

Abstract
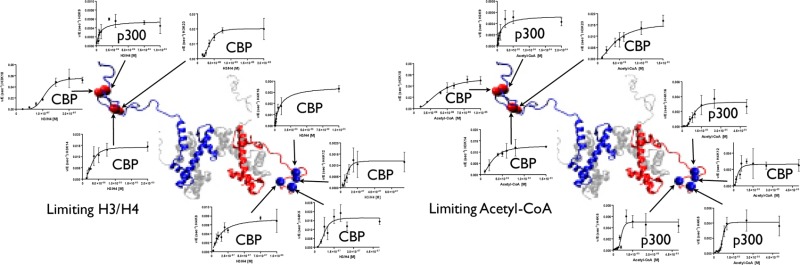
Although p300 and CBP lysine acetyltransferases are often treated interchangeably, the inability of one enzyme to compensate for the loss of the other suggests unique roles for each. As these deficiencies coincide with aberrant levels of histone acetylation, we hypothesized that the key difference between p300 and CBP activity is differences in their specificity/selectivity for lysines within the histones. Utilizing a label-free, quantitative mass spectrometry based technique, we determined the kinetic parameters of both CBP and p300 at each lysine of H3 and H4, under conditions we would expect to encounter in the cell (either limiting acetyl-CoA or histone). Our results show that while p300 and CBP acetylate many common residues on H3 and H4, they do in fact possess very different specificities, and these specificities are dependent on whether histone or acetyl-CoA is limiting. Steady-state experiments with limiting H3 demonstrate that both CBP and p300 acetylate H3K14, H3K18, H3K23, with p300 having specificities up to 1010-fold higher than CBP. Utilizing tetramer as a substrate, both enzymes also acetylate H4K5, H4K8, H4K12, and H4K16. With limiting tetramer, CBP displays higher specificities, especially at H3K18, where CBP specificity is 1032-fold higher than p300. With limiting acetyl-CoA, p300 has the highest specificity at H4K16, where specificity is 1018-fold higher than CBP. This discovery of unique specificity for targets of CBP- vs p300-mediated acetylation of histone lysine residues presents a new model for understanding their respective biological roles and possibly an opportunity for selective therapeutic intervention.
Access to DNA is regulated through post-transcriptional modifications to the histones around which DNA is wrapped. One of the most common of these modifications is acetylation, which occurs on 10–20 lysines per histone. Lysine acetyltransferases (KATs) are responsible for this modification, adding an acetyl group to specific lysine residues on the histone. Acetylation of the histones results in an increase in negative charge and a decrease in DNA interaction, making the DNA accessible to proteins required to initiate transcription, DNA replication, or repair.1−5 As such, histone acetylation must be carefully regulated to prevent changes in chromatin structure and gene expression.6
CBP and p300 are both prolific lysine acetyltransferases, involved in several biological pathways including neurological development,7 gene activation,8 and the DNA damage response.9−11 There are a number of similarities between the two proteins: both proteins are regulators of RNA polymerase II-mediated transcription. Both CBP and p300 are large proteins (∼300 kDa) and are structural homologues, sharing high sequence identity in several structured regions. These regions include the histone acetyltransferase (HAT) domain and the bromodomain, an acetyl-lysine binding domain common to many KATs.12 Sequence alignments of these HATs reveal an ∼90% homology in the KAT domain, with an ∼93% homology in the bromodomain. Outside of these highly conserved domains, however, homology is much lower. Additionally, both KATs have been shown to acetylate multiple residues on each of the four core histones,12,13 and both are important to healthy human growth and development.
Accumulating evidence suggest that there are unique roles for CBP and p300 in the cell. In mice, heterozygous inactivation of p300 leads to more severe abnormalities in heart, lung, and small intestine formation than inactivation of CBP.14,15 Heterozygous inactivation of CBP, however, leads to growth retardation and craniofacial abnormalities.12 Human diseases arising from deficiencies of CBP and p300 also implicate discrete function. Mutations in CBP have been linked to Rubinstein-Taybi syndrome,7,16,17 a congenital neurodevelopmental disorder, and fetal alcohol syndrome,18 while deficiencies in p300 have been linked to aberrant levels of acetylation in multiple cancers.19−23 Although both proteins are expressed in almost all tissues, the inability of p300 to compensate for the loss of CBP and vice versa suggests an important divergence in function between CBP and p300. Therefore, distinguishing the activities of these two proteins from one another is an important step in understanding and treating the diseases that they cause.
Both CBP and p300 have been shown to acetylate multiple lysines on various histones (on histone H3: K14, K18, K23, and on histone H4: K5, K8, and K12),12,13,24−26 suggesting the difference between the two enzymes lies in their specificity/selectivity for the residues they acetylate: specificity is defined as how likely one enzyme is to acetylate one position relative to another on the same histone, while selectivity is how likely one enzyme versus another is to acetylate a specific residue. Differences in specificity and selectivity could account for both the biological requirement for both proteins,12,14,15 and the difference in diseases caused by mutation or loss of either CBP or p300.16−23 In order to test the hypothesis that CBP and p300 exhibit different specificities, we employed a label-free, quantitative mass spectrometry-based method already established in our lab.27 Building on this assay, we have also added the ability to monitor H4, thus allowing us to determine the specificity of residues in H4, which in turn enables us to observe how the formation of H3/H4 (or (H3/H4)2) alters specificity. This assay allows us to monitor each individual acetylation site on histone H3 and H4, enabling us to quantitate and compare the acetylation of all lysines simultaneously. Our mass spectrometry approach is unique because it allows us to see all acetylation events, even if they occur on sites where p300 and CBP acetylation had not previously been reported. Additionally, the high throughput nature of this assay lends itself to the determination of kinetic parameters for each target site, allowing us to determine the kcat/K1/2 of either protein for a given site. Thus, the wealth of information that these assays provide allows us to characterize these proteins in a way that was not previously possible.
Elucidating the differences between p300 and CBP is important to understanding how to treat the diseases that their deficiencies cause. Therefore, the goal of this study is to characterize the histone acetylation patterns of both p300 and CBP in order to determine in what ways they are similar and, importantly, how they differ. By developing methods to detect acetylation of histone H4, this research expands on the label-free mass spectrometry method already established in our lab, which allowed us to study each individual acetylated site on histone H3. This enables us to simultaneously analyze the acetylation of multiple lysine residues of the H3/H4 tetramer by CBP or p300 in order to compare their histone acetylation patterns. The results of this study provide greater insight into how CBP and p300 differentially regulate histone acetylation and will help us to understand why p300 cannot compensate for deficiencies in CBP and vice versa. Importantly, understanding the kinetics of CBP and p300 and their specific targets on the histone tetramer will provide valuable insight into treating the cancers and neurodegenerative disorders that arise from mutations in CBP and p300.
Experimental Procedures
Reagents
All Chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher (Pittsburgh, PA), and the purity is the highest commercial grade or meets LC/MS grade. Ultrapure water was generated from a Millipore Direct-Q 5 ultrapure water system (Bedford, MA). Recombinant histone H3 and H4 were purified and provided from the Protein Purification Core at Colorado State University. Acetyl-CoA was obtained from Sigma-Aldrich. Synthetic peptides (acetylated and propionylated) of high purity (>98%) were purchased from JPT peptide technologies (Acton, MA) and Anaspec (Fremont, CA).
Sequence Alignment
Sequences were obtained from the NCBI protein database. Sequence alignments for human p300 (accession: Q09472), human CBP (Q92793), as well as the p300 HAT domain (3BIY_A) and p300 bromodomain (3I3J_A) were performed using CLC Sequence Viewer 6.
Protein Purification
The sequence for human p300 and human CBP, containing an N-terminal His tag, and C-terminal Strep2 and FLAG tags, was synthesized and cloned by Genewiz (Cambridge, MA) into the pVL1393 vector for baculovirus expression. This was done to optimize codon expression for Sf9 cells and reduce the amount of RNA secondary structure. Utilizing the BD (Franklin Lake, NJ) BaculoGold transfection system, the plasmid was transfected into Sf9 cells. After successful transfection and virus amplification, p300 was expressed in Sf9 cells and purified using a GE Healthcare (Piscataway, NJ) HiTrap column. The protein identity and purity were confirmed through protein staining with Coomassie dye. The p300 construct was graciously provided by Karolin Luger (Colorado State University).
Enzymatic Kinetics Assays for p300 and CBP
Steady-state kinetics for H3 and the H3/H4 tetramer were performed under identical buffer conditions (100 mM ammonium bicarbonate and 50 mM HEPES buffer (pH 7.8) at 37 °C. Steady-state assays contained from 1 to 50 nM p300 or 0.5 to 22.5 nM CBP, varying either H3 (0.25–15 μM), H3/H4 (0.05–20 μM), or acetyl-CoA (1–200 μM). Assays were quenched using 4 vol of trichloroacetic acid (TCA). The precipitate was then washed twice with 150 μL of acetone (−20 °C).28 Samples were dried, 2 μL of propionic anhydride was added, and ammonium hydroxide was used to quickly adjust the pH to ∼8.29 Samples were then incubated at 51 °C for 1 h followed by trypsin digestion (overnight at 37 °C).
UPLC-MS/MS Analysis
A Waters Acquity H-class UPLC (Milford, MA) coupled to a Thermo TSQ Quantum Access (Waltham, MA) triple quadrupole (QqQ) mass spectrometer was used to quantify acetylated H3/H4 peptides. The digested H3/H4 peptides were injected into an Acquity BEH C18 column (2.1 × 50 mm; particle size 1.7 μm) with 0.2% formic acid (FA) aqueous solution (solution A) and 0.2% FA in acetonitrile (solution B). Peptides were eluted over 11 min at 0.6 mL/min and 60 °C, and the gradient was programmed from 95% solution A and 5% solution B and down to 80% solution A and 20% solution B in 11 min. Selected reaction monitoring (SRM) was used to monitor the elution of the acetylated and propionylated H3/H4 peptides. The detailed transitions of H3 have previously been reported.27 The accuracy of theoretical mass transitions for SRMs was confirmed utilizing acetylated H4 peptides. The transitions for H4 are shown in Table 1.
Table 1. Detection Parameters of Tryptic Peptids from Histone H4.
| precursor ion (m/z) | product ions (m/z) | collision energy (eV) | retention time (min) | |
|---|---|---|---|---|
| GKaGGKaGLGKaGGAKaR | 719.910 | 530.304521 | 25 | 3.70 |
| 757.431513 | 25 | |||
| 1211.685496 | 25 | |||
| GKpGGKpGLGKpGGAKaR | 740.929 | 530.304521 | 25 | 5.70 |
| 771.447163 | 25 | |||
| 1239.716796 | 25 | |||
| GKpGGKpGLGKaGGAKpR | 740.931 | 544.320171 | 25 | 5.70 |
| 771.447163 | 25 | |||
| 1239.716796 | 25 | |||
| GKpGGKaGLGKpGGAKpR | 740.935 | 544.320171 | 25 | 5.70 |
| 785.462813 | 25 | |||
| 1239.716796 | 25 | |||
| GKaGGKpGLGKpGGAKpR | 740.933 | 544.320171 | 25 | 5.70 |
| 785.462813 | 25 | |||
| 1253.732446 | 25 | |||
| GKGGKGLGKGGAKRa | 733.926 | 1225.701146 | 25 | 5.00 |
| GKpGGKpGLGKaGGAKaR | 733.926 | 757.431513 | 25 | 5.00 |
| GKGGKGLGKGGAKRa | 733.926 | 530.304521 | 25 | 5.00 |
| GKGGKGLGKGGAKRa | 733.926 | 771.447163 | 25 | 5.00 |
| GKGGKGLGKGGAKRa | 733.926 | 544.320171 | 25 | 5.00 |
| GKGGKGLGKGGAKRa | 733.926 | 1239.716796 | 25 | 5.00 |
| GKaGGKaGLGKpGGAKpR | 733.926 | 785.462813 | 25 | 5.00 |
| GKpGGKaGLGKaGGAKaR | 726.914 | 530.304521 | 25 | 4.30 |
| 757.431513 | 25 | |||
| 1211.685496 | 25 | |||
| GKaGGKpGLGKaGGAKaR | 726.916 | 530.304521 | 25 | 4.30 |
| 757.431513 | 25 | |||
| 1225.701146 | 25 | |||
| GKaGGKaGLGKpGGAKaR | 726.920 | 530.304521 | 25 | 4.30 |
| 771.447163 | 25 | |||
| 1225.701146 | 25 | |||
| GKaGGKaGLGKaGGAKpR | 726.918 | 544.320171 | 25 | 4.30 |
| 771.447163 | 25 | |||
| 1225.701146 | 25 | |||
| GKpGGKpGLGKpGGAKpR | 747.941 | 544.320171 | 25 | 6.40 |
| 785.462813 | 25 | |||
| 1253.732446 | 25 | |||
| DNIQGITKaPAIR | 853.979 | 385.244546 | 29 | 11.70 |
| 904.561464 | 29 | |||
| 1150.628892 | 29 | |||
| DNIQGITKpPAIR | 691.394 | 741.46175 | 24 | 8.40 |
| 854.545814 | 24 | |||
| 911.567278 | 24 | |||
| GVLKaVFLENVIR | 714.932 | 743.441014 | 24 | 11.73 |
| 890.509428 | 24 | |||
| 989.577842 | 24 | |||
| GVLKpVFLENVIR | 721 | 743.441014 | 25 | 11.70 |
| 890.509428 | 25 | |||
| 989.577842 | 25 | |||
| DAVTYTEHAKaR | 666 | 553.320505 | 23 | 1.56 |
| 651.298432 | 23 | |||
| 946.474106 | 23 | |||
| DAVTYTEHAKpR | 673 | 567.336155 | 23 | 0.40 |
| 651.298432 | 23 | |||
| 960.489756 | 23 | |||
| KaTVTAMDWYALKaR | 839 | 371.228896 | 28 | 11.70 |
| 890.545814 | 28 | |||
| 1136.613242 | 28 | |||
| KpTVTAMDWYALKaR | 846.971 | 385.244546 | 28 | 11.70 |
| 890.545814 | 28 | |||
| 1136.613242 | 28 | |||
| KaTVTAMDWYALKpR | 846.973 | 371.228896 | 28 | 11.70 |
| 904.561464 | 28 | |||
| 1150.628892 | 28 | |||
| KpTVTAMDWYALKpR | 853 | 385.244546 | 29 | 11.70 |
| 904.561464 | 29 |
These transitions are indicative of a double acetylation but cannot be distinguished by the precursor ion alone. In these cases, the product ion is utilized for deconvolution.30
QqQ MS Data Analysis
Each acetylated and/or propionylated peak was identified by retention time and specific transitions (Table 1). The resulting peak integration was done using Xcalibur software (version 2.1, Thermo). The fraction of a specific peptide (Fp) is calculated by eq 1, where Is is the intensity of a specific peptide state and Ip is the intensity of any state of that peptide, and analyzed as previously described.27,30
| 1 |
Data Analysis
All models were fit to the data-using Prism (version 5.0d). The initial rates (v) of acetylation were calculated from the linear increase in acetylation as a function of time prior to 10% of the sum of acetylated residues. To measure steady-state parameters for acetyl-CoA, the initial rates were calculated based on time points where less than 10% of the acetyl-CoA was consumed (based on a coupled assay31) and where the acetylated H3 or H3/H4 fraction is less than 0.1 times the fraction of unacetylated H3 or H3/H4. kcat, K1/2, and the Hill coefficient (nH) were determined by fitting the equation:
| 2 |
where [S] is the concentration of substrate (either H3, H3/H4, or acetyl-CoA), and [E] is the concentration of enzyme (either CBP or p300). The nH was assumed to be one unless the data dictated otherwise, in which case the nH was confirmed by the equation:
| 3 |
where f is the normalized change in v/[E].
Results
Both CBP and p300 acetylate many of the same positions on histone H3,12,13,32 but little is known about how specific or selective these enzymes are. Both of these factors can be quantified by the specificity constant or kcat/Km for systems displaying Michaelis–Menten type kinetics and kcat/K1/2nH for more complex systems.33 This level of understanding requires not only knowing which sites are acetylated but also quantitating acetylation at each individual site. To do this, we employed an assay capable of monitoring all of the acetylation sites on histone H3, that we have previously used to characterize Gcn5,27 and expanded the assay to include sites of H4. This assay has the advantage of using full-length proteins (over peptides) and measuring both the location and amount of acetylation in each location. Briefly, by using a QqQ mass spectrometer with selective reaction monitoring (SRM), we are able to distinguish different modifications on the histone fragments and can use this to quantitate the amount of acetylation of a given lysine. This method allows us to quantitate the fraction of a residue that is acetylated, and the utilization of multiple SRMs allows us to observe every lysine residue of histone H3 and H4 simultaneously.
Steady-State Acetylation of Histone H3 by CBP and p300
To understand how the KAT activity of p300 and CBP differ, and so that these results could be compared with our previous findings on Gcn5, we characterized the acetylation patterns of p300 and CBP on histone H3. Before performing the kinetic analysis, we first determined which sites of H3 and H4 could be acetylated by p300 and CBP, under any conditions, even at low levels. We allowed the reaction to proceed for long incubation times (24 h) to detect sites that may only be acetylated at low levels/frequency. We found that both proteins acetylated K9, K14, K18, and K23 on H3. We observed low levels of p300 acetylation of H3K27 and CBP acetylation of H3K9; however, these sites did not appear in our steady-state experiments because they are not acetylated before 10% of the total substrate is acetylated. Specifically, this means that although they are detectable at long time points, kinetic parameters could not be determined for these sites.
Specificity constants for CBP and p300 for each targeted lysine provide a means to quantify and compare the preference of these enzymes for each site. Previously, we have shown that under steady-state conditions, an enzyme that is capable of acetylating two or more locations or residues on the same substrate will have an impact on the ability to obtain an accurate measurement of Km or kcat, but kcat/Km (or kcat/K1/2nH) is the correct value to quantify targeting of a given site.27 To obtain these parameters, we performed steady-state assays to determine the specificity constants of p300 and CBP for each of the acetylated lysines in which [enzyme] ≪ [substrate]. These experiments used saturating acetyl-CoA (200 μM) and excess substrate to enzyme. We limited our analysis to time points when the total fraction of acetylated histone (the sum of the fraction acetylated for every residue) was less than 10% of the initial substrate concentration. This approach also allows us to observe if sites such as K9 and K14, and K18 and K23, which are on the same tryptic peptide, are acetylated on the same histone. Under steady-state conditions, one would not expect to observe two acetylated residues on the same histone unless the enzyme was acting processively or that one acetylation site greatly increases the specificity of another. In fact, we did observe a small amount (<0.1%) of K23ac after K18ac in p300-mediated acetylation but chose not to include this fraction in the analysis, as it had little impact on the parameters measured. We carried out a series of time course experiments to determine v/E, which was then plotted as a function of substrate.
Under steady-state conditions (for H3), both KATs acetylated several residues on the histone tail region of H3 (H3K9, K14, K18, and K23) (Figure 1AB, Figure S1). The order of the specificity constants for CBP is K14 ≈ K18 > K23, and the constants for K14 and K18, was ∼8-fold higher than K23 (Figure 1C). In addition to these residues, p300 also acetylated K9, as well as displayed a sigmoidal dependence on substrate concentration, with a Hill of 2–3. The specificity, based on kcat/K1/2m, and not taking into account the Hill coefficients for p300, reveals a preference for K18 > K14 > K23 > K9, with selectivity differences up to ∼2-fold. However, when we consider that the Hill coefficient is potentially critical for understanding selectivity between sites, the more accurate constant is kcat/K1/2nH. This changes the order to K14 > K18 > K9 > K23, and the difference is up to ∼1015-fold (Figure 1C). It is also noteworthy that in the presence of p300, H3K9 acetylation levels are detectable before 10% of the substrate is acetylated, thus allowing for kinetic analysis of this site. The same is not true with CBP, where H3K9 levels are not detectable under these conditions. While CBP is observed to be more enzymatically active, with kcat values up to 3-fold higher than p300, this is likely because p300 is acetylating an additional site (K9). Comparing the selectivity of CBP and p300, we see that p300 has a specificity that is a factor of ∼1010 higher than CBP for K14 and K18, while on K23 CBP has a specificity that is ∼3.5 fold higher than p300, while the advantage for K9 is undetermined (Figure 1D). The kinetic parameters for these experiments are summarized in Table 2. By comparing the catalytic efficiency (kcat/K1/2 or kcat/K1/2nH) to the nonenzymatic rate of acetylation (knE), we can calculate the catalytic proficiency,27 or how well the enzyme will acetylate a specific residue compared to the highest rate of any residue getting acetylated nonenzymatically.27 The catalytic proficiency values measured for p300 and CBP range from ∼106 to ∼1018. This is compared to Gcn5, which comes in at 106 under the same conditions for H3K14 (the site for which Gcn5 has the highest specificity).27
Figure 1.

Determination of steady-state kinetic parameters of CBP- and p300-mediated acetylation of histone H3 when titrating H3. Experiments were performed at 37 °C in 100 mM ammonium bicarbonate and 50 mM HEPES buffer (pH 7.8) at 37 °C. Assays contained from 1 to 150 nM p300 or 0.5 to 10 nM CBP, with varying concentrations of H3 (0.25–15 μM) and constant (200 μM) acetyl-CoA. Experiments were quenched with 4 vol of TCA and boiled at 95 °C for 5 min. Sites displaying the highest specificity (kcat/KmnH) for either CBP or p300 where chosen for representative graphs. (A) Nonlinear fit of CBP acetylation of histone H3K18. (B) Nonlinear fit of p300 acetylation of histone H3K14. (C) Comparison of the specificity constants (kcat/KmnH) of CBP (black) and p300 (gray) on H3K9, H3K14, H3K18, and H3K23. (D) The log of the ratio of specificity (CBP/p300) between CBP and p300 at each site of H3. All quantified sites can be found in Supplemental Figure 1. The apparent kinetic parameters are summarized in Table 2.
Table 2. Steady-State Parameters of H3 for p300- and CBP-Mediated Acetylation of H3.
| kcat (× 10–3 s–1) | K1/2 (× 10–6 M) | kcat/K1/2 (× 103 M–1 s–1) | nH (Hill coefficient) | kcat/K1/2nH (M–nH s–1) | |
|---|---|---|---|---|---|
| p300 H3 | |||||
| H3K9 | 5.24 ± 0.39 | 3.52 ± 0.54 | 1.49 ± 0.25 | 1.65 ± 0.25 | (5.03 ± 1.63) × 106 |
| H3K14 | 12.27 ± 0.47 | 3.33 ± 0.16 | 3.68 ± 0.23 | 3.10 ± 0.40 | (1.11 ± 0.23) × 1015 |
| H3K18 | 51.89 ± 1.14 | 2.07 ± 0.10 | 25.10 ± 1.34 | 2.82 ± 0.25 | (5.22 ± 1.62) × 1014 |
| H3K23 | 9.14 ± 0.83 | 2.23 ± 0.39 | 4.10 ± 0.81 | n.a. | n.a. |
| CBP H3 | |||||
| H3K14 | 31.58 ± 1.35 | 0.03 ± 0.07 | 95.35 ± 20.00 | n.a. | n.a. |
| H3K18 | 106.50 ± 4.05 | 1.12 ± 0.15 | 94.84 ± 13.34 | n.a. | n.a. |
| H4K16 | 19.38 ± 2.21 | 1.53 ± 0.40 | 12.67 ± 3.65 | n.a. | n.a. |
Next we measured the steady-state parameters for acetyl-CoA under saturating histone H3 concentrations (10–15 μM) (Figures 2A,B and S2). These experiments are more complicated in the fact that, for concentrations of acetyl-CoA that are less than the concentration of H3, we have to measure total acetylation less than 0.1 times the total concentration of acetyl-CoA times the concentration of histone (see ref (27) for details). These experiments allowed us to determine if limiting the amount of acetyl-CoA available to either CBP or p300 would affect their specificity. The amount of acetyl-CoA used for these experiments falls well within previously reported ranges for cellular acetyl-CoA concentrations.34 Interestingly, the order of specificity changed from what we observed in titrating H3. For CBP we found that the order of kcat/K1/2 is K18 > K14 > K23 with a range of 7–40-fold difference in specificity. The Hill coefficient changes this order again, where the order of specificity based on kcat/K1/2nH is K14 > K23 > K18 (Figure 2C); the movement of K18 from the first position to last is due to the large Hill coefficient for K14 (∼6). While we did not observe a change in the order of the kcat/K1/2 for p300 (K18 > K14 > K23 > K9), when we compare the kcat/K1/2nH to that of the H3 titration, we observe a change in the order for the last two positions to (K14 > K18 > K23 > K9) (Figure 2C). This results in a difference in specificity of up to 1042-fold. This is in contrast with what we previously observed for Gcn5, where the order of acetylation was unchanged for either limiting acetyl-CoA or H3.27 Catalytic proficiency or ((kcat/K1/2nH)/knE) for p300 goes as high as 1045 and 1032 for CBP. The differences in specificity between CBP and p300 are also more exaggerated: with acetyl-CoA p300 has an advantage of ∼1015-fold for K14 and K18, and CBP has a ∼105-fold advantage for K23 (Figure 2D). This value is too large to know for K9. The kinetic parameters for these experiments are summarized in Table 3.
Figure 2.

Determination of steady-state kinetic parameters of CBP- and p300-mediated acetylation of histone H3 when titrating acetyl-CoA. Experiments were performed at 37 °C in 100 mM ammonium bicarbonate and 50 mM HEPES buffer (pH 7.8) at 37 °C. Assays for p300 contained 50 nM p300, 17.5 μM H3, and varying concentrations of acetyl-CoA (1–200 μM). Assays for CBP contained 7 nM CBP, 7.5 μM H3, and varying concentrations of acetyl-CoA (1–200 μM). Experiments were quenched with 4 vol of TCA and boiled at 95 °C for 5 min. Sites displaying the highest specificity (kcat/KmnH) for either CBP or p300 where chosen for representative graphs. (A) Nonlinear fit of CBP acetylation of histone H3K14. (B) Nonlinear fit of p300 acetylation of histone H3K14. (C) Comparison of the specificity constants (kcat/KmnH) of CBP (black) and p300 (gray) on H3K9, H3K14, H3K18, and H3K23. (D) The log of the ratio of specificity (CBP/p300) between CBP and p300 at each site of H3. All quantified sites can be found in Supplemental Figure 2. The apparent kinetic parameters are summarized in Table 3.
Table 3. Steady-State Parameters of Acetyl-CoA for p300- and CBP-Mediated Acetylation of H3.
| kcat (× 10–3 s–1) | K1/2 (× 10–6 M) | kcat/K1/2 (× 103 M–1 s–1) | nH (Hill coefficient) | kcat/K1/2nH (M–nH s–1) | |
|---|---|---|---|---|---|
| p300 H3 | |||||
| H3K9 | 4.99 ± 0.30 | 17.70 ± 3.53 | 0.28 ± 0.06 | n.a. | n.a. |
| H3K14 | 12.53 ± 0.65 | 7.84 ± 0.20 | 1.60 ± 0.09 | 9.28 ± 2.17 | (2.97 ± 0.19) x1044 |
| H3K18 | 47.06 ± 2.32 | 5.98 ± 0.17 | 7.87 ± 0.45 | 6.33 ± 1.00 | (5.70 ± 0.55) × 1030 |
| H3K23 | 8.32 ± 0.44 | 6.97 ± 0.51 | 1.19 ± 0.11 | 3.23 ± 0.66 | (3.77 ± 0.52) × 1013 |
| CBP H3 | |||||
| H3K14 | 40.23 ± 1.91 | 4.94 ± 0.27 | 8.15 ± 0.59 | 5.81 ± 1.67 | (2.59 ± 0.18) × 1029 |
| H3K18 | 115.90 ± 4.20 | 7.34 ± 0.29 | 15.79 ± 0.84 | 3.48 ± 0.38 | (8.96 ± 2.02) × 1016 |
| H3K23 | 22.88 ± 1.06 | 6.06 ± 0.35 | 3.78 ± 0.28 | 4.00 ± 0.74 | (1.60 ± 0.20) × 1018 |
Table 4. Steady-State Parameters of H3 and H4 for p300- and CBP-Mediated Acetylation of H3/H4.
| kcat (× 10–3 s–1) | K1/2 (× 10–6 M) | kcat/K1/2 (× 103 M–1 s–1) | nH (Hill coefficient) | kcat/K1/2nH (M–nH s–1) | |
|---|---|---|---|---|---|
| p300 H3/H4 | |||||
| H3K9 | 0.55 ± 0.03 | 0.52 ± 0.11 | 1.07 ± 0.24 | n.a. | n.a. |
| H3K14 | 4.56 ± 0.23 | 0.27 ± 0.06 | 16.99 ± 4.01 | n.a. | n.a. |
| H3K18 | 12.92 ± 0.65 | 0.73 ± 0.14 | 17.81 ± 3.46 | n.a. | n.a. |
| H3K23 | 4.24 ± 0.23 | 0.26 ± 0.05 | 16.41 ± 3.60 | n.a. | n.a. |
| H4K5 | 6.08 ± 0.45 | 1.16 ± 0.23 | 5.24 ± 1.11 | n.a. | n.a. |
| H4K8 | 5.92 ± 0.43 | 0.60 ± 0.14 | 9.92 ± 2.41 | n.a. | n.a. |
| H4K12 | 3.77 ± 0.27 | 0.24 ± 0.07 | 15.49 ± 4.50 | n.a. | n.a. |
| H4K16 | 0.74 ± 0.05 | 0.19 ± 0.05 | 3.90 ± 1.06 | n.a. | n.a. |
| CBP H3/H4 | |||||
| H3K14 | 14.22 ± 0.74 | 2.91 ± 0.21 | 4.88 ± 0.43 | 2.75 ± 0.42 | (2.23 ± 0.43) × 1012 |
| H3K18 | 55.80 ± 2.13 | 0.11 ± 0.01 | 503.61 ± 25.43 | 5.49 ± 0.78 | (8.83 ± 0.78) × 1035 |
| H3K23 | 20.21 ± 0.90 | 3.62 ± 0.20 | 5.58 ± 0.39 | 3.34 ± 0.49 | (3.02 ± 0.51) × 1015 |
| H4K5 | 16.51 ± 1.11 | 0.08 ± 0.01 | 217.15 ± 24.78 | 3.23 ± 0.90 | (1.74 ± 0.17) × 1021 |
| H4K8 | 8.42 ± 0.57 | 0.17 ± 0.03 | 50.33 ± 8.72 | n.a. | n.a. |
| H4K12 | 1.19 ± 0.10 | 0.08 ± 0.01 | 15.53 ± 2.29 | 3.23 ± 1.13 | (1.18 ± 0.12) × 1020 |
| H4K16 | 3.58 ± 0.20 | 0.83 ± 0.09 | 4.33 ± 0.54 | n.a. | n.a. |
Table 5. Steady-State Parameters of Acetyl-CoA for p300- and CBP-Mediated Acetylation of H3/H4.
| kcat (× 10–3 s–1) | K1/2 (× 10–6 M) | kcat/K1/2 (× 103 M–1 s–1) | nH (Hill coefficient) | kcat/K1/2nH (M–nH s–1) | |
|---|---|---|---|---|---|
| p300 H3/H4 | |||||
| H3K9 | 0.55 ± 0.03 | 11.67 ± 1.95 | 0.05 ± 0.01 | n.a. | n.a. |
| H3K14 | 3.61 ± 0.24 | 2.24 ± 0.52 | 1.61 ± 0.39 | n.a. | n.a. |
| H3K18 | 10.30 ± 0.37 | 2.39 ± 0.34 | 4.30 ± 0.63 | n.a. | n.a. |
| H3K23 | 2.22 ± 0.12 | 10.72 ± 1.43 | 0.21 ± 0.03 | n.a. | n.a. |
| H4K5 | 4.11 ± 0.16 | 6.84 ± 0.25 | 0.60 ± 0.03 | 6.11 ± 1.00 | (1.46 ± 0.13) × 1029 |
| H4K8 | 5.02 ± 0.26 | 6.24 ± 0.41 | 0.80 ± 0.07 | 5.43 ± 1.62 | (8.86 ± 0.65) × 1025 |
| H4K12 | 2.55 ± 0.19 | 12.64 ± 1.48 | 0.20 ± 0.03 | 2.31 ± 0.40 | (4.97 ± 1.16) × 108 |
| H4K16 | 0.68 ± 0.04 | 4.38 ± 0.15 | 0.16 ± 0.01 | 6.70 ± 1.27 | (5.63 ± 0.48) × 1032 |
| CBP H3/H4 | |||||
| H3K14 | 13.16 ± 1.07 | 2.89 ± 0.37 | 4.56 ± 0.69 | 2.00 ± 0.35 | (1.50 ± 0.36) × 109 |
| H3K18 | 54.38 ± 1.62 | 3.63 ± 0.17 | 14.99 ± 0.83 | 2.38 ± 0.22 | (4.53 ± 1.62) × 1011 |
| H3K23 | 21.31 ± 1.94 | 7.50 ± 1.47 | 2.84 ± 0.61 | n.a. | n.a. |
| H4K5 | 16.70 ± 1.06 | 13.16 ± 1.10 | 1.27 ± 0.13 | 2.65 ± 0.42 | (1.37 ± 0.30) × 1011 |
| H4K8 | 7.12 ± 0.72 | 12.42 ± 2.73 | 0.57 ± 0.14 | n.a. | n.a. |
| H4K12 | 2.63 ± 0.14 | 3.11 ± 0.40 | 0.85 ± 0.12 | 2.41 ± 0.65 | (4.65 ± 0.61) × 1010 |
| H4K16 | 3.24 ± 0.17 | 8.90 ± 0.65 | 0.36 ± 0.03 | 3.24 ± 0.59 | (7.60 ± 1.17) × 1013 |
Expanding the Assay To Include Histone H4
Having characterized the acetylation pattern of p300 and CBP on H3, we wished to expanded our analysis to the H3/H4 tetramer. We hypothesized that formation of the tetramer could alter the accessibility of certain residues on H3 to CBP or p300, and thus could alter the specificity of these proteins. Additionally, both KATs are known to acetylate H4 residues. The addition of alternative targets for these enzymes could also potentially alter their specificities. In order to effectively characterize the activity on tetramer, however, we needed to develop new SRMs for the detection of H4 acetylation. The details on these SRMs can be found in the Experimental Procedures, and the detailed transitions are summarized in Table 1.
Steady-State Analysis of p300 and CBP Specificity on the H3/H4 Tetramer
H4 alone aggregates at low concentrations (<1 μM) and thus will not function as a proper substrate on its own. Therefore, these experiments were performed utilizing the H3/H4 tetramer. As before, prior to performing experiments under steady-state conditions, we allowed reactions with either p300 or CBP with the H3/H4 substrate to occur for 24 h. We then determined that both p300 and CBP are capable of acetylating four lysine residues on H4: K5, K8, K12, and K16 (data not show). Also as before, we observed a small amount (<0.1%) of double acetylation events (K5/K8, and K12/K16), this time in p300- and CBP-mediated acetylation and chose not to include this fraction in the analysis, although doing so had little impact on the parameters measured. We began our steady-state experiments with p300 and CBP by varying the histone (H3/H4) substrate concentration (Figures 3A,B and S3). Starting with CBP, looking at just the kcat/K1/2 without consideration for cooperativity, on H3 we see a specificity of K18 > K14 > K23. On the tetramer, we see that H3K18 is significantly higher (503.61 × 103 M–1 s–1) than either H3K14 or H3K23 (50.33 and 15.53 × 103 M–1 s–1, respectively). The order of specificity for p300 mimics that of CBP (K18 > K14 > K23). However, comparing the acetylation pattern of p300 to CBP on the H3/H4 tetramer, we see a stark contrast between the two. While p300 specificity is almost evenly distributed between K14, 18, and 23 (with a difference of less than 1.1-fold between K18 and K23), CBP highly prefers K18 (with a difference of 103-fold between K18 and K23).
Figure 3.

Determination of steady-state kinetic parameters of CBP- and p300-mediated acetylation of histone H3/H4 when titrating H3/H4. Experiments were performed at 37 °C in 100 mM ammonium bicarbonate and 50 mM HEPES buffer (pH 7.8) at 37 °C. Assays contained from 1 to 50 nM p300 or 1 to 22.5 nM CBP, with varying concentrations of H3/H4 (0.2–10 μM) and constant (200 μM) acetyl-CoA. Experiments were quenched with 4 vol of TCA and boiled at 95 °C for 5 min. Sites displaying the highest specificity (kcat/KmnH) for either CBP or p300 where chosen for representative graphs. (A) Nonlinear fit of CBP acetylation of histone H3K18. (B) Nonlinear fit of p300 acetylation of histone H3K18. (C) Comparison of the specificity constants (kcat/KmnH) of CBP (black) and p300 (gray) on H3K9, H3K14, H3K18, and H3K23 and H4K5, H4K8, H4K12, and H4K16. (D) The difference in change in free energy (ΔΔG) between CBP and p300 at each site of H3 and H4. The Y-axis is inverted to more clearly show favorable (−ΔG) changes. All quantified sites can be found in Supplemental Figures 3 and 4. The apparent kinetic parameters are summarized in Table 4.
When considering cooperativity, the difference between p300 and CBP becomes even more pronounced (Figure 3C). On the tetramer, CBP demonstrates cooperativity for H3 acetylation while p300 does not. Taking this into consideration, the (kcat/K1/2nH) for K18 is 1034 times higher for CBP than p300 (Figure 3D). The specificity of CBP is 1011 and 1014 times higher than p300, respectively, for H3K14 and H3K23 (Figure 3D).
The same samples that were analyzed for H3 acetylation on H3/H4 were simultaneously analyzed for H4 acetylation (Figures 3 and S4). Focusing on H4, we observe several differences between the activity of p300 and CBP. CBP shows a strong preference for H4K5 (with a kcat/K1/2 of 217.15 × 103 M–1 s–1), followed by K8, K12, and K16, with a difference of ∼50-fold between K5 and K16. The kcat/K1/2 for p300 are closer to each other than for CBP, with the order of specificity being K12 (15.49 × 103 M–1 s–1) > K8 > K5 > K16, with a difference in specificity between K12 and K16 being ∼4-fold. We noted that CBP demonstrates cooperativity on H4K5 and H4K12, while p300 does not display cooperativity on H4. This results in an ∼1015-fold advantage for CBP when aceylating H4K12 (Figure 3D). Taking all of this information into consideration, when we look at the kcat/K1/2nH, we see that the order of specificity for CBP on all sites of the tetramer is H3K18 ≫ H4K5 ≫ H4K12 > H2K23 > H2K14 > H4K8 > H4K16 (Figure 3C). This is compared to p300, which demonstrates no cooperativity, with an order of specificity of H3K18 ≈ K3H14 ≈ H3K23 > H4K12 > H4K8 > H4K5 > H4K16 > H3K9 (Figure 3C). The kinetic parameters for these experiments are summarized in Table 3.
As with H3, we also performed acetyl-CoA titrations where H3/H4 tetramer concentrations and enzyme concentrations were kept constant (Figure 4A,B, Figures S5 and S6). Under these conditions, the order of specificity, not taking into consideration cooperativity, for CBP is K18 > K14 > K23, which is the same as the H3/H4 substrate titration. Despite the order being the same, the preference for K18 acetylation is not as pronounced when titrating acetyl-CoA, with only an ∼3-fold preference over the second highest site, K14. When titrating acetyl-CoA, p300 displays the same order of specificity as CBP (but with the addition of K9), K18 > K14 > K23 > K9. However, under these conditions we observe a stronger preference of p300 for H3K18, with the kcat/k1/2 being over 2.5-fold higher than second highest site, K14, instead of being approximately equal.
Figure 4.

Determination of steady-state kinetic parameters of CBP- and p300-mediated acetylation of histone H3/H4 when titrating acetyl-CoA. Experiments were performed at 37 °C in 100 mM ammonium bicarbonate and 50 mM HEPES buffer (pH 7.8) at 37 °C. Assays for p300 contained 50 nM p300, 7.5 μM H3/H4, and varying concentrations of acetyl-CoA (1–200 μM). Assays for CBP contained 20 nM CBP, 10 μM H3/H4, and varying concentrations of acetyl-CoA (1–200 μM). Experiments were quenched with 4 volumes of TCA and boiled at 95 °C for 5 min. Sites displaying the highest specificity (kcat/KmnH) for either CBP or p300 where chosen for representative graphs. (A) Nonlinear fit of CBP acetylation of histone H4K16. (B) Nonlinear fit of p300 acetylation of histone H4K16. (C) Comparison of the specificity constants (kcat/KmnH) of CBP (black) and p300 (gray) on H3K9, H3K14, H3K18, and H3K23 and H4K5, H4K8, H4K12, and H4K16. (D) The log of the ratio of specificity (CBP/p300) between CBP and p300 at each site of H3 and H4. All quantified sites can be found in Supplemental Figures 5 and 6. The apparent kinetic parameters are summarized in Table 5.
Under these conditions we note that CBP still shows cooperativity for most sites (excluding H3K23 and H4K8). p300, which demonstrated no cooperativity when substrate was limiting, now displays cooperativity on the H4 sites. Thus we see a change in the order of kcat/K1/2nH for both CBP and p300. CBP preferentially acetylates: H4K16 > H3K18 > H4K5 > H4K12 > H3K14 > H3K23 > H4K8 (Figure 4C). The movement of H4K16 from the last position to the first is due to its Hill coefficient being 0.6 higher than any other site. When titrating acetyl-CoA, the order of specificity for p300 changes significantly from the H3/H4 substrate titration and becomes: H4K16 > H4K5 > H4K8 > H4K12 > H3K18 > H3K14 > H3K23 > H3K9 (Figure 4C). The increase in specificity for the H4 sites are because p300 demonstrates cooperativity at these sites, but not on the H3 sites of the tetramer. Because of this cooperativity, we see that there is a higher specificity for p300 on H4K5, H4K12, and H4K16 ranging from 1018 to 1023-fold higher than CBP (Figure 4D). Meanwhile, CBP has an advantage acetylating H3K14 (∼106-fold), K18 (∼108-fold), and K23 (∼13-fold) (Figure 4D).
Comparison of p300 and CBP Acetylation of H3 Compared to H3/H4
Finally, using the data obtained from these experiments, we sought to determine how specificity of CBP and p300 changed on the tetramer compared to H3 alone. To do so, we examined the ratio of (H3/H4)/H3, which are themselves calculated from the kcat/K1/2nH. Because we have no point of comparison for H4 alone (as it aggregates), only sites of H3 were considered. When we analyze the substrate titrations involving CBP, it is clear that CBP has a much higher preference for H3/H4 (Figure 5A). Acetylation by CBP on K18 of the H3/H4 tetramer is ∼1031-fold more favorable than on H3 alone. K14 is ∼108-fold more favorable, while K23 is ∼1012-fold more favorable. The opposite, however, is true when acetyl-CoA is limiting (Figure 5C). H3 is favored over H3/H4 on all three sites, ranging from ∼105–1020-fold more favorable on H3.
Figure 5.

Comparison of specificities of CBP and p300 on H3 and H3/H4. (A) The log of the difference of (H3/H4)/H3 for CBP when substrate is limiting. (B) The log of the difference of (H3/H4)/H3 for p300 when substrate is limiting. (C) The log of the difference of (H3/H4)/H3 for CBP when acetyl-CoA is limiting. (D) The log of the difference of (H3/H4)/H3 for p300 when acetyl-CoA is limiting. (E) Summary of specificities (kcat/K1/2nH(app)) of CBP on H3 when substrate (black) or acetyl-CoA (light gray) is limited, or on H3/H4 when substrate (dark gray) or acetyl-CoA (dark gray border) is limited. (F) Summary of specificities (kcat/K1/2nH(app)) of p300 on H3 when substrate (black) or acetyl-CoA (light gray) is limited, or on H3/H4 when substrate (dark gray) or acetyl-CoA (dark gray border) is limited.
For p300, the H3 substrate is almost always preferred to H3/H4 (Figure 5B,D). The exception is under conditions of limiting substrate on H3K23, where there is an ∼4.5-fold advantage on the H3/H4 tetramer. When substrate is titrated, the preference for H3 alone compared to H3/H4 as a substrate ranges from ∼104-fold (K9) to ∼1010-fold (K14 and K18). There is an even stronger preference for H3 alone when acetyl-CoA is limiting, with the highest preference at 1020-fold for K14.
The observed cooperativity of histone acetylation changes with the histone complex. When substrate is limiting, we observe cooperative dependence on histone H3 with p300, but not CBP, while the dependence on H3/H4 displays cooperativity with CBP but not p300. This cooperativity plays a large part in the preference of CBP for H3/H4 substrate and in the preference of p300 for H3. Overall we see that changing the substrate from H3 to H3/H4 tetramer or varying whether we limit acetyl-CoA levels or substrate has a marked affect on the ability of both p300 and CBP to acetylate the residues of H3 and H4 (Figure 5E,F). It is interesting to note that while the specificity for some sites decrease, others increase. The potential importance of these changes is explored further in the discussion.
Discussion
Here we have observed significant differences in the specificity of CBP and p300 histone acetylation. We have shown that although both preferentially acetylate similar residues they have very different specificities. Using our label-free, quantitative method, we were also able to determine the kinetics for these proteins at several other sites of histone H3 and H4. We have also shown that these selectivities are affected by acetyl-CoA levels and can be altered by the formation of histone complexes. Understanding how the activities of these two enzymes differ is the first step in understanding why they cannot compensate for each other in an organism deficient in either protein, while determining how these specificities can be alter is important in deciphering how the histone epigenetic code is written.
These steady state experiments reveal several differences in specificity between p300 and CBP. The large increases in specificity (up to 1031-fold) are mostly a result of an increase in the apparent cooperativity, which results in a much larger denominator when calculating the specificity constant (kcat/K1/2nH). The origins of cooperativity in this system are likely complicated, but some possibilities are precluded by our data: if cooperativity were truly a function of either histone or acetyl-CoA binding in a specific complex, resulting in a specific residue being acetylated, then one would expect that one particular site would begin to out compete others. This would result in what would appear to be product inhibition at certain residues, or in other words as the v/E increased for the site with the higher Hill coefficient, the v/E would begin to decrease for sites with a smaller Hill coefficient. This is not what we observe under conditions where we detect cooperativity for one site and not another site; we see no signs of the major v/E decrease that we would expect from this type of mechanism. Another possibility is that we are observing the dimerization of H3 or H3/H4, resulting in the appearance of cooperativity. This has been seen before with Nap1 binding H3/H4,35 but if this were the case we would expect to see a similar Hill coefficient for all sites where the catalytic efficiency is enhance by dimerization. However, it is possible that certain sites are less or more sensitive to the dimer form of the substrate, which we cannot rule out. Another interesting possibility is that the enzyme is in multiple conformations or isomerization states, all of which are catalytically active at different rates but are slow compared to the catalytic reaction,36 with the rate of isomerization being influenced by substrate. This model would make biological sense in the fact that proteins could bind p300 or CBP to act as allosteric regulators, altering their specificity for particular lysines. Together, these observations may suggest mechanisms by which both the chromatin conformation and factors interacting with p300 or CBP could alter the residues acetylated, opening a wide field of investigation into factors that influence enzyme specificity. Regardless of the mechanism behind this observed cooperativity, we believe that it is an important factor in determining the specificity of p300 and CBP, and as such will focus our discussion on our calculations that take into consideration the Hill coefficient.
In analyzing the specificity data, it is important to first draw attention to the magnitude of the differences in selectivity between CBP and p300. As we mentioned previously, different diseases arise due to mutations in either CBP or p300, suggesting that one protein is incapable of fully substituting for the other. As we see that both proteins target the same residues, it is likely that it is the ratio of acetylation that is the important marker of these proteins’ activities. For CBP, acetylation of H3K18 is much greater than any other site on H3, with a specificity that is a factor of 1014 greater than the second most abundant site (H4K5). For p300, though, the specificity for each site is much closer, with only a tiny ∼1.05-fold difference between K18 and the next most abundant site (H3K14). Additionally, the acetylation of K9 is much higher for p300, with K9 not detectable for CBP before 10% of the histone is acetylated. All of these factors combined mean that p300 and CBP, despite targeting the same sites, will do so to different degrees of efficacy; while p300 will acetylate K9, 14, 18, and 23 in a more evenly distributed fashion, CBP will heavily favor K18 acetylation, to the detriment of the other sites, just as we see with H3K9. Because of these differences, we can speculate that it is these more subtle activities that are ultimately used in the cell to distinguish KATs from each other. Indeed, it has been shown that p300 plays a role in acetylation of H3K9 in vivo and is important to maintaining the balance between methylation and acetylation at this site.37 Meanwhile, more evidence points to the importance of CBP in maintaining levels of H3K18 acetylation: recent work has shown that inactivation of CBP by phosphorylation leads to a marked decrease in H3K18 acetylation.38
Though weak, it is likely that the acetylation of H3K9 is still an important part of the activity of p300 and potentially CBP. The relevance of this site is emphasized by the fact that K9 hypoacetylation has been noted in a number of human cancers.39,40 Our results suggest that because p300 targets H3K9, it could potentially be used by the cell to compensate for decreases in H3K9 acetylation. Indeed, it has been shown that the histone deacetylase inhibitor valproic acid (VPA) leads to a p300 associated increase in K9 acetylation in embryonic stem cells.41 Additionally, VPA has been shown to be successful in treating several cancer types, including cervical cancer.42,43
In addition to weak H3K9 acetylation, we noted that if we allowed p300 to acetylate well past steady-state conditions (hours instead of minutes), we were able to detect low levels of H3K27 acetylation (as has been previously reported in vivo(44)). However, we observe that the level of acetylation of H3K27 was significantly lower than the other sites we have detected (data not shown). Additionally, under steady-state conditions, no acetylation is detected at this site before 10% of the histone is acetylated. Considering the noted presence of the H3K27 mark in vivo,44,45 it is likely that some cofactor (possibly a histone chaperone (46)) is influencing p300 acetylation of this site.
When comparing the steady-state experiments where either H3/H4 is limiting or acetyl-CoA is limiting, it is worth noting that the specificity of both CBP and p300 changes. For CBP, when titrating H3/H4 the order of specificity for H3 acetylation is K18 > K23 > K14. Meanwhile, for p300, titrating H3/H4 leads to almost identical specificities for K18, K23, and K14. When titrating acetyl-CoA, however, a clear order emerges of K18 > K14 > K23 for both KATs. Similarly, we see a change in the acetylation order of H4 both with CBP and p300 when comparing H3/H4 titrations versus acetyl-CoA titrations (Figure 5). In addition, the difference in specificity from the highest to the lowest residue decreases when acetyl-CoA is limited, compared to when histone is limited. For CBP, specificity varies by a factor of 1032 when titrating H3/H4 compared to a factor of 1011 when titrating acetyl-CoA. These changes that are observed when acetyl-CoA are limited could be indicative of a mechanism for altering histone acetylation patterns when, for example, nutrient intake is limited. Such a change could potentially correlate with the upregulation/downregulation of certain genes in response to limited energetic intake. Previous studies have already found a link between metabolism and histone acetylation.47 It is an interesting possibility that metabolism influences histone acetylation, which in turn affects gene expression, a mechanism with the potential to aid survival under less than ideal metabolic conditions.
Similarly, we also note the relative increase in H4 specificity between substrate and acetyl-CoA titrations with both p300 and CBP (Figures 4 and 5). When acetyl-CoA is limited, the distinct possibility exists that not all sites that can be targeted by these proteins will be acetylated. The higher specificity for H4 sites would ensure that these sites are preferentially targeted when acetyl-CoA is limited. In this way, H4 marks would be preserved even in adverse metabolic conditions, which implies an importance for these marks. We also note that even under limited acetyl-CoA conditions, CBP maintains a high specificity for H3K18 (Figure 5C), further stressing the role of CBP in maintaining this histone mark.
Histone complexes/conformation can also influence CBP and p300 specificity; we observe changes to the specificity of p300 and CBP depending on whether the substrate is the H3/H4 tetramer or just H3 alone. Under conditions of limiting histone and p300, specificity for H3K18 decreases slightly (by 1.4-fold) when the tetramer is formed, but the kcat/K1/2 for H3K14 and H3K23 both increase on the tetramer (∼4.5-fold for each). Observing a higher specificity for a site when H3 is alone is not necessarily surprising, as without the additional H4 sites for p300 to target, we would expect to see an increase in acetylation of the H3 sites. Seeing a higher specificity for a site on the H3/H4 tetramer (as is the case for K14 and K23) is less expected. While it is unclear why there would be this drop on H3, it is possible that confirmation changes as a result of the formation of the H3/H4 tetramer could make K14 a sterically more viable target. This study, as well as previous work from our lab, noted that H3K4 was rarely targeted for acetylation, possibly due to its ability to more readily sample different conformations. In other words, while it is important for a residue to be accessible to the KAT, too much conformational freedom could be detrimental to KAT binding (and therefore acetylation). This could also explain the change in behavior of p300 for K14: it is possible that formation of the tetramer limits the conformational freedom of K14, which could account for the increase in acetylation of this site on the H3/H4 tetramer.
With the information currently available in the field, it is difficult to know exactly what the biological reason is for the change in specificity of CBP to be more uniform on histone H3 alone when substrate is limited. We believe that the increase in specificity that we observe for H3K14 and H3K23 is important to newly synthesized histones, or that they could be important to histone assembly. The reason for this is that, in the cell, the most readily available free H3 would be from newly synthesized histone that has yet to be assembled into a nucleosome. Therefore, it is under these conditions that an increase of H3K14 and H3K23 acetylation would be the most relevant. Supporting this idea is the fact that in Drosophila, K14 and K23 acetylation is detected on newly synthesized H3,48 although these modifications can vary from organism to organism. An alternative explanation is that instead of high levels of K14 and K23 acetylation being an important epigenetic marker, it could be key to not have disproportionally high levels of K18 during histone assembly, and that is why the acetylation pattern of CBP is more evenly distributed on H3 alone, although this has yet to be seen.
Investigating the acetylation pattern of p300 and CBP, it is interesting to observe how many sites on H3 (and H4) are efficiently acetylated, as compared to previous published data on Gcn5, which has a single, highly preferred site of acetylation, followed by acetylation of secondary sites. A protein that targets fewer sites is likely to have less utility as it can perform fewer functions. At the same time, though, as with Gcn5, having fewer targets means that the site that is targeted has a stronger signal that stands out significantly compared to other sites that it targets. Understanding why some KATs have such high specificity for a particular site while others are more evenly distributed across several residues could be key in unraveling the histone code. An important component to doing so is elucidating the mechanism behind how KAT specificity is regulated. Seeing the disparity in specificity between such highly homologous proteins as CBP and p300 may hold the key to understanding the factors within a protein that determine specificity.
In summary, this study has revealed several important facts about the specificity of p300 and CBP and how it is regulated. Although both KATs are capable of acetylating the same residues of H3 and H4, p300 and CBP both display unique specificities for each lysine residue, under a variety of conditions. Changing the histone substrate from H3 alone to the H3/H4 tetramer clearly influences this order of specificity, although the targeted sites remain the same. Limiting acetyl-CoA concentrations also affects the specificity of these proteins and also alters their order of specificity on H3 and H4. The results presented here distinguishing the targets and specificity of CBP and p300 provide valuable insight into how these enzymes differentially acetylate the histone. Such knowledge could be invaluable for treating the cancers and neurodegenerative disorders that arise from mutations in either CBP or p300. Ultimately, if we can understand how to better manipulate p300 KAT activity to mimic that of CBP and vice versa, we may be able to overcome some of the detrimental effects that result from mutations in either.
Acknowledgments
We gratefully thank Dr. Karolin Luger at Colorado State University for the generous gift of purified histone H3 and H4. We also thank Dr. Jeff Neitz and Dr. James Hougland for helpful comments and discussion in the preparation of this manuscript.
Glossary
Abbreviations
- acetyl-CoA
acetyl coenzyme A
- CBP
CREB-binding protein
- HAT
histone acetyltransferase
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- KAT
lysine acetyltransferase
- PTM
post-translational modification
- SRM
selected reaction monitoring
- TCA
trichloroacetic acid
- UPLC
ultra performance liquid chromatography
Supporting Information Available
Secondary plots for each site of H3 and H3/H4 for steady-state experiments with CBP and p300, when either histone or acetyl-CoA is limiting. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This work was supported by the WW Smith Trust and a grant from the Pennsylvania Department of Health. The Pennsylvania Department of Health specifically disclaims responsibility for any analysis, interpretations, or conclusions. R.A.H. was supported by NIH Training Grant 2T32 CA-009037.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Davie J. R. (1998) Covalent modifications of histones: expression from chromatin templates. Curr. Opin. Genet. Dev. 8, 173–178. [DOI] [PubMed] [Google Scholar]
- Unnikrishnan A.; Gafken P. R.; Tsukiyama T. (2010) Dynamic changes in histone acetylation regulate origins of DNA replication. Nat. Struct. Mol. Biol. 17, 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamburini B. A.; Tyler J. K. (2005) Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol. Cell. Biol. 25, 4903–4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo N.; Goodman R. H. (2001) CREB-binding protein and p300 in transcriptional regulation. J. Biol. Chem. 276, 13505–13508. [DOI] [PubMed] [Google Scholar]
- Capell B. C.; Berger S. L. (2013) Genome-wide epigenetics. J. Invest. Dermatol. 133, e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Beekum O.; Kalkhoven E. (2007) Aberrant forms of histone acetyltransferases in human disease. Subcell. Biochem. 41, 233–262. [PubMed] [Google Scholar]
- Roelfsema J. H.; White S. J.; Ariyurek Y.; Bartholdi D.; Niedrist D.; Papadia F.; Bacino C. A.; den Dunnen J. T.; van Ommen G. J.; Breuning M. H.; Hennekam R. C.; Peters D. J. (2005) Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 76, 572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesena T. I.; Cui T. X.; Piwien-Pilipuk G.; Kaplani J.; Calinescu A. A.; Huo J. S.; Iniguez-Lluhi J. A.; Kwok R.; Schwartz J. (2007) Multiple mechanisms of growth hormone-regulated gene transcription. Mol. Genet. Metab. 90, 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vempati R. K. (2012) DNA damage in the presence of chemical genotoxic agents induce acetylation of H3K56 and H4K16 but not H3K9 in mammalian cells. Mol. Biol. Rep. 39, 303–308. [DOI] [PubMed] [Google Scholar]
- Hasan S.; Hassa P. O.; Imhof R.; Hottiger M. O. (2001) Transcription coactivator p300 binds PCNA and may have a role in DNA repair synthesis. Nature 410, 387–391. [DOI] [PubMed] [Google Scholar]
- Hasan S.; Stucki M.; Hassa P. O.; Imhof R.; Gehrig P.; Hunziker P.; Hubscher U.; Hottiger M. O. (2001) Regulation of human flap endonuclease-1 activity by acetylation through the transcriptional coactivator p300. Mol. Cell 7, 1221–1231. [DOI] [PubMed] [Google Scholar]
- Kalkhoven E. (2004) CBP and p300: HATs for different occasions. Biochem. Pharmacol. 68, 1145–1155. [DOI] [PubMed] [Google Scholar]
- Schiltz R. L.; Mizzen C. A.; Vassilev A.; Cook R. G.; Allis C. D.; Nakatani Y. (1999) Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J. Biol. Chem. 274, 1189–1192. [DOI] [PubMed] [Google Scholar]
- Tanaka Y.; Naruse I.; Maekawa T.; Masuya H.; Shiroishi T.; Ishii S. (1997) Abnormal skeletal patterning in embryos lacking a single Cbp allele: a partial similarity with Rubinstein-Taybi syndrome. Proc. Natl. Acad. Sci. U. S. A. 94, 10215–10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T. P.; Oh S. P.; Fuchs M.; Zhou N. D.; Ch’ng L. E.; Newsome D.; Bronson R. T.; Li E.; Livingston D. M.; Eckner R. (1998) Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 93, 361–372. [DOI] [PubMed] [Google Scholar]
- Lopez-Atalaya J. P.; Gervasini C.; Mottadelli F.; Spena S.; Piccione M.; Scarano G.; Selicorni A.; Barco A.; Larizza L. (2012) Histone acetylation deficits in lymphoblastoid cell lines from patients with Rubinstein-Taybi syndrome. J. Med. Genet. 49, 66–74. [DOI] [PubMed] [Google Scholar]
- Viosca J.; Lopez-Atalaya J. P.; Olivares R.; Eckner R.; Barco A. (2010) Syndromic features and mild cognitive impairment in mice with genetic reduction on p300 activity: Differential contribution of p300 and CBP to Rubinstein-Taybi syndrome etiology. Neurobiol. Dis. 37, 186–194. [DOI] [PubMed] [Google Scholar]
- Guo W.; Crossey E. L.; Zhang L.; Zucca S.; George O. L.; Valenzuela C. F.; Zhao X. (2011) Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS One 6, e19351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santer F. R.; Hoschele P. P.; Oh S. J.; Erb H. H.; Bouchal J.; Cavarretta I. T.; Parson W.; Meyers D. J.; Cole P. A.; Culig Z. (2011) Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol. Cancer Ther. 10, 1644–1655. [DOI] [PubMed] [Google Scholar]
- Iyer N. G.; Ozdag H.; Caldas C. (2004) p300/CBP and cancer. Oncogene 23, 4225–4231. [DOI] [PubMed] [Google Scholar]
- Gayther S. A.; Batley S. J.; Linger L.; Bannister A.; Thorpe K.; Chin S. F.; Daigo Y.; Russell P.; Wilson A.; Sowter H. M.; Delhanty J. D.; Ponder B. A.; Kouzarides T.; Caldas C. (2000) Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 24, 300–303. [DOI] [PubMed] [Google Scholar]
- Bryan E. J.; Jokubaitis V. J.; Chamberlain N. L.; Baxter S. W.; Dawson E.; Choong D. Y.; Campbell I. G. (2002) Mutation analysis of EP300 in colon, breast and ovarian carcinomas. Int. J. Cancer 102, 137–141. [DOI] [PubMed] [Google Scholar]
- Ozdag H.; Batley S. J.; Forsti A.; Iyer N. G.; Daigo Y.; Boutell J.; Arends M. J.; Ponder B. A.; Kouzarides T.; Caldas C. (2002) Mutation analysis of CBP and PCAF reveals rare inactivating mutations in cancer cell lines but not in primary tumours. Br. J. Cancer 87, 1162–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebben W. R.; Sharma N.; Nyborg J. K. (2010) Nucleosome eviction and activated transcription require p300 acetylation of histone H3 lysine 14. Proc. Natl. Acad. Sci. U. S. A. 107, 19254–19259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Wang L.; Zhao K.; Thompson P. R.; Hwang Y.; Marmorstein R.; Cole P. A. (2008) The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature 451, 846–850. [DOI] [PubMed] [Google Scholar]
- Xue K.; Song J.; Wei H.; Chen L.; Ma Y.; Liu S.; Li Y.; Dai Y.; Zhao Y.; Li N. (2010) Synchronous behaviors of CBP and acetylations of lysine 18 and lysine 23 on histone H3 during porcine oocyte first meiotic division. Mol. Reprod. Dev. 77, 605–614. [DOI] [PubMed] [Google Scholar]
- Kuo Y. M.; Andrews A. J. (2013) Quantitating the specificity and selectivity of gcn5-mediated acetylation of histone h3. PLoS One 8, e54896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. C.; Chen Y.; Mirza S.; Xu Y.; Lee J.; Liu P.; Zhao Y. (2006) A clean, more efficient method for in-solution digestion of protein mixtures without detergent or urea. J. Proteome Res. 5, 3446–3452. [DOI] [PubMed] [Google Scholar]
- Garcia B. A.; Mollah S.; Ueberheide B. M.; Busby S. A.; Muratore T. L.; Shabanowitz J.; Hunt D. F. (2007) Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2, 933–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. M.; Gafken P. R.; Zhang Z.; Gottschling D. E.; Smith J. B.; Smith D. L. (2003) Mass spectrometric quantification of acetylation at specific lysines within the amino-terminal tail of histone H4. Anal. Biochem. 316, 23–33. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Tanner K. G.; Denu J. M. (2000) A continuous, nonradioactive assay for histone acetyltransferases. Anal. Biochem. 280, 308–314. [DOI] [PubMed] [Google Scholar]
- Giles R. H.; Peters D. J.; Breuning M. H. (1998) Conjunction dysfunction: CBP/p300 in human disease. Trends Genet. 14, 178–183. [DOI] [PubMed] [Google Scholar]
- Cornish-Bowden A.; Cardenas M. L. (2010) Specificity of non-Michaelis-Menten enzymes: necessary information for analyzing metabolic pathways. J. Phys. Chem. B 114, 16209–16213. [DOI] [PubMed] [Google Scholar]
- Gilibili R. R.; Kandaswamy M.; Sharma K.; Giri S.; Rajagopal S.; Mullangi R. (2011) Development and validation of a highly sensitive LC-MS/MS method for simultaneous quantitation of acetyl-CoA and malonyl-CoA in animal tissues. Biomed. Chromatogr. 25, 1352–1359. [DOI] [PubMed] [Google Scholar]
- Andrews A. J.; Downing G.; Brown K.; Park Y. J.; Luger K. (2008) A thermodynamic model for Nap1-histone interactions. J. Biol. Chem. 283, 32412–32418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish-Bowden A.; Cardenas M. L. (1987) Co-operativity in monomeric enzymes. J. Theor. Biol. 124, 1–23. [DOI] [PubMed] [Google Scholar]
- Ohzeki J.; Bergmann J. H.; Kouprina N.; Noskov V. N.; Nakano M.; Kimura H.; Earnshaw W. C.; Larionov V.; Masumoto H. (2012) Breaking the HAC Barrier: histone H3K9 acetyl/methyl balance regulates CENP-A assembly. EMBO J. 31, 2391–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Xing Z.-b.; Fang Y. (2013) Akt kinase targets the association of CBP with histone H3 to regulate the acetylation of lysine K18. FEBS Lett. 587, 847–853. [DOI] [PubMed] [Google Scholar]
- Fahrner J. A.; Eguchi S.; Herman J. G.; Baylin S. B. (2002) Dependence of histone modifications and gene expression on DNA hypermethylation in cancer. Cancer Res. 62, 7213–7218. [PubMed] [Google Scholar]
- Chen M. W.; Hua K. T.; Kao H. J.; Chi C. C.; Wei L. H.; Johansson G.; Shiah S. G.; Chen P. S.; Jeng Y. M.; Cheng T. Y.; Lai T. C.; Chang J. S.; Jan Y. H.; Chien M. H.; Yang C. J.; Huang M. S.; Hsiao M.; Kuo M. L. (2010) H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 70, 7830–7840. [DOI] [PubMed] [Google Scholar]
- Hezroni H.; Sailaja B. S.; Meshorer E. (2011) Pluripotency-related, valproic acid (VPA)-induced genome-wide histone H3 lysine 9 (H3K9) acetylation patterns in embryonic stem cells. J. Biol. Chem. 286, 35977–35988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C.; Leslie J. S.; Franko-Tobin L. G.; Prasnal M. C.; Yang T.; Vienna Mackey L.; Fuselier J. A.; Coy D. H.; Liu M.; Yu C.; Sun L. (2013) Valproic acid suppresses cervical cancer tumor progression possibly via activating Notch1 signaling and enhances receptor-targeted cancer chemotherapeutic via activating somatostatin receptor type II. Arch. Gynecol. Obstet. 288, 393–400. [DOI] [PubMed] [Google Scholar]
- Witt D.; Burfeind P.; von Hardenberg S.; Opitz L.; Salinas-Riester G.; Bremmer F.; Schweyer S.; Thelen P.; Neesen J.; Kaulfuss S. (2013) Valproic acid inhibits the proliferation of cancer cells by re-expressing cyclin D2. Carcinogenesis 34, 1115–1124. [DOI] [PubMed] [Google Scholar]
- Jin Q.; Yu L. R.; Wang L.; Zhang Z.; Kasper L. H.; Lee J. E.; Wang C.; Brindle P. K.; Dent S. Y.; Ge K. (2011) Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 30, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D.; Malatesta M.; Jung H. R.; Walfridsson J.; Willer A.; Olsson L.; Skotte J.; Wutz A.; Porse B.; Jensen O. N.; Helin K. (2010) Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 38, 4958–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recht J.; Tsubota T.; Tanny J. C.; Diaz R. L.; Berger J. M.; Zhang X.; Garcia B. A.; Shabanowitz J.; Burlingame A. L.; Hunt D. F.; Kaufman P. D.; Allis C. D. (2006) Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc. Natl. Acad. Sci. U. S. A. 103, 6988–6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen K. E.; Hatzivassiliou G.; Sachdeva U. M.; Bui T. V.; Cross J. R.; Thompson C. B. (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel R. E.; Cook R. G.; Perry C. A.; Annunziato A. T.; Allis C. D. (1995) Conservation of deposition-related acetylation sites in newly synthesized histones H3 and H4. Proc. Natl. Acad. Sci. U. S. A. 92, 1237–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.