

Abstract
Background
Harstfield syndrome is the rare and unique association of holoprosencephaly (HPE) and ectrodactyly, with or without cleft lip and palate, and variable additional features. All the reported cases occurred sporadically. Although several causal genes of HPE and ectrodactyly have been identified, the genetic cause of Hartsfield syndrome remains unknown. We hypothesised that a single key developmental gene may underlie the co-occurrence of HPE and ectrodactyly.
Methods
We used whole exome sequencing in four isolated cases including one case-parents trio, and direct Sanger sequencing of three additional cases, to investigate the causative variants in Hartsfield syndrome.
Results
We identified a novel FGFR1 mutation in six out of seven patients. Affected residues are highly conserved and are located in the extracellular binding domain of the receptor (two homozygous mutations) or the intracellular tyrosine kinase domain (four heterozygous de novo variants). Strikingly, among the six novel mutations, three are located in close proximity to the ATP's phosphates or the coordinating magnesium, with one position required for kinase activity, and three are adjacent to known mutations involved in Kallmann syndrome plus other developmental anomalies.
Conclusions
Dominant or recessive FGFR1 mutations are responsible for Hartsfield syndrome, consistent with the known roles of FGFR1 in vertebrate ontogeny and conditional Fgfr1-deficient mice. Our study shows that, in humans, lack of accurate FGFR1 activation can disrupt both brain and hand/foot midline development, and that FGFR1 loss-of-function mutations are responsible for a wider spectrum of clinical anomalies than previously thought, ranging in severity from seemingly isolated hypogonadotropic hypogonadism, through Kallmann syndrome with or without additional features, to Hartsfield syndrome at its most severe end.
Keywords: Clinical genetics, Developmental, Genetics
Introduction
Holoprosencephaly (HPE) results from impaired midline cleavage of the embryonic forebrain. It varies in severity from alobar HPE (a monoventricular cerebrum that lacks interhemispheric division) to microform HPE (such as single central maxillary incisor). Milder cerebral midline defects including isolated corpus callosum agenesis or arhinencephaly (the absence of olfactory bulbs and tracts) have also been classified as falling within the HPE spectrum, at least in some instances.1 Ectrodactyly, also known as split-hand/foot malformation, is a congenital limb malformation characterised by a median cleft of the hand and/or foot due to the absence of the central rays.2 It encloses a broad spectrum of malformations, from shortening of the central digit to reduction of the hand/foot to a single ray, and may be variable even between the limbs of an affected individual. Several genes have been involved in non-syndromic HPE (including SHH, ZIC2, SIX3, GLI2, and TGIF) or ectrodactyly (such as P63, or the 10q24 duplication), with incomplete penetrance and variable expressivity.1–3
HPE and ectrodactyly can occur, separately, as part of numerous syndromes, but the co-occurrence of these two malformations, known as Harstfield syndrome (OMIM 300571), has only been reported in 14 males and three females (see online supplementary table S1).4–9 In addition to HPE and ectrodactyly, patients with Hartsfield syndrome show developmental defects of variable severity, ranging from one mildly affected individual with isolated hypogonadotropic hypogonadism (IHH), central diabetes insipidus, borderline low intelligence, and no facial dysmorphism to patients showing multiple congenital anomalies such as cleft lip and palate, malformed ears, hypo- or hypertelorism. There are also anecdotal reports of skull defects, vertebral anomalies, radial aplasia, eye anomalies or cardiac malformation (see online supplementary table S1). Targeted sequencing of HPE or ectrodactyly genes in selected patients has failed to identify mutations, and no convincing copy number variants were found.5–7 9 Despite the variable expressivity of Hartsfield syndrome, we postulated that mutations in a single key developmental gene underlie the co-occurrence of HPE and ectrodactyly.
Methods
Patients
We selected six patients with Hartsfield syndrome (patients 1–6), and one female fetus with the association of HPE, ectrodactyly, and additional severe malformations. Patients 1, 3, 5, and 6 were previously described.5 Patients 1–7 detailed phenotypes are described in table 1. Pictures of faces and hands of patients 1, 5, and 6 demonstrating the phenotypic range of facial dysmorphism and ectrodactyly found in patients with Harstfield syndrome are shown in figure 1. All patient samples were obtained and handled in agreement with the guidelines set out by the Université Libre de Bruxelles Hôpital Erasme ethics committee. Written informed consent was obtained from all participants (or guardians), except for patient 7, for whom parents gave verbal consent.
Table 1.
Detailed phenotypic description of the seven tested patients
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| FGFR1 mutation | c.494T>C p.L165S* |
c.572T>C p.L191S* |
c.1468G>C p.G490R |
c.1867G>T p.D623Y |
c.1884 T>G p.N628K |
c.2174 G>A p.C725Y |
– |
| Sex | M | M | M | F | M | M | F |
| Previous report5 | Patient 3 | – | Patient 5 | – | Patient 2 | Patient 4 | – |
| Consanguinity | + | −† | − | − | − | − | − |
| Brain | |||||||
| HPE | AL | L | SL | L | SL | L | AL |
| CCA | + | + | nr | Partial | Partial | Partial | nr |
| Pituitary | Normal | nr | nr | nr | Normal | Normal | nr |
| Diminished cortical thickness | + | + | − | − | − | − | +‡ |
| Face | |||||||
| CLP | Median | – | Bilateral | – | Bilateral | – | Cleft palate only |
| Eye | Hypotelorism | Hypotelorism | hypertelorism | Normal | Normal | Normal | Hypertelorism§ |
| Hands | |||||||
| Ectrodactyly | + | + | + | − | + | + | + |
| Digit number (right/left) | 2/2 | 3/3 | 3/3 | 5/5 | 4/4 | 5/5 | 2/3 |
| Other | 6 metacarpal bones on the left side, with partial fusion of the 4th and 5th | Bifurcation of the thumbs | Fused 2nd and 3rd metacarpal bones | Forearm hypoplasia | |||
| Feet | |||||||
| Ectrodactyly | + | + | + | +¶ | + | + | − |
| Digit number (right/left) | 1/1 | 2/2 | 2/2 | 5/5 | 2/3 | 4/3 | 5/5 |
| Other | Equinovarus deformity | ||||||
| Pituitary insufficiency | nr | nr | nr | CDI, HH, normal GH secretion, low response to TRH | CDI, HH | CDI, HH, normal GH secretion | nr |
| Genitalia | Normal | nr | Micropenis, cryptorchidism | Normal | Micropenis, cryptorchidism | Micropenis, cryptorchidism | Normal |
| Growth retardation | + | + | + | + Good response to GH treatment |
+ 160 cm (target: 176.5±8.5 cm) |
+ 161.6 cm (target: 172.5±8.5 cm) |
nr |
| DD/ID | Severe | Severe | Severe | Mild | Moderate | Mild | na |
| other | Generalised hypertonia, no smile, seizures (grand mal) | No language, spasticity | No language, wheelchair bound | Wheelchair bound (spastic paraplegia) | IQ 63 (Stanford-Binet score), at 6 years 8 months | ||
| Follow-up | Died at the age of 5 years | Died at the age of 4 years (respiratory infection) | Mainstream school with support | Lives in an institution | Works in a sheltered workshop | TOP | |
Positions of the mutations refer to coding DNA reference sequence CCDS6107.2 and Uniprot protein sequence P11362-1.
*Homozygous mutations.
†Low level consanguinity could not be assessed, the parents being lost to follow-up.
‡Patient 7 has severe microcephaly (head circumference of 15 cm at 20 weeks), hydrocephaly, and severe disruption of the telencephalic architecture.
§Patient 7 has severe facial anomalies: absence of nasal wing on the right side, right microphthalmia and eye defect.
¶Patient 4: Left foot: fusion of first and second toes, large gap between second and third rays, syndactyly of toes 3–5, absence of the third phalange of digits 3 and 4. Right foot: central large gap with partial syndactyly of toes 3–5, absence of the third phalange of digits 2 and 3.
AL, alobar; CCA, corpus callosum agenesis; CDI, central diabetes insipidus; CLP, cleft lip and palate; DD, developmental delay; F, female; GH, growth hormone; HH, hypogonadotropic hypogonadism; HPE, holoprosencephaly; ID, intellectual disability; L, lobar; M, male; na, not applicable; nr, not reported; SL, semilobar; TOP, termination of pregnancy; TRH, thyrotropin releasing hormone.
Figure 1.

FGFR1 mutations are found in patients with Hartsfield syndrome. (A) Pictures of three patients diagnosed with Hartsfield syndrome, showing the wide range of disease severity. (B) Identification of the N628K mutation in patient 5. The upper part shows the exome sequencing reads (horizontal grey bars with mismatching bases highlighted) aligned to chromosome 8. Vertical bars above the reads represent the total number of reads covering a specific position (visualisation from Integrative Genomics Viewer37). The identified mutation is covered by 83 reads. The lower part shows the corresponding Sanger sequencing chromatogram. (C) Schematic representation of a FGFR1 dimer bound with FGF1 and the positions of Hartsfield syndrome mutations. Homozygous mutations are marked with an asterisk. (D) Sanger sequencing of patients 1–6. Chromatograms show FGFR1 mutations in patients 1–6 with Hartsfield syndrome, along with their parents. Parents of patient 2 were unavailable. For each patient, the reference sequence from human genome GRCh37 surrounding the mutated position is shown on top, and the sequence from Sanger sequencing is shown below.
Exome sequencing
Exomes were captured using the TruSeq capture kit (Illumina) and paired-end sequenced over 100 bp on a Illumina HiSeq2000 sequencer by a third party provider (AROS applied biotechnology). For exome analysis, we followed the guidelines from the Genome Analysis ToolKit (GATK) best practice recommendations v3 to process an average of 118.7 million paired-end reads per sample.10 We aligned the reads to the human genome GRCh37/hg19 for each of the six samples independently with the Burrows-Wheeler Alignment tool,11 and removed duplicate reads using Picard MarkDuplicates, and used GATK for local realignment around indels and base quality score recalibration. We used GATK over the six samples together to call single nucleotide polymorphisms (SNPs) and indels. Our data were stored and processed from raw reads to variant calling using InSilicoDB.12 Variant annotation was done with SNPeff and depth of coverage was computed using BEDtools coveragebed13 and in-house perl and R scripts, over all coding exons from Ensembl release 66. Variants were filtered using GATK, SnpSift and in-house perl scripts. First, only non-synonymous coding or splice site variants were selected. Second, variants were evaluated for technical quality with GATK. SNPs were processed with ‘variant quality score recalibration’, a Gaussian mixture model using Hapmap and 1000 genomes Omni 2.5 M SNP chip arrays as a training. Indels were processed with direct filtering. Third, we selected de novo variants in the trio by restricting to variants with heterozygous genotype in the patient and homozygous reference in the parents, and unknown in dbSNP v135. Additional filtering was performed by adapting a described procedure.14 Specifically, we removed heterozygous variants in the child when >70% of reads were reference, discarded cases where >10% non-reference reads in a parent matched the child's call, removed calls where the offspring depth was <10% of the parents total depth, and retained only variants with genotype quality ≥20 for the three samples. Fourth, we searched for genes where variants were present in all four patients, and for which the variant found in the patient of the case-parents trio was de novo. Parameters used in the programmes are listed in online supplementary table S6.
Sanger sequencing
For the patients analysed through whole exome sequencing (WES) (patients 1, 3, 5, 6), the known mutated exons were amplified and sequenced using the Sanger method to verify the exome results. Whole sequencing of FGFR1 was performed for the three additional patients (patients 2, 4, 7) (table 1). To include all potential splice variants, a ‘merged transcript’ was considered, containing all coding positions on the 19 FGFR1 protein coding transcripts described in Ensembl release 66 (Ensembl gene ENSG00000077782). Primers have been designed with ExonPrimer (see online supplementary table S2). The PCR reaction was performed with 50 ng DNA, 3 pmol primers F and R, 2 mM MgCl2, 0.2 μL Taq and H2O to 20 μL. The PCR programme comprised 94°C for 3 min, 94°C for 30 s, 20 touchdown cycles 65°C to 55°C, 20 cycles 55°C for 30 s, 72°C for 1 min, and 72°C for 5 min. Purification of PCR product for sequencing was realised with ExoSap-IT (USB products, Affymetrix) and Sanger sequencing was initiated with the PCR primers of the corresponding amplicons.
Results
We performed WES in a series of four unrelated patients (patients 1, 3, 5, 6, table 1). Given the sporadic occurrence of cases of Hartsfield syndrome, we included one case-parent trio (patient 5), to focus our analysis on de novo variants. FGFR1 was selected as the best candidate, being the only gene where unknown variants were identified for the four patients, and patient 5's FGFR1 variant being a de novo mutation (ie, not found in his parents’ exome sequence). We used Sanger sequencing of PCR products from genomic DNA to confirm the FGFR1 variants identified through exome sequencing and to sequence the parents when available (figure 1). To support the above results, we performed Sanger sequencing of all coding exons and exon–intron boundaries of FGFR1 for two other patients with Harstfield syndrome (patients 2 and 4) and for a fetus with Hartsfield syndrome and severe additional features (patient 7) (table 1). We identified mutations in patients 2 and 4, bringing to six the number of Hartsfield patients carrying an FGFR1 mutation (figure 1). For patient 7, we could not find any FGFR1 coding or splice site mutation using Sanger sequencing, or pathogenic copy number variations (CNVs) using an Agilent 60K comparative genomic hybridisation (CGH) array.
FGFR1 (Uniprot P11362) is a member of the receptor tyrosine kinase superfamily. It is composed of an extracellular ligand binding domain that contains three immunoglobulin (Ig)-like domains (D1–D3), a single transmembrane helix, and a cytoplasmic domain responsible for tyrosine kinase activity (figure 1).15 The fibroblast growth factor (FGF) signalling pathway is a major player in embryonic development. Notably, in mice, the conditional lack of Fgfr1 expression in developing telencephalon results in loss of cerebral commissures,16 and also in the absence of olfactory bulbs,17 as early emergence of gonadotropin releasing hormone (GnRH) neurons from the embryonic olfactory placode is dependent on FGFR1 activation by FGF8.18 During mouse autopod patterning, depending on the time and localisation of conditional gene inactivation, an Fgfr1 insufficiency results in a variety of limb defects including loss restricted to the central digit or monodactyly.19
A wide phenotypic spectrum is observed in humans with FGFR1 loss-of-function mutations, ranging from apparently asymptomatic carrier, IHH, typical Kallmann syndrome (KS) (the association of hypogonadotropic hypogonadism and anosmia, the latter due to the absence or hypoplasia of olfactory bulbs and tracts), to KS with associated features, mainly cleft lip and palate, and oligodontia (see online supplementary table S3). Identical FGFR1 mutations may vary in phenotypic severity.20–22 FGFR1 mutations have also been reported in patients with phenotypes reminiscent of Hartsfield syndrome, such as the association of combined pituitary hormone deficiency, mild expression of HPE (corpus callosum agenesis, single central incisor), hand anomalies (brachydactyly, or fusion of metacarpal bones), and eye defects.20 22 23
In two of our patients with Hartsfield syndrome, we identified homozygous mutations affecting amino acid residues located in the extracellular ligand binding domain D2 of FGFR1: L165S, in patient 1, with a severe phenotype, and L191S, in patient 2, with a moderate phenotype (table 1, figure 1). Both parents of patient 1 were heterozygous for the L165S mutation, and were reported to be asymptomatic and spontaneously fertile. Parents of patient 2 were not available for testing. Mapping of these mutations on available FGFR1 structures from the RCSB Protein Data Bank shows that L165S is likely to affect FGF binding; the effect for L191S is less clear (figure 2). One previous KS patient has been described with a homozygous FGFR1 A167S mutation.20 This patient had KS, cleft palate, corpus callosum agenesis, vertebral anomalies, unilateral fusion of fourth and fifth metacarpal bones, and bilateral oligodactyly of feet (four digits).24
Figure 2.
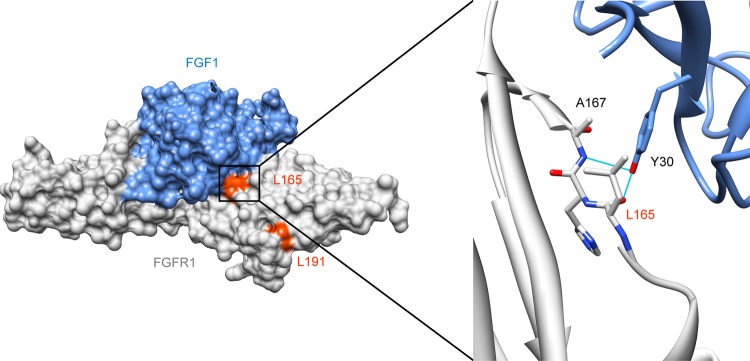
Mapping of mutations L165S and L191S on crystal structure. Protein Data Bank structure 3OJV38 showing the extracellular Ig-like domains 2 and 3 of FGFR1 (amino acids 147–359) bound to FGF1 in surface representation, and detail around leucine 165 in ribbon representation. FGFR1 is shown in grey and FGF1 in blue. Leucines 165 and 191 are coloured in orange red. The detailed view is highlighting the interface between FGFR1 and FGF1 around leucine 165. Tyrosine 30 on FGF1 forms hydrogen bonds with leucine 165 and alanine 167.39 Substitution of the leucine 165 by a serine should affect FGF binding. These pictures were made using UCSF Chimera.40
We identified three heterozygous mutations affecting amino acid residues located in the ATP binding pocket of the intracellular tyrosine kinase domain (TKD, amino acids 478–767): G490R (patient 3, moderate phenotype), D623Y (patient 4, mild phenotype), and N628K (patient 5, moderate phenotype). Analysis of the available crystallographic structures shows that the mutated amino acids are in close proximity to the ATP and the coordinating magnesium, suggesting impairment of FGFR1 kinase activity (figure 3). In support of this hypothesis, D623 is known to be required for catalysis.25 A tyrosine substitution would prevent it from fulfilling its role as a proton acceptor for the substrate. Adjacent mutations H621R, R622G, R622Q, and R622X provoke syndromic KS, some patients having corpus callosum agenesis (H621R), digit number anomalies (H621R) or fusion of metacarpal bones (R622G) (figure 4, see online supplementary table S3).20–22 26
Figure 3.

Mapping of mutations G490R, D623Y, N628K, and C725Y on FGFR1 tyrosine kinase domain crystal structure. Protein Data Bank structure 3GQI41 showing the intracellular kinase domain of FGFR1 (residues 464–770) in ribbon representation. The lower left part shows the details of the crystal structure surrounding the ATP binding pocket in the intracellular kinase domain of FGFR1. G490, D623, and N628 are in close proximity to the ATP's phosphates or coordinating magnesium. The lower right part shows the involvement of cysteine 725 in the positioning of the αG-containing segment, along with T726, P722, and K721. Substitution of the cysteine 725 by a tyrosine will likely affect the conformation of this region.27 The ATP analogue (AMPPCP) and wild-type residues of positions 490, 623, 628 and 721, 722, 725 and 726 are pictured in stick representation. Nitrogen, oxygen, phosphorus, and magnesium atoms are coloured blue, red, orange, and green, respectively. These pictures were made using UCSF Chimera.40
Figure 4.

Mapping of FGFR1 mutations on known crystallographic structures. Idiopathic hypogonadotropic hypogonadism and Kallmann syndrome (KS) variants are depicted in black, KS with orofacial features variants are depicted in dark blue, more syndromic KS variants are depicted in magenta, and Hartsfield syndrome variants are depicted in orange red with the wild-type side chain in stick representation. Variants from the phenotypic three most severe categories are labelled. The most severe phenotype was considered if the same mutation was identified in several patients. Asterisk indicates a homozygous mutation. (A) Protein Data Bank (PDB) structure 3OJV,38 showing extracellular immunoglobulin (Ig)-like domains 2 and 3, from amino acids 147–359. (B) PDB structure 3GQI,41 showing the intracellular kinase domain, from residue 464–770. These pictures were made using UCSF Chimera.40
We located one other heterozygous mutation, C725Y, in the intracellular C-terminal loop of the TKD (patient 6, mild phenotype). Mutations of neighbouring residues (P722S, P722H, and N724K) have been previously suggested to alter the conformation of this region (figures 3 and 4, see online supplementary table S3) and shown to decrease kinase activity.27 28
To our knowledge, none of these FGFR1 mutations have been previously reported in dbSNP, Exome Variant Server (http://evs.gs.washington.edu/EVS/) or the scientific literature, and all heterozygous mutations have occurred de novo. The substitutions involve amino acids highly conserved in mammals (L191), vertebrates (L165, C725) and eukaryotes (G490, D623, and N628) (see online supplementary figure S1). All mutations are predicted to be deleterious by SIFT and Polyphen 2.29 30
Discussion
Our study shows that FGFR1 is responsible for Hartsfield syndrome, which is consistent with the known roles of FGFR1 in vertebrate ontogeny, human diseases, and observations of brain and digits anomalies in conditional Fgfr1 deficient mice.
FGFR1 mutations also cause KS. From the cases reported in literature and in this study (see online supplementary table S4), it is not possible to confirm that KS is systematically part of Hartsfield syndrome. Nevertheless, we suggest performing arhinencephaly, endocrinology and olfactory evaluations in patients with ectrodactyly, with or without additional malformations.
Our six FGFR1 mutated patients show mild to severe Hartsfield syndrome. Variable expressivity and incomplete penetrance is well known in HPE, ectrodactyly and KS suggesting the role of additional factors and tissue specific sensitivity.31 32 We explored the possibility of oligogenic inheritance33–36 in the four patients screened by WES, as well as the potential role of distinct FGFR1 isoforms, but no obvious pattern could be found (see online supplementary table S5, supplementary figure S2). These questions should be addressed by the study of a large cohort of patients with IHH, KS, and Hartsfield syndrome.
We observe that FGFR1 mutations responsible for Hartsfield syndrome occur in several clusters in important functional domains (figure 4): homozygous mutations in the ligand binding domain D2; heterozygous substitutions in the TKD core. Only the C725Y mutation lies alone at the TKD C-terminal extremity, among mutations reported in patients with IHH/KS with orofacial features. It will be interesting to see if this clustering will resist the addition of new Hartsfield mutations.
The above work represents substantial evidence that FGFR1 is the most prevalent, if not the sole gene causing Hartsfield syndrome. The six FGFR1 mutated patients described here represent a homogeneous phenotype of HPE, ectrodactyly, with or without cleft lip and palate, and pituitary deficiency, although each of the features observed vary in severity. We however found no FGFR1 mutation or large FGFR1 deletion in a female fetus with severe brain malformation (HPE with severe disruption of the telencephalic architecture, heterotopies and diminished cortical thickness), ectrodactyly, bilateral forearm hypoplasia, cleft palate, hypertelorism, eye defect, and orbital hypoplasia on the right side (patient 7, table 1). This phenotype substantially deviates from the spectrum of clinical features observed in patients with Hartsfield syndrome and FGFR1 mutations, and might represent another diagnostic entity. Whether Hartsfield syndrome is a genetically homogeneous affliction will need further study.
Depending on the localisation of the amino acid substitution, Hartsfield syndrome can have an autosomal dominant or autosomal recessive mode of inheritance. With no recurrence of Hartsfield syndrome having been reported so far, the intrafamilial variability in clinical manifestations is unknown. The main challenge to improve genetic counselling will be to decipher the genetic and environmental factors responsible for the wide variability of the FGFR1 mutations disease spectrum.
In conclusion, our findings demonstrate that Hartsfield syndrome is part of a wide spectrum of developmental anomalies caused by FGFR1 loss-of-function mutations. This spectrum included unaffected carrier, seemingly IHH, isolated KS, and KS with additional features (including anomalies of digits falling out of the definition of ectrodactyly, or mild expression of HPE, such as corpus callosum agenesis or central incisor), and septo-optic-like dysplasia.20–22 The clinical entity known as Hartsfield syndrome now sits at its most severe end. In consequence, any patient with hand/foot midline defects (even mild ones) and affected by central diabetes insipidus, hypogonadotropic hypogonadism, anosmia or HPE should have their FGFR1 gene sequenced.
Supplementary Material
Acknowledgments
We thank Cedric Govaerts for help with the structure analysis, Dr Sharman Rajindrajith for referring patient 1 to DCdS, Alain Verloes for contact with BK and DCdS. The authors declare no competing interests.
Footnotes
Contributors: GS and CV designed the study strategy. NS performed analysis of next generation sequencing data, conservation and structure analysis. IM, NL and CP performed Sanger sequencing and analysis. CV, GS, DCdS, BD, CH, BK, GM, SJ, and GVV recruited subjects, gathered clinical data and contributed DNA samples. IM, PL, GC and MA contributed technical support and discussions. NS, GS and CV wrote the manuscript. All authors reviewed the manuscript.
Funding: This work was supported by the Fondation Robert Dubois (Department of Pediatrics, ULB). MA is supported by the FNRS and the Fonds Erasme. MA and CV are supported by the Fondation Lippens.
Competing interests: NS is a postdoctoral researcher, IM is a research associate, and NL is a clinician-scientist from the Fonds de la Recherche Scientifique (FNRS).
Patient consent: Obtained.
Ethics approval: ULB Hôpital Erasme ethics committee.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: Data used in this study are available upon request.
References
- 1.Cohen MM., Jr Holoprosencephaly: clinical, anatomic, and molecular dimensions. Birth Defects Res A Clin Mol Teratol 2006;76:658–73 [DOI] [PubMed] [Google Scholar]
- 2.Duijf PH, van Bokhoven H, Brunner HG. Pathogenesis of split-hand/split-foot malformation. Hum Mol Genet 2003;12(Spec No 1):R51–60 [DOI] [PubMed] [Google Scholar]
- 3.de Mollerat XJ, Gurrieri F, Morgan CT, Sangiorgi E, Everman DB, Gaspari P, Amiel J, Bamshad MJ, Lyle R, Blouin JL, Allanson JE, Le Marec B, Wilson M, Braverman NE, Radhakrishna U, Delozier-Blanchet C, Abbott A, Elghouzzi V, Antonarakis S, Stevenson RE, Munnich A, Neri G, Schwartz CE. A genomic rearrangement resulting in a tandem duplication is associated with split hand-split foot malformation 3 (SHFM3) at 10q24. Hum Mol Genet 2003;12:1959–71 [DOI] [PubMed] [Google Scholar]
- 4.Hartsfield J, Bixler D, DeMeyer W. Hypertelorism associated with holoprosencephaly and ectrodactyly. J Clin Dysmorphol 1984:27–31 [Google Scholar]
- 5.Vilain C, Mortier G, Van Vliet G, Dubourg C, Heinrichs C, de Silva D, Verloes A, Baumann C. Hartsfield holoprosencephaly-ectrodactyly syndrome in five male patients: further delineation and review. Am J Med Genet A 2009;149A:1476–81 [DOI] [PubMed] [Google Scholar]
- 6.Zechi-Ceide RM, Ribeiro LA, Raskin S, Bertolacini CD, Guion-Almeida ML, Richieri-Costa A. Holoprosencephaly, ectrodactyly, and bilateral cleft of lip and palate: exclusion of SHH, TGIF, SIX3, GLI2, TP73L, and DHCR7 as candidate genes. Am J Med Genet A 2009;149A:1277–9 [DOI] [PubMed] [Google Scholar]
- 7.Keaton AA, Solomon BD, van Essen AJ, Pfleghaar KM, Slama MA, Martin JA, Muenke M. Holoprosencephaly and ectrodactyly: report of three new patients and review of the literature. Am J Med Genet C Semin Med Genet 2010;154C:170–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalil KA Metwalley, Fargalley HS. Holoprosencephaly in an Egyptian baby with ectrodactyly-ectodermal dysplasia-cleft syndrome: a case report. J Med Case Rep 2012;6:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takenouchi T, Okuno H, Kosaki R, Ariyasu D, Torii C, Momoshima S, Harada N, Yoshihashi H, Takahashi T, Awazu M, Kosaki K. Microduplication of Xq24 and Hartsfield syndrome with holoprosencephaly, ectrodactyly, and clefting. Am J Med Genet A 2012;158A:2537–41 [DOI] [PubMed] [Google Scholar]
- 10.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coletta A, Molter C, Duque R, Steenhoff D, Taminau J, de Schaetzen V, Meganck S, Lazar C, Venet D, Detours V, Nowe A, Bersini H, Solis DY Weiss. InSilico DB genomic datasets hub: an efficient starting point for analyzing genome-wide studies in GenePattern, Integrative Genomics Viewer, and R/Bioconductor. Genome Biol 2012;13:R104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y, Lewis L, Han Y, Voight BF, Lim E, Rossin E, Kirby A, Flannick J, Fromer M, Shakir K, Fennell T, Garimella K, Banks E, Poplin R, Gabriel S, DePristo M, Wimbish JR, Boone BE, Levy SE, Betancur C, Sunyaev S, Boerwinkle E, Buxbaum JD, Cook EH, Jr, Devlin B, Gibbs RA, Roeder K, Schellenberg GD, Sutcliffe JS, Daly MJ. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012;485:242–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groth C, Lardelli M. The structure and function of vertebrate fibroblast growth factor receptor 1. Int J Dev Biol 2002;46:393–400 [PubMed] [Google Scholar]
- 16.Tole S, Gutin G, Bhatnagar L, Remedios R, Hebert JM. Development of midline cell types and commissural axon tracts requires Fgfr1 in the cerebrum. Dev Biol 2006;289:141–51 [DOI] [PubMed] [Google Scholar]
- 17.Hebert JM, Lin M, Partanen J, Rossant J, McConnell SK. FGF signaling through FGFR1 is required for olfactory bulb morphogenesis. Development 2003;130:1101–11 [DOI] [PubMed] [Google Scholar]
- 18.Chung WC, Moyle SS, Tsai PS. Fibroblast growth factor 8 signaling through fibroblast growth factor receptor 1 is required for the emergence of gonadotropin-releasing hormone neurons. Endocrinology 2008;149:4997–5003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verheyden JM, Lewandoski M, Deng C, Harfe BD, Sun X. Conditional inactivation of Fgfr1 in mouse defines its role in limb bud establishment, outgrowth and digit patterning. Development 2005;132:4235–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pecheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-van de Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 2003;33:463–5 [DOI] [PubMed] [Google Scholar]
- 21.Pitteloud N, Meysing A, Quinton R, Acierno JS, Jr, Dwyer AA, Plummer L, Fliers E, Boepple P, Hayes F, Seminara S, Hughes VA, Ma J, Bouloux P, Mohammadi M, Crowley WF., Jr Mutations in fibroblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol Cell Endocrinol 2006;254–255:60–9 [DOI] [PubMed] [Google Scholar]
- 22.Dode C, Fouveaut C, Mortier G, Janssens S, Bertherat J, Mahoudeau J, Kottler ML, Chabrolle C, Gancel A, Francois I, Devriendt K, Wolczynski S, Pugeat M, Pineiro-Garcia A, Murat A, Bouchard P, Young J, Delpech M, Hardelin JP. Novel FGFR1 sequence variants in Kallmann syndrome, and genetic evidence that the FGFR1c isoform is required in olfactory bulb and palate morphogenesis. Hum Mutat 2007;28:97–8 [DOI] [PubMed] [Google Scholar]
- 23.Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, Sykiotis GP, Gregory LC, Diaczok D, Tziaferi V, Elting MW, Padidela R, Plummer L, Martin C, Feng B, Zhang C, Zhou QY, Chen H, Mohammadi M, Quinton R, Sidis Y, Radovick S, Dattani MT, Pitteloud N. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab 2012;97:E694–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarzabek K, Wolczynski S, Lesniewicz R, Plessis G, Kottler ML. Evidence that FGFR1 loss-of-function mutations may cause variable skeletal malformations in patients with Kallmann syndrome. Adv Med Sci 2012;57:314–21 [DOI] [PubMed] [Google Scholar]
- 25.Lew ED, Furdui CM, Anderson KS, Schlessinger J. The precise sequence of FGF receptor autophosphorylation is kinetically driven and is disrupted by oncogenic mutations. Sci Signal 2009;2:ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zenaty D, Bretones P, Lambe C, Guemas I, David M, Leger J, de Roux N. Paediatric phenotype of Kallmann syndrome due to mutations of fibroblast growth factor receptor 1 (FGFR1). Mol Cell Endocrinol 2006;254–255:78–83 [DOI] [PubMed] [Google Scholar]
- 27.Pitteloud N, Acierno JS, Jr, Meysing A, Eliseenkova AV, Ma J, Ibrahimi OA, Metzger DL, Hayes FJ, Dwyer AA, Hughes VA, Yialamas M, Hall JE, Grant E, Mohammadi M, Crowley WF., Jr Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci U S A 2006;103:6281–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trarbach EB, Costa EM, Versiani B, de Castro M, Baptista MT, Garmes HM, de Mendonca BB, Latronico AC. Novel fibroblast growth factor receptor 1 mutations in patients with congenital hypogonadotropic hypogonadism with and without anosmia. J Clin Endocrinol Metab 2006;91:4006–12 [DOI] [PubMed] [Google Scholar]
- 29.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4:1073–81 [DOI] [PubMed] [Google Scholar]
- 30.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkie AO. Bad bones, absent smell, selfish testes: the pleiotropic consequences of human FGF receptor mutations. Cytokine Growth Factor Rev 2005;16:187–203 [DOI] [PubMed] [Google Scholar]
- 32.Hebert JM. FGFs: neurodevelopment's jack-of-all-trades—how do they do it? Front Neurosci 2011;5:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S, Dwyer AA, Quinton R, Hall JE, Gusella JF, Seminara SB, Crowley WF, Jr, Pitteloud N. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci U S A 2010;107:15140–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaaralahti K, Raivio T, Koivu R, Valanne L, Laitinen EM, Tommiska J. Genetic overlap between holoprosencephaly and Kallmann Syndrome. Mol Syndromol 2012;3:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, Pitteloud N. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest 2008;118:2822–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roessler E, Velez JI, Zhou N, Muenke M. Utilizing prospective sequence analysis of SHH, ZIC2, SIX3 and TGIF in holoprosencephaly probands to describe the parameters limiting the observed frequency of mutant genexgene interactions. Mol Genet Metab 2012;105:658–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol 2011;29:24–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beenken A, Eliseenkova AV, Ibrahimi OA, Olsen SK, Mohammadi M. Plasticity in interactions of fibroblast growth factor 1 (FGF1) N terminus with FGF receptors underlies promiscuity of FGF1. J Biol Chem 2012;287:3067–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plotnikov AN, Hubbard SR, Schlessinger J, Mohammadi M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 2000;101:413–24 [DOI] [PubMed] [Google Scholar]
- 40.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605–12 [DOI] [PubMed] [Google Scholar]
- 41.Bae JH, Lew ED, Yuzawa S, Tome F, Lax I, Schlessinger J. The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell 2009;138:514–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.