

Abstract
The pituitary gland produces hormones that play important roles in both the development and homeostasis of the body. Ontogeny of the anterior and posterior pituitary is orchestrated by inputs from neighboring tissues, cellular signaling molecules and transcription factors. Disruption of expression or function of these factors has been implicated in the etiology of combined pituitary hormone deficiency (CPHD). These include the transcription factors HESX1, PROP1, POU1F1, LHX3, LHX4, OTX2, SOX2, SOX3 and GLI2. This review focuses on summarizing most recent mutations in LHX4 and OTX2 responsible for pituitary hormone deficiency. In both genetic defects of LHX4 and OTX2, there is high variability in clinical manifestations even in the same family. In addition, there is no clear phenotype-genotype correlation. These findings indicate that the other genetic and/or environmental factors influence the phenotype. In addition, the variability might reflect a plasticity during pituitary development and maintenance. Over the past two decades, a genetic basis for pituitary hormone deficiency and the mechanism of pituitary development have been clarified. It should be kept in mind that this review is not comprehensive, and defects of other transcriptional factors have been described in patients with CPHD. Furthermore, the causes in many patients with CPHD have not yet been determined. Therefore, continuing efforts for the clarification of the etiology are necessary.
Keywords: combined pituitary hormone deficiency, OTX2, LHX4, mutation
Introduction
The anterior pituitary gland is the primary site of endocrine regulation in the control of growth, reproduction and the stress response. The anterior pituitary gland develops from a midline structure contiguous with the primordium of the ventral diencephalon, and the cells undergo a highly selective determination and differentiation in a distinct spatial and temporal fashion (1, 2). Numerous cells in the pituitary gland are specialized to produce and secrete specific hormones, such as growth hormone (GH), prolactin (PRL), thyroid stimulating hormone (TSH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), and adrenocorticotropic hormone (ACTH). These processes are controlled by the actions of pituitary-specific and pituitary-enriched transcription factors and signaling molecules (1,2,3,4). The identification and characterization of pituitary developmental factors in vivo and in vitro has enabled us to clarify a genetic basis for combined pituitary hormone deficiency (CP HD) in humans (1,2,3,4). These factors include HESX1, LHX3, LHX4, POU1F1, PROP1, SIX6, OTX2, PTX2, GLI2, SOX2, and SOX3 (3,4,5). Numerous studies demonstrate that mutations and deletions of these transcription factor genes cause a wide range of pituitary phenotypes including severe life-threatening CPHD and isolated GH deficiency (GHD) (3,4,5). However, as the frequency of reported mutations of these transcription factors remains low, it appears that other multiple genes remain to be identified in human CPHD patients.
This review will focus on the clinical findings and molecular basis of patients who had mutations of the genes encoding for the transcription factors of LHX4 and OTX2.
LHX4
The human LHX4, located on chromosomal position 1q25 with six exons encoding 390 amino acids, is a member of the LIM homeodomain of transcription factors. LHX4 contains two LIM domains in its N-terminus and a DNA-binding homeodomain (6, 7). In murine studies, Lhx4 is expressed in the developing neural tube, hindbrain, Rathke’s pouch, and pituitary gland (6, 8, 9). Pituitary development in homozygous Lhx4 knockout mice proceeds normally until Rathke’s pouch rudiment is formed, but demonstrated diminished cell numbers, resulting in a hypoplastic anterior lobe compared with the wild type due to the increased cell apoptosis (8, 9).
In humans, the first patient had c.607-1G>C in intron 4 of LHX4 (10). Several affected members in this family have CPHD with deficiencies in GH, TSH and ACTH. Gonadotropins and PRL levels have not been reported; however, one patient was fertile. MRI analyses of affected members have shown hypoplasia of the pituitary, a small sella turcica, and chiari malformation. In vitro analysis demonstrated that wild-type LXH4 could bind a proximal promoter of another transcription factor, POU1F1, and activate its promoter activity, but the mutant identified by Machinis et al. (11) could not. Pfaeffle et al. (12) reported three heterozygous missense mutations (p.R84C, p.L190R, and p.A210P) of LHX4. p.R84C was located in the LIM domain, and the other two mutations were in the homeodomain. Two mutations of p.L190R and p.A210P had impaired DNA binding, and could not activate αGSU, POU1F1, and TSHβ promoters in a luciferase assay at all. In contrast, p.R84C had normal DNA biding capacity, but had a small amount of activity in a promoter assay. A p.A210P was found in a familial case. Whereas a sister was diagnosed with CPHD, a father and one sister harboring the identical mutation had isolated GHD. Another patient with a p.L190R mutation had GH, ACTH, and TSH, and the remaining patient with p.R84C had GH, TSH and gonadotropin deficiency. MRI findings in this study showed that the four cases exhibited hypoplastic anterior pituitaries but that one case with isolated GHD had a normal sized anterior pituitary. Regarding the posterior pituitary, three showed a normal position, and two showed an ectopic position. No one showed other brain malformations or a small sella turcica. It is of note that two siblings with p.A210P had a pituitary cyst.
Another familial mutation was a one-base insertion (c293_294insC), resulting in frame shift and producing a premature stop codon (p.Thr99fsX53) (13). Functional studies of the mutant LHX4 demonstrated a complete loss of transcriptional activity on the POU1F1 promoter. Two brothers showed GH and TSH deficiency with pituitary hypoplasia and a poorly developed sella turcica. The youngest brother also had corpus callosum hypoplasia and an ectopic posterior lobe. Their father, who also harbored the identical mutation, had only GHD with pituitary hyperplasia. Dateki et al. (14) identified the first patient with a de novo 0.5-megabase heterozygous deletion including LHX4. This patient had a small anterior pituitary, ectopic posterior lobe and underdeveloped sella turcica. Whereas GH, TSH, LH, and FSH were deficient, ACTH secretion was retained when he was evaluated at the age of 17 yr. Filges et al. (15) reported a 1.5-megabase heterozygous deletion in 1q25.2-q25.3 including LHX4. In this patient, severe respiratory distress, cardiac insufficiency, hypoglycemia, and heart failure were found soon after birth. She also had minor anomalies such as a short nose, short and broad forehead, and nail hypoplasia. Endocrinological evaluation showed complete defect of GH, TSH, LH, FSH and ACTH. Hypoplastic anterior pituitary gland, ectopic posterior lobe and a poorly formed sella were observed by MRI. Her cardiac insufficiency was completely reversed by hormonal replacement, and psychomotor development was within the normal range at 9 mo of age. It is of note that this 1.5-megabase deletion was inherited from an apparently healthy mother. Most recently, Takagi et al. (16) reported two patients with LHX4 mutations. One patient with p. V75I showed GH, TSH, and gonadotropin deficiency. The other patient harboring c.249-1G>A was diagnosed as having isolated GHD at 5 yr of age. Although this patient did not show episodes of adrenal insufficiency, longitudinal follow-up showed that her peak blood cortisol and plasma ACTH levels after insulin tolerance test decreased gradually with age. This mutation was also identified in his apparently normal father and siblings.
We reported two mutations (p.P389T and p.V101A) of LHX4 in two Japanese patients with CPHD (17, 18). One patient with P389T showed severe respiratory distress and hypoglycemia soon after birth and had defects of all anterior pituitary hormones. These symptoms were improved by hormone replacement. MRI demonstrated a hypoplastic anterior pituitary, ectopic posterior lobe and a poorly developed sella turcica. The second patient with p.V101A in the LIM domain in exon 3 also had a small anterior pituitary and ectopic posterior lobe but had a normal sized sella turcica. In vitro functional analysis demonstrated that this mutation could not activate the POU1F1 and FSHβ subunit gene promoter (18).
Mutations of LHX4 and clinical characteristics are summarized in Fig. 1 and Table 1. As mentioned above, hormone deficiency is variable even in familial cases. Some patients show partial GHD, but some had deficits of all pituitary hormones and developed life-threatening symptoms soon after birth. However, there existed apparently normal individuals with identical mutations in familial cases. The range of pituitary malformations in most cases comprises hypoplasia of the anterior pituitary. However, an enlarged or normal sized anterior pituitary has also been described. An early report suggested that a poorly developed sella turcica was a characteristic feature caused by the LHX4 mutations (10, 17). In about half of the reported cases, the sella turcica is poorly developed. Therefore, although the finding of a poorly developed sella turcica may distinguish CPHD patients with LHX4 defects from those reported with mutations in other genes, this finding is not a universal feature of patients with LHX4 mutations.
Fig. 1.

Schema of LHX4 genomic organization and protein structure. The location of reported mutations is shown. Mutations of introns are indicated by the arrows. Missense mutations and a frameshift mutation are indicated by black dots. Numbers next to the protein indicate amino acid positions of domain boundaries. White boxes, exon; LIM, LIM domains; HD homeodomain.
Table 1 . Clinical characteristics and mutations of LHX4.
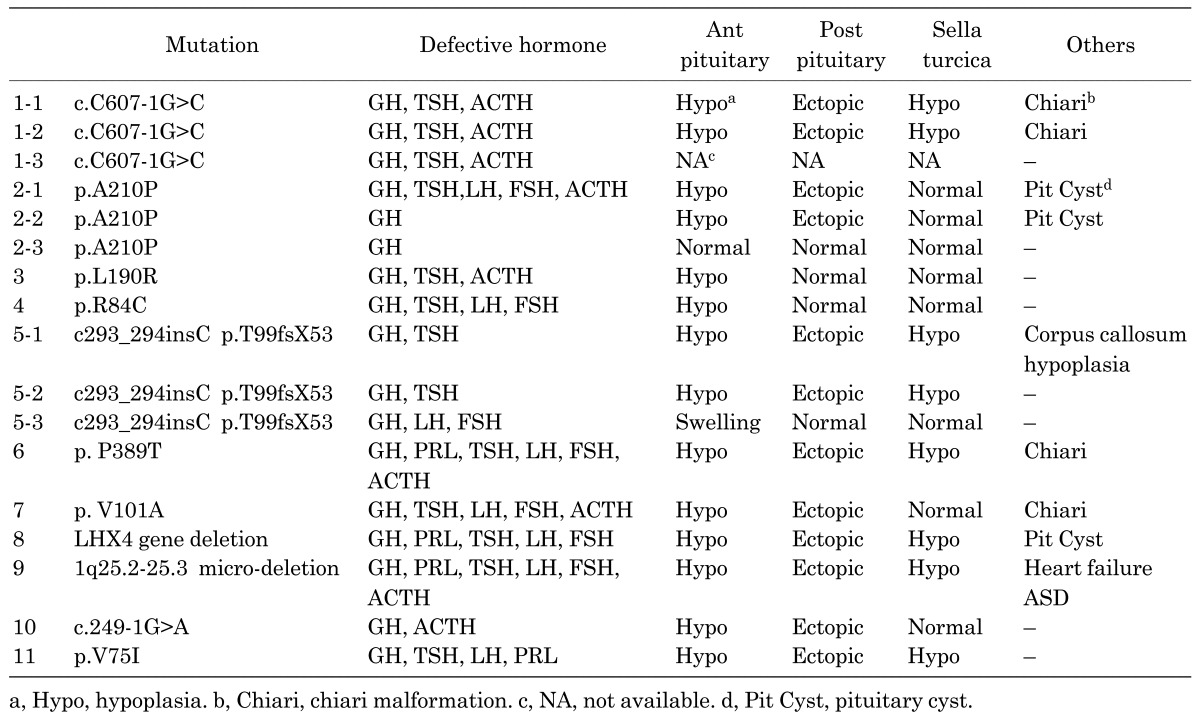
All mutations and deletions of LHX4 reported to date are heterozygous. In familial cases, the inheritance is autosomal dominant. Moreover, several in vitro studies have shown that mutants did not have a dominant negative effect. Taken together, haploinsufficiency of LHX4 is a probable mechanism for the development of the disease.
OTX2
OTX2 (MIM 600037), a bicoid-type homeodomain gene, is a vertebrate ortholog of the Drosophila gene orthodenticle (Otd), and is located in chromosome 14q and has five exons of which three are coding. There are two known isoforms: a and b; isoform b is the major product of the gene and has eight fewer amino acids than isoform a. Both proteins consist of a homeodomain, and transactivation domains at both the C and N terminal region (19, 20). Mouse Otx2 is expressed in developing neural and sensory structures, including the brain, ear, nose and eye and has a pivotal role in the development of the brain, face, and skull (20,21,22,23). Homozygous Otx2 knockout mice die at midgestation with severe brain anomalies (21,22,23). On the other hand, heterozygous knockout mice reveal variable phenotypes ranging from anencephaly, micrognathia, anophthalmia, and microphthalmia to normal, depending on the genetic background (24).
Consistent with these findings, heterozygous mutations of OTX2 were first reported in patients with severe ocular malformations and/or brain anomalies, seizures and developmental delay without craniofacial anomalies (25). In this report, none of the patients were subjected to evaluation of pituitary function. However, in humans, a deletion in the 14q22-23 region, including OTX2, was reported to cause anophthalmia and hypothalamic-pituitary anomalies (26, 27). Thus, a mutation in the OTX2 may be the cause of the ophthalmologic anomaly observed in patients with CPHD.
Diaczok et al. (28) were the first to report a novel missense mutation (p.N233S) of OTX2 in two unrelated patients without eye malformation. These patients showed deficiency of GH, TSH, LH, FSH, and ACTH. MRI demonstrated a hypoplastic anterior pituitary gland, and ectopic posterior lobe. Because OTX2 binding has been shown on the HESX1 promoter, which is also involved in pituitary development (29), functional analysis of p.N233S was performed using this element. As a result, this mutant was proved to have a dominant negative effect on wild-type OTX2. Dateki et al. (30) showed that OTX2 is expressed in the human pituitary and that a de novo frameshift OTX2 mutation (c. 402dupC, S135fsX) causes bilateral anophthalmia and partial isolated GHD with normal MRI findings. This patient showed developmental delay. The same group of researchers also studied 94 Japanese patients with various ocular or pituitary abnormalities. In this study, heterozygous p.K74fsX103 and p.G188X and a 2.8MB microdeletion involving OTX2 were identified (31). They showed that p.K74fsX103 and p.G188X lost the activation function in the GNRH1, HESX1, POU1F1, and IRBP (interstitial retinoid-binding protein) gene promoters compared with wild-type OTX2. Two patients with p.K74fsX103 and a 2.8 MB microdeletion showed isolated GHD, and one patient with p.G188X showed CPHD. These three patients had ocular abnormalities and developmental delay. They also reported one patient with p.T178S who had CPHD without ocular abnormality (31). The functional consequence of this amino acid substitution was not determined.
Our group reported p.S136fsX178 mutation in the third translated exon in a patient with severe CPHD, developmental delay, and bilateral anopthalmia (32). MRI demonstrated a small anterior pituitary gland and ectopic posterior lobe. Ashkenazi-Hoffnung et al. (33) reported a novel missense mutation (p.R90S) in a patient with unilateral anopthalmia and isolated GHD and a normal MRI. The father of this patient carried the same mutation but had a normal eye structure and a normal height. Henderson et al. (34) screened OTX2 mutations in 142 patients with eye anomalies, and identified one de novo heterozygous mutation (p.S138X) in a patient with isolated GHD and normal MRI findings. The patient had bilateral retinal dystrophy. Schilter et al. (35) screened 52 patients with anophthalmia and/or microphthalmia and identified mutations in four families. Among them, two had pituitary abnormalities. One patient with a p.W152X mutation had GH, TSH and ACTH deficiency. The other patient had a c.556–557insTATA mutation (p.S186IfsX187). Whereas the defective hormone in this patient was not described in the literature, MRI showed a small anterior pituitary and absent posterior pituitary glands. Two mutations occurred de novo. Two patients also had severe developmental delay.
Most recently, Del Blanco et al. (36) screened 92 patients with CPHD and identified a novel missense mutation, p.P134R, of OTX2. This patient had deficiency of GH, TSH, LH, FSH, and ACTH. MRI showed a normal anterior pituitary, invisible pituitary stalk, ectopic posterior lobe, and underdeveloped left optic nerve. Moreover, the patient had behavioral problems. In vitro analysis demonstrated that this was a second dominant negative mutant on wild-type OTX2. In this family, whereas the patient’s father also had an identical mutation, he had no clinical symptoms including hormone deficiency.
Mutations of OTX2 of patients associated with abnormal pituitary function and structure are shown in Fig. 2. Defective hormones, MRI findings and clinical features in these patients are summarized in Table 2. Mutations of OTX2 are present throughout the entire gene, and all mutations and a single gene deletion were heterozygous. Most of the reported mutations and the single gene deletion occurred de novo. In familial cases, mutations were transmitted from asymptomatic parents. It is of note that two amino acid substitutions of p.N233S and p.T178S were identified in patients with CPHD without eye malformation. These two mutations are located in the OTX2 family-specific domain. It is possible that any missense mutation in this domain may cause only the pituitary phenotype. To prove this hypothesis, more patients with CPHD are necessary to screen for OTX2 mutations.
Fig. 2.

Schema of OTX2 genomic organization and protein structure. Reported OTX2 gene mutations associated with human pituitary abnormalities or pituitary hormone deficiency are shown. Missense and frameshift mutations are indicated by black dots. The two missense mutations enclosed in boxes show the dominant negative effect on wild-type OTX2. Numbers next to the protein indicate amino acid positions of domain boundaries. NRS, nuclear retention signal; OTX, OTX family domain; white boxes, exon; HD, homeodomain. The closed triangles represent two tandem repeat conserved transactivation motifs.
Table 2. Clinical characteristics and mutations of OTX2.
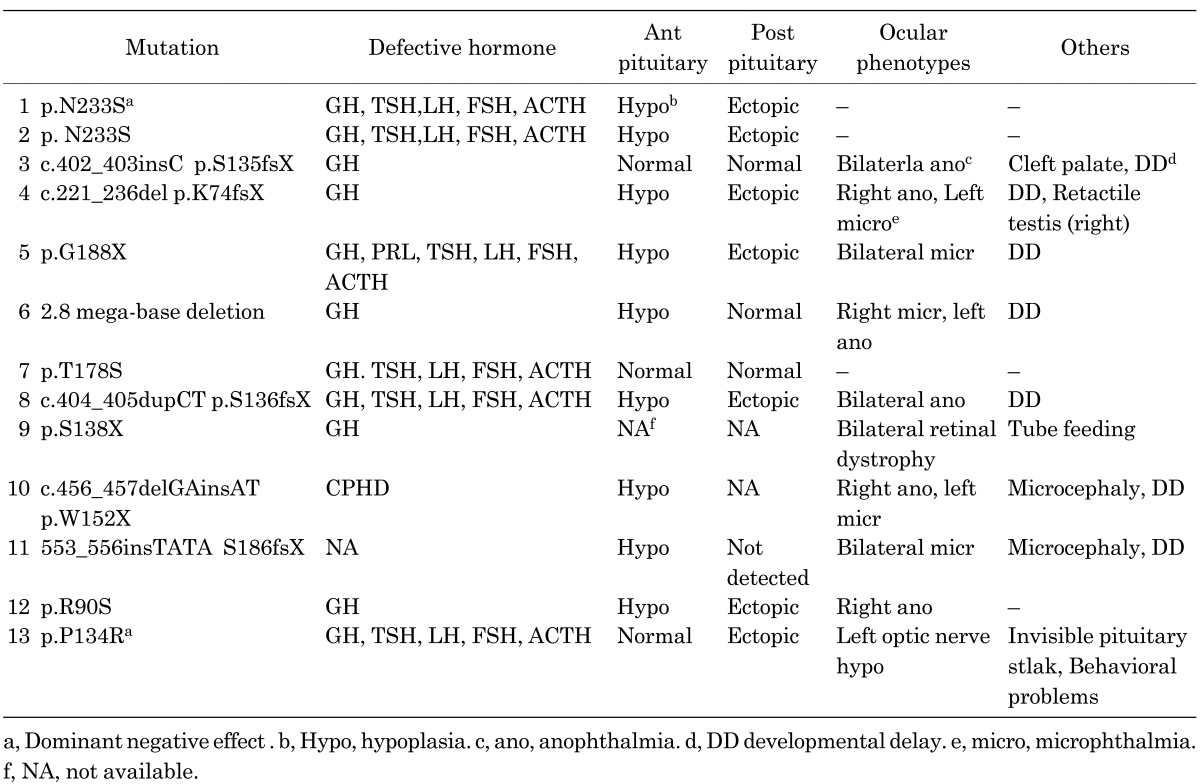
As summarized in Table 2, ocular phenotypes range from bilateral anophthalmia to nearly normal eye development. In regard to hormone deficiency, although several patients showed CPHD, five patients had isolated GHD. This suggests that GH is the most vulnerable pituitary hormone in OTX2 mutations. Moreover, eight patients showed developmental delay or behavioral problem. These neurological findings suggest that OTX2 is important for the development of the central nervous system.
Conclusion
In recent yr, identification of mutations in pituitary transcription factor genes has markedly advanced our understanding of the mechanisms of pituitary development and congenital pituitary hormone deficiency. The focus of this review is hypopituitarism caused by genetic defects of LHX4 and OTX2. As mentioned, hypopituitarism of these patients ranges from normal to life-threatening severe hypopituitarism. The phenotypic variability may be explained by other environmental factors and modifier genes. Such variability might also reflect a plasticity during pituitary development and maintenance.
Because defects of some transcription factors show a gene-specific phenotype such as a small sella turcica in LHX4 mutations and ocular abnormalities in OTX2 mutations, specific gene mutations may be speculated as causes of CPHD, and gene analysis is useful for diagnosis and genetic counseling. Moreover, as many studies indicate that hormone deficiencies caused by transcriptional factor defects may appear with age, regular evaluation of other pituitary hormones is necessary for early recognition of hormone deficiency.
In conclusion, understanding the molecular mechanism of defects of transcriptional factors results in improvement of morbidity in patients with CPHD. Because the causes in many patients with CPHD have not yet been determined, continuing efforts for clarification of the etiology are necessary.
References
- 1.Cushman LJ, Showalter AD, Rhodes SJ. Genetic defects in the development and function of the anterior pituitary gland. Ann Med 2002;34:179–91 [PubMed] [Google Scholar]
- 2.Kelberman D, Dattani MT. The role of transcription factors implicated in anterior pituitary development in the aetiology of congenital hypopituitarism. Ann Med 2006;38:560–77 10.1080/07853890600994963 [DOI] [PubMed] [Google Scholar]
- 3.Cohen LE. Genetic disorders of the pituitary. Curr Opin Endocrinol Diabetes Obes 2012;19:33–9Review [DOI] [PubMed] [Google Scholar]
- 4.Pfäffle R, Klammt J. Pituitary transcription factors in the aetiology of combined pituitary hormone deficiency. Best Pract Res Clin Endocrinol Metab 2011;25:43–60 10.1016/j.beem.2010.10.014 [DOI] [PubMed] [Google Scholar]
- 5.Prince KL, Walvoord EC, Rhodes SJ. The role of homeodomain transcription factors in heritable pituitary disease. Nat Rev Endocrinol 2011;7:727–37 10.1038/nrendo.2011.119 [DOI] [PubMed] [Google Scholar]
- 6.Yamashita T, Moriyama K, Sheng HZ, Westphal H. Lhx4, a LIM homeobox gene. Genomics 1997;44:144–6 10.1006/geno.1997.4852 [DOI] [PubMed] [Google Scholar]
- 7.Howard PW, Maurer RA. Identification of a conserved protein that interacts with specific LIM homeodomain transcription factors. J Biol Chem 2000;275:13336–42 10.1074/jbc.275.18.13336 [DOI] [PubMed] [Google Scholar]
- 8.Sheng HZ, Moriyama K, Yamashita T, Li H, Potter SS, Mahon KA, et al. Multistep control of pituitary organogenesis. Science. 1997;278:1809–12 10.1126/science.278.5344.1809 [DOI] [PubMed] [Google Scholar]
- 9.Raetzman LT, Ward R, Camper SA. Lhx4 and Prop1 are required for cell survival and expansion of the pituitary primordia. Development 2002;129:4229–39 [DOI] [PubMed] [Google Scholar]
- 10.Machinis K, Pantel J, Netchine I, Léger J, Camand OJ, Sobrier ML, et al. Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet 2001;69:961–8 10.1086/323764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Machinis K, Amselem S. Functional relationship between LHX4 and POU1F1 in light of the LHX4 mutation identified in patients with pituitary defects. J Clin Endocrinol Metab 2005;90:5456–62 10.1210/jc.2004-2332 [DOI] [PubMed] [Google Scholar]
- 12.Pfaeffle RW, Hunter CS, Savage JJ, Duran-Prado M, Mullen RD, Neeb ZP, et al. Three novel missense mutations within the LHX4 gene are associated with variable pituitary hormone deficiencies. J Clin Endocrinol Metab 2008;93:1062–71 10.1210/jc.2007-1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castinetti F, Saveanu A, Reynaud R, Quentien MH, Buffin A, Brauner R, et al. A novel dysfunctional LHX4 mutation with high phenotypical variability in patients with hypopituitarism. J Clin Endocrinol Metab 2008;93:2790–9 10.1210/jc.2007-2389 [DOI] [PubMed] [Google Scholar]
- 14.Dateki S, Fukami M, Uematsu A, Kaji M, Iso M, Ono M, et al. Mutation and gene copy number analyses of six pituitary transcription factor genes in 71 patients with combined pituitary hormone deficiency: identification of a single patient with LHX4 deletion. J Clin Endocrinol Metab 2010;95:4043–7 10.1210/jc.2010-0150 [DOI] [PubMed] [Google Scholar]
- 15.Filges I, Bischof-Renner A, Röthlisberger B, Potthoff C, Glanzmann R, Günthard J, et al. Panhypopituitarism presenting as life-threatening heart failure caused by an inherited microdeletion in 1q25 including LHX4. Pediatrics 2012;129:e529–34 10.1542/peds.2010-3849 [DOI] [PubMed] [Google Scholar]
- 16.Takagi M, Ishii T, Inokuchi M, Amano N, Narumi S, Asakura Y, et al. Gradual loss of ACTH due to a novel mutation in LHX4: Comprehensive mutation screening in Japanese patients with congenital hypopituitarism. PLoS One. 2012;7:e46008 10.1371/journal.pone.0046008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tajima T, Hattori T, Nakajima T, Okuhara K, Tsubaki J, Fujieda K. A novel missense mutation (P366T) of the LHX4 gene causes severe combined pituitary hormone deficiency with pituitary hypoplasia, ectopic posterior lobe and a poorly developed sella turcica. Endocr J 2007;54:637–41 10.1507/endocrj.K06-200 [DOI] [PubMed] [Google Scholar]
- 18.Tajima T, Yorifuji T, Ishizu K, Fujieda K. A novel mutation (V101A) of the LHX4 gene in a Japanese patient with combined pituitary hormone deficiency. Exp Clin Endocrinol Diabetes 2010;118:405–9 10.1055/s-0029-1225612 [DOI] [PubMed] [Google Scholar]
- 19.Kastury K, Druck T, Huebner K, Barletta C, Acampora D, Simeone A, et al. Chromosome locations of human EMX and OTX genes. Genomics 1994;22:41–5 10.1006/geno.1994.1343 [DOI] [PubMed] [Google Scholar]
- 20.Chatelain G, Fossat N, Brun G, Lamonerie T. Molecular dissection reveals decreased activity and not dominant negative effect in human OTX2 mutants. J Mol Med 2006;84:604–15 10.1007/s00109-006-0048-2 [DOI] [PubMed] [Google Scholar]
- 21.Acampora D, Mazan S, Lallemand Y, Avantaggiato V, Maury M, Simeone A, et al. Forebrain and midbrain regions are deleted in Otx2−/− mutants due to a defective anterior neuroectoderm specification during gastrulation. Development 1995;121:3279–90 [DOI] [PubMed] [Google Scholar]
- 22.Ang SL, Jin O, Rhinn M, Daigle N, Stevenson L, Rossant J. A targeted mouse Otx2 mutation leads to severe defects in gastrulation and formation of axial mesoderm and to deletion of rostral brain. Development 1996;122:243–52 [DOI] [PubMed] [Google Scholar]
- 23.Kurokawa D, Kiyonari H, Nakayama R, Kimura-Yoshida C, Matsuo I, Aizawa S, et al. Regulation of Otx2 expression and its functions in mouse forebrain and midbrain. Development 2004;131:3319–31 10.1242/dev.01220 [DOI] [PubMed] [Google Scholar]
- 24.Hide T, Hatakeyama J, Kimura-Yoshida C, Tian E, Takeda N, Ushio Y, et al. Genetic modifiers of otocephalic phenotypes in Otx2 heterozygous mutant mice. Development 2002;129:4347–57 [DOI] [PubMed] [Google Scholar]
- 25.Ragge NK, Brown AG, Poloschek CM, Lorenz B, Henderson RA, Clarke MP, et al. Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet 2005;76:1008–22 10.1086/430721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elliott J, Maltby EL, Reynolds B. A case of deletion 14(q22.1→q22.3) associated with anophthalmia and pituitary abnormalities. J Med Genet 1993;30:251–2 10.1136/jmg.30.3.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nolen LD, Amor D, Haywood A, St Heaps L, Willcock C, Mihelec M, et al. Deletion at 14q22-23 indicates a contiguous gene syndrome comprising anophthalmia, pituitary hypoplasia, and ear anomalies. Am J Med Genet A 2006;140:1711–8 [DOI] [PubMed] [Google Scholar]
- 28.Diaczok D, Romero C, Zunich J, Marshall I, Radovick S. A novel dominant negative mutation of OTX2 associated with combined pituitary hormone deficiency. J Clin Endocrinol Metab 2008;93:4351–9 10.1210/jc.2008-1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spieler D, Baumer N, Stebler J, Koprunner M, Reichman-Fried M, Teichmann U, et al. Involvement of Pax6 and Otx2 in the forebrain-specific regulation of the vertebrate homeobox gene ANF/Hesx1. Dev Biol 2004;269:567–9 10.1016/j.ydbio.2004.01.044 [DOI] [PubMed] [Google Scholar]
- 30.Dateki S, Fukami M, Sato N, Muroya K, Adachi M, Ogata T. OTX2 mutation in a patient with anophthalmia, short stature, and partial growth hormone deficiency: functional studies using the IRBP, HESX1, and POU1F1 promoters. J Clin Endocrinol Metab 2008;93:3697–702 10.1210/jc.2008-0720 [DOI] [PubMed] [Google Scholar]
- 31.Dateki S, Kosaka K, Hasegawa K, Tanaka H, Azuma N, Yokoya S, et al. Heterozygous orthodenticle homeobox 2 mutations are associated with variable pituitary phenotype. J Clin Endocrinol Metab 2010;95:756–64 10.1210/jc.2009-1334 [DOI] [PubMed] [Google Scholar]
- 32.Tajima T, Ohtake A, Hoshino M, Amemiya S, Sasaki N, Ishizu K, et al. OTX2 loss of function mutation causes anophthalmia and combined pituitary hormone deficiency with a small anterior and ectopic posterior pituitary. J Clin Endocrinol Metab 2009;94:314–9 10.1210/jc.2008-1219 [DOI] [PubMed] [Google Scholar]
- 33.Ashkenazi-Hoffnung L, Lebenthal Y, Wyatt AW, Ragge NK, Dateki S, Fukami M, et al. A novel loss-of-function mutation in OTX2 in a patient with anophthalmia and isolated growth hormone deficiency. Hum Genet 2010;127:721–9 10.1007/s00439-010-0820-9 [DOI] [PubMed] [Google Scholar]
- 34.Henderson RH, Williamson KA, Kennedy JS, Webster AR, Holder GE, Robson AG, et al. A rare de novo nonsense mutation in OTX2 causes early onset retinal dystrophy and pituitary dysfunction. Mol Vis 2009;15:2442–7 [PMC free article] [PubMed] [Google Scholar]
- 35.Schilter KF, Schneider A, Bardakjian T, Soucy JF, Tyler RC, Reis LM, et al. OTX2 microphthalmia syndrome: four novel mutations and delineation of a phenotype. Clin Genet 2011;79:158–68 10.1111/j.1399-0004.2010.01450.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gorbenko Del Blanco D, Romero CJ, Diaczok D, de Graaff LC, Radovick S, Hokken-Koelega AC. A novel OTX2 mutation in a patient with combined pituitary hormone deficiency, pituitary malformation, and an underdeveloped left optic nerve. Eur J Endocrinol 2012;167:441–52 10.1530/EJE-12-0333 [DOI] [PubMed] [Google Scholar]