

Abstract
Peptide nucleic acids containing thymidine and 2-aminopyridine (M) nucleobases formed stable and sequence selective triple helices with double stranded RNA at physiologically relevant conditions. The M-modified PNA displayed unique RNA selectivity by having two orders of magnitude higher affinity for the double stranded RNAs than for the same DNA sequences. Preliminary results suggested that nucleobase-modified PNA could bind and recognize double helical precursors of microRNAs.
Keywords: sequence selective RNA recognition, peptide nucleic acid, PNA, triple helix, modified nucleobases
Compared to DNA, molecular recognition of double stranded RNA has received relatively little attention. Until the early 90’s, RNA was viewed as a passive messenger in the transfer of genetic information from DNA to proteins. However, since the discovery that RNA can catalyze chemical reactions, the number and variety of non-coding RNAs and the important roles they play in biology have been growing steadily.[1] Currently, the functional importance of most RNA transcripts is still unknown and it is fairly safe to predict that we will discover many more regulatory RNAs in the near future. The ability to selectively recognize and control the function of such RNAs will be highly useful for both fundamental research and practical applications. However, recognition of double helical RNA by sequence selective ligand binding is a formidable challenge.[2, 3]
Recently, we proposed that biologically relevant double helical RNAs could be recognized by major groove triple helix formation using peptide nucleic acid (PNA).[4–6] We showed that PNAs as short as hexamers formed stable and sequence selective Hoogsteen triple helices with RNA duplexes (Ka > 107 M−1) at pH 5.5.[4] A limitation of triple helical recognition was the requirement for long homopurine tracts, as only the Hoogsteen T(U)*A-T(U) and C+*G-C triplets could be used (Figure 1). We found that modification of PNA with 2-pyrimidinone[7] and 3-oxo-2,3-dihydropyridazine (E)[8] nucleobases allowed efficient and selective recognition of isolated C-G and U-A inversions, respectively, in polypurine tracts of double helical RNA at pH 6.25.[5, 6] However, the high affinity of PNA at pH 5.5 was greatly reduced at pH 6.25 and no binding could be observed at physiologically relevant salt and pH 7.4.[5] The remaining problem was the unfavorable protonation of cytosine, which was required for formation of the Hoogsteen C+*G-C triplets (Figure 1). Because its pKa = 4.5, cytosine is hardly protonated under physiological pH, which greatly decreases the stability of the triple helix. Herein we provide an efficient solution to this problem and demonstrate that sequence selective recognition of the RNA duplex can be achieved at physiologically relevant conditions by replacing cytosine with a more basic (pKa = 6.7[9]) heterocycle, 2-aminopyridine (abbreviated as M in Figure 1).
Figure 1.
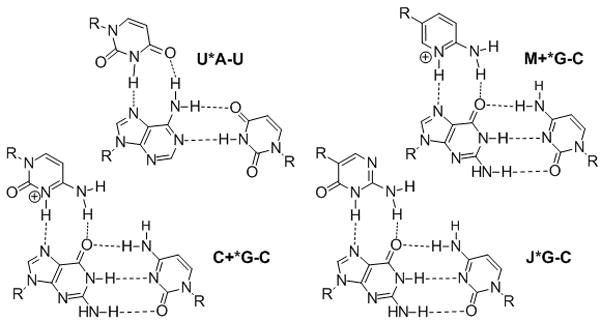
Standard and modified Hoogsteen triplets.
Povsic and Dervan pioneered the chemical modulation of the cytosine pKa by showing that triple helices containing 5-methylcytosine were more stable at higher pH than those of unmodified DNA.[10] More recently, derivatives of 2-aminopyridine have been used to increase the stability of DNA triple helices at high pH.[11–14] Before our studies, this approach had not been used in PNA.
An alternative approach has used neutral nucleobases that mimic the hydrogen-bonding scheme of protonated cytosine. The most notable examples are pseudoisocytosine (abbreviated as J in Figure 1) by Kan and co-workers,[15] methyloxocytosine by McLaughlin and co-workers,[16, 17] and a pyrazine derivative by von Krosigk and Benner.[18] The J base is widely used in PNA to alleviate the pH dependency of PNA-DNA triplexes.[19, 20] The use of modified heterocycles to recognize double stranded RNA at physiologically relevant conditions had no precedent before our studies.
We started by comparing the binding of unmodified PNA1, J-modified PNA2 and M-modified PNA3 to HRPA (Figure 2), an A-rich RNA hairpin similar to the model systems used previously by us.[4, 5] Fmoc-protected J monomer 1 was synthesized according to the literature procedure.[21] The M monomer 2 was synthesized from Fmoc-protected PNA backbone 3 and the known carboxylic acid 4 using DCC mediated coupling followed by deprotection of the allyl group as previously described by us (Scheme 1).[5] All PNAs were synthesized using a standard PNA protocol on an Expedite 8909 DNA synthesizer, purified by HPLC and characterized by mass spectroscopy as previously reported.[4–6]
Figure 2.
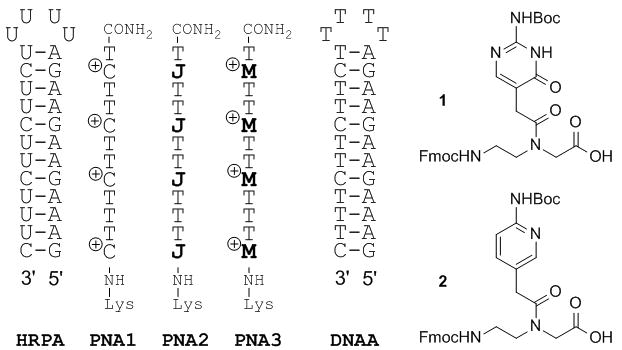
RNA, PNA and DNA sequences used to compare C, J and M nucleobases.
Scheme 1.
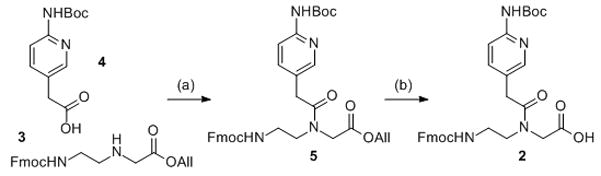
Synthesis of M PNA monomer: a) DCC, 3-Hydroxy-1,2,3-benzotriazin-4(3H)-one, DMF, rt, overnight, 57%; b) Pd(PPh3)4, N-ethylaniline, THF, rt, 2h, 79%.
Following the same approach as in our previous studies,[4–6] we used isothermal titration calorimetry (ITC) and UV thermal melting to characterize the binding of PNA to RNA hairpins. ITC directly measures the enthalpy of binding and, through fitting of the binding data, provides binding affinity (association constant Ka in M−1) and stoichiometry (the ratio of PNA to RNA in the final complex).[22] Due to operational simplicity, reliability and rich thermodynamic data, ITC is one of the best methods to study ligand binding to RNA. The unmodified PNA1 formed a stable triplex with HRPA at pH 5.5 (Table 1) in sodium acetate buffer at 25 °C. As expected, because of the unfavorable protonation of cytosine at higher pH, the affinity decreased significantly when the pH of the buffer was increased to 7 and we could not observe any binding in phosphate buffer mimicking the physiological conditions at 37 °C.
Table 1.
Binding of C, J and M containing PNA to RNA HRPA.[a]
| PNA | Acetate pH 5.5[b] | Acetate pH 7.0[b] | Phosphate pH 7.4[c] |
|---|---|---|---|
| PNA1 (C) | 0.76 | 0.06 | NB |
| PNA2 (J) | - | 0.41 | 0.17 |
| PNA3 (M) | - | 36.5 | 1.8 |
Association constants Ka × 107 M−1, NB – no binding, Ka < 103.
100 mM sodium acetate buffer at 25 °C
2 mM MgCl2, 90 mM KCl, 10 mM NaCl, 50 mM potassium phosphate at 37 °C.
We used the affinity of PNA1 at pH 5.5 as a benchmark to gauge the effect of J and M modifications on PNA affinity at higher pH. The affinity of J-modified PNA2 for HRPA in acetate buffer at pH 7 was lower than the affinity of PNA1 at pH 5.5 and decreased even more under the more demanding physiological conditions (Table 1). Nielsen and co-workers[20] reported that the affinity of an unmodified PNA 15mer (having 5 isolated cytosines) for a DNA duplex dropped by three orders of magnitude (Kd changed from 2 nM to 2.2 mM) when changing the pH from 5.5 to 7.2. Substitution of all five cytosines by J base increased the affinity only about tenfold (Kd = 0.15 mM).[20] Thus, our result was qualitatively consistent with that reported by Nielsen, only smaller in magnitude, and suggested that the positive charge on cytosine contributed significantly to stability of the Hoogsteen triplet, presumably via electrostatic attraction to the negatively charged nucleic acid. Consequently, an ideal design for recognition of G-C pairs would include both a correct hydrogen bonding scheme and a positive charge on the heterocycle. Because unmodified PNA containing cytosine (pKa = 4.5) forms a stable triple helix at pH 5.5, we hypothesized that PNA modified with 2-aminopyridine M (pKa = 6.7) would form at least equally strong triple helices at physiological pH 7.4 (due to a similar pH/pKa difference).
Confirming our hypothesis ITC showed that M modification strongly enhanced the binding affinity of PNA3. In acetate buffer at pH 7 M-modified PNA3 had about two orders of magnitude higher affinity (Ka = 3.7 × 108) for HRPA than the J-modified PNA2 (Figure 2 and Table 1). Under physiologically relevant conditions PNA3 bound to HRPA with Ka = 1.8 × 107, which was an order of magnitude higher than the affinity of PNA2 at the same conditions and twice that of unmodified PNA1 at pH 5.5. The larger drop in affinity for PNA3 compared to PNA2 going from acetate to phosphate buffer is most likely due to screening of the electrostatic interactions (that are more important for the charged M) by higher salt concentration and the presence of MgCl2 in the physiologically relevant buffer. Most remarkably, binding of PNA3 to the matched DNA hairpin (DNAA) in physiological phosphate buffer was about two orders of magnitude weaker (Ka = 3 × 105) than binding to RNA HRPA. This result suggested that the M-modified PNA might have unique selectivity for triple helical recognition of RNA over DNA. For all experiments at physiologically relevant conditions, fitting of ITC titration curves gave a 1:1 PNA:RNA stoichiometry (see Table S1 in Supporting Information) consistent with the triple helix formation.
UV thermal melting experiments (Figure S15) confirmed the ITC results. Consistent with our previous observations,[4] the complexes of HRPA and high affinity PNAs melted in one transition of triple helix to single strands without an intermediate duplex. In phosphate buffer at pH 7.4 adding PNA2 had little effect on the stability of HRPA: tm = 75 °C for HPRA alone and 74 °C for a 1:1 complex of HRPA-PNA2. Consistent with the higher Ka observed in the ITC experiments, the thermal stability of a 1:1 complex of HRPA-PNA3 was significantly higher at 80 °C. Taken together, the results confirmed our hypothesis that the charged M would have an advantage over the neutral J for triple helical recognition of RNA.
Next we probed the sequence specificity of M-modified PNA using a model system from our previous studies (Figure 3).[4, 5] Table 2 shows that PNA5 (four M modifications) had high affinity for the matched HRP1 at physiologically relevant conditions while maintaining excellent sequence specificity. The binding affinity of M-modified PNA5 at pH 7.4 was somewhat lower than that of unmodified PNA4 at pH 5.5.[4] This was in contrast to a similar comparison of M-modified PNA3 and unmodified PNA1 in Table 1. The discrepancy might be related to electrostatic repulsion of adjacent charged nucleobases, which has been reported to have a negative effect on affinity,[23] and may affect PNA5 more than PNA4. Nevertheless, the strong and highly selective RNA binding by PNA5 at physiologically relevant conditions was extremely encouraging. UV thermal melting (Figure S25) of the matched PNA5-HRP1 complex showed a broad and relatively weak transition at ~ 55 °C that might be assigned to triplex melting preceding the melting of the HRP1 hairpin at ~ 100 °C. Similar transitions unique to the matched PNA5-HRP1 complex were also observed in CD melting plots (Figure S26). Consistent with the high sequence selectivity we could not observe any transitions above 30 °C that could be assigned to triplex melting of the mismatched complexes. Confirming the unique RNA selectivity observed for PNA3, PNA5 showed little, if any binding to its matched DNA hairpin HRP5 (Figure S24).
Figure 3.
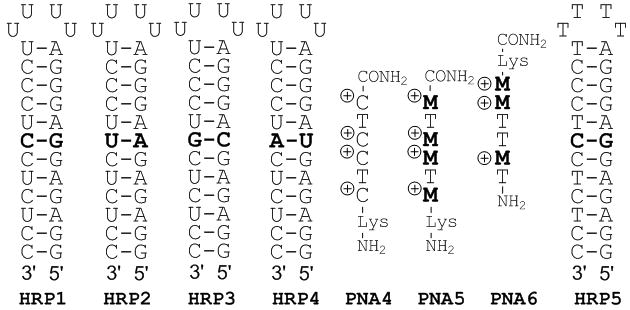
PNA, RNA and DNA used in sequence specificity study.
Table 2.
Binding of M-modified PNA to RNA hairpins.[a]
| PNA | HRP1 (G-C) | HRP2 (A-U) | HRP3 (C-G) | HRP4 (U-A) |
|---|---|---|---|---|
| PNA4[b] | 8.4 | 0.04 | 0.05 | 0.02 |
| PNA5[c] | 2.0 | <0.001[d] | NB[e] | NB[e] |
| PNA6[c] | NB[e] | 0.4 | NB[e] | NB[e] |
Association constants Ka × 107 M−1.
From ref. 4, in 100 mM sodium acetate buffer, pH 5.5 at 25 °C.
In phosphate buffer, pH 7.4 at 37 °C.
Highest estimate; the low binding prevented more accurate curve fit.
NB – no binding, Ka < 103.
PNA6 (three M modifications) had five times lower affinity for the matched HRP2 than PNA5 for HRP1, which was consistent with a higher stability of triplets involving G-C base pairs and the notion that the positive charges are important for high binding affinity. As expected, PNA6 showed excellent sequence specificity. The PNA-RNA stoichiometry was 1:1 in all Table 2 experiments (see Table S1).
Finally, we chose microRNA-215, which is implicated in cancer development and drug resistance,[24, 25] as an initial target to check if M-modified PNA could bind to biologically relevant double helical RNA. MicroRNAs (miRNAs) are transcribed as long hairpin structures, pri-miRNAs, which are processed into mature miRNA duplexes (~22 nt) by Drosha and Dicer endonucleases. It is common to find stretches of eight and more contiguous purines interrupted by one or two pyrimidines in pri-miRNA hairpins.[26] Triple helical binding to such sites could be used to detect miRNAs and interfere with their function, which would find broad applications in fundamental science, medicine and biotechnology. We chose HRP6 as a model that contains the purine rich recognition site present in pri-miRNA-215.[26] HRP6 has a stretch of nine purines interrupted by a uridine and features several non-canonical base pairs, which are hallmarks of pri-miRNA hairpins. For recognition of the uridine interruption we used nucleobase E (Figure 4) that was originally designed for thymidine recognition in DNA[8] and later adopted by us for uridine recognition in RNA.[5] PNA7 having three M and one E modification was prepared using monomers 2 and the previously reported[5] 6 (Figure 4).
Figure 4.
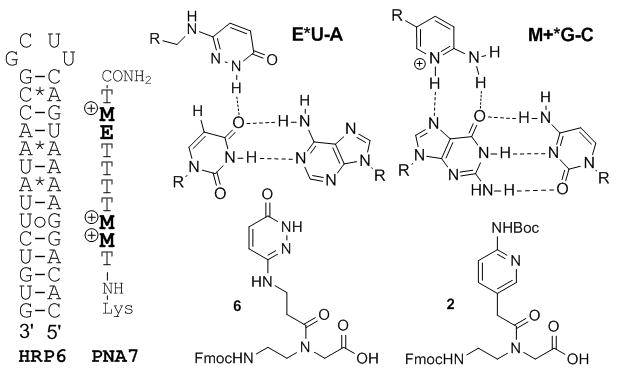
Binding of E- and M-modified PNA7 to HRP6 modeling pri-miRNA-215 hairpin structure.
Consistent with results obtained with other M-modified PNAs, PNA7 recognized HRP6 with high affinity (Ka = 1.2 × 107) and 1:1 stoichiometry (Table S1) under physiologically relevant conditions. Remarkably, the non-canonical C*A and A*A and the wobble UoG base pairs did not prevent formation of the PNA-RNA complex.
In summary, we have demonstrated that modification of PNA with 2-aminopyridine (M) nucleobases allows formation of stable and sequence selective triple helices with double stranded RNA at physiologically relevant conditions. For triple helical RNA recognition, modulation of nucleobase basicity (c.f., pKa = 6.7 for M with 4.5 for C) was a more efficient approach than using the neutral J base. The M-modified PNAs exhibited unique RNA selectivity and had two orders of magnitude higher affinity for the double stranded RNAs than for the same DNA sequences. It is conceivable that the deep and narrow major groove of RNA presented a better steric fit for the PNA ligands than the wider major groove of DNA. In preliminary experiments nucleobase-modified PNA recognized a purine rich model sequence of a double helical miRNA precursor with high affinity at physiologically relevant conditions. While this is a relatively new area of research, Beal and co-workers[27] have already demonstrated the potential of targeting pri-miRNAs using helix-threading peptides. Taken together our results suggest that PNA may have unique and previously underappreciated potential for triple helical recognition of biologically relevant RNA. Low stability at pH 7.4 has been a long-standing problem for practical applications of triple helices. The excellent performance of M-modified PNAs at pH 7.4 observed herein provide efficient solution to this problem that should open the door for new approaches to detection and interference with the function of double stranded RNA molecules.
Experimental Section
See supporting information for details
Supplementary Material
Footnotes
We thank Binghamton University and NIH (R01 GM071461) for support of this research. The Regional NMR Facility (600 MHz instrument) at Binghamton University is supported by NSF (CHE-0922815).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Thomas Zengeya, Department of Chemistry, Binghamton University, The State University of New York, Binghamton, New York 13902, USA, Fax: (+) 1-607-777-4478.
Dr. Pankaj Gupta, Department of Chemistry, Binghamton University, The State University of New York, Binghamton, New York 13902, USA, Fax: (+) 1-607-777-4478
Dr. Eriks Rozners, Email: erozners@binghamton.edu, Department of Chemistry, Binghamton University, The State University of New York, Binghamton, New York 13902, USA, Fax: (+) 1-607-777-4478, http://www2.binghamton.edu/chemistry/people/rozners/rozners.html
References
- 1.Sharp PA. Cell. 2009;136:577–580. doi: 10.1016/j.cell.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Thomas JR, Hergenrother PJ. Chem Rev. 2008;108:1171–1224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 3.Guan L, Disney MD. ACS Chem Biol. 2012;7:73–86. doi: 10.1021/cb200447r. [DOI] [PubMed] [Google Scholar]
- 4.Li M, Zengeya T, Rozners E. J Am Chem Soc. 2010;132:8676–8681. doi: 10.1021/ja101384k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta P, Zengeya T, Rozners E. Chem Commun. 2011;47:11125–11127. doi: 10.1039/c1cc14706d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta P, Muse O, Rozners E. Biochemistry. 2012;51:63–73. doi: 10.1021/bi201570a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buchini S, Leumann CJ. Angew Chem. 2004;116:4015–4018. doi: 10.1002/anie.200460159. [DOI] [PubMed] [Google Scholar]; Buchini S, Leumann CJ. Angew Chem, Int Ed. 2004;43:3925–1224. doi: 10.1002/anie.200460159. [DOI] [PubMed] [Google Scholar]
- 8.Eldrup AB, Dahl O, Nielsen PE. J Am Chem Soc. 1997;119:11116–11117. [Google Scholar]
- 9.Stewart LR, Harris MG. J Org Chem. 1978;43:3123–3126. [Google Scholar]
- 10.Povsic TJ, Dervan PB. J Am Chem Soc. 1989;111:3059–3061. [Google Scholar]
- 11.Hildbrand S, Blaser A, Parel SP, Leumann CJ. J Am Chem Soc. 1997;119:5499–5511. [Google Scholar]
- 12.Cassidy SA, Slickers P, Trent JO, Capaldi DC, Roselt PD, Reese CB, Neidle S, Fox KR. Nucleic Acids Res. 1997;25:4891–4898. doi: 10.1093/nar/25.24.4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rusling DA, Powers VEC, Ranasinghe RT, Wang Y, Osborne SD, Brown T, Fox KR. Nucleic Acids Res. 2005;33:3025–3032. doi: 10.1093/nar/gki625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen DL, McLaughlin LW. J Org Chem. 2000;65:7468–7474. doi: 10.1021/jo000754w. [DOI] [PubMed] [Google Scholar]
- 15.Ono A, Ts’o POP, Kan LS. J Am Chem Soc. 1991;113:4032–4033. [Google Scholar]
- 16.Xiang G, Soussou W, McLaughlin LW. J Am Chem Soc. 1994;116:11155–11156. [Google Scholar]
- 17.Xiang G, Bogacki R, McLaughlin LW. Nucleic Acids Res. 1996;24:1963–1970. doi: 10.1093/nar/24.10.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.von Krosigk U, Benner SA. J Am Chem Soc. 1995;117:5361–5362. [Google Scholar]
- 19.Egholm M, Christensen L, Dueholm KL, Buchardt O, Coull J, Nielsen PE. Nucleic Acids Res. 1995;23:217–222. doi: 10.1093/nar/23.2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hansen ME, Bentin T, Nielsen PE. Nucleic Acids Res. 2009;37:4498–4507. doi: 10.1093/nar/gkp437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudson RHE, Wojciechowski F. Can J Chem. 2008;86:1026. [Google Scholar]
- 22.Feig AL. Methods Enzymol. 2009;468:409–422. doi: 10.1016/S0076-6879(09)68019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Priestley ES, Dervan PB. J Am Chem Soc. 1995;117:4761–4765. [Google Scholar]
- 24.Song B, Wang Y, Titmus MA, Botchkina G, Formentini A, Kornmann M, Ju J. Molecular Cancer. 2010;9:1–10. [Google Scholar]
- 25.Jin Z, Selaru FM, Cheng Y, Kan T, Agarwal R, Mori Y, Olaru AV, Yang J, David S, Hamilton JP, Abraham JM, Harmon J, Duncan M, Montgomery EA, Meltzer SJ. Oncogene. 2011;30:1577–1585. doi: 10.1038/onc.2010.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. Nucleic Acids Res. 2008;36:D154–D158. doi: 10.1093/nar/gkm952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krishnamurthy M, Simon K, Orendt AM, Beal PA. Angew Chem. 2007;119:7174–7177. doi: 10.1002/anie.200702247. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2007;46:7044–7047. doi: 10.1002/anie.200702247. [DOI] [PubMed] [Google Scholar]; Gooch BD, Beal PA. J Am Chem Soc. 2004;126:10603–10610. doi: 10.1021/ja047818v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.