

Abstract
Vascular endothelia growth factor VEGF (VEGF-A or VEGF165) is a potent angiogenic factor that also signals neuroprotection through activation of its cognate receptor VEGFR-2. In this capacity, VEGF signaling can rescue neurons from the damage induced by stressful stimuli many of which elicit oxidative stress. However, the regulatory role that VEGFR-2 plays in providing neuroprotection remains elusive. Therefore, we investigated the effects of VEGFR-2 inhibition on primary cultures of mature hippocampal neurons undergoing nutritional stress. We found that neurons cultured under nutritional stress had increased expression of VEGF and its receptors, VEGFR-1, VEGFR-2 and NP-1 as well as enhanced levels of VEGFR-2 phosphorylation. These neurons also showed increased activation of the prosurvival pathways for MEK/ERK1/2 and PI3K/Akt, enhanced phosphorylation (inactivation) of the pro-apoptotic BAD and higher levels of the anti-apoptotic protein Bcl-xL, all of which were augmented by treatments with exogenous VEGF and blocked by VEGFR-2 inhibition. The blockade of VEGFR-2 function also elicited a cytotoxicity that was accompanied by caspase-3 activation, induction of hemeoxygenase-1 (HO-1), oxidative stress and a collapse in the mitochondrial membrane potential (Δψlm). Knockdown of VEGFR-2 by siRNA generated a similar pattern of redox change and mitochondrial impairment. Pretreatments with VEGF, VEGF-B or the antioxidant N-acetyl-cysteine (NAC) rescued SU1498 or siRNA treated neurons from the mitochondrial dysfunction and oxidative stress induced by VEGFR-2 inhibition in a timely fashion. These findings suggested that VEGF or VEGF-B can provide neuroprotection by signaling through an alternate VEGF receptor. Together, our findings suggest that VEGF signaling through VEGFR-2 plays a critical regulatory role in protecting stressed hippocampal neurons from the damaging effects of an oxidative insult. These findings also implicate VEGFR-1 or NP-1 as compensatory receptors that mediate neuroprotection when VEGFR-2 function is blocked.
Introduction
Oxidative stress and mitochondrial dysfunction contribute to the pathogenesis of a number of neurodegenerative disorders including Alzheimer’s and Parkinson’s disease [1]. One approach for protecting neurons from oxidative insults is to administer neurotrophic growth factors. One such factor is VEGF (VEGF-A or VEGF165) which is a mitogen that stimulates angiogenesis and neuroprotection through autocrine or paracrine mechanisms [2, 3]. In addition, VEGF can stimulate axonal outgrowth, and rescue rat mesencephalic neurons or hippocampal cells from death induced by serum withdrawal, ischemia, hypoxia and glutamate-induced toxicity [4–6] and elicit neuroprotection via angiogenesis and neurogenesis [7–9]. Delineating this neuroprotective mechanism is complex since VEGF can undergo cell-surface interactions with different cognate tyrosine kinase receptors such as VEGFR-1, VEGFR-2 and the non-tyrosine kinase members of the neuropilin family of class 3 semaphorin receptors Neuropilin-1 and -2 (NP-1, NP-2) receptors [10]. While VEGF mediates most biological effects through VEGFR-2, it can interact with NP-1 as a co-receptor that enhances VEGF signaling. Ligand binding to VEGFR-1 and VEGFR-2 results in receptor dimerization followed by autophosphorylation and activation of downstream signaling cascades [3]. In neurons, NP-1 is a cell-surface receptor for both VEGF and class 3 semaphorins and plays a functional role in axonal pathfinding, retraction and collapse during development. While the function of VEGFR-1 remains unclear, it is implicated as a decoy receptor that sequesters VEGF from activating VEGFR-2. VEGF-B, which is a ligand for VEGFR-1 but not VEGFR-2, is poorly angiogenic in the brain but can protect against mitochondrial membrane permeabilization [11], pro-apoptotic gene expression [12], and is coexpressed with genes encoding mitochondrial proteins [13]. Thus, VEGF-B may protect against mitochondrial dysfunction independent of angiogenesis.
Oxidative insults are implicated as causative factors of neuronal damage in several different neurological disorders and increasing evidence from in vitro and in vivo studies shows that VEGF signaling protects neurons from insults which are known to induce oxidative stress [14, 15]. In this context, VEGF overexpression has been shown to delay the onset of neuronal death in an animal model of amyotrophic lateral sclerosis (ALS) where oxidative stress is a contributing factor [16]. VEGF has been shown to mediate neuroprotection under stress conditions through the downstream activation of the phosphatidylinositol 3-kinase (PI3K)/Akt and mitogen-activated protein kinase MEK/ERK1/2 pathways [6, 17–19] and by suppressing caspase activation [20]. While several of these studies show that VEGF activates VEGFR-2 to mediate survival, the protective capacity of VEGFR-2 signaling remains unclear. Therefore, the aim of these studies is to determine the impact of VEGFR-2 signaling on the survival of mature hippocampal neurons by blocking its activation using pharmacological and gene silencing methods.
In previous studies we used a neuronal model of serum deprived SK-N-SH cells to show that VEGF signaling through VEGFR-2 prevents a caspase-dependent cell death [21]. Therefore, we addressed whether VEGF signaling through VEGFR-2 protected rat hippocampal neurons from the harmful effects of nutritional stress. Depriving cultured neurons from vital nutrients provides a viable model to identify the molecular basis of neuronal insults that would occur under pathological conditions in vivo. Our findings show that a blockade of VEGFR-2 function in hippocampal neurons leads to a rapid loss in viability that is manifested by induction of markers of oxidative stress, mitochondrial dysfunction and a loss in the activation of prosurvival pathways. Notably, the inclusion of exogenous VEGF or VEGF-B mediates a time-dependent rescue from this response, suggesting that a molecular switch to an alternate receptor can provide neuroprotection when VEGFR-2 activity is blocked. Our findings establish a link between VEGFR-2 signaling and mitochondrial function in differentiated rat neurons and provide insight on oxidative stress-related pathways that mediate neuronal damage and how exogenous VEGF or VEGF-B may counteract these events.
Materials and methods
Materials
Recombinant human vascular endothelial growth factor 165 (VEGF165) and VEGF-B were obtained from PeproTech Inc (Rocky Hill, NJ). The inhibitors of VEGFR-2 (SU1498) and PI3K/Akt (Worthmannin) were obtained from EMD Biosciences Inc (San Diego, CA). The inhibitor of MEK1/2 (U0126) was obtained from Promega Corporation (Madison, WI). The antioxidant N-acetyl-cysteine (NAC) was purchased from Sigma-Aldrich (St. Louis, MO).
Primary Cell Culture and Treatments
All animal studies were performed in accordance with the National Institutes of Health (NIH) Guidelines for the Care and Use of Laboratory Animals and the approval of the Institutional Animal Care Committee at Hunter College of the City University of New York. Rat hippocampal neurons were prepared from Sprague-Dawley (Charles River Laboratories) 1 day postnatal (P1) rat pups using a modification of a previously described procedure [4]. Briefly, hippocampi were isolated and dissected aseptically in ice-cold Ca2+/Mg2+-free Hank’s balance salt solution (HBSS, Invitrogen/Life Technologies). Following the removal of the meninges, the hippocampal tissue was minced, digested in 0.25% trypsin (Invitrogen/Life Technologies) for 15 minutes at 37°C and then rinsed at 37°C with Ca2+/Mg2+-free HBSS. The tissue was triturated in DMEM/10% FBS (Invitrogen/Life Technologies) through pasteur pipettes and then passed through a 40 μm mesh strainer. Isolated cells were then resuspended in DMEM/10% FBS, and plated into poly-D-lysine (100 μg/ml) coated plates as follows: 96-well microtiter plates (2 X 104 cells/well) for viability studies, 6-well plates (5 X 105 cells/well) for western blot analyses, 8-well chamber slides (1 X 105 cells/well) for immunofluoresence staining, ROS and TMRE detection and 10 cm plates (5 X 106 cells/well) for RT-PCR. The medium was replaced after 2 hr with Neurobasal (Invitrogen/Life Technologies) medium, L-glutamine (GlutaMAX™ Invitrogen/Life Technologies) and penicillin + streptomycin (Antibiotic-Antimycotic; Invitrogen/Life Technologies) (NB) supplemented with B27 (NB/B27) with 2 μM cytosine arabinoside (AraC) to inhibit the proliferation of glial cells. At day 4, one-half of NB/B27 was removed and replaced with a glial-conditioned medium (GCM). Neurons were then cultured in NB/B27/GCM (1:1 ratio) for 13 days in vitro (13 DIV), replacing one-half of the medium with NB/B27/GCM (1:1 ratio) every 4 days. Cells were then incubated in NB/B27 or NB and cultured for 48 hr (15 DIV). Where indicated, exogenous VEGF or VEGF-B (100 ng/ml) was administered at 0 time and replenished at 24 hr of the 48 hr incubation period. Neurons were also pretreated with NAC (5 mM) for the final 24 hr as indicated. For inhibitor studies, neurons were treated with predetermined concentrations of each selective agent. Incubations with SU1498 (10 μM) were at different times as indicated within the 48 hr timeframe while treatments with Wortmannin (100 nM) and U0126 (10 μM) were added for the final 2 hr of incubation. DMSO treated neurons served as the control for cells incubated with SU1498, Wortmannin and U0126. Culturing the neurons for 15 DIV allowed adequate time for neuronal maturation, network formation and the development of synapses. At this time, neurons consistently represent >90 to 99% of the cell population as stained with the dendritic marker microtubule-associated protein (MAP-2). The level of glial contamination usually ranges from 1–10% which is consistent with that previously reported [4, 20, 22].
Protein Extraction and Western Blotting
Total cell lysates from 15 DIV neurons were harvested in a lysis buffer as described previously [23] with the exception that the NP40 concentration was 0.3%. Protein concentrations were determined with a bicinchoninic acid assay (BCA) according to manufacturer’s instructions (Pierce). Equal amounts of protein (30 μg) from each lysate were resolved by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Blots were then blocked and incubated overnight at 40C with the primary antibodies for the phosphorylated (Y996) and total forms of VEGFR-2 (Santa Cruz), Akt (T308 and S473, Cell Signaling), BAD (S112, Cell signaling), phosphorylated ERK1/2 (T202/Y204, Cell Signaling) and total ERK1/2 (Santa Cruz) as well as Bcl-xL (Cell Signaling) and HO-1 (Santa Cruz). Actin (Sigma-Aldrich) was used as a loading control. Immunoreactive bands were detected with the corresponding anti-mouse and anti-rabbit secondary antibodies conjugated to horseradish peroxidase and visualized with the SuperSignal West Pico Chemiluminescent Substrate (Pierce Endogen). Data were quantified using the ImageJ program from NIH.
RNA Isolation and Semi-quantitative RT-PCR
Total RNA was isolated using the RNeasy RNA isolation Kit (Qiagen) according to the manufacturer’s protocol. RNA (2–4 μg) was reverse transcribed at 50°C for 30 min and amplified for 40 cycles using the Qiagen OneStep RT-PCR Kit following the manufacturer’s instruction. Amplifications were performed using previously described rat primers for VEGF, VEGFR-1, VEGFR-2, NP-1 and GAPDH [24]. PCR products were visualized by electrophoresis on 2% agarose gel and quantified using ImageJ.
Cell Viability
Cell viability was determined using a colorimetric MTS assay (Promega Corp.) and the amount of MTS reduction by viable cells was quantified according to manufacturer’s instruction. Survival measurements are expressed as the percent of the untreated or vehicle control.
Oxidative Stress
The live cell assay using the fluorescent dye 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFCA) was employed as an indicator of the redox status of the cell following the manufacturer’s instructions (Invitrogen/Life Technologies). Briefly, neurons were cultured in NB for 48 hr without and with VEGF (100 ng/ml), VEGF-B (100 ng/ml) or NAC (5 mM) alone or in the presence of SU1498 (10 μM) for the final 2 hr of incubation. Cells were then incubated with 25 μM carboxy-H2DCFCA which indicates oxidative stress by its conversion to an oxidized product that emits a green fluorescence. Images were immediately taken using a Nikon Eclipse TE200 inverted epifluorescence scope. Cells treated with tert-butyl hydroperoxide (THBP; 100 μM) for 90′ were used as positive control for oxidative stress. Data are representative of experiments that were repeated at least three times.
Mitochondrial Membrane Potential
Loss of mitochondrial membrane potential mitochondrial (Δψm) was assessed using a live cell assay with the fluorescent lipophilic cationic dye tetramethylrhodamin methyl (TMRE) (Invitrogen/Life Technologies). This dye is incorporated into mitochondria with an intact transmembrane potential and its release serves as an indicator of a loss of the inner mitochondrial membrane potential. Neurons (13 DIV) were cultured in NB for 48 hr under the same conditions as described for the protocol to measure oxidative stress. Neurons were then changed into fresh NB with 200 nM TMRE for 30 min. Cells were washed with fresh medium and fluorescent images were immediately taken using a Nikon Eclipse TE200 inverted epifluorescence scope. A 30 min incubation with carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP; 20 μM), an uncoupler of electron transport and oxidative phosphorylation, served as a positive control for disruption of the mitochondrial membrane potential. Data are representative of experiments that were repeated at least three times.
RNA Interference
Cultured neurons were transfected with 50 nM of Flk-1/KDR (VEGFR-2) SMARTpool® siRNA duplexes (Dharmacon RNA Technologies, Lafayette, CO) using BLOCK-iT Transfection Kit (Invitrogen/Life Technologies) according to manufacturer’s directions. The BLOCK-iT fluorescein-labeled oligo (Invitrogen/Life Technologies) which is not homologous to any known genes was used as negative control and indicator for transfection efficiency. After 4 hr, cells were changed to NB and incubated for a total of 48 hr in the absence and presence of VEGF (100 ng/ml), VEGF-B (100 ng/ml) as described above.
Immunofluorescence
Neurons were transfected with siRNA and cultured in NB for 48 with VEGF (100 ng/ml) as described above. Cells were then fixed in 3.7% formaldehyde in NB for 20 min at 37°C, permeabilized with 0.1% saponin for 20 min and blocked with 1% BSA in PBS with 0.25% Tween detergent for 30 min. Cells were incubated overnight with primary antibodies against VEGFR-1, VEGFR-2 (Santa Cruz) and MAP-2 (Millipore). Antigen detection was performed using fluorescein-conjugated FITC or Texas-Red secondary antibodies (Invitrogen). Images were captured at 63X using a Leica TCS SP2 Laser Scanning Spectral Confocal Microscope. Data are representative of experiments that were repeated at least three times.
Statistical Analyses
Data are expressed as the mean + SEM of experiments that were replicated at least three times. Statistical significance was assessed by a one-way ANOVA followed by pairwise contrasts (Bonferroni analysis). A difference resulting in P<0.05 is considered significant.
Results
VEGFR-2 signals survival in mature hippocampal neurons
We showed previously that VEGFR-2 elicits neuroprotection in serum starved neuroblastoma cells through autocrine and paracrine mechanisms and a blockade of receptor function leads to a loss in cell viability [21, 25]. Whether VEGFR-2 plays a similar role in primary cultures of mature hippocampal neurons remains unclear. In these studies, we hypothesized that VEGF signaling through VEGFR-2 alone plays a significant role in protecting mature hippocampal neurons from oxidative stress induced by suboptimal culture conditions. To test this hypothesis, cells were cultured in Neurobasal medium (NB) without the optimized culture conditions provided by the supplement B27. B27 contains biological antioxidants which would sequester free radical production, thus, complicating the identification of signaling mechanisms that protect against oxidative stress [26]. In addition, the high level of insulin in B27 would interfere with the identification of downstream effector pathways associated with VEGF/VEGFR-2 signaling since both growth factors utilize overlapping pathways to promote survival. Therefore, mature hippocampal neurons were cultured in NB alone in the absence and presence of VEGF or VEGF-B at 100 ng/ml for 48 hr. This concentration was shown to mediate significant levels of survival or neurite growth over time for cultured neurons [4, 27, 28].
To establish this model system, 13 DIV neurons were cultured in NB or NB/B27 for 48 hr (15 DIV) and then compared for their viability, expression of VEGF and its receptors and morphological characteristics. Bright field images show that neurons cultured under both conditions exhibited the expected morphology with extensive dendritic branching (Figure 1A). Semi-quantitative RT-PCR analyses revealed that the gene expression of VEGF and its receptors VEGFR-1, VEGFR-2 and NP-1 was greater in NB cultured cells when compared to that obtained with NB/B27 (Figure 1B). Whereas the omission of B27 reduced cell viability significantly by 25%, VEGF also elicited a significant stimulatory effect on survival both in NB (90%) and NB/B27 cultured cells (Figure 1C). Moreover, treatments with the VEGFR-2 inhibitor SU1498 decreased the cell viability of neurons cultured in NB and NB/B27 over time that reached approximately 50% survival up to 24 hr (Figure 1D). However, the removal of B27 sensitized NB cultured neurons to a significant loss in viability at 15 min of a 2 hr exposure to SU1498. These findings suggested that VEGF mediates neuroprotection through VEGFR-2 in the absence and presence of exogenous VEGF. Consistent with this notion, NB cultured cells showed a level of VEGFR-2 phosphorylation at Tyrosine 996 (Y996) that was augmented by 48 hr pretreatments with exogenous VEGF and attenuated by 24 hr treatments with the selective inhibitor SU1498 independent of exogenous VEGF (Figure 1E). Therefore, the increase in VEGF gene expression in NB together with SU1498-induced loss in viability without VEGF suggests that paracrine and autocrine VEGF signaling mediates survival in NB cultured cells.
Figure 1. VEGF mediates neuronal survival in NB cultured cells via endogenous and exogenous signaling.

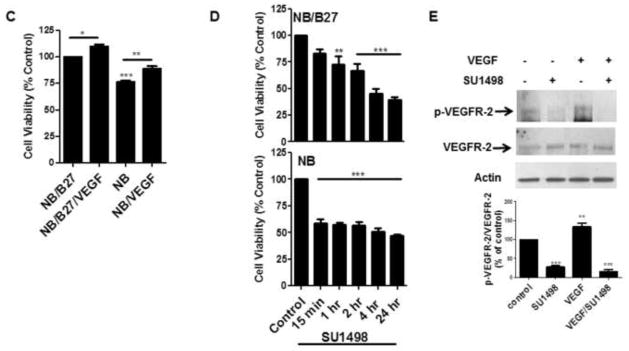
(A), Cells were incubated in NB/B27 or NB for 48 hr. Bright field images were taken by using a Nikon Eclipse TE200 inverted scope. Scale bar: 50 μm. (B), Total RNA was isolated from neurons cultured in NB and NB/B27 for 48 hr and analyzed by semi-quantitative RT-PCR for VEGF, VEGFR-2, VEGFR-1, NP-1 as described in “Materials and methods”. (C), Neurons were cultured as in (A) without and with 100 ng/ml VEGF and cell viability was determined using a MTS assay as described in “Materials and methods”. Results represent the percent cell viability relative to the vehicle treated control ± S.E.M. from at least three independent experiments. *(P<0.05) indicates a significance between NB/B27/VEGF versus NB/B27. ** (P<0.01) indicates a significant difference between NB versus NB/VEGF. *** (P<0.001) indicates a significant difference between NB versus NB/B27. (D), Cell viability was measured in cells cultured as in (A) with SU1498 (10 μM) added for the final 15′, 1, 2, 4 and 24 hr of the 48 hr incubation period. **(P<0.01) indicates a significant difference between 1 hr SU1498 versus control (NB/B27). ***(P<0.001) indicates a significant difference between 2, 4, and 24 hr SU1498 versus the NB/B27 control and between 15′, 1, 2, 4, 24 hr SU1498 versus the NB control. (E), Neurons were cultured in NB without and with VEGF in the absence and presence of 24 hr treatments with SU1498 (10 μM). Lysates were analyzed by western blotting for the levels of phosphorylated (p-VEGFR-2) and total VEGFR-2 using antigen specific antibodies. Actin served as the loading control. The levels of phosphorylation are normalized to their respective total protein control and then quantified by ImageJ relative to the DMSO treated control (100%) ± S.E.M. from at least three independent experiments. **(P<0.01) indicates a significant difference between VEGF versus control; ***(P<0.001) indicates a significant difference between SU1498 or VEGF/SU1498 versus control.
VEGFR-2 inhibition induces an oxidative stress that can be hindered by NAC, VEGF or VEGF-B
To evaluate whether VEGFR-2 inhibition was associated with oxidative stress and/or apoptosis, NB cultured cells were treated with SU1498 for 24 hr and analyzed for the induction of HO-1, a biomarker of oxidative insults, and caspase-3 cleavage, respectively. The results showed that a 24 hr exposure to SU1498 induced a 3-fold increase in caspase-3 cleavage and HO-1 induction (Figure 2A, lane 3) and a 50% loss in viability (Figure 2B) that were rescued by pretreatments with the N-acetly-cysteine (NAC), a precursor of the cellular thiol glutathione which in turn maintains redox homeostasis by sequestering free radicals through a thiol/disulfide exchange (Figure 2A, lane 4; Figure 2B). To determine whether VEGF signaled protection exclusively through VEGFR-2, the effects of the 48 hr pretreatment with the growth factor were evaluated on the cell viability in SU1498 treated neurons following a short (2 hr) and long (24 hr) time frame of exposure (Figure 3A). Interestingly, VEGF rescued SU1498 treated cells from a loss in cell viability following 2 but not 24 hr of exposure to the inhibitor. Subsequent analyses showed that SU1498 induced a significant increase in HO-1 induction and caspase-3 cleavage that was suppressed by VEGF following the same 2 hr timeframe of VEGFR-2 inhibition (Figure 3B, compare lanes 2 and 4). Although VEGF elicited protection under these conditions, western blot analyses confirmed that VEGFR-2 phosphorylation was completely blocked by the 2 hr treatment with SU1498 (data not shown). These findings raised the possibility that VEGF activated an alternate receptor (VEGFR-1 or NP-1) as a compensatory pathway for survival when VEGFR-2 function was blocked. Indeed, pretreatments with VEGF-B, which signals through VEGFR-1 or NP-1 but not VEGFR-2, mediated a significant increase in the viability of neurons exposed to SU1498 for 2h (Figure 3C). Given these findings, subsequent studies were then performed in neurons exposed to VEGFR-2 inhibition for 2 hr.
Figure 2. VEGFR-2 inhibition induces oxidative stress and apoptosis that are prevented by treatments with NAC.
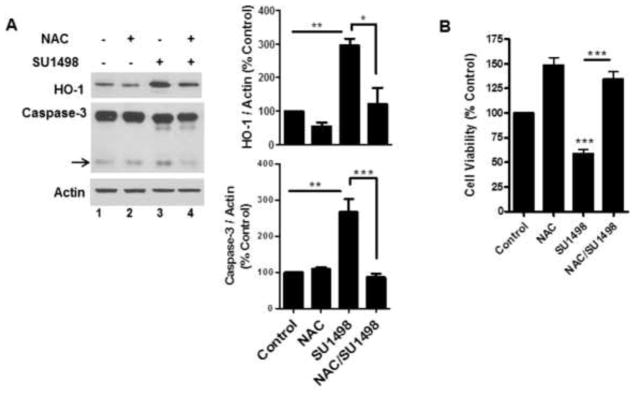
(A), NB cultured neurons were treated without and with SU1498 (10 μM) alone or in combination with NAC during the final 24 hr of the 48 hr incubation. Lysates were analyzed by western blotting for the levels of HO-1 and cleaved caspase-3 (arrow). Data are quantified relative to the vehicle control. *(P<0.05) indicates a significant difference between SU1498 versus NAC/SU1498 for HO-1 and **(P<0.01) indicates a significant difference between SU1498 versus control or NAC for HO-1 and Caspase-3. ***(P<0.001) indicates a significant difference between SU1498 versus NAC/SU1498 for Caspase-3. (B), Neurons cultured as in (A) were analyzed for cell viability as described in Figure 1C. ***(P<0.001) indicates a significant difference between SU1498 versus control or NAC/SU1498.
Figure 3. VEGF and VEGF-B protect neurons from the effects of a short but not long timeframe of VEGFR-2 inhibition.

(A), Cell viability was measured in neurons cultured in NB without and with VEGF following a 2 and 24 hr exposure to SU1498 (10 μM). *(P<0.05) indicates a significant difference between VEGF versus control and ***(P<0.001) indicates a significant difference between SU1498 2hr versus VEGF/SU1498 2 hr in A; (B), Lysates were analyzed by western blotting for the levels of HO-1 and cleaved caspase-3 (arrow) as in Figure 2A. **(P<0.01) and *(P<0.05) indicates a significant difference between SU1498 versus control or VEGF or VEGF/SU1498 for HO-1 and caspase-3, respectively. (C), Cell viability was measured in neurons cultured in NB without and with VEGF-B following a 2 hr exposure to SU1498 (10 μM). **(P<0.01) indicates a significant difference between VEGF-B versus control and ***(P<0.001) indicates a significant difference between SU1498 versus control and SU1498 versus VEGF-B/SU1498.
Selective inhibition or gene silencing of VEGFR-2 induces oxidative stress and a collapse of the mitochondrial membrane potential
The finding that VEGFR-2 inhibition leads to HO-1 induction and caspase-3 cleavage suggested that the receptor signals downstream pathways that protect against apoptosis and oxidative stress. To address this possibility, a live cell assay was employed to evaluate changes in the intracellular redox status and mitochondrial dysfunction following a 2 hr exposure to SU1498 in cells pretreated without and with VEGF, VEGF-B or NAC (Figure 4). For these assays, oxidative stress is measured using 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate which is converted to a green-fluorescent form by oxidation (removal) of the acetate groups by free radicals. To measure mitochondrial dysfunction, cells were also loaded with the fluorescent lipophilic cationic dye tetramethylrhodamin methyl (TMRE; red) which is only retained if the mitochondrial membrane potential (Δψm) is intact. Cells were counterstained with the nuclear dye DAPI to show the number of neurons per field. The results in Figure 4 showed that SU1498 induced oxidative stress (green fluorescence; compare panels A & B) and a collapse in the mitochondrial membrane potential (TMRE, loss of red fluorescence; compare panels A & B) within a 2 hr timeframe that can be prevented by pretreatments with exogenous VEGF (compare panels B & F), VEGF-B (compare panels B & H) or NAC (compare panels B & D). Similarly, an attenuation of VEGFR-2 expression levels by siRNA led to oxidative stress and a loss of Δψm (Figure 5A, panels A and B) that were inhibited by treatments with VEGF or VEGF-B (panels C and D). Since VEGF and VEGF-B are ligands for VEGFR-1, these results suggest that both factors may signal protection through VEGFR-1 when VEGFR-2 function is blocked. Double immunofluorescent studies for VEGFR-1 or VEGFR-2 with the dendritic marker MAP-2 showed the presence of both receptors in NB cultured cells (Figure 5B, panel A). Interestingly, VEGF increased localization of VEGFR-2 along dendrites (Figure 5B, upper panel C) while siRNA VEGFR-2 reduced both the basal (upper panel B) and dendritic levels of the receptor (upper panel D). In contrast, the expression levels of VEGFR-1 remained unchanged by VEGF or siRNA VEGFR-2 (Figure 5B, lower panels B, C and D) and were localized in the soma of neurons.
Figure 4. VEGF and VEGF-B protect against oxidative stress and mitochondrial dysfunction induced by SU1498.
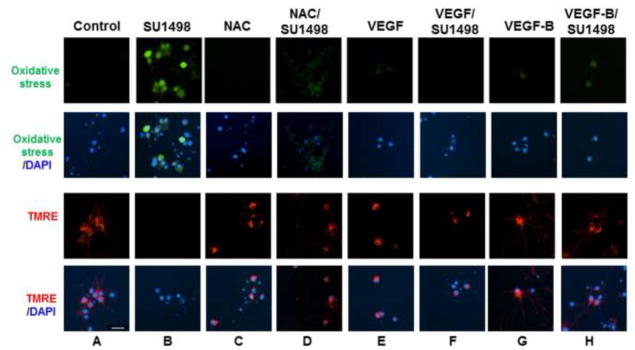
Neurons were cultured in NB for 48 hr in the absence and presence of NAC (5 mM, final 24 hr), VEGF or VEGF-B (100 ng/ml for 48 hr, replenished after 24 hr). After 46 hr of incubation, cells were treated with SU1498 as indicated and labeled with either carboxy-H2DCFCA to detect oxidative stress (green) or TMRE (red) to measure the Δψm as described in “Materials and methods”. Nuclei were counterstained with DAPI. Scale bar: 50 μm. Data are representative of experiments that were repeated at least three times.
Figure 5. VEGF and VEGF-B protect against oxidative stress and mitochondrial dysfunction induced by siRNA VEGFR-2.
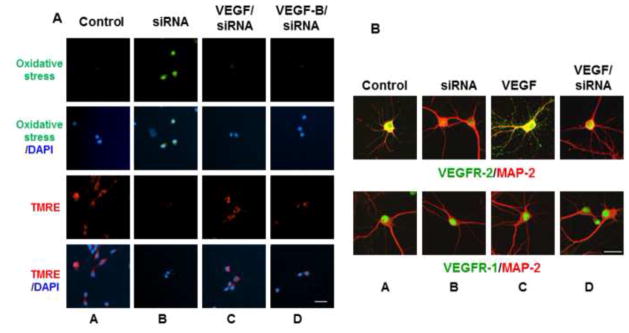
(A), Neurons were transfected with siVEGFR-2 as described in “Materials and methods” and then cultured without and with VEGF or VEGF-B for oxidative stress and TMRE measurements as described in Figure 4. Scale bar: 50 μm. (B), Cells were double labeled for fluorescence with MAP-2 (red) and either VEGFR-2 (green; upper panels) or VEGFR-1 (green; lower panels) as described in “Materials and methods”. Scale bar: 100 μm. Data are representative of experiments that were repeated at least three times.
The MEK/ERK1/2 and PI3K/Akt pathways are downstream effectors of VEGFR-2 mediated protection
To delineate the downstream effector pathways activated by VEGFR-2 to mediate survival, we examined the effects of SU1498 on the activation of Akt and ERK1/2 by their upstream kinases, phosphatidyl inositol 3′ kinase (PI3K) and mitogen-activated protein kinase (MEK), respectively. Both pathways have been shown to signal VEGF-mediated neuroprotection against harmful insults [4, 22]. Consistent with these reports, VEGFR-2 inhibition prevented both the basal and VEGF-stimulated levels of phosphorylated ERK1/2 (T202/Y204) and Akt at the T308 and S473 activation sites (Figure 6A, compare lanes 1 & 2 to lanes 3 & 4) and selective inhibition of either PI3K (Figure 6A, Wortmannin (Wort), lane 5) or MEK (U0126, lane 6) induced a significant loss in viability (Figure 6B). These results show that VEGF signaling through VEGFR-2 leads to the downstream activation of the MEK/ERK1/2 and PI3K/Akt pathways as survival mechanisms in our paradigm of oxidative stress.
Figure 6. VEGF signaling through VEGFR-2 leads to activation of the PI3K/Akt and MEK/ERK1/2 pathways.
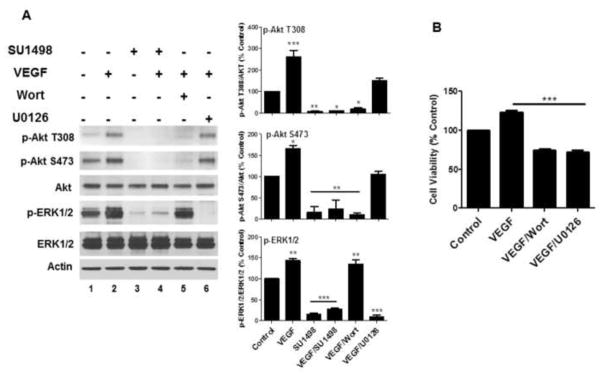
(A), Neurons were cultured 48 hr in NB without and with VEGF as in Figure 1 and treated at 46 hr with SU1498 (10 μM), Wortmannin (Wort; 100 nM) or U0126 (10 μM). Lysates from each condition were then analyzed by western blotting for detection of the phosphorylated and total forms of Akt and ERK1/2 as indicated. Data were quantified relative to the vehicle control. *** (P<0.001) indicates a significant difference between VEGF versus control for p-Akt (T308); a significanct difference between SU1498, VEGF/SU1498 or VEGF/U0126 versus control or VEGF for p-ERK1/2. ** (P<0.01) indicates a significant difference between SU1498 versus control for p-Akt (T308); a significant difference between SU1498, VEGF/SU1498 or VEGF/Wort versus control or VEGF for p-Akt (S473); a significant difference between VEGF or VEGF/Wort versus control for pERK. * (P<0.05) indicates a significant difference between VEGF/SU1498 or VEGF/Wort versus control or VEGF for p-Akt (T308); a significant difference between VEGF versus control for p-Akt (S473). (B), Cell viability was determined in VEGF cultured cells without or with 2 hr incubations with Wort or U0126. ***(P<0.001) indicates a significant difference between VEGF, VEGF/Wort or VEGF/U0126 versus control.
BAD phosphorylation and the protein levels of Bcl-xL are regulated by VEGFR-2
Mitochondrial function is tightly regulated by a phosphorylation-dependent inactivation of the pro-apoptotic protein BAD by Akt and ERK1/2 at S136 and S112, respectively, to prevent interaction with the anti-apoptotic protein Bcl-xL [29]. Inhibitor studies revealed that VEGF mediated significant increases in BAD phosphorylation at S112 and Bcl-xL protein levels (Figure 7A, lane 2) that were suppressed following 2 hr treatments with SU1498 (lanes 3 and 4), but not with Wortmannin or U0126 (lanes 5 and 6). These results suggest that VEGFR-2 signals both events through a mechanism that is independent of the Akt and ERK1/2 pathways. Nevertheless, co-incubations of SU1498 with VEGF elicited a partial rescue in BAD and Bcl-xl phosphorylation. Recent studies show that a Bcl-xL knockdown increased oxidant-induced apoptosis in cardiomyocytes, suggesting that Bcl-xL protein levels are redox regulated [30]. To test whether Bcl-xL is redox sensitive in our paradigm of oxidative stress, we examined the effects of 24 hr pretreatments with NAC on neurons incubated with SU1498 for 2 hr. Indeed, NAC stimulated an increase in Bcl-xL protein content (Figure 7B, lane 2) that was reduced significantly to control levels by co-incubation with SU1498 (lane 3).
Figure 7. VEGF enhances BAD phosphorylation (inactivation) while VEGF and NAC increase Bcl-xL protein expression.

(A, B), Cells were cultured as described in Figure 6A and treated with Wortmannin (Wort) or U0126 in (A) and NAC (5 mM) in (B) and subjected to western blot analyses for the phosphorylated and total form of BAD and Bcl-xL. Data were quantified relative to the vehicle control. *(P<0.05) indicates a significant difference between VEGF versus control and SU1498 versus VEGF/SU1498 for Bcl-xL in A. **(P<0.01) indicates a significant difference between VEGF versus control for p-BAD in A; a significant difference between NAC versus NAC/SU1498 for Bcl-xL in B. ***(P<0.001) indicates a significant difference between SU1498 versus control for p-BAD and Bcl-xL in A and a significant difference between NAC versus control for Bcl-xL in B. ### (P<0.001) indicates a significant difference between VEGF versus VEGF/SU1498 for p-BAD in A.
Discussion
In this study we examined the effects of VEGFR-2 inhibition on the survival of nutritionally deprived hippocampal neurons. We found that neuronal viability was reduced only slightly in NB cultured neurons when compared to those cultured in NB supplemented with B27 and survival under both conditions was enhanced by the inclusion of exogenous VEGF and inhibited by VEGFR-2 inhibition. We attribute survival without B27 to an autocrine signaling by endogenous VEGF through VEGFR-2 based on the observation that NB cultured neurons show increased gene expression of VEGF and its receptors, phosphorylation of VEGFR-2 and a greater sensitivity to decreased viability by VEGFR-2 inhibition compared to NB/B27 cultured cells. Also, ELISA assays showed that VEGF secretion was also greater from NB cultured neurons (data not shown). In support of these findings, autocrine VEGF signaling was shown to promote survival in retinal epithelial cells and cortical neurons in response to oxidative stress and hypoxia, respectively [2, 31]. In addition, activated VEGFR-2 was shown to protect endothelial cells against oxidative stress [32] and a reduction in VEGFR-2 protein expression induced cell death in motor neurons exposed to hypoxia [33]. The finding that VEGFR-2 mediates survival through the activation of the PI3K/Akt and MEK/ERK1/2 pathways is also consistent with previous demonstrations that either one or both of these mechanisms elicit neuroprotection depending on the cell type and stressor employed [4, 6, 17–19]. Furthermore, VEGF signaling through VEGFR-2 also mediates neurite outgrown in cortical neurons through the downstream activation of Akt and ERK1/2 [28]. Our results implicate essential roles for the PI3K/Akt and MEK/ERK1/2 pathways in promoting the survival of hippocampal neurons exposed to oxidative stress.
The mitochondrial cell death pathway is tightly regulated by a critical balance of interactions between the Bcl-2 family of proteins, namely, the anti-apoptotic members (e.g., Bcl-2 and Bcl-xL) and pro-apoptotic proteins (e.g., BAD). BAD normally exists in the cytosol in a phosphorylated (inactivated) form. However, Bad dephosphorylation (activation) induces its translocation to the mitochondrial membrane where it forms an inhibitory complex with Bcl-xL. This event in turn leads to oxidative stress with a collapse of the Δψm and a release of cytochrome c from permeabilized mitochondria to the cytoplasm that triggers caspase activation [29]. Therefore, the findings that a blockade of VEGFR-2 by pharmacological inhibition or gene silencing with siRNA induces a loss in the mitochondrial membrane potential with a concomitant increase in oxidative stress implicates VEGFR-2 signaling as a critical mediator of protection against mitochondrial dysfunction in hippocampal neurons. This implication is further strengthened by our demonstration that these events are accompanied by caspase-3 activation, HO-1 induction, BAD dephosphorylation and decreased levels of Bcl-xL and previous studies showing that VEGF promotes survival in hypoxic neurons by suppressing the activation of caspase-3 [20]. Interestingly, our findings are also consistent with evidence that cancer therapeutics that target tyrosine kinase receptors such as VEGFR-2 or ErbB2 induce a cardiotoxicity that is manifested by oxidative stress, a loss in the Δψm, and decreased levels of BAD phosphorylation and Bcl-xL protein expression [34, 35]. Moreover, previous studies from our laboratory showed that VEGFR-2 inhibition induced a loss in Bcl-xL protein expression and cell death in serum starved SK-N-SH neuroblastoma cells and viability was restored by overexpression of Bcl-xL or ERK1/2 [21]. The demonstration that NAC and VEGF stimulate an increase in Bcl-xL that is attenuated by treatments with SU1498 further supports the notion that VEGFR-2 signaling regulates both mitochondrial integrity and redox homeostasis in hippocampal neurons. However, the downstream mechanism(s) that modulate Bcl-xL protein levels and BAD phosphorylation and the exact pathway(s) linking VEGFR-2 signaling with the induction of oxidative stress or mitochondrial dysfunction in our studies are unclear and require further investigation. In this regard, inhibitor studies revealed that activated VEGFR-2 signals phosphorylation of BAD and increased protein levels of Bcl-xL in hippocampal neurons through a downstream mechanism that is independent of Akt or ERK1/2. Although ERK1/2 is usually the upstream kinase for BAD phosphorylation at S112, it is possible that kinases such as the MAPK-activated kinase, p90(RSK) via a PKC dependent pathway [36] or mitochondrial bound PKA [37] mediate this event in our paradigm of VEGF directed neuroprotection. Neurons are highly susceptible to oxidative damage due to their high oxidative energy metabolism and their moderate levels of antioxidant systems. Therefore, the imbalance created by excessive levels of free radicals would overwhelm the antioxidant capacity of the cell to cause oxidative modifications to lipids, proteins, and nucleic acids that are neurotoxic. In our studies, the induction of HO-1 protein expression serves as a reliable marker of this redox imbalance. HO-1 is maintained at low levels in the brain but is sensitive to induction by a wide array of toxic insults that mediate oxidative damage and inflammation [38] as found in neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease [39]. However, the role of HO-1 in neurodegeneration is questionable since it can provide neuroprotection against oxidative stress [40] or enhance neural injury through excess iron production [41]. Our demonstration that enhanced levels of HO-1 accompany mitochondrial dysfunction, oxidative stress and a loss in viability within a 2 hr exposure to SU1498 suggests that hippocampal neurons undergo a potent oxidative insult when VEGFR-2 function is blocked. Nevertheless, pretreatments with the antioxidant NAC provides protection against the oxidative insult induced by a prolonged period (24 hr, Figure 2) of VEGFR-2 inhibition. Similarly, pretreatments with VEGF or VEGF-B also mediate protection against changes in the redox state, Δψm collapse, caspase-3 activation and HO-1 induction but only within the initial 2 hr of VEGFR-2 inhibition. These findings suggest that VEGF or VEGF-B can provide protection when VEGFR-2 activity is blocked by signaling through an alternate receptor. Two possible receptors for this molecular switch are VEGFR-1 and NP-1 which are known to bind both VEGF and VEGF-B as ligands [3]. It is postulated that VEGFR-1 functions as a decoy mechanism that sequesters VEGF from binding to VEGFR-2 while NP-1 serves as a co-receptor for VEGF/VEGF receptor complexes to enhance VEGF signaling [3]. Our demonstration that the gene expression of VEGFR-1 and NP-1 are upregulated in NB cultured neurons raises the possibility that VEGF or VEGF-B signals through VEGFR-1 alone or in a complex with NP-1 to compensate for the neuroprotection lost by VEGFR-2 inhibition. It is likely that this response involves VEGFR-1 but this line of reasoning requires further investigation since recent findings show that NP-1 can promote neuronal survival independent of interactions with VEGFR-2 [42]. A model of a molecular switch from VEGFR-2 to VEGFR-1 signaling is shown in Figure 8. There is considerable support for VEGFR-1 signaling as a prosurvival mechanism in neuronal and nonneuronal cells. VEGFR-1 activation by VEGF-B was shown to elicit neuroprotection in spinal cord motor neurons [27], dopaminergic neurons [43] and in sensory neurons by maintaining the Δψm [11]. In cardiomyocytes, VEGF-B prevents ischemia and oxidative stress through VEGFR-1 [44, 45] and is coexpressed with genes encoding mitochondrial proteins in endothelial cells [13]. In our studies, VEGFR-1 is detected in the soma, raising the possibility that the receptor localizes in the nucleus as previously demonstrated in endothelial cells [46]. In contrast, treatments with exogenous VEGF stimulate VEGFR-2 to localize along dendrites, suggesting that activated VEGFR-2 resides in close proximity to synapses. In addition, VEGF can stimulate the nuclear localization of PGC-1 alpha, a key regulator of mitochondrial biogenesis, resulting in the increased expression of nuclear encoded mitochondrial gene expression and survival [47]. Moreover, the partial rescue of Bcl-xL by VEGF in SU1498 treated cells suggests that signaling through an alternate VEGF receptor can serve as a compensatory mechanism that modulates Bcl-xL levels at the mitochondrial membrane to prevent permeabilization. In this context, Bcl-xL was shown to delay mitochondrial membrane permeabilization in response to calcium overload [48]. In addition, Bcl-xL can increase mitochondrial biomass in neurons [49] or interact directly with either the mitochondrial F1F0 ATP synthase to enhance energy production [50] or with VDAC, an outer mitochondrial membrane protein, to regulate metabolite trafficking [51].
Figure 8. A model of VEGFR-2 mediated neuroprotection in mature hippocampal neurons.

VEGF mediated signaling through VEGFR-2 promotes survival through the downstream activation of PI3K/Akt and MEK/ERK1/2 pathways. A blockade of VEGFR-2 of signaling leads to a loss in Akt and ERK1/2 activation that is accompanied by a mitochondrial dysfunction involving a loss in the Δψm, oxidative stress and caspase-3 activation together with HO-1 induction, BAD activation and decreased levels of Bcl-xL. However, treatments with exogenous VEGF or VEGF-B can elicit a time dependent protection against the HO-1 induction, caspase-3 activation, mitochondrial dysfunction and the change in redox state induced by VEGFR-2 inhibition (in box) by signaling through an alternate receptor such as VEGFR-1.
Evidence suggests that neurons depend on ATP production from mitochondrial-derived oxidative phosphorylation rather than glycolysis as an energy source [52] and mitochondria are transported from the soma to axonal regions and dendritic synaptic terminals to meet the metabolic demand of neural transmission [53]. It is likely that the apparent loss in Δψm and elevated oxidative stress induced by VEGFR-2 inhibition would lead to a disruption in ATP production that severally compromise neuronal function. Thus, VEGFR-2 signaling may protect stressed hippocampal neurons against ATP depletion and oxidative stress induced cellular damage that would hinder synaptic plasticity. In this regard, oxidative stress is functionally linked to the oxidative DNA damage that leads to cognitive impairment in Alzheimer’s disease [54]. Together, these findings implicate VEGFR-2 signaling as an essential mechanism for maintaining mitochondrial integrity and protecting against oxidative insults in hippocampal neurons.
Conclusions
In conclusion, we show that a blockade of VEGFR-2 function by pharmacological inhibition or gene silencing triggers oxidative stress and mitochondrial dysfunction in mature hippocampal neurons. These events are manifested by an induction of HO-1, caspase-3 cleavage with a parallel loss in both cell viability and the activation of prosurvival Akt and ERK1/2. These findings also implicate activated BAD and decreased Bcl-xL levels as participants in the mitochondrial dysfunction and oxidative stress induced by VEGFR-2 inhibition. Treatments with exogenous VEGF or VEGF-B can signal through an alternate receptor to delay the onset of neuronal damage induced by VEGFR-2 inhibition. These findings implicate VEGFR-2 signaling in hippocampal neurons as a prosurvival mechanism that protects against mitochondrial dysfunction and oxidative stress.
Highlights.
1- Nutritional stress upregulates VEGF and its receptors in mature hippocampal neurons
2- Activated VEGFR-2 signals neuroprotection through the PI3K/Akt and ERK1/2 pathways
3- VEGFR-2 inactivation decreased viability with mitochondrial dysfunction and ROS
4- VEGF and VEGF-B signaling or NAC counteract the insult induced by VEGFR-2 inhibition
Acknowledgments
This investigation was funded by a grant from the National Center for Research Resources [RR003037], a component of the National Institutes of Health (NIH) and a NINDS/NIH grant [SC1NS066033]. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 2.Ogunshola OO, Antic A, Donoghue MJ, Fan SY, Kim H, Stewart WB, Madri JA, Ment LR. Paracrine and autocrine functions of neuronal vascular endothelial growth factor (VEGF) in the central nervous system. The Journal of biological chemistry. 2002;277:11410–11415. doi: 10.1074/jbc.M111085200. [DOI] [PubMed] [Google Scholar]
- 3.Ruiz de Almodovar C, Lambrechts D, Mazzone M, Carmeliet P. Role and therapeutic potential of VEGF in the nervous system. Physiological reviews. 2009;89:607–648. doi: 10.1152/physrev.00031.2008. [DOI] [PubMed] [Google Scholar]
- 4.Matsuzaki H, Tamatani M, Yamaguchi A, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: signal transduction cascades. Faseb J. 2001;15:1218–1220. [PubMed] [Google Scholar]
- 5.Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:10242–10247. doi: 10.1073/pnas.97.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2002;22:6401–6407. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:11946–11950. doi: 10.1073/pnas.182296499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao L, Jiao X, Zuzga DS, Liu Y, Fong DM, Young D, During MJ. VEGF links hippocampal activity with neurogenesis, learning and memory. Nature genetics. 2004;36:827–835. doi: 10.1038/ng1395. [DOI] [PubMed] [Google Scholar]
- 9.Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, Greenberg DA. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. The Journal of clinical investigation. 2003;111:1843–1851. doi: 10.1172/JCI17977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neufeld G, Cohen T, Shraga N, Lange T, Kessler O, Herzog Y. The neuropilins: multifunctional semaphorin and VEGF receptors that modulate axon guidance and angiogenesis. Trends in cardiovascular medicine. 2002;12:13–19. doi: 10.1016/s1050-1738(01)00140-2. [DOI] [PubMed] [Google Scholar]
- 11.Dhondt J, Peeraer E, Verheyen A, Nuydens R, Buysschaert I, Poesen K, Van Geyte K, Beerens M, Shibuya M, Haigh JJ, Meert T, Carmeliet P, Lambrechts D. Neuronal FLT1 receptor and its selective ligand VEGF-B protect against retrograde degeneration of sensory neurons. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25:1461–1473. doi: 10.1096/fj.10-170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Zhang F, Nagai N, Tang Z, Zhang S, Scotney P, Lennartsson J, Zhu C, Qu Y, Fang C, Hua J, Matsuo O, Fong GH, Ding H, Cao Y, Becker KG, Nash A, Heldin CH, Li X. VEGF-B inhibits apoptosis via VEGFR-1-mediated suppression of the expression of BH3-only protein genes in mice and rats. The Journal of clinical investigation. 2008;118:913–923. doi: 10.1172/JCI33673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hagberg CE, Falkevall A, Wang X, Larsson E, Huusko J, Nilsson I, van Meeteren LA, Samen E, Lu L, Vanwildemeersch M, Klar J, Genove G, Pietras K, Stone-Elander S, Claesson-Welsh L, Yla-Herttuala S, Lindahl P, Eriksson U. Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature. 2010;464:917–921. doi: 10.1038/nature08945. [DOI] [PubMed] [Google Scholar]
- 14.Zachary I. Neuroprotective role of vascular endothelial growth factor: signalling mechanisms, biological function, and therapeutic potential. Neuro-Signals. 2005;14:207–221. doi: 10.1159/000088637. [DOI] [PubMed] [Google Scholar]
- 15.Storkebaum E, Carmeliet P. VEGF: a critical player in neurodegeneration. The Journal of clinical investigation. 2004;113:14–18. doi: 10.1172/JCI200420682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Mao XO, Xie L, Banwait S, Marti HH, Greenberg DA, Jin K. Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:304–307. doi: 10.1523/JNEUROSCI.4433-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kilic U, Kilic E, Jarve A, Guo Z, Spudich A, Bieber K, Barzena U, Bassetti CL, Marti HH, Hermann DM. Human vascular endothelial growth factor protects axotomized retinal ganglion cells in vivo by activating ERK-1/2 and Akt pathways. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26:12439–12446. doi: 10.1523/JNEUROSCI.0434-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kilic E, Kilic U, Wang Y, Bassetti CL, Marti HH, Hermann DM. The phosphatidylinositol-3 kinase/Akt pathway mediates VEGF’s neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2006;20:1185–1187. doi: 10.1096/fj.05-4829fje. [DOI] [PubMed] [Google Scholar]
- 19.Jin KL, Mao XO, Nagayama T, Goldsmith PC, Greenberg DA. Induction of vascular endothelial growth factor receptors and phosphatidylinositol 3′-kinase/Akt signaling by global cerebral ischemia in the rat. Neuroscience. 2000;100:713–717. doi: 10.1016/s0306-4522(00)00331-6. [DOI] [PubMed] [Google Scholar]
- 20.Jin K, Mao XO, Batteur SP, McEachron E, Leahy A, Greenberg DA. Caspase-3 and the regulation of hypoxic neuronal death by vascular endothelial growth factor. Neuroscience. 2001;108:351–358. doi: 10.1016/s0306-4522(01)00154-3. [DOI] [PubMed] [Google Scholar]
- 21.Gomes E, Papa L, Hao T, Rockwell P. The VEGFR2 and PKA pathways converge at MEK/ERK1/2 to promote survival in serum deprived neuronal cells. Mol Cell Biochem. 2007;305:179–190. doi: 10.1007/s11010-007-9542-2. [DOI] [PubMed] [Google Scholar]
- 22.Jin K, Mao XO, Zhu Y, Greenberg DA. MEK and ERK protect hypoxic cortical neurons via phosphorylation of Bad. Journal of neurochemistry. 2002;80:119–125. doi: 10.1046/j.0022-3042.2001.00678.x. [DOI] [PubMed] [Google Scholar]
- 23.Rockwell P, Martinez J, Papa L, Gomes E. Redox regulates COX-2 upregulation and cell death in the neuronal response to cadmium. Cell Signal. 2004;16:343–353. doi: 10.1016/j.cellsig.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 24.Onofri C, Theodoropoulou M, Losa M, Uhl E, Lange M, Arzt E, Stalla GK, Renner U. Localization of vascular endothelial growth factor (VEGF) receptors in normal and adenomatous pituitaries: detection of a non-endothelial function of VEGF in pituitary tumours. J Endocrinol. 2006;191:249–261. doi: 10.1677/joe.1.06992. [DOI] [PubMed] [Google Scholar]
- 25.Edelstein J, Hao T, Cao Q, Morales L, Rockwell P. Crosstalk between VEGFR2 and muscarinic receptors regulates the mTOR pathway in serum starved SK-N-SH human neuroblastoma cells. Cellular signalling. 2011;23:239–248. doi: 10.1016/j.cellsig.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. Journal of neuroscience research. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 27.Poesen K, Lambrechts D, Van Damme P, Dhondt J, Bender F, Frank N, Bogaert E, Claes B, Heylen L, Verheyen A, Raes K, Tjwa M, Eriksson U, Shibuya M, Nuydens R, Van Den Bosch L, Meert T, D’Hooge R, Sendtner M, Robberecht W, Carmeliet P. Novel role for vascular endothelial growth factor (VEGF) receptor-1 and its ligand VEGF-B in motor neuron degeneration. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:10451–10459. doi: 10.1523/JNEUROSCI.1092-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenstein JM, Mani N, Khaibullina A, Krum JM. Neurotrophic effects of vascular endothelial growth factor on organotypic cortical explants and primary cortical neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:11036–11044. doi: 10.1523/JNEUROSCI.23-35-11036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nature reviews. Neuroscience. 2009;10:481–494. doi: 10.1038/nrn2665. [DOI] [PubMed] [Google Scholar]
- 30.Gallogly MM, Shelton MD, Qanungo S, Pai HV, Starke DW, Hoppel CL, Lesnefsky EJ, Mieyal JJ. Glutaredoxin regulates apoptosis in cardiomyocytes via NFkappaB targets Bcl-2 and Bcl-xL: implications for cardiac aging. Antioxidants & redox signaling. 2010;12:1339–1353. doi: 10.1089/ars.2009.2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Byeon SH, Lee SC, Choi SH, Lee HK, Lee JH, Chu YK, Kwon OW. Vascular endothelial growth factor as an autocrine survival factor for retinal pigment epithelial cells under oxidative stress via the VEGF-R2/PI3K/Akt. Investigative ophthalmology & visual science. 2010;51:1190–1197. doi: 10.1167/iovs.09-4144. [DOI] [PubMed] [Google Scholar]
- 32.Mathews MT, Berk BC. PARP-1 inhibition prevents oxidative and nitrosative stress-induced endothelial cell death via transactivation of the VEGF receptor 2. Arteriosclerosis, thrombosis, and vascular biology. 2008;28:711–717. doi: 10.1161/ATVBAHA.107.156406. [DOI] [PubMed] [Google Scholar]
- 33.Shiote M, Nagano I, Ilieva H, Murakami T, Narai H, Ohta Y, Nagata T, Shoji M, Abe K. Reduction of a vascular endothelial growth factor receptor, fetal liver kinase-1, by antisense oligonucleotides induces motor neuron death in rat spinal cord exposed to hypoxia. Neuroscience. 2005;132:175–182. doi: 10.1016/j.neuroscience.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 34.Chen MH, Kerkela R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation. 2008;118:84–95. doi: 10.1161/CIRCULATIONAHA.108.776831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mellor HR, Bell AR, Valentin JP, Roberts RR. Cardiotoxicity associated with targeting kinase pathways in cancer. Toxicological sciences : an official journal of the Society of Toxicology. 2011;120:14–32. doi: 10.1093/toxsci/kfq378. [DOI] [PubMed] [Google Scholar]
- 36.Tan Y, Ruan H, Demeter MR, Comb MJ. p90(RSK) blocks bad-mediated cell death via a protein kinase C-dependent pathway. The Journal of biological chemistry. 1999;274:34859–34867. doi: 10.1074/jbc.274.49.34859. [DOI] [PubMed] [Google Scholar]
- 37.Harada H, Becknell B, Wilm M, Mann M, Huang LJ, Taylor SS, Scott JD, Korsmeyer SJ. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Molecular cell. 1999;3:413–422. doi: 10.1016/s1097-2765(00)80469-4. [DOI] [PubMed] [Google Scholar]
- 38.Schipper HM. Heme oxygenase-1: transducer of pathological brain iron sequestration under oxidative stress. Annals of the New York Academy of Sciences. 2004;1012:84–93. doi: 10.1196/annals.1306.007. [DOI] [PubMed] [Google Scholar]
- 39.Schipper HM, Song W, Zukor H, Hascalovici JR, Zeligman D. Heme oxygenase-1 and neurodegeneration: expanding frontiers of engagement. Journal of neurochemistry. 2009;110:469–485. doi: 10.1111/j.1471-4159.2009.06160.x. [DOI] [PubMed] [Google Scholar]
- 40.Lin Q, Weis S, Yang G, Weng YH, Helston R, Rish K, Smith A, Bordner J, Polte T, Gaunitz F, Dennery PA. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. The Journal of biological chemistry. 2007;282:20621–20633. doi: 10.1074/jbc.M607954200. [DOI] [PubMed] [Google Scholar]
- 41.Yuan Y, Guo JZ, Zhou QX. The homeostasis of iron and suppression of HO-1 involved in the protective effects of nimodipine on neurodegeneration induced by aluminum overloading in mice. European journal of pharmacology. 2008;586:100–105. doi: 10.1016/j.ejphar.2008.02.033. [DOI] [PubMed] [Google Scholar]
- 42.Cariboni A, Davidson K, Dozio E, Memi F, Schwarz Q, Stossi F, Parnavelas JG, Ruhrberg C. VEGF signalling controls GnRH neuron survival via NRP1 independently of KDR and blood vessels. Development. 2011;138:3723–3733. doi: 10.1242/dev.063362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Falk T, Zhang S, Sherman SJ. Vascular endothelial growth factor B (VEGF-B) is up-regulated and exogenous VEGF-B is neuroprotective in a culture model of Parkinson’s disease. Molecular neurodegeneration. 2009;4:49. doi: 10.1186/1750-1326-4-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thirunavukkarasu M, Juhasz B, Zhan L, Menon VP, Tosaki A, Otani H, Maulik N. VEGFR1 (Flt-1+/-) gene knockout leads to the disruption of VEGF-mediated signaling through the nitric oxide/heme oxygenase pathway in ischemic preconditioned myocardium. Free radical biology & medicine. 2007;42:1487–1495. doi: 10.1016/j.freeradbiomed.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zentilin L, Puligadda U, Lionetti V, Zacchigna S, Collesi C, Pattarini L, Ruozi G, Camporesi S, Sinagra G, Pepe M, Recchia FA, Giacca M. Cardiomyocyte VEGFR-1 activation by VEGF-B induces compensatory hypertrophy and preserves cardiac function after myocardial infarction. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24:1467–1478. doi: 10.1096/fj.09-143180. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Z, Neiva KG, Lingen MW, Ellis LM, Nor JE. VEGF-dependent tumor angiogenesis requires inverse and reciprocal regulation of VEGFR1 and VEGFR2. Cell death and differentiation. 2010;17:499–512. doi: 10.1038/cdd.2009.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wright GL, Maroulakou IG, Eldridge J, Liby TL, Sridharan V, Tsichlis PN, Muise-Helmericks RC. VEGF stimulation of mitochondrial biogenesis: requirement of AKT3 kinase. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2008;22:3264–3275. doi: 10.1096/fj.08-106468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tornero D, Posadas I, Cena V. Bcl-x(L) blocks a mitochondrial inner membrane channel and prevents Ca2+ overload-mediated cell death. PloS one. 2011;6:e20423. doi: 10.1371/journal.pone.0020423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB, 3rd, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, Hardwick JM. Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. The Journal of cell biology. 2009;184:707–719. doi: 10.1083/jcb.200809060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, Chen Y, Messerli SM, Mariggio MA, Rahner C, McNay E, Shore GC, Smith PJ, Hardwick JM, Jonas EA. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nature cell biology. 2011;13:1224–1233. doi: 10.1038/ncb2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arbel N, Ben-Hail D, Shoshan-Barmatz V. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. The Journal of biological chemistry. 2012;287:23152–23161. doi: 10.1074/jbc.M112.345918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kann O, Kovacs R. Mitochondria and neuronal activity. American journal of physiology. Cell physiology. 2007;292:C641–657. doi: 10.1152/ajpcell.00222.2006. [DOI] [PubMed] [Google Scholar]
- 53.Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nature reviews. Neuroscience. 2012;13:77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lovell MA, Markesbery WR. Oxidative damage in mild cognitive impairment and early Alzheimer’s disease. Journal of neuroscience research. 2007;85:3036–3040. doi: 10.1002/jnr.21346. [DOI] [PubMed] [Google Scholar]