

Abstract
Aim
Ischemia-reperfusion injury is associated with reduced bioavailability of nitric oxide and microvascular dysfunction. One emerging mechanism behind reduced nitric oxide bioavailability is upregulation of arginase which metabolizes the nitric oxide synthase substrate L-arginine. This study investigated the effects of arginase inhibition on coronary flow velocity and infarct size during reperfusion.
Methods
Anaesthetized rats, subjected to 30 min coronary artery ligation and reperfusion up to 8 days, were treated with vehicle or the arginase inhibitor Nω-hydroxy-nor-L-arginine (nor-NOHA; 100 mg/kg) intravenously 15 min before ischemia. Coronary flow velocity was determined repeatedly during reperfusion.
Results
Arginase activity in the ischemic-reperfused myocardium was increased already at 20 min of reperfusion and maintained at 8 days. Infarct size was reduced by arginase inhibition at 2 h (39 ± 3% of the area at risk vs. 51 ± 2% in the vehicle group, P<0.01) and at 8 days of reperfusion (13 ± 2% of the left ventricle vs. 22 ± 2%, P<0.05). Basal coronary flow velocity was higher during reperfusion in the group given nor-NOHA and it correlated inversely to infarct size (P<0.01, r=−0.60). Hyperemic coronary flow velocity was also increased in the nor-NOHA treated group compared to vehicle at 24 h and at day 8 (P<0.05).
Conclusion
It is concluded that arginase activity is increased already during early reperfusion. Arginase inhibition increases coronary flow velocity and reduces infarct size that is sustained 8 days after reperfusion. Inhibition of arginase may thus be a promising therapeutic target to prevent the development of microvascular dysfunction and myocardial injury following ischemia-reperfusion.
Keywords: Arginase, Coronary Artery, Microvascular function, Nitric Oxide, Reperfusion
Introduction
Current treatment of acute myocardial infarction due to coronary artery occlusion is aimed at initiating reperfusion to minimize myocardial damage. Early revascularization by percutaneous coronary intervention (PCI) rescues the myocardium at risk and limits infarct size, which improves the outcome for the patient (Shah et al. 2007). However, reperfusion per se may contribute to myocardial injury beyond that induced by ischemia, the so called reperfusion injury (Park & Lucchesi. 1999, Yellon & Hausenloy. 2007). Several factors contribute to the development of myocardial ischemia-reperfusion injury including endothelial and microvascular dysfunction, pro-inflammatory activation and oxidative stress. Furthermore, despite an open infarct-related epicardial artery, obstruction of coronary microvasculature can markedly reduce blood flow to the infarct zone. This effect is known to contribute to the no-reflow phenomenon and to the reperfusion injury (Ito. 2006). It has been demonstrated that the no-reflow phenomenon during reperfusion is associated with an unfavorable prognosis after PCI (Ito. 2006, Dong-bao et al. 2010). Determination of and interventions targeting myocardial perfusion and coronary flow is therefore of value both in the acute phase and in the long term after established reperfusion.
A central phenomenon in the development of ischemia-reperfusion injury is endothelial dysfunction with reduced bioavailability of nitric oxide (NO) (Cohen et al. 2006). Recently, upregulation of arginase has emerged as an important factor contributing to reduced production of NO by competing with endothelial NO synthase (eNOS) for their common substrate L-arginine (Durante et al. 2007). Thus, increased arginase activity induced by factors including oxygen-derived free radicals, proinflammatory cytokines and hypoxia may reduce the pool of L-arginine available for NO production (Durante et al. 2007). Accordingly, recent data from our group demonstrate that inhibition of arginase reduces myocardial infarct size measured up to 4 hours of reperfusion via a mechanism related to increased NO production (Jung et al. 2010, Gonon et al. 2012). It remains unknown, however, whether the effect of arginase inhibition in these short-term studies is due to a delayed development of the reperfusion injury or a true sustained reduction in infarct size. Furthermore, considering the impact of coronary microvascular function for the development of reperfusion injury it appeared to be of interest to evaluate the effect of arginase inhibition on coronary flow during reperfusion and its relation to myocardial infarct size. With this background, the present study was designed to investigate the acute and long-term effects of reperfusion on arginase activity and the effect of arginase inhibition on coronary flow velocity and infarct size in an acute and chronic rat model of reperfusion following ischemia.
Material and Methods
Animals
The study was approved by the Ethics Committee for laboratory animal experiments at Gothenburg University. Male Wistar rats (Charles River, Germany) with body weight 300–350 g were used. All animals were housed at constant temperature (21°C) with 12-hour dark-light cycles and had free access to tap water and chow diet (Lactamin, Sweden).
Experimental protocol
The rats were anesthetized with isoflurane (Abbott Scandinavia AB, Solna, Sweden), intubated and connected to a ventilator (2–2.5% of isoflurane, stroke volume 3.5 ml/min and 65 strokes/min, Ugo Basile Rodent Ventilator, Stoelting Co, Wood Dale, Illinois, US) using a mixture of oxygen (0.2 l/min) and air (0.8 l/min). The rats were placed on a ventilated heated bench. Rectal temperature was kept at 37.5–38°C using a thermo-regulating lamp. In the acute protocol (see below), the left carotid artery was cannulated for registration of arterial pressure and heart rate (equipment made in-house at Astra Zeneca, Mölndal, Sweden). Additional catheters were inserted into the right jugular vein or the tail vein for drug administration. All rats underwent left thoracotomy and a suture (7-0 silk suture Perma-hand, Johnson & Johnson AB, Sollentuna, Sweden) was placed under the left coronary artery. Marcain (1 mg/kg) was injected subcutaneous before the incision. The rats were subjected to 30 min of coronary artery occlusion followed by reperfusion for 2 h (acute protocol) or 8 days (chronic protocol). In each protocol, rats were randomized to saline (acute protocol n=12, chronic protocol n=10) or the arginase inhibitor Nω-hydroxy-nor-L-arginine (nor-NOHA, Bachem, Bubendorf, Switzerland; 100 mg/kg) (acute protocol n=12, chronic protocol n=10) administered as intravenous bolus injections 15 min prior to ischemia. The dose of nor-NOHA was based on previous experience (Jung et al. 2010). In the acute protocol the chest was closed with sutures immediately after start of ischemia and prepared for coronary flow velocity measurements (see description below) during reperfusion. In the chronic protocol the incision was sutured after initiation of reperfusion. The lungs were re-inflated and the rats were allowed to wake up in a warm cage. Fentanyl was given after completion of the surgical procedure and the following day 1 (50 and 25 μg/kg subcutaneous injections, respectively) to reduce postoperative pain.
An additional group of rats (n=6) were anaesthetized and subjected to coronary artery ligation and reperfusion as above. After 20 min of reperfusion, cardiac arrest was induced by 1 ml of 15% potassium chloride injected intravenously. The heart was rapidly excised, the ischemic and non-ischemic areas were separated and stored frozen at −80°C for subsequent analysis of arginase activity.
Microvascular function
After removal of hair from the chest, a non-invasive linear Color Doppler-guided 15-MHz probe (Entos CL15-7, Philips Medical Systems, Bothell, Washington) connected to an ultrasound system (ATL-HDI5000, Philips Medical Systems) was used for investigation of coronary flow velocity during the different time points of reperfusion in the two protocols.
Acute protocol
Measurement of basal coronary flow velocity was performed at 5, 10, 15, 20 min and at 2 h of reperfusion. At 20 min and at 2 h reperfusion measurements of coronary hyperemic flow velocity was performed by infusion of adenosine (ITEM Development AB, Stocksund, Sweden) via the tail vein at doses of 60, 100 and 140 μg/kg/min for 2–3 minutes per dose based on a previous study (Gronros et al. 2011). Each dose was separated by one minute. The mean velocity (Vmean) was calculated from the delineated diastolic phase of the flow profile at baseline and the hyperemia obtained during the 140 μg/kg/min dose using a software analysis program (Image Arena 2.9.1, Tomtec Imaging Systems, GmbH, Unterschleißheim, Germany). Coronary flow velocity reserve (CFVR) is expressed as the ratio between the hyperemic blood flow velocity and baseline blood flow velocity. Measurements of CFVR have been described previously in detail (Wikstrom et al. 2005).
Chronic protocol
The rats were re-anaesthetized and the tail vein catheterized as described above on day 1 and day 8 following initiation of reperfusion. Measurements of basal and hyperemic coronary flow velocity were performed according to above mentioned protocol.
Determination of infarct size
In the acute protocol, the left coronary artery was re-occluded at 2 h of reperfusion and 2 ml of 2% Evans Blue solution (Sigma-Aldrich Sweden, Stockholm, Sweden) was injected via the jugular vein for area at risk (AAR) determination. To induce cardiac arrest, 1 ml of 15% potassium chloride was injected intravenously. The heart was extirpated and the atria and right ventricle were removed. The left ventricle was immediately frozen at −20°C. The heart was cut into 2 mm thick slices using a plastic device (AgnTho’s AB, Lidingo, Sweden) and two razor blades. The slices were scanned to determine the AAR and subsequently put in 0.8% triphenyltetrazolium chloride (TTC, Sigma-Aldrich Sweden, Stockholm, Sweden) for 18 min at 37°C to distinguish the viable myocardium from the necrotic. After 24 h of incubation in 4% formaldehyde, slices were scanned from both sides and the extent of myocardial necrosis and the area at risk were determined using an image analyze program (Bio Pix iQ 2.1.8, BioPix AB, Gothenburg, Sweden). In the chronic protocol, the rats were anaesthetized 8 days after reperfusion, the heart extirpated and the left ventricle frozen, sliced and stained with TTC and necrotic parts analyzed as mentioned above. The third slice from the apex was stored frozen at −80°C for analysis of arginase activity. The infarct size tracings were performed in a blinded fashion by one investigator (JG).
Arginase activity
Arginase activity is expressed in urea concentration (μmol/mg protein/min) and was determined by using a modified colorimetric assay previously described (Gonon et al. 2012, Berkowitz et al. 2003).
Statistics
Statistical comparisons between two groups and within the same group were made with unpaired and paired t-tests, respectively, or Wilcoxon signed rank test where appropriate (Prism™ 4 software, GraphPad Inc., San Diego, CA, USA). A p-value < 0.05 was considered to be significant. Data are presented as mean±SE.
Results
Infarct size
Acute protocol
There was no difference in AAR between the two groups treated with vehicle or nor-NOHA (Fig. 1A). Infarct size in relation to AAR was significantly smaller in the nor-NOHA treated group compared to vehicle (39 ± 3% vs. 51 ± 2%, respectively, P<0.01, Fig. 1B). Furthermore, infarct size in relation to left ventricle (LV) was significantly reduced in the group given nor-NOHA compared to controls (Fig. 1C).
Figure 1.
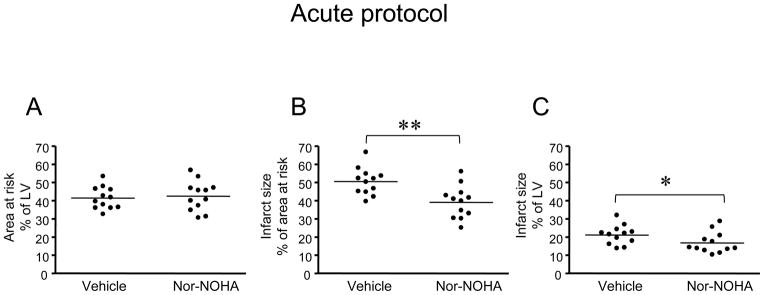
(A) Area at risk expressed in percent of the left ventricle (LV), (B) infarct size expressed in percent of the area at risk and (C) infarct size expressed in percent of the LV in rats included in the acute protocol (30 min ischemia and 2 h reperfusion). The groups were randomized to treatment with vehicle or the arginase inhibitor nor-NOHA. Significant differences from vehicle are shown; *P<0.05, **P<0.01.
Chronic protocol
Infarct size in relation to the total LV was markedly decreased after 8 days of reperfusion in the group given nor-NOHA compared to the vehicle group (13 ± 2% vs. 22 ± 2%, respectively, P<0.01, Fig. 2).
Figure 2.

Infarct size expressed in percent of the left ventricle (LV) in rats included in the chronic protocol (30 min ischemia and 8 days reperfusion). The groups were randomized to treatment with vehicle or the arginase inhibitor nor-NOHA. Significant differences from vehicle are shown; **P<0.01.
Coronary flow velocity
Acute protocol
Basal coronary flow velocity was significantly higher in the nor-NOHA treated group than in the vehicle group between 15 min and 2 h of reperfusion (P<0.05, Fig. 3A). The response to adenosine was small during the first two h of reperfusion and no significant difference was found between the two groups. There were significant correlation between the average coronary flow velocity from all early time points (average from 5, 10, 15 and 20 min in each animal compared to MI size) of reperfusion and the myocardial infarct size after 2 h of reperfusion (Fig. 4).
Figure 3.

Basal and hyperemic coronary flow velocity at different time points of reperfusion. (A) 15 min and 2 h reperfusion in the acute protocol and (B) 1 and 8 days reperfusion in the chronic protocol. The groups were randomized to treatment with vehicle or the arginase inhibitor nor-NOHA. Data are presented as means and SE, n=10–12. Significant differences from vehicle are shown; *P<0.05, **P<0.01, ***P<0.001.
Figure 4.
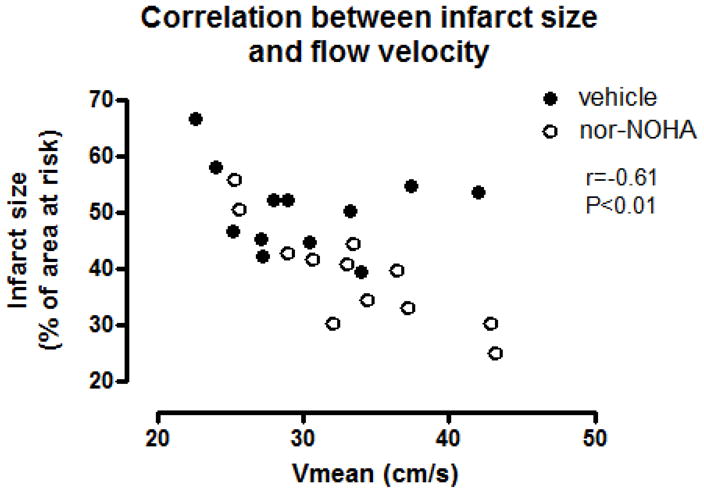
Correlation between the average coronary flow velocity from time points 5, 10, 15 and 20 min of reperfusion and infarct size after 2 h reperfusion (P<0.01, Pearsons r = −0.60).
Chronic protocol
An increase in basal flow velocity in the nor-NOHA treated group in comparison with the vehicle group was observed at day 1 and day 8 of reperfusion (Fig. 3B). The hyperemic flow velocity was also increased in the nor-NOHA treated group compared to vehicle at day 1 (P<0.01) and at day 8 (P<0.05, Fig. 3B). There was no significant difference in CFVR between the groups at day 1 or day 8 since both basal and hyperemic flow velocity was increased by nor-NOHA (data not shown).
Arginase activity
Arginase activity was significantly elevated in the ischemic-reperfused myocardium compared to the non-ischemic myocardium already at 20 min of reperfusion (332+226 vs. 24+6 μmol urea/mg protein/min; n=6; P<0.05) This increase was maintained at 2 h and 8 days of reperfusion in the vehicle groups (Fig. 5). On the other hand, there was no increase in arginase activity in the groups treated with nor-NOHA.
Figure 5.

Arginase activity in non-ischemic and ischemic myocardium from rats from the two protocols i.e. subjected to 30 min ischemia and 2 h (upper) or 8 days reperfusion (lower). Data are presented as means and SE, n=4–6. Significant differences from non-ischemic myocardium are shown; *P<0.05, **P<0.01.
Hemodynamics
Mean arterial pressure, heart rate and rate pressure product was analyzed in the acute protocol before and after drug administration, before ischemia and during reperfusion. No significant difference was found between the groups at any time point (Table 1). Heart rate in the chronic protocol, determined from the ultrasound measurements, was 404±11 bpm and 423±8 bpm at baseline and 397±13 bpm and 420±14 bpm during hyperemia on day 1 in the vehicle and nor-NOHA treated rats, respectively. Heart rate at day 8 was 395±9 bpm and 414±7 bpm at baseline and 386±11 bpm and 417±10 bpm during hyperemia in the vehicle and nor-NOHA treated rats, respectively.
Table 1.
Hemodynamic parameters in the acute protocol
| Group | Parameter | Pre drug | Before ischemia | Before rep | 10 min rep | 20 min rep | Hyperemia 20 min rep | 2h rep | Hyperemia 2h rep |
|---|---|---|---|---|---|---|---|---|---|
| Vehicle | MAP (mmHg) | 74±3 | 80±2 | 74±1 | 70±1 | 70±1 | 65±1 | 71±2 | 67±2 |
| HR (bpm) | 355±8 | 375±6 | 372±7 | 357±7 | 359±8 | 356±7 | 364±6 | 355±5 | |
| RPP (mmHg x bpm) | 26217±1331 | 30099±799 | 27947±839 | 24957±749 | 25167±810 | 23099±699 | 25882±1028 | 23798±786 | |
| Nor-NOHA | MAP (mmHg) | 78±2 | 81±2 | 75±2 | 72±1 | 72±1 | 65±2 | 75±2 | 66±2 |
| HR (bpm) | 349±5 | 365±8 | 366±6 | 342±5 | 350±6 | 344±6 | 355±5 | 346±5 | |
| RPP (mmHg x bpm) | 27331±635 | 29797±1184 | 27447±879 | 24618±707 | 25279±690 | 22221±832 | 26621±925 | 22889±1101 |
Data are presented as mean and SE, n=12 in each group. There were no significant differences between the groups. Abbreviations: MAP, mean arterial pressure; HR, heart rate; RPP, rate pressure product.
Discussion
The present study demonstrates that arginase activity is markedly increased during early reperfusion and this increase is still present up to 8 days of reperfusion. Inhibition of arginase reduces infarct size both in the acute and chronic model of ischemia-reperfusion, demonstrating a sustained cardioprotective effect. Furthermore, basal coronary flow velocity was significantly increased following arginase inhibition both during early and late reperfusion. These data suggest that inhibition of arginase induces long term improvement of microvascular function and limitation myocardial injury following ischemia-reperfusion.
The present study demonstrates that arginase activity is rapidly increased in the ischemic-reperfused myocardium. Arginase activity seems to play an important role in the development of ischemia-reperfusion injury by reducing the bioavailability of NO (Jung et al. 2010, Hein et al. 2003). Accordingly, the cardioprotective effect of arginase inhibition is critically dependent on NOS activity and NO bioavailability (Jung et al. 2010, Gonon et al. 2012). The present study in addition shows that the early increase in arginase activity is maintained for up to 8 days after reperfusion. Furthermore, the cardioprotective effect of nor-NOHA is maintained when infarct size is determined 8 days after reperfusion. This is of importance since it demonstrates that single administration of nor-NOHA does not only delay the development of reperfusion injury but clearly inhibits the development of myocardial injury following ischemia-reperfusion. It is of interest that administration of nor-NOHA did not only inhibit the upregulation of arginase activity in the acute protocol but also in myocardial tissue sampled 8 days after drug administration in the chronic protocol. It is unlikely that this is due to accumulation of the arginase inhibitor due to its brief half-life following single intravenous injection in rats (Hroch et al. 2012). An alternative possibility may be that inhibition of arginase during early reperfusion translates into reduced activity also 8 days after reperfusion via interference with the acute development of reperfusion injury.
Since myocardial microvascular dysfunction is closely related to reperfusion injury and of prognostic importance following PCI (Dong-bao et al. 2010) we determined coronary flow velocity during various time points following reperfusion. It is a well-known phenomenon that the degree of no-reflow during early reperfusion predicts the infarct expansion in the experimental and clinical settings (Ito. 2006, Ambrosio & Tritto. 1999, Reffelmann & Kloner. 2002). Reffelmann et al. (Reffelmann et al. 2003) found that impaired regional myocardial perfusion determined using radioactive microspheres 4 weeks after a 60 min coronary occlusion was a predictor of infarct expansion in rats. However, less is known regarding the magnitude of coronary flow at different time points during prolonged reperfusion by repeated measurements of coronary flow and the influence of treatment targeting vascular function on infarct size. In the present study we found that arginase inhibition resulted in increased basal coronary flow velocity during early reperfusion in the acute protocol. This effect was sustained when measured repeatedly at day 1 and day 8 in the chronic protocol. Since coronary flow velocity in the absence of epicardial artery stenosis reflects microvascular flow (Wikstrom et al. 2005, Iwakura et al. 1996, Yamamoto et al. 2002), the present findings suggest improvement of myocardial microvascular flow following arginase inhibition. Interestingly, there was a significant inverse relationship between coronary flow velocity during early reperfusion and infarct size. This may be of importance considering the prognostic impact of microvascular flow in patients with acute myocardial infarction (Ito. 2006, Reffelmann & Kloner. 2002).
The effect on coronary flow velocity is in line with our previous study in type 2 diabetic rats treated with nor-NOHA. In that study we showed that arginase inhibition shunts the substrate towards the NOS pathway instead of the arginase pathway resulting in increased NO production and thereby improved coronary microvascular function as measured with the ultrasound technique (Gronros et al. 2011). Accordingly, the cardioprotective effect of nor-NOHA has been shown to be dependent on NOS activity and NO bioavailability (Jung et al. 2010, Gonon et al. 2012). It is therefore likely that the improved flow velocity is a sign of improved endothelial function, which protects the myocardium from ischemia-reperfusion injury in our model. However, it cannot from the present study be excluded that the beneficial effect of nor-NOHA on coronary microvascular function is the result of a primary protective effect on the myocardium limiting the reperfusion injury.
The hyperemic response to adenosine during the first two measurements in the early reperfusion phase and on day 1 of reperfusion was low in both the control and nor-NOHA treated groups. This is consistent with previous findings in patients (Ishihara et al. 1993), mice (Tian et al. 2009) and dogs (Parker et al. 1975) with a reduced reactivity of the coronary arteries early after an ischemic event. Basal flow velocity might therefore be a better predictor of the microvascular function and myocardial injury during early reperfusion (Ito. 2006).
There are some limitations of the study that deserves commenting. Isoflurane induces vasodilatation (Larach & Schuler. 1991) and may mediate a preconditioning-like effect to the myocardium that makes the myocardium more tolerant against ischemia (Mullenheim et al. 2002). The reason isoflurane was used is that it is suitable for chronic experiments when animals should recover quickly from anesthesia. Pentobarbital is preferable in acute experiments due to its lack of preconditioning effects. However, it is unpractical in chronic experiments due to its depressive effect on respiration and long duration. We therefore chose to use isoflurane in both protocols. Moreover, it is of importance that in our previous study (Jung et al. 2010) we could show that nor-NOHA reduced infarct size also in a model of pentobarbital anesthesia. This demonstrates that arginase inhibition protects from ischemia-reperfusion injury via a mechanism that is independent of that involving isoflurane.
In addition, adenosine may also protect against reperfusion injury (Donato & Gelpi. 2003). However, any such effect would underestimate the protective effect of arginase inhibition. Furthermore, adenosine was not given until 20 min of reperfusion. Differences in basal coronary flow velocity were already observed before this time point. Cardiac function using echocardiography was not assessed in the present study. Left ventricular function investigated with ultrasound would preferably be analyzed by using contrast agent, multi-segment analysis or velocity vector imaging. Such investigations were not feasible to perform in the present protocol that was focused on determination of coronary flow velocity and its effect on infarct size. Future interventional studies are needed to address the effect of arginase inhibition on left ventricular function post infarct.
Conclusion
The present study demonstrates that arginase activity is increased early during reperfusion and that this increase is sustained for up to 8 days. Inhibition of arginase activity causes long-term improvement in coronary microvascular function and limitation of infarct size. Inhibition of arginase may thus be a promising therapeutic target to prevent the development of microvascular dysfunction and myocardial injury following ischemia-reperfusion.
Acknowledgments
This work was supported by grants from the Research Council of Sweden, the Swedish Heart and Lung Foundation, European Association for the Study of Diabetes, Novo Nordisk Foundation, Karolinska Institute/Stockholm County Council Strategic Cardiovascular Program, Gustav V and Queen Victoria Foundation, and Astra Zeneca R&D, Mölndal, Sweden. We are grateful to Mrs Marita Wallin for excellent technical assistance.
Footnotes
Conflict of interest
Author no 3 (Malin Palmér) is employed by Astra Zeneca R&D. No conflict of interest.
References
- Ambrosio G, Tritto I. Reperfusion injury: experimental evidence and clinical implications. Am Heart J. 1999;138:S69–75. doi: 10.1016/s0002-8703(99)70323-6. [DOI] [PubMed] [Google Scholar]
- Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, Burke S, Shoukas AA, Nyhan D, Champion HC, Hare JM. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003;108:2000–2006. doi: 10.1161/01.CIR.0000092948.04444.C7. [DOI] [PubMed] [Google Scholar]
- Cohen MV, Yang XM, Downey JM. Nitric oxide is a preconditioning mimetic and cardioprotectant and is the basis of many available infarct-sparing strategies. Cardiovasc Res. 2006;70:231–239. doi: 10.1016/j.cardiores.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Donato M, Gelpi RJ. Adenosine and cardioprotection during reperfusion--an overview. Mol Cell Biochem. 2003;251:153–159. [PubMed] [Google Scholar]
- Dong-bao L, Qi H, Zhi L, Shan W, Wei-ying J. Predictors and long-term prognosis of angiographic slow/no-reflow phenomenon during emergency percutaneous coronary intervention for ST-elevated acute myocardial infarction. Clin Cardiol. 2010;33:E7–12. doi: 10.1002/clc.20634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durante W, Johnson FK, Johnson RA. Arginase: a critical regulator of nitric oxide synthesis and vascular function. Clin Exp Pharmacol Physiol. 2007;34:906–911. doi: 10.1111/j.1440-1681.2007.04638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonon AT, Jung C, Katz A, Westerblad H, Shemyakin A, Sjoquist PO, Lundberg JO, Pernow J. Local Arginase Inhibition during Early Reperfusion Mediates Cardioprotection via Increased Nitric Oxide Production. PLoS One. 2012;7:e42038. doi: 10.1371/journal.pone.0042038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronros J, Jung C, Lundberg JO, Cerrato R, Ostenson CG, Pernow J. Arginase inhibition restores in vivo coronary microvascular function in type 2 diabetic rats. Am J Physiol Heart Circ Physiol. 2011;300:H1174–81. doi: 10.1152/ajpheart.00560.2010. [DOI] [PubMed] [Google Scholar]
- Hein TW, Zhang C, Wang W, Chang CI, Thengchaisri N, Kuo L. Ischemia-reperfusion selectively impairs nitric oxide-mediated dilation in coronary arterioles: counteracting role of arginase. FASEB J. 2003;17:2328–2330. doi: 10.1096/fj.03-0115fje. [DOI] [PubMed] [Google Scholar]
- Hroch M, Havlinova Z, Nobilis M, Chladek J. HPLC determination of arginases inhibitor N-(omega)-hydroxy-nor-L-arginine using core-shell particle column and LC-MS/MS identification of principal metabolite in rat plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;880:90–99. doi: 10.1016/j.jchromb.2011.11.022. [DOI] [PubMed] [Google Scholar]
- Ishihara M, Sato H, Tateishi H, Kawagoe T, Yoshimura M, Muraoka Y. Impaired coronary flow reserve immediately after coronary angioplasty in patients with acute myocardial infarction. Br Heart J. 1993;69:288–292. doi: 10.1136/hrt.69.4.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H. No-reflow phenomenon and prognosis in patients with acute myocardial infarction. Nat Clin Pract Cardiovasc Med. 2006;3:499–506. doi: 10.1038/ncpcardio0632. [DOI] [PubMed] [Google Scholar]
- Iwakura K, Ito H, Takiuchi S, Taniyama Y, Nakatsuchi Y, Negoro S, Higashino Y, Okamura A, Masuyama T, Hori M, Fujii K, Minamino T. Alternation in the coronary blood flow velocity pattern in patients with no reflow and reperfused acute myocardial infarction. Circulation. 1996;94:1269–1275. doi: 10.1161/01.cir.94.6.1269. [DOI] [PubMed] [Google Scholar]
- Jung C, Gonon AT, Sjoquist PO, Lundberg JO, Pernow J. Arginase inhibition mediates cardioprotection during ischaemia-reperfusion. Cardiovasc Res. 2010;85:147–154. doi: 10.1093/cvr/cvp303. [DOI] [PubMed] [Google Scholar]
- Larach DR, Schuler HG. Direct vasodilation by sevoflurane, isoflurane, and halothane alters coronary flow reserve in the isolated rat heart. Anesthesiology. 1991;75:268–278. doi: 10.1097/00000542-199108000-00015. [DOI] [PubMed] [Google Scholar]
- Mullenheim J, Ebel D, Frassdorf J, Preckel B, Thamer V, Schlack W. Isoflurane preconditions myocardium against infarction via release of free radicals. Anesthesiology. 2002;96:934–940. doi: 10.1097/00000542-200204000-00022. [DOI] [PubMed] [Google Scholar]
- Park JL, Lucchesi BR. Mechanisms of myocardial reperfusion injury. Ann Thorac Surg. 1999;68:1905–1912. doi: 10.1016/s0003-4975(99)01073-5. [DOI] [PubMed] [Google Scholar]
- Parker PE, Bashour FA, Downey HF, Kechejian SJ, Williams AG. Coronary hemodynamics during reperfusion following acute coronary ligation in dogs. Am Heart J. 1975;90:593–599. doi: 10.1016/0002-8703(75)90223-9. [DOI] [PubMed] [Google Scholar]
- Reffelmann T, Hale SL, Dow JS, Kloner RA. No-reflow phenomenon persists long-term after ischemia/reperfusion in the rat and predicts infarct expansion. Circulation. 2003;108:2911–2917. doi: 10.1161/01.CIR.0000101917.80668.E1. [DOI] [PubMed] [Google Scholar]
- Reffelmann T, Kloner RA. The “no-reflow” phenomenon: basic science and clinical correlates. Heart. 2002;87:162–168. doi: 10.1136/heart.87.2.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah SR, Hochberg CP, Pinto DS, Gibson CM. Reperfusion strategies for ST-elevation myocardial infarction. Curr Cardiol Rep. 2007;9:281–288. doi: 10.1007/BF02938376. [DOI] [PubMed] [Google Scholar]
- Tian F, Zhou X, Wikstrom J, Karlsson H, Sjoland H, Gan LM, Boren J, Akyurek LM. Protein disulfide isomerase increases in myocardial endothelial cells in mice exposed to chronic hypoxia: a stimulatory role in angiogenesis. Am J Physiol Heart Circ Physiol. 2009;297:H1078–86. doi: 10.1152/ajpheart.00937.2008. [DOI] [PubMed] [Google Scholar]
- Wikstrom J, Gronros J, Bergstrom G, Gan L. Functional and Morphologic Imaging of Coronary Atherosclerosis in Living Mice Using High-Resolution Color Doppler Echocardiography and Ultrasound Biomicroscopy. Journal of the American College of Cardiology. 2005;46:720–727. doi: 10.1016/j.jacc.2005.04.053. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Ito H, Iwakura K, Kawano S, Ikushima M, Masuyama T, Ogihara T, Fujii K. Two different coronary blood flow velocity patterns in thrombolysis in myocardial infarction flow grade 2 in acute myocardial infarction: insight into mechanisms of microvascular dysfunction. J Am Coll Cardiol. 2002;40:1755–1760. doi: 10.1016/s0735-1097(02)02486-5. [DOI] [PubMed] [Google Scholar]
- Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]