of CDC mediated by ch14.18 and hu14.18K322A
To determine the capacity of ch14.18 and hu14.18K322A to mediate CDC, neuroblastoma cells were incubated with varying concentrations of ch14.18 or hu14.18K322A in the presence of 0.5% human complement serum. In the absence of antibody, there was very little cell lysis (Fig. 2). The antibody ch14.18 displayed a concentration dependent CDC activity with 20.2% lysis or 2–3 times basal levels at 0.1 μg/ml. At the highest concentration, (10 μg/ml), 64.3% specific lysis of the target cells was observed. In marked contrast, hu14.18K322A showed no appreciable CDC activity at 0.1 and 1 μg/ml compared to the level seen with no antibody. At the highest concentration of hu14.18K322A, only 20.5% lysis was observed, equivalent to that seen with 100 times less ch14.18. Thus, the mutant hu14.18K322A had greatly diminished capacity to mediate CDC, as compared to ch14.18.
Fig. 2.
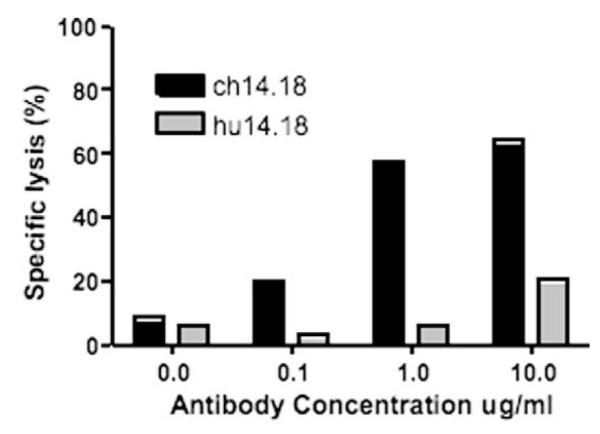
Complement-dependent cytotoxicity in the presence of varying concentrations of ch14.18 or hu14.18K322A was determined in a 2 h Europium-TDA release assay. In the presence of 0.5% human serum complement ch14.18 elicits a much stronger CDC than does hu14.18K322A.
3.3. In vivo experiments
After demonstrating that hu14.18K322A maintained the ability to mediate ADCC by natural killer cells and displayed reduced capacity to fix complement in vitro, we compared hu14.18 to ch14.18 in vivo, by injecting one or the other antibody into the rat-tail vein and testing mechanical thresholds.
3.4. Comparison of allodynia caused by ch14.18 and hu14.18K322A
Prior to injection, rats typically had 50% probability withdrawal thresholds close to cut-off (15.1 g) and mean thresholds among the four treatment groups were similar (range 14.2 g ± 0.7 to 14.8 g ± 0.2). After IV injection of saline, thresholds were essentially unchanged for the next 48 h. Percent allodynia averaged 6 ± 3 for the first 4.5 h. In contrast, injection of ch14.18 (1 mg/kg), caused thresholds to fall precipitously (Fig. 3A). At 30 min post-injection, mean mechanical threshold was 6.5 g ± 1.5 and mean threshold remained reduced and stayed between 4.3–6.8 g for the next 4 h. Although thresholds had started to recover, they remained significantly depressed at 24 h as compared to both baseline and to saline treated animals. Full recovery and total loss of allodynia was not observed until 48 h post-injection. These results duplicate those seen in earlier studies [15,31]. Treatment with either 1 or 3 mg/kg of hu14.18K322A also resulted in a steep decrease in mechanical withdrawal threshold over the first hour, similar, but with a tendency to be smaller than that observed for ch14.18. For the first 2 h, the general pattern of allodynia was the same for all three antibody treatments; ANOVA for the areas over the curve (AOC) for the four groups for this period yielded a p value ≥0.0001 and post hoc tests indicated that all three treatments resulted in allodynia. Although, there was a tendency towards greater allodynia following ch14.18, this was not significant for the AOCs compared to either the 1 or 3 mg/kg dose of hu14.18K322A (p = 0.15 and 0.13, respectively; Fig. 3B). However, after 2 h, pain behavior was significantly more prominent in animals treated with ch14.18 than those treated with hu14.18K322A. Animals injected with either dose of hu14.18K322A displayed AOCs for this period that were no different than those injected with saline and thresholds approached baseline after 4 h.
Fig. 3.

(A) Intravenous injection of 1 mg/kg of ch14.18 in Sprague–Dawley rats results in a prolonged decrease in mechanical withdrawal thresholds, which was still significant 24 h post-injection. In contrast, hu14.18K322A injected at 1 or 3 mg/kg resulted in a slightly less severe decrease in threshold that began to resolve 2 h post-injection and was totally gone by 3.5 h. Thresholds of animals injected with saline are shown for comparison. (B) Areas over the curve (increasing numbers indicate more allodynia) are shown for the first 2 h post-injection and 2–4.5 h post-injection. Ch14.18 = 1 mg/kg ch14.18, hu 1 and hu 3 = 1 and 3 mg/kg hu14.18K322A, respectively. Thresholds of hu 1 and hu 3 rats were not different from each other during any time period.
3.5.1. Allodynic actions of ch14.18 in rats with compromised complement systems
In order to confirm that loss of allodynia in the hu14.18K322A treated animals was due to loss of CDC activity rather than to the small decrement in ADCC activity, we performed two different experiments to eliminate specific components of the complement cascade. First, we used animals that were pretreated with a receptor antagonist to block the actions of complement factor C5a, which leaves both the upstream C3a and downstream membrane attack complex systems unaffected. Rats pretreated with the antagonist displayed a total lack of response to injection of 1 mg/kg ch14.18 (Fig. 4); percent AOC was 4 ± 2% for the first 2 h and 5 ± 2% for the 2.5–5.0 h after the tail injection. In comparison, percent AOCs were 37 ± 9% and 40 ± 5%, respectively, for simultaneously tested animals given saline as a pre-treatment.
Fig. 4.
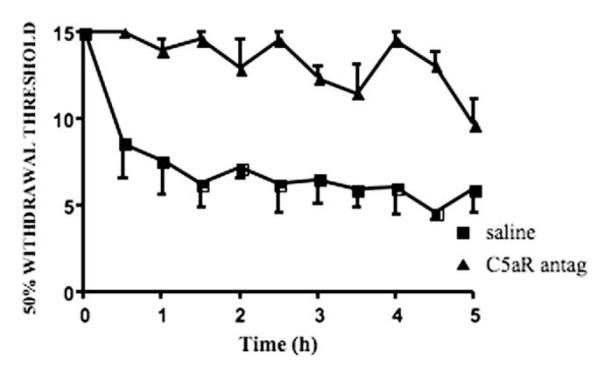
Blockade of complement fragment C5a prevents development of ch14.18-induced allodynia. Animals were pretreated with either subcutaneous saline or C5a receptor antagonist, 1 h later intravenous GD2 antibody induced a robust allodynia in the saline pretreated animals, but no pain behavior in animals with antagonist.
Next we injected ch14.18, hu14.18K322A and saline (not shown) into PVG (C−) rats with non-functional C6 complement and ch14.18 into their wild type (PVG C+) counterparts (Fig. 5). Baseline withdrawal thresholds of both PVG strains were virtually identical with that seen for Sprague–Dawley rats. The PVG (C+) rats, with intact complement cascades, developed allodynia following injection of the ch14.18 at the same rate and of the same magnitude as did the Sprague–Dawley rats and it was maintained for the entire observation period. Mean thresholds remained depressed until the end of the experiment and the percent AOC was 42 ± 3% for the time period between 2 and 4.5 h. Analysis showed no significant difference between PVG (C+) and Sprague–Dawley rats, injected with 1 mg/kg ch14.18 in the previous experiment, for either the early or the maintained phase of allodynia (0–2 h, p ≥ 0.19; 2–4.5 h, p ≥ 0.38).
Fig. 5.

(A) Intravenous injection of 1 mg/kg of ch14.18 in PVG (C+) rats results in a prolonged decrease in mechanical withdrawal thresholds that were comparable to that seen in Sprauge–Dawley rats. In marked contrast, the same injection in PVG (C−) rats resulted in much less severe allodynia that varied significantly from both the Sprague–Dawley and the PVG (C+) rats injected with ch14.18. Injection of hu14.18K322A into C− rats resulted in a strong tendency towards even less allodynia. (B) Areas over the curve indicate the sum of activity over the first 2 h post-injection and the period from 2.5–4.5 h post-injection. C− rats injected with either dose of hu were no different than saline injected animals. *p 6 0.001–0.02 compared to C+ch1; #p 6 0.01 compared to C−ch1.
In marked contrast, PVG (C−) rats developed minimal allodynia. At 1 h post-injection, thresholds of the PVG (C−) rats had fallen only to 9.5 g ± 1.2 from a mean baseline threshold of 14.49 g ± 0.4. In comparison, at 1 h post-injection, Sprague–Dawley and PVG (C+) animals treated with ch14.18 had mean withdrawal thresholds of 5.2 g ± 1.2 and 6.3 g ± 1.7, respectively despite having had similar basal thresholds. Withdrawal thresholds of all three strains given ch14.18 appeared to plateau from about 2.5–3 h post-injection when C6 deficient rats began what looked like an early recovery. Comparison of the area over the curve for the early (0–2 h) and this later period (2.0–4.5 h) indicated substantial differences between the C6 deficient rats (PVG (C−) and those with functioning complement cascades (compared to PVG(C+) p ≥ 0.02 and p ≥ 0.002 for early and later period, respectively; or to Sprague–Dawley rats p ≥ 0.000 and p ≥ 0.0001, respectively) with larger differences being present for the second portion of the experiment.
3.5.2. Comparison of allodynia caused by ch14.18 and hu14.18K322A in rats with deficient complement factor C6
We also injected PVG (C−) rats with 1 or 3 mg/kg of hu14.18K322A. The rationale behind this portion of the experiment was that while the PVG (C−) rats were incapable of generating activated complement factors downstream of C6, the complement cascade upstream of C6, including both C3a and C5a could still be activated. Differences in percent allodynia between PVG (C−) rats injected with ch14.18 and hu14.18K322A should be due to complement activation upstream of complement factor C6, presumably most of its activity evoked by generation of C5a. PVG (C−) rats injected with either dose of the mutated antibody exhibited very little allodynia (Fig. 5) and there was a tendency over the first 2.5 h towards slightly higher withdrawal thresholds compared to PVG (C−) rats injected with ch14,18. The difference between these two sets of animals as indicated by their AOCs was significant for rats treated with 1 mg/kg hu14.18K322A (p ≥ 0.01; ch14.18 had more allodynia) compared to ch14.18, but did not reach significance for the 3 mg/kg dose. During the second portion of the test period, administration of either dose of the mutated antibody resulted in allodynia no different from that elicited by ch14.18, although again there was a tendency towards lower values and values were also not different from those achieved by saline (not shown).
4. Discussion
Our results confirm earlier findings demonstrating profound mechanical allodynia resulting from intravenous ch14.18 treatment, an antibody that has recently been shown to improve the outcome of patients with high risk neuroblastoma in a phase III clinical trial. Our principle novel finding is that magnitude and especially duration of ch14.18-induced mechanical sensitization are due in great part to the ability of this antibody to fix complement. Animals treated with an antibody variant with reduced capacity to fix complement (hu14.18K322A), displayed a tendency towards reduced allodynia for the first 2 h post-injection. More importantly there was a much faster recovery from allodynia back to basal mechanical withdrawal threshold values; differences were readily apparent just a few hours after injection. This decrease occurred despite the fact that hu14.18K322A retained ADCC activity.
The complement system consists of serum and membrane bound proteins that interact with each other and with immune cell molecules in a stylized cascade and serves to amplify the original signal. Many complement proteins act as receptor-specific activator molecules [1]. Although there are three major complement pathways, the classical pathway activated by antigen–antibody complexes, is most likely to be activated by anti-GD2 antibody. This results in formation of complement factors C3a and C5a as well as membrane attack complex (MAC), which consists of complement factors C5b joined to C6–C9. The macromolecule thus formed is also known as terminal complement complex.
Complement activation is acknowledged to be a major factor in several systemic inflammatory disorders and autoimmune diseases such as reperfusion injury, rheumatoid arthritis and Guillain Barré Syndrome [21]. There is also a precedent for involvement of complement in local inflammation generated by antibody-antigen reactions and pain. Complement factors C1, C1q, C3, C3d and C9 are released into the spinal dorsal horn following nerve injury [25] and, although pain behavior was not measured in these studies, it is likely to have occurred as the degree of localized spinal glial activation generated in these experiments is frequently associated with spinal sensitization and hyperalgesia. More recently, three models of neuropathic pain were all shown to induce increases in mRNA for complement factors C1q, C3, C4 and C5 specificallyin microglia [16,22]. Increases in complement factor mRNA peaked 3–7 days after the nerve injury.
Complement receptor 1 is a high affinity receptor for C3b and C4b, it regulates complement activation and prevents activation of C3 and C5 convertase, thus, blocking formation of complement components C3a and C5a as well as membrane attack complex [1]. Spinal administration of the antagonist, soluble complement receptor 1, totally reverses already established mechanical allodynia due to neuroinflammation or to nerve injury [34]. It is proposed that this reversal was due to blockade of MAC formation, although actions through complement factor C5a are also likely. Our data support their conclusion that, complement fixation, followed by generation of both C5a and MAC, is involved in neuropathic pain.
In our study, animals pretreated with a complement factor C5a receptor antagonist displayed no mechanical allodynia throughout the experiment. This contrasts with our observation that hu14.18K322A injection, with limited complement activation, resulted in allodynia for the first 2 h. This difference between animals given the C5a receptor antagonist and those given the mutated antibody must be due to the residual complement activation of the hu14.18K322A and its actions through generation of C5a and point to a unique role for this factor in initiating the pain state. In the periphery, intradermal injection of the C5a complement fragment, elicits mechanical hyperalgesia peaking 20 min after injection [23]. Complement fragment C5a, as well as the other anaphylatoxic peptide C3a, induce the release of histamine and other inflammatory mediators from mast cells [20] [24]. However, previous experiments in our laboratory using rats pretreated for several days with compound 48/80, a potent degranulator of mast cells, followed by administration of ch14.18, resulted in only a trend towards reduction in the magnitude of mechanical allodynia as compared to rats with control pre-treatment, and no change in duration of the pain behavior (Sorkin and Yu, unpublished results). These data indicate that mast cell degranulation, at best, contributes only a small component of the observed ch14.18-evoked allodynia. Nanomolar concentrations of complement-derived C5a have a chemotactic effect on neutrophils [30] and other inflammatory cells, and they also induce upregulation of adhesion molecules and cause increased vascular permeability [21]. Subsequent activation of infiltrating immune cells results in release of oxygen free radicals and lysosomal proteases. Peripheral blockade of the C5a receptor by systemic administration of a receptor-specific antagonist reduces, but importantly does not totally block induction of, mechanical sensitization associated with paw incision. Antagonist pre-treatment also reduces acute local production of several pro-inflammatory cytokines [8]. These data further support the hypothesis that C5a contributes to induction and maintenance of pain behavior.
While there is no doubt that C5a elicits peripheral inflammation, infiltration of neutrophils and hyperalgesia on its own [23], that is probably not the only complement-dependent mechanism by which binding of ch14.18 to GD2 ganglioside along the nerve trunk causes allodynia and ectopic activity. In these experiments, we observed that lack of C6 in the PVG (C−) animals, downstream of presumably intact C5a formation, is sufficient to significantly reduce the anti-GD2-induced pain behavior in the induction phase (0–2 h) and particularly in the later portions (after 2 h) of the observation period. The most likely mechanism of this reduction was loss of MAC formation. This loss was less complete than that seen following pre-treatment with C5a receptor antagonist. Injection of hu14.18K322A into C6 deficient animals had a strong tendency to result in less allodynia than did injection of ch14.18, although this was significant only for the lower dose during the first post-injection period (0–2 h). As the difference between these two antibodies is their ability to fix complement and both sets of animals were unable to form MAC, the disparity must be due to complement mediated factor upstream of C6, most likely complement factor C5a.
Insertion of MAC into cell membranes results in the formation of transmembrane pores. Depending on MAC concentration, potential outcomes range from calcium influx and excitation (activation) to cell lysis and death [19,27]. Sublytic levels of MAC produce protein kinase C activation, production of diacylglycerol, ceramide and arachidonic acid metabolites as well as activation of mitogen-activated kinase pathways in various cell types [19,26,27]. Recently, Koski and colleagues [9] demonstrated that MAC induces increase in Jun N-ternial Kinase (JNK) activity in Schwann cell cultures within a time course roughly corresponding to onset of ch14.18-evoked pain behavior. Membrane attack complex is found in serum and cerebrospinal fluid and on Schwann cells in rats with experimental allergic neuritis [18] as well as in patients with Guillain–Barré syndrome. In these patients, presence of MAC correlates with progression of clinical signs [28]. In animals with experimental allergic neuritis, inhibition of complement activation delays and or totally suppress demyelination and disease onset [10].
An interesting study from Anderson and colleagues indicates that C6 deficient rats exhibit milder motor deficits and faster recovery than wild type controls following spinal cord contusion, suggesting that MAC contributes to spinal cord excitotoxicity [13]. These animals were not tested for pain behavior.
However, results from Costigan and Woolf’s group show that nerve injury-elicited pain behavior was dependent, in part, on C5a activation in the spinal cord and not on factors including or down stream of C6 [16]. The lack of MAC involvement was thoroughly demonstrated using C6 deficient rats similar to those that we obtained, and by a reversal of allodynia with intrathecal administration of the C5a receptor antagonist. There are several important differences between the two studies. We examined responses to innocuous mechanical pressure within hours of the insult. Indeed, maximal allodynia was achieved within 30–45 min. Woolf examined cold allodynia and hyperalgesia in response to pinprick [16] 3 days post injury. The injuries, although both clinically relevant, are quite different with respect to time of onset and reversibility. Overt surgical injury to a nerve vs. administration of an antibody that acts along the axon to elicit ectopic activity [36] and is likely to predominantly activate the classical complement pathway. Anti-GD2 induced-pain is completely resolved in children within 2–24 h after completion of treatment [35]. Thus, it should not be surprising if the mechanisms differ, despite complement involvement in both.
In summary, our findings demonstrated that the mutant hu14.18K322A displayed greatly reduced capacity to mediate CDC while retaining significant ADCC activity, as compared to ch14.18. In animal studies, the first set of experiments showed that anti-GD2 induced allodynia is reduced by use of an antibody with a point mutation that reduces complement activation. Similar reductions in allodynia were observed following injection of the original antibody in an animal with deficient C6 complement, a deficit that would prevent the formation of MAC. Complete abrogation of allodynia was achieved with antagonism of complement receptor C5a. Thus, we propose that complement activation, including formation of both C5a and MAC are components of anti-GD2 antibody-induced allodynia.
Acknowledgements
We would like to thank Damon McCumber for performing the blinded tail vein injections and Julie Nguyen for secretarial support. This work was supported by the Cindy Matters Fund (L.S.S.) and NIH NS048563-04 (L.S.S.), and in part by a grant from FDA, FD-R-002319 (A.L.Y.).
Abbreviations
- ADCC
antibody-dependent cellular cytotoxicity
- CDC
complement-dependent cytotoxicity
- APC
allophycocyan
- ANOVA
analysis of variance
- AOC
areas over the curve
- MAC
membrane attack complex
- JNK
Jun N-terminal Kinase
- PKC
phospho kinase C
- mAb
monoclonal antibody
Footnotes
None of the authors has any financial arrangements that represent a conflict of interest.
References
- [1].Abbas AK, Lichtman Ah. Cellular and molecular immunology. Elsevier; Philadelphia: 2005. [Google Scholar]
- [2].Barker E, Mueller BM, Handgretinger R, Herter M, Yu AL, Reisfeld RA. Effect of a chimeric anti-ganglioside GD2 antibody on cell-mediated lysis of human neuroblastoma cells. Cancer Res. 1991;51:144–9. [PubMed] [Google Scholar]
- [3].Blomberg K, Hautala R, Lovgren J, Mukkala VM, Lindqvist C, Akerman K. Time-resolved fluorometric assay for natural killer activity using target cells labelled with a fluorescence enhancing ligand. J Immunol Methods. 1996;193:199–206. doi: 10.1016/0022-1759(96)00063-4. [DOI] [PubMed] [Google Scholar]
- [4].Brauer RB, Baldwin WM, 3rd, Daha MR, Pruitt SK, Sanfilippo F. Use of C6-deficient rats to evaluate the mechanism of hyperacute rejection of discordant cardiac xenografts. J Immunol. 1993;151:7240–8. [PubMed] [Google Scholar]
- [5].Chacur M, Milligan ED, Gazda LS, Armstrong C, Wang H, Tracey KJ, Maier SF, Watkins LR. A new model of sciatic inflammatory neuritis (SIN): induction of unilateral and bilateral mechanical allodynia following acute unilateral perisciatic immune activation in rats. Pain. 2001;94:231–44. doi: 10.1016/S0304-3959(01)00354-2. [DOI] [PubMed] [Google Scholar]
- [6].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- [7].Cheung NK, Lazarus H, Miraldi FD, Abramowsky CR, Kallick S, Saarinen UM, Spitzer T, Strandjord SE, Coccia PF, Berger NA. Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma [see comments] J Clin Oncol. 1987;5:1430–40. doi: 10.1200/JCO.1987.5.9.1430. [DOI] [PubMed] [Google Scholar]
- [8].Clark JD, Qiao Y, Li X, Shi X, Angst MS, Yeomans DC. Blockade of the complement C5a receptor reduces incisional allodynia, edema, and cytokine expression. Anesthesiology. 2006;104:1274–82. doi: 10.1097/00000542-200606000-00024. [DOI] [PubMed] [Google Scholar]
- [9].David S, Hila S, Fosbrink M, Rus H, Koski CL. JNK1 activation mediates C5b-9-induced P0 mRNA instability and P0 gene expression in Schwann cells. J Peripher Nerv Syst. 2006;11:77–87. doi: 10.1111/j.1085-9489.2006.00067.x. [DOI] [PubMed] [Google Scholar]
- [10].Davoust N, Nataf S, Reiman R, Holers MV, Campbell IL, Barnum SR. Central nervous system-targeted expression of the complement inhibitor sCrry prevents experimental allergic encephalomyelitis. J Immunol. 1999;163:6551–6. [PubMed] [Google Scholar]
- [11].Dixon W. Efficient analysis of experimental observations. Ann Rev Pharmacol Toxicol. 1980;20:441–62. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- [12].Finch AM, Wong AK, Paczkowski NJ, Wadi SK, Craik DJ, Fairlie DP, Taylor SM. Low-molecular-weight peptidic and cyclic antagonists of the receptor for the complement factor C5a. J Med Chem. 1999;42:1965–74. doi: 10.1021/jm9806594. [DOI] [PubMed] [Google Scholar]
- [13].Galvan MD, Luchetti S, Anderson AJ. Deficiency in complement C6 improves locomotor recovery after SCI. Neurosci Abs. 2006 #645.17. [Google Scholar]
- [14].Gillies SD, Lo KM, Wesolowski J. High-level expression of chimeric antibodies using adapted cDNA variable region cassettes. J Immunol Methods. 1989;125:191–202. doi: 10.1016/0022-1759(89)90093-8. [DOI] [PubMed] [Google Scholar]
- [15].Gillin S, Sorkin LS. Gabapentin reverses the allodynia produced by the administration of anti-GD2 ganglioside, an immunotherapeutic drug. Anesth Analg. 1998;86:111–6. doi: 10.1097/00000539-199801000-00022. [DOI] [PubMed] [Google Scholar]
- [16].Griffin RS, Costigan M, Brenner GJ, Ma CH, Scholz J, Moss A, Allchorne AJ, Stahl GL, Woolf CJ. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J Neurosci. 2007;27:8699–708. doi: 10.1523/JNEUROSCI.2018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Handgretinger R, Baader P, Dopfer R, Klingbiel T, Reuland P, Treuner J, Reisfeld R. A phase I study of neuroblastoma with the anti-ganglioside GD2 antibody 14G2a. Cancer Immunol Immunother. 1992;35:199–204. doi: 10.1007/BF01756188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hartung HP, Jung S, Stoll G, Zielasek J, Schmidt B, Archelos JJ, Toyka KV. Inflammatory mediators in demyelinating disorders of the CNS and PNS. J Neuroimmunol. 1992;40:197–210. doi: 10.1016/0165-5728(92)90134-7. [DOI] [PubMed] [Google Scholar]
- [19].Hila S, Soane L, Koski CL. Sublytic C5b-9-stimulated Schwann cell survival through PI 3-kinase-mediated phosphorylation of BAD. Glia. 2001;36:58–67. doi: 10.1002/glia.1095. [DOI] [PubMed] [Google Scholar]
- [20].Johnson A, Hugli T, Muller-Eberhard H. Release of histamine from mast cells by complement peptides C3a and C5a. Immunology. 1975;28:1067. [PMC free article] [PubMed] [Google Scholar]
- [21].Kirschfink M. Targeting complement in therapy. Immunol Rev. 2001;180:177–89. doi: 10.1034/j.1600-065x.2001.1800116.x. [DOI] [PubMed] [Google Scholar]
- [22].Levin ME, Jin JG, Ji RR, Tong J, Pomonis JD, Lavery DJ, Miller SW, Chiang LW. Complement activation in the peripheral nervous system following the spinal nerve ligation model of neuropathic pain. Pain. 2008;137:182–201. doi: 10.1016/j.pain.2007.11.005. [DOI] [PubMed] [Google Scholar]
- [23].Levine J, Gooding J, Donatoni P, Borden L, Goetzl E. The role of the polymorphonuclear leukocyte in hyperalgesia. J Neurosci. 1985;5:3025–9. doi: 10.1523/JNEUROSCI.05-11-03025.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li M, Peake PW, Charlesworth JA, Tracey DJ, Moalem-Taylor G. Complement activation contributes to leukocyte recruitment and neuropathic pain following peripheral nerve injury in rats. Eur J Neurosci. 2007;26:3486–500. doi: 10.1111/j.1460-9568.2007.05971.x. [DOI] [PubMed] [Google Scholar]
- [25].Liu L, Tornqvist E, Mattsson P, Eriksson NP, Persson JK, Morgan BP, Aldskogius H, Svensson M. Complement and clusterin in the spinal cord dorsal horn and gracile nucleus following sciatic nerve injury in the adult rat. Neuroscience. 1995;68:167–79. doi: 10.1016/0306-4522(95)00103-p. [DOI] [PubMed] [Google Scholar]
- [26].Nicholson-Weller A, Halperin JA. Membrane signaling by complement C5b-9, the membrane attack complex. Immunol Res. 1993;12:244. doi: 10.1007/BF02918256. [DOI] [PubMed] [Google Scholar]
- [27].Niculescu F, Rus H, Shin S, Lang T, Shin ML. Generation of diacylglycerol and ceramide during homologous complement activation. J Immunol. 1993;150:214–24. [PubMed] [Google Scholar]
- [28].Sanders ME, Koski CL, Robbins D, Shin ML, Frank MM, Joiner KA. Activated terminal complement in cerebrospinal fluid in Guillain–Barre syndrome and multiple sclerosis. J Immunol. 1986;136:4456–9. [PubMed] [Google Scholar]
- [29].Schultz G, Cheresh D, Varki N, Yu A, Staffileno L, Reisfeld R. Detection of ganglioside GD2 in tumor tissues and sera neuroblastoma patients. Cancer Res. 1984;44:5914–20. [PubMed] [Google Scholar]
- [30].Shin HS, Snyderman R, Friedman E, Mellors A, Mayer MM. Chemotactic and anaphylatoxic fragment cleaved from the fifth component of guinea pig complement. Science. 1968;162:361–3. doi: 10.1126/science.162.3851.361. [DOI] [PubMed] [Google Scholar]
- [31].Slart R, Yu AL, Yaksh TL, Sorkin LS. An animal model of pain produced by systemic administration of an immunotherapeutic anti-ganglioside antibody. Pain. 1997;69:119–25. doi: 10.1016/s0304-3959(96)03247-2. [DOI] [PubMed] [Google Scholar]
- [32].Sorkin LS, Yu AL, Junger H, Doom CM. Antibody directed against GD(2) produces mechanical allodynia, but not thermal hyperalgesia when administered systemically or intrathecally despite its dependence on capsaicin sensitive afferents. Brain Res. 2002;930:67–74. doi: 10.1016/s0006-8993(01)03408-4. [DOI] [PubMed] [Google Scholar]
- [33].Svennerholm L, Boström K, Fredman P, Jungbejer B, Lekman A, Månsson J-E. Gangliosides and allied glycosphyngolipids in human peripheral nerve and spinal cord. Biochem Biophys Acta. 1994;1214:115–23. doi: 10.1016/0005-2760(94)90034-5. [DOI] [PubMed] [Google Scholar]
- [34].Twining CM, Sloane EM, Schoeniger DK, Milligan ED, Martin D, Marsh H, Maier SF, Watkins LR. Activation of the spinal cord complement cascade might contribute to mechanical allodynia induced by three animal models of spinal sensitization. J Pain. 2005;6:174–83. doi: 10.1016/j.jpain.2004.11.011. [DOI] [PubMed] [Google Scholar]
- [35].Wallace MS, Lee J, Sorkin L, Dunn JS, Yaksh T, Yu A. Intravenous lidocaine: effects on controlling pain after anti-GD2 antibody therapy in children with neuroblastoma – a report of a series. Anesth Analg. 1997;85:794–6. doi: 10.1097/00000539-199710000-00014. [DOI] [PubMed] [Google Scholar]
- [36].Xiao WH, Yu AL, Sorkin LS. Electrophysiological characteristics of primary afferent fibers after systemic administration of anti-GD2 ganglioside antibody. Pain. 1997;69:145–51. doi: 10.1016/s0304-3959(96)03280-0. [DOI] [PubMed] [Google Scholar]
- [37].Yu AL, Gilman MF, Ozkaynak WB, London S, Kreissman H, Chen KK, Mathay SL, Cohn JMM, Sondel P. Phase III randomized study of chimeric antibody 14.18 (Ch14.18) in high risk neuroblastoma following myeloablative therapy and autologous stem cell rescue. J Clin Oncol. 2009;27:15s. [Google Scholar]
- [38].Yu AL, Uttenreuther-Fischer MM, Huang CS, Tsui CC, Gillies SD, Reisfeld RA, Kung FH. Phase I trial of a human-mouse chimeric anti-disialoganglioside monoclonal antibody ch14.18 in patients with refractory neuroblastoma and osteosarcoma. J Clin Oncol. 1998;16:2169–80. doi: 10.1200/JCO.1998.16.6.2169. [DOI] [PubMed] [Google Scholar]