

Abstract
We report a microfluidic approach for de novo protein structure determination via crystallization screening and optimization, as well as on-chip X-ray diffraction data collection. The structure of phosphonoacetate hydrolase (PhnA) has been solved to 2.11Å via on-chip collection of anomalous data that has an order of magnitude lower mosaicity than what is typical for traditional structure determination methods.
Knowledge of the three dimensional structure of a protein is critical because it provides insight into its operation, as well as into potential sites for drug targeting.1–3 Recent efforts in structural biology have produced not only a large number of protein structures, but have helped to reduce bottlenecks through robotics and/or microfluidics,4,5 leading to improved, and often automated, high throughput methodologies for crystallization, and X-ray diffraction analysis of novel protein targets.6–15 Even with these improvements, the harvesting and mounting of protein crystals for X-ray analysis remains the only manual step in the structural biology pipeline, thus making collection of data from small/fragile crystals cumbersome.10,16–18
Current data collection strategies require harvesting single protein crystals from the crystallization droplet in which it was grown; a process that takes skill, and can result in damage to the often small and fragile crystals. Cryocooling is then used to minimize radiation damage during data collection, yet this process may introduce structural artifacts.15 Despite these challenges, the success of single-crystal, cryo-crystallography has led to protein structural analysis at biologically relevant temperatures largely being abandoned.
During the collection of X-ray diffraction data only intensity is recorded while the phase information necessary to obtain meaningful electron density maps and structural information is lost. Computational methods for phasing, such as molecular replacement, are not suited for novel proteins for which homologous structural information is not available.19,20 Phase information is most commonly obtained experimentally by measuring anomalous diffraction signals, either by single- or multi-wavelength anomalous dispersion (SAD and MAD).20,21 These anomalous diffraction measurements require the collection of symmetry-related pairs of data to observe the small anomalous signal arising from one or more heavy atoms. Thus, obtaining diffraction data with high signal-to-noise ratio, and minimal radiation-induced damage and crystal non-isomorphism is critical for such measurements. As a result of these stringent requirements, anomalous data effectively must be collected from a single crystal under cryogenic conditions,15,20 even though a few examples of anomalous data collected from multiple crystals at room temperature have been reported.21–24
Compared to larger scales, microfluidic approaches for crystallization provide more precise control over transport phenomena as well as over the composition and kinetic parameters of an experiment.25–37 On-chip X-ray analysis has been reported for both simple microfluidic chips18,26,27,29,30,36,38,39 and trimmed sections of multi-layer chips.31,40,41 However, the majority of materials used to date to fabricate microfluidic chips are incompatible with the requirements for high quality X-ray diffraction analysis.25,31,33,37,42
Here we report a microfluidic approach for the crystallization and de novo X-ray structure determination of novel targets via on-chip anomalous X-ray diffraction analysis. The focus of this paper is the complete demonstration of the capabilities of our microfluidic array chips, from (i) initial crystallization screening, and (ii) subsequent optimization yielding high quality protein crystals, to (iii) X-ray data collection. Furthermore, we demonstrate a strategy for the collection of high quality, on-chip anomalous X-ray diffraction data at room temperature with sufficient signal-to-noise to successfully extract phasing information. Thus, these chips serve as a one-stop, room temperature, de novo structure determination platform that eliminates the manual handling of crystals entirely.
Metering of protein and reagents, mixing, and incubation of crystallization trials is performed in an integrated, two-layer microfluidic chip (Figure 1) designed to have a minimal X-ray cross-section (>75% transmission for λ =1Å or 12.4 keV).43 We fabricated 24- and 96-well microfluidic array chips from cyclic olefin copolymer (COC), polydimethylsiloxane (PDMS), and Duralar® (Graphix Arts) with automated fluid handling capabilities for screening crystallization conditions, and subsequent structure determination (Figure 1a,b). For crystallization screening and optimization, the chip was designed such that the ratio of protein-to-precipitant varies for each precipitant solution tested (screening phase, Figure 1b). Next, a chip that reproduces the identified optimal crystallization condition in all wells is used to allow growth of a large number of isomorphous crystals for X-ray data collection.
Figure 1.
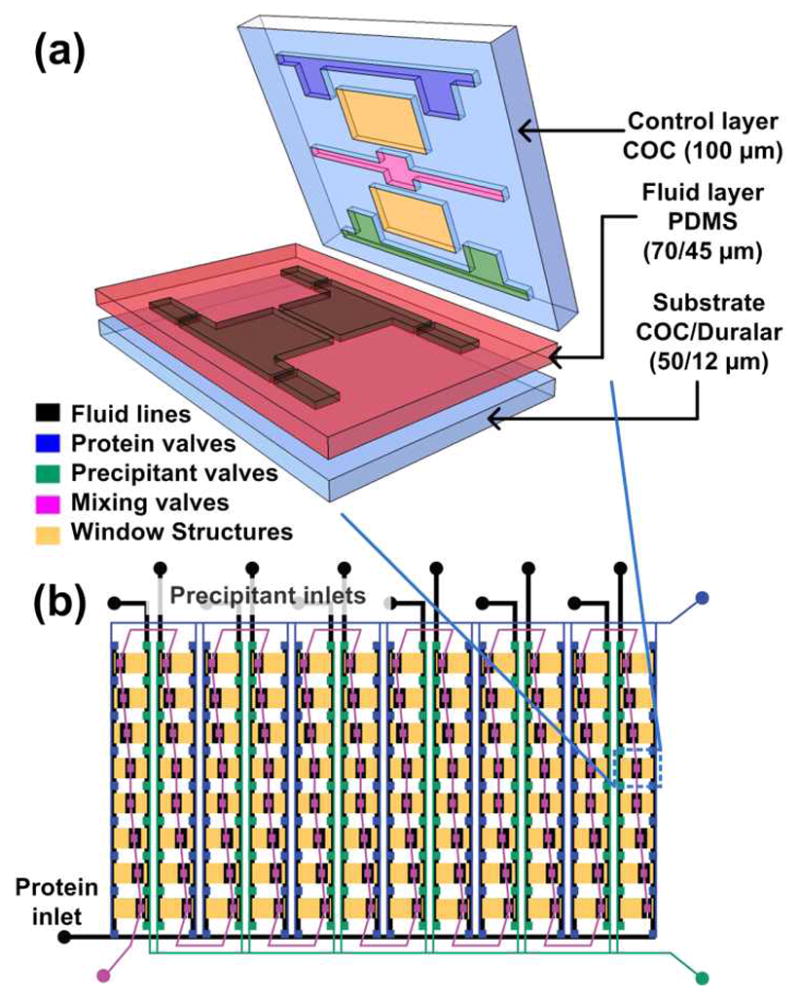
(a) 3D exploded view showing the different materials used for various layers of the hybrid device. (b) Schematic design of a 96-well array chip showing the various valve lines for filling in different components as well as the protein and precipitant chambers. The ratio of the protein to precipitant chambers varies from 4:1 to 1:4 to allow for screening of a larger number of conditions.
After successful validation using several model proteins,43 we embarked on testing the platform for de novo structure determination. We screened a selenomethionine (SeMet) derivative of PhnA from Sinorhizobium meliloti (10 mg/ml) for crystallization conditions using the 96-condition Index screen (Hampton Research) on a 96-well chip in which the ratio of protein-to-precipitant was varied (4:1 to 1:4, Figure 1b). This approach to screen a larger sample space can be achieved automatically with these chips, whereas implementation would be much more cumbersome using traditional well plates. The entire screening process utilized only 6 μL of protein (SeMet derivative of PhnA) per 96-well microfluidic chip and a total of 8 chips and yielded hits in 21 out of the 96 conditions tested (see ESI). In comparison, just 3 hits were obtained off-chip using traditional well plate methods. Whereas this first round of screening only produced showers of tiny crystals, diffracting to 3Å with poor signal-to-noise, subsequent optimization of the crystallization conditions and the chip geometry resulted in significant improvement in both data quality and resolution (Figure 2).
Figure 2.

(a) Optical micrograph of 24-well hybrid array chip mounted on the 21ID-G beamline at LS-CAT. Optimization of crystal quality on-chip: (b) Initial crystals of PhnA grown with the I-80 condition of the Index Screen. Crystals diffracted to > 3 Å and were on the order of 30–40 μm in size. (c) Crystals grown after optimizing the crystallization condition, crystals diffracted to < 2 Å and were on the order of 100–150 μm in size. Scale bars correspond to 200 μm.
Our approach to X-ray data collection involves accumulating small wedges of data from a large number of crystals at room temperature. This method has been used previously to obtain structural information from tiny or fragile crystals, as well as crystals that suffer from excessive radiation damage.44,45 The advantage of applying this small wedge strategy to crystals grown in a microfluidic chip is the ease by which a large number of crystals can be generated and analyzed directly on-chip. The fine control of transport possible at the microfluidic scale also allows for improved reproducibility both in crystal quality and isomorphism, as evidenced by the extremely low mosaicity and the minimal coefficient of variation (standard deviation divided by the mean) in the unit cell dimensions of 0.06% for a and b, and 0.13% for c among crystals of PhNA grown in different wells or in different chips.
Anomalous diffraction experiments require the comparison of the diffraction signals from symmetry-related pairs of data, necessitating an inverse-beam mode of data collection.46 The crystal was rotated 180° after the collection of 5 frames (1 second, 1° oscillation, 19 crystals) to collect data on the corresponding Bijvoet pairs. Our strategy helped to minimize radiation damage due to extended exposure to the X-ray beam, and maintain crystal quality during data collection of symmetry-related signals. The SeMet PhnA crystals diffracted to a resolution limit of 1.80Å. The wedges of data were merged to obtain a complete dataset and then used to solve the structure of PhnA to a resolution of 2.11Å using only on-chip data (Figure 3a; Table 1). The anomalous signal is clearly visible beyond 2.75 Å (Figure 3c), indicated by the difference in χ2 for Friedel mates treated as equivalent and independent. Other anomalous indicators including Rmerge and <ΔF±>/<σ(F)> showed similar patterns.
Figure 3.

Electron density map and theoretical model of a portion of the PhnA active site determined from (a) traditional, cryogenic, single-crystal data and (b) merged diffraction data collected on-chip. The structure clearly shows three key residues (Asp211, His215, and His377) in the active site. The PhnA carbon atoms are shown in green in stick representation. Superimposed is a 2Fo-Fc Fourier electron density map (contoured at 2.0σ). (c) χ2 values from merging procedure in SCALEPACK for Friedel mates treated as independent (filled squares) and equivalent (open squares). Data shown is to the resolution of 2.75Å as used in phasing.
Table 1.
Summary of crystallographic statistics for PhnA crystals grown on-chip
| Parameter | SeMeta |
|---|---|
|
| |
| Data Collection | |
|
| |
| Unit Cell Dimensions | a = b = 113.19 Å c = 73.87 Å |
| Space Group | P43212 |
| Total Observations | 412,491 |
| Unique Observations | 28,002 |
| Resolution | 50 Å – 2.11 Å |
| Rsym | 0.111 (0.508) |
| Mosaicity | 0.04° |
| Redundancy | 7.9 (6.7) |
| Figure of Merit | 0.394 |
| Completeness | 99.8% (99.8%) |
| I/σ | 15.4 (6.6) |
| # of Frames | 188 |
|
| |
| Refinement | |
|
| |
| R (Rfree) | 0.176 (0.211) |
|
| |
| Ramachandran Statistics | |
|
| |
| Most Favored | 95.61% (392) |
| Allowed | 2.68% (11) |
| Disallowed | 1.71% (7) |
Selenomethionine derivative of PhnA for phasing. Reported values are for all hkls. Values shown in parenthesis represent the value for the highest resolution shell except where indicated. For the Ramachandran statistics, the number in parenthesis indicates the number of residues in a given region. R-factor = Σ(|Fobs|-k|Fcalc|)/Σ |Fobs|and Rfree is the R value for a test set of reflections consisting of a random 5% of the diffraction data not used in refinement. Rsym = Σ |(Ii − <Ii> | Σ Ii where Ii = intensity of the ith reflection and <Ii> = mean intensity.
This is the first report of PhnA structure determination using a SeMet derivative (Table 1), and the data obtained here is in good agreement both with the structure obtained from analysis of a single crystal grown in a traditional well plate as evidenced by a RMSD < 0.4 Å when aligned in PyMOL (Figure 3a,b),47 and with the structural characterization of other PhnA derivatives published by our collaborators (see ESI).48 However, a detailed structural analysis is beyond the scope of this work.
The merged dataset was complete, indicative of random orientation of crystals grown on chip. Variations in Rsym and I/σ were within a range that is typical for good diffraction data, and structural refinement parameters such as R/Rfree and Ramachandran statistics demonstrate the high quality of our data. Interestingly, the mosaicity of the data collected on-chip at room temperature is nearly one order of magnitude lower compared to the single-crystal cryogenic data (see ESI). This measure of the intrinsic disorder within a crystal describes, along with resolution, the quality of the observed diffraction and can impact the quality of the resultant electron density maps. We attribute this key benefit to the lack of physical handling, manual harvesting and multi-crystal data collection method when using the on-chip approach reported here.
In summary, we successfully validated the microfluidic approach for de novo structure determination with the on-chip crystallization screening, optimization, and collection of SAD diffraction data for PhnA, resulting in a 2.11Å resolution structure. The microfluidic approach presented here enables the entire process of X-ray structure determination for a novel protein to be performed without manual handling of the crystals, starting from a purified protein sample. After on-chip screening for suitable crystallization conditions and subsequent optimization of those conditions, tens to hundreds of isomorphous crystals can be grown in 24–96 well array chips, and then, without crystal handling or harvesting, diffraction data wedges of each of those crystals can be obtained. The electron density map and structural information can then be obtained by merging these high quality wedges of data, yielding a complete dataset. We also demonstrated the ability to collect anomalous diffraction data on-chip, a critical input for structure determination efforts of novel proteins for which no homologous structural information is available. This microfluidic approach represents a simple, low-cost alternative to crystallization robots, as it requires only access to vacuum for filling of the chip, a microscope for visualization of crystals, and very small amounts of protein (~5 μL of protein solution per 96 wells that are each less than 20 nL in volume).
Looking ahead, elimination of the need for manual harvesting and cryo-cooling makes this microfluidic approach especially suitable for those instances where traditional methods yield protein crystals that may be too small or fragile for use, even after extensive optimization. Furthermore, the microfluidic platform reported here could also be used for dynamic structural studies to shed light on changes in structure during protein function, ranging from proteins that respond to stimuli such as light, pH, or the presence of ligands, to those that respond to temperature or electrical changes. These strategies have the potential to facilitate a variety of previously inaccessible biological studies.
Supplementary Material
Acknowledgments
This work was funded by NIH (R01 GM086727) and a NIH NIBIB Kirschstein Predoctoral Fellowship to SLP (F31 EB008330). Use of the Advanced Photon Source (APS), Argonne National Laboratory, was supported by the US Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357 as well as UIUC. We would like to thank Dr. Joseph Brunzelle, Dr. Keith Brister, and Dr. Spencer Anderson from LS-CAT at APS for help in data collection at the LS-CAT beamlines.
Footnotes
Electronic Supplementary Information (ESI) available: [detailed materials and methods, X-ray diffraction data analysis and PhnA screening data]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Service RF. Science. 2008;321:1758–1761. doi: 10.1126/science.321.5897.1758. [DOI] [PubMed] [Google Scholar]
- 2.Arinaminpathy Y, Khurana E, Engelman DM, Gerstein MB. Drug Discovery Today. 2009;14:1130–1135. doi: 10.1016/j.drudis.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Overington JP, Al-Lazikani B, Hopkins AL. Nature Reviews Drug Discovery. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 4.Hanson DK, Mielke DL, Laible PD. In: Current Topics in Membranes. 63. DeLucas L, editor. Academic Press; 2009. pp. 51–82. [Google Scholar]
- 5.Newby ZER, O’Connell JD, Gruswitz F, Hays FA, Harries WEC, Harwood IM, Ho JD, Lee JK, Savage DF, Miercke LJW, Stroud RM. Nature Protocols. 2009;4:619–637. doi: 10.1038/nprot.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mancia F, Love J. J Struct Biol. 2010;172:85–93. doi: 10.1016/j.jsb.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewinson O, Lee AT, Rees DC. Journal of Molecular Biology. 2008;377:62–73. doi: 10.1016/j.jmb.2007.12.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stevens RC. Current Opinion in Structural Biology. 2000;10:558–563. doi: 10.1016/s0959-440x(00)00131-7. [DOI] [PubMed] [Google Scholar]
- 9.Cherezov V, Hanson MA, Griffith MT, Hilgart MC, Sanishvili R, Nagarajan V, Stepanov S, Fischetti RF, Kuhn P, Stevens RC. Journal of The Royal Society Interface. 2009;6:S587–S597. doi: 10.1098/rsif.2009.0142.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cherezov V, Peddi A, Muthusubramaniam L, Zheng YF, Caffrey M. Acta Crystallogr D. 2004;60:1795–1807. doi: 10.1107/S0907444904019109. [DOI] [PubMed] [Google Scholar]
- 11.Koszelak-Rosenblum M, Krol A, Mozumdar N, Wunsch K, Ferin A, Cook E, Veatch CK, Nagel R, Luft JR, DeTitta GT, Malkowski MG. Protein Science. 2009;18:1828–1839. doi: 10.1002/pro.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caffrey M, Cherezov V. Nature Protocols. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobe B, Guss M, Huber T, editors. Methods in Molecular Biology: Structural Proteomics - High Throughput Methods. Humana Press; Totowa, NJ: 2008. [DOI] [PubMed] [Google Scholar]
- 14.Fogg MJ, Wilkinson AJ. Biochemical Society Transactions. 2008;036:771–775. doi: 10.1042/BST0360771. [DOI] [PubMed] [Google Scholar]
- 15.Juers DH, Matthews BW. Quarterly Reviews of Biophysics. 2004;37:105–119. doi: 10.1017/s0033583504004007. [DOI] [PubMed] [Google Scholar]
- 16.Hansen C, Quake SR. Current Opinion in Structural Biology. 2003;13:538–544. doi: 10.1016/j.sbi.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 17.Viola R, Carman P, Walsh J, Frankel D, Rupp B. J Struct Funct Genomics. 2007;8:145–152. doi: 10.1007/s10969-007-9031-6. [DOI] [PubMed] [Google Scholar]
- 18.Kisselman G, Qiu W, Romanov V, Thompson CM, Lam R, Battaile KP, Pai EF, Chirgadze NY. Acta Crystallogr D. 2011;67:533–539. doi: 10.1107/S0907444911011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans P, McCoy A. Acta Crystallogr D. 2008;64:1–10. doi: 10.1107/S0907444907051554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor GL. Acta Crystallogr D. 2010;66:325–338. doi: 10.1107/S0907444910006694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Q, Zhang Z, Hendrickson WA. Acta Crystallogr D. 2011;67:45–59. doi: 10.1107/S0907444910046573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clemons WM, Brodersen DE, McCutcheon JP, May JLC, Carter AP, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Journal of Molecular Biology. 2001;310:827–843. doi: 10.1006/jmbi.2001.4778. [DOI] [PubMed] [Google Scholar]
- 23.Ji XY, Sutton G, Evans G, Axford D, Owen R, Stuart DI. Embo J. 2010;29:505–514. doi: 10.1038/emboj.2009.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerdts CJ, Tereshko V, Yadav MK, Dementieva I, Collart F, Joachimiak A, Stevens RC, Kuhn P, Kossiakoff A, Ismagilov RF. Angewandte Chemie-International Edition. 2006;45:8156–8160. doi: 10.1002/anie.200602946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng B, Tice JD, Roach LS, Ismagilov RF. Angewandte Chemie-International Edition. 2004;43:2508–2511. doi: 10.1002/anie.200453974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerdts CJ, Elliott M, Lovell S, Mixon MB, Napuli AJ, Staker BL, Nollert P, Stewart L. Acta Crystallogr D. 2008;64:1116–1122. doi: 10.1107/S0907444908028060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steinert CP, Mueller-Dieckmann J, Weiss M, Roessle M, Zengerle R, Koltay P. Miniaturized and highly parallel protein crystallization on a microfluidic disc. 2007. [Google Scholar]
- 28.Lau BTC, Baitz CA, Dong XP, Hansen CL. Journal of the American Chemical Society. 2007;129:454–455. doi: 10.1021/ja065855b. [DOI] [PubMed] [Google Scholar]
- 29.Ng JD, Clark PJ, Stevens RC, Kuhn P. Acta Crystallogr D. 2008;64:189–197. doi: 10.1107/S0907444907060064. [DOI] [PubMed] [Google Scholar]
- 30.Dhouib K, Malek CK, Pfleging W, Gauthier-Manuel B, Duffait R, Thuillier G, Ferrigno R, Jacquamet L, Ohana J, Ferrer JL, Theobald-Dietrich A, Giege R, Lorber B, Sauter C. Lab on a Chip. 2009;9:1412–1421. doi: 10.1039/b819362b. [DOI] [PubMed] [Google Scholar]
- 31.Hansen CL, Classen S, Berger JM, Quake SR. Journal of the American Chemical Society. 2006;128:3142–3143. doi: 10.1021/ja0576637. [DOI] [PubMed] [Google Scholar]
- 32.Hansen CL, Skordalakes E, Berger JM, Quake SR. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:16531–16536. doi: 10.1073/pnas.262485199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perry SL, Roberts GW, Tice JD, Gennis RB, Kenis PJA. Crystal Growth & Design. 2009;9:2566–2569. doi: 10.1021/cg900289d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gavira JA, Toh D, Lopez-Jaramillo J, Garcia-Ruiz JM, Ng JD. Acta Crystallogr D. 2002;58:1147–1154. doi: 10.1107/s0907444902006959. [DOI] [PubMed] [Google Scholar]
- 35.Ng JD, Gavira JA, Garcia-Ruiz JM. Journal of Structural Biology. 2003;142:218–231. doi: 10.1016/s1047-8477(03)00052-2. [DOI] [PubMed] [Google Scholar]
- 36.Sauter C, Dhouib K, Lorber B. Crystal Growth and Design. 2007;7:2247–2250. [Google Scholar]
- 37.Li L, Ismagilov RF. Annual Review of Biophysics. 2010 doi: 10.1146/annurev.biophys.050708.133630. [DOI] [PubMed] [Google Scholar]
- 38.Barrett R, Faucon M, Lopez J, Cristobal G, Destremaut F, Dodge A, Guillot P, Laval P, Masselon C, Salmon JB. Lab on a Chip. 2006;6:494–499. doi: 10.1039/b517055a. [DOI] [PubMed] [Google Scholar]
- 39.Emamzadah S, Petty TJ, De Almeida V, Nishimura T, Joly J, Ferrer JL, Halazonetis TD. Acta Crystallogr D. 2009;65:913–920. doi: 10.1107/S0907444909021489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson MJ, Hansen CL, Quake SR. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16746–16751. doi: 10.1073/pnas.0605293103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson MJ, DeLaBarre B, Raghunathan A, Palsson BO, Brunger AT, Quake SR. Biochemistry. 2007;46:5722–5731. doi: 10.1021/bi700096p. [DOI] [PubMed] [Google Scholar]
- 42.Greaves ED, Manz A. Lab on a Chip. 2005;5:382–391. doi: 10.1039/b415836a. [DOI] [PubMed] [Google Scholar]
- 43.Guha S, Perry SL, Pawate AS, Kenis PJA. Sensors and Actuators B: Chemical. 2012;174:1–9. doi: 10.1016/j.snb.2012.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, IJzerman AP, Stevens RC. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hendrickson WA, Ogata CM. In: Methods in Enzymology. Carter Charles W., Jr, editor. Vol. 276. Academic Press; 1997. pp. 494–523. [DOI] [PubMed] [Google Scholar]
- 47.Schrödinger, LLC. 2010 [Google Scholar]
- 48.Agarwal V, Borisova SA, Metcalf WW, van der Donk WA, Nair SK. Chemistry & Biology. 2011;18:1230–1240. doi: 10.1016/j.chembiol.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.