

Abstract
Background
Diet is known to have an important impact on cardiovascular health. n-3 FAs, found in high quantity in fish oil, have demonstrated beneficial effects in patients with coronary artery disease. The role of n-6 FAs remains more controversial. The objective of this study was to examine the effect of arachidonic acid (AA), an n-6 FA, and eicosapentanoic acid (EPA), an n-3 FA, on the interaction between monocytes and endothelial cells.
Design
We used a cellular model of endothelial cells (EA.hy.926) and monocytes (human leukemic myelomonocytic U937). Confluent endothelial cells were treated with AA or EPA, in the presence of TNF-α or vehicle-alone for either 4 or 24 hours. Adhesion of monocytes to the endothelial monolayer was performed. For gene expression, reverse transcription, followed by real-time quantitative polymerase chain reaction, were performed.
Results
There was a significant increase in adhesion of monocytes to the endothelial monolayer in the presence of n-6 FAs, both in the presence or absence of TNF-α at 4 and 24 hours. The adhesion of monocytes to the endothelial monolayer was decreased with n-3 FAs at 24 hours. ICAM-1, VCAM, E-Selectin, IL-6 and TNF-α were significantly increased in endothelial cells treated with n-6 FAs.
Conclusions
We conclude that AA increases inflammation and enhances the ability of endothelial cells to bind monocytes in vitro. EPA leads to a decrease in the ability of EA.hy.926 to bind monocytes, although the effect appears more modest. Taken together, these data indicate that the n-6 FA AA could potentiate inflammation and early events of atherosclerosis.
Keywords: Endothelial cells, Monocytes, Polyunsaturated fatty acids, Inflammation, Adhesion molecules
INTRODUCTION
There is significant evidence that polyunsaturated fatty acids (PUFAs), particularly n-3 fatty acids (FAs), affect coronary artery disease (CAD)1–3 and possibly other atherosclerotic syndromes, such as peripheral arterial disease (PAD)4. Secondary prevention trials have established that fish oil and n-3 FA supplementation lead to a decrease in total mortality, cardiovascular death, sudden cardiac death and non-fatal cardiovascular events in patients with CAD1–3,5. Mechanisms postulated for their clinical benefits include their actions as endogenous HMG-CoA reductase and ACE enzyme inhibitors, anti-arrhythmics, anti-hypertensive, anti-atherosclerotic, anti-inflammatory, cytoprotective and cardioprotective agents6. It is unclear whether n-6 FAs play a similar protective role, or conversely, may lead to atherosclerosis7.
Atherosclerosis is an inflammatory disease process in which arterial walls become thickened with lipid, due to accumulation of lipid-loaded macrophages and smooth muscle cells8,9. One of the earliest events in atherosclerosis is binding and infiltration of monocytes through the endothelium. Monocyte-endothelial interactions are therefore key to the development of atherosclerosis. Monocyte binding is facilitated by adhesion molecule expression (VCAM-1, ICAM-1, and selectins) on the endothelial cells (ECs) which occurs as a result of inflammatory activation of the endothelium10. It is likely that n-3 and n-6 FAs affect the binding of monocytes to the endothelium, but several studies report discordant results11. Furthermore, the effects of PUFAs on adhesion molecule expression remain controversial, leaving a significant gap in knowledge as to the specific effects of n-3 and n-6 FAs on endothelial activation and subsequent monocyte binding11.
We studied the effects of n-3 and n-6 FAs on ECs with regard to: 1) inflammatory activation, 2) adhesion molecules expression, and 3) monocyte binding, using a cellular model of EA.hy.926 ECs and U937 monocytes. We hypothesized that n-6 FAs would lead to an increase in inflammation, adhesion molecules expression and subsequently, monocyte binding, and that these effects are mediated through the cyclooxygenase pathway. We further hypothesized that n-3 FAs would have the opposite effects to those of n-6 FAs in view of their overall beneficial clinical effects. This experimental design would allow us to better understand the effects of PUFAs on ECs and possibly uncover some of the molecular and cellular mechanisms involved in their cardiovascular actions.
MATERIALS AND METHODS
Materials
Dulbecco’s Modified Eagle Medium (DMEM) and RPMI-1640 medium were purchased from Fisher Scientific (Pittsburg, PA). Penicillin-Streptomycin and L-Glutamine were purchased from Mediatech (Herdon, VA). RNase inhibitor, MultiScribe Reverse Transcriptase and SyBr green master mix were purchased from Applied Biosystems (Foster City, CA). Oligonucleotides were purchased from Operon Technologies, Inc. (Alameda, CA). Fetal Bovine Serum (FBS) was obtained from Hyclone (Logan, UT). TNFα was purchased from R&D systems (Minneapolis, MN). AA (n-6 FA) and EPA (n-3 FA) were obtained from Cayman Chemical (Ann Arbor, MI). Vybrant cell adhesion assay kit and Hoescht stain for nuclear staining were obtained from Invitrogen (Eugene, OR). Albumin was purchased from Sigma (St. Louis, MO). Phosphate saline buffer solution (PBS) was from the University of California San Francisco Cell Culture Facility (San Francisco, CA).
Cell culture
The cellular model used in this experiment included EA.hy.926 (for ECs) and U937 monocytes. EA.hy.926 cells are a fusion of human umbilical vein endothelial cells (HUVEC) and the A549 epidermal carcinoma line, that retains many of the characteristics of primary endothelial cells12. The EA.hy.926 cells were a kind gift from Dr. Cora-Jean S. Edgell from the University of North Carolina, Chapel Hill12. U937 cells are a human monocyte cell line and were obtained from the American Type Cell Collection13. The ECs were maintained in a 37°C/5% CO2 incubator in DMEM with 10% FBS, 2mM L-glutamine, 1% (v/v) penicillin-streptomycin-neomycin antibiotic mix, 4.5 g/L D-Glucose and 1% (v/v) Amphotericin. The monocytes were maintained in a 37°C/5% CO2 incubator in RPMI-1640 medium containing 10% FBS, 2 mM L-glutamine, 1% (v/v) penicillin-streptomycin-neomycin antibiotic mix. Medium was changed every 2 days. Cell concentration was adjusted to 5×105 cells/ml at each medium change. Cell counts were performed with a hemacytometer (Improved Neubauer, Reichert, NY).
Treatment of endothelial cells for adhesion assays
Confluent ECs were trypsinized and plated at 5000 cells per well in a 96-well plate, then grown to confluency prior to experimental treatments. FA and cytokine treatments were performed as follows: ECs were incubated with either 5μg/ml EPA, 5μg/ml AA, 100ng/ml TNFα, vehicle alone, or a combination of these for 4 or 24 hours prior to the adhesion assay. The dose of TNF-α was selected based on dose-response curves leading to the greatest activation of the ECs based on adhesion molecule expression. The doses of FAs were selected based on the human physiological dose range as well as dose-response curves performed in our laboratory. Vehicle contained DMEM with 2% FBS, 2mM L-glutamine, 1% (v/v) penicillin-streptomycin-neomycin antibiotic mix, 4.5 g/L D-Glucose, 1% (v/v) Amphotericin, and 1.25mg/ml FA-free albumin. Monocytes were labeled with Calcein-AM14 according to the Molecular Probes Vybrant cell adhesion assay kit manufacturer’s instructions and washed twice with Phosphate saline buffer solution. Labeled monocytes were resuspended in vehicle with 0.1% FBS and incubated for 20 minutes with ECs. The 96-well plate was gently washed 1–2 times with Phosphate Saline Buffer solution. Fluorescence per well (Ex 488 nm, Em 515 nm) was measured using a Fluoroscan II plate reader.
In order to investigate a possible pathway for the actions of PUFAs (i.e. the cyclooxygenase pathway), experiments with the n-6 FA AA were performed in the presence of a COX-2 inhibitor. For pre-treatment with the COX-2 inhibitor, adhesion assays were repeated using the same conditions mentioned above, with pre-treatment for 1 hour with indomethacin (50 uM) prior to treatment with the n-6 FA.
RNA isolation
RNA was isolated using RNeasy™ Mini kit (QIAGEN, Valencia, CA) according to the manufacturer’s protocol. For RNeasy™ Mini kit RNA isolation, cells were seeded in 6-well plates with DMEM media supplemented with 10%, 2mM L-glutamine, 1% (v/v) penicillin-streptomycin-neomycin antibiotic mix, 4.5 g/L D-Glucose and 1% (v/v) Amphotericin until they reached confluence. Once confluent, media was changed to DMEM supplemented with 2% FBS while all other supplements remained the same. The cells were then treated with the different FAs and TNFα [5μg/ml EPA, 5μg/ml AA, 100ng/ml TNFα, a combination of AA and TNF-α or vehicle alone] for a period of 4 hours. Upon removal of media, the ECs were washed two times with PBS and then 350ul of buffer RLT (supplied in kit) were added to each well. Cells were scrapped off the plate with a cell lifter. The lysate was then placed into a QIA shredder homogenizer (QIAGEN, Valencia, CA) and the flow through was isolated using the Qiagen RNeasy™ Mini kit and processed as per manufacturer’s instruction. The samples were then stored at −80°C until further analysis.
Reverse Transcription (RT)
RNA (0.3ug) was added to 30ul reverse transcriptase (RT) reaction buffer containing 5 mM MgCl2, 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 4 mM dNTPs, 2.5 μM oligo d(T) primer, 2.5 U/μl of MultiScribe, and 20 U/μl of RNase inhibitor. The RT reaction was incubated at room temperature for 10 minutes, 42°C for 30 minutes, inactivated at 99°C for 5 minutes, and cooled at 5°C for 5 minutes.
Real-time Quantitative RT-PCR (qRTPCR)
cDNA (2 μl) from the RT reaction was added to 20 μl real-time quantitative polymerase chain reaction (qPCR) mixture containing 10 μl of 2x SYBR® Green PCR Master Mix (Applied Biosystems, Foster City, CA) and 12 pmol oligonucleotide primers. PCRs were carried out in a Bio-Rad MyiQ Single-Color Real-Time PCR Detection System (Bio-Rad, Hercules, CA). The thermal profile was 50°C for 2 minutes and 95°C for 10 minutes to activate the Taq polymerase, followed by 50 amplification cycles, consisting of denaturation at 95°C for 1 minute 40 seconds, annealing at 63°C for 1 minute 10 seconds and elongation at 72°C for 1 minute 40 seconds. Fluorescence was measured and used for quantitative purposes. At the end of the amplification period, melting curve analysis was performed to confirm the specificity of the amplicon. RNA samples were normalized to cyclophilin (CPHI) internal standard. Relative quantification of gene expression was calculated by using the 2−(Ct gene T − Ct CPHI T)−(Ct gene 0hr −Ct CPHI 0hr) equation, where “Ct gene T” represents the calculated threshold cycle (Ct) of a time point of each sample other than 0 hour, or each treatment other than control. Relative gene abundance was calculated using 2(Ct gene T − Ct CPHI T). Some primer sequences have been previously used15–17-and are presented in Table 1. All data derived using qRTPCR were from independent biological samples (n= 4).
Table 1.
Primers used for qRTPCR in Endothelial Cells
| Primer | Forward Primer 5′->3′ | Reverse Primer 5′->3′ |
|---|---|---|
| E-selectin | ACCTCCACGGAAGCTATGACT | CAGACCCACACATTGTTGACTT |
| ICAM1 | GTGGTAGCAGCCGCAGTC | GGCTTGTGTGTTCGGTTTCA |
| IL-6 | GCTGAAAAAGATGGATGCTT | GGCTTGTTCCTCACTACTCTC |
| TNFα | TCAGATCATCTTCTCGAACCCC | ATCTCTCAGCTCCACGCCAT |
| VCAM | AATGGGAATCTACAGCACCTTT | ATATCCGTATCCTCCAAAAACT |
Fluorescence microscopy
Confluent ECs were tryptinized and plated on a coverslips in 96-well plates, then grown to confluency. FAs and cytokine treatments were performed as described above for the 96-well plate assay. Calcein-AM labeled monocytes were incubated with ECs for 30 minutes, before the coverslips were gently dipped in PBS 4 times. ECs and monocytes were imaged using Zeiss Axioscop Fluorescent Microscope (Carl Zeiss, Germany) and an Orca-ER CCD camera (Hamamatsu Corporation, Bridgewater, NJ).
Statistical considerations
Data were assessed for normality and outliers. Means and standard deviations are presented. ANOVAs were used to compare the means of the different treatment groups. The p-values presented were corrected for multiple-hypothesis testing using the Bonferroni method. Statistical analyses were performed using Stata/SE 12 (StataCorp, College Station, TX).
RESULTS
n-6 increases while n-3 decreases the adhesion of monocytes to the endothelial cells
In order to test the hypothesis that PUFAs affect the adhesion of monocytes to the endothelial monolayer, we studied the effects of 4 and 24 hours of exposure of n-3 and n-6 PUFAs on monocyte adhesion to ECs. Four hours of FA treatment led to significant changes in adhesion of monocytes to ECs, both in the non-activated (without TNF-α; p<0.0001 for the overall group effect) and activated conditions (with TNF-α; p<0.0001 for the overall group effect) (Figure 1a). In the non-activated state, treatment with n-6 FA led to a significant increase in the adhesion of monocytes to ECs compared to baseline (p<0.001). Although the effect of n-3 FA on adhesion was not significant at baseline, the difference between n-3 and n-6 FA responses on monocytes adhesion was significant (p<0.001). In the activated state, there was also a significant increase in the adhesion of monocytes to ECs with n-6 compared to baseline (p<0.001). There was no significant difference between n-3 FAs and TNF-α compared to TNF-α alone (p=0.98). Significant response difference persisted for ECs treated with n-3 and n-6 FA in the presence of TNF-α (p<0.001).
Figure 1. Monocyte Adhesion After Treatment of Endothelial cells with n-3 and n-6 Fatty Acids for 4 hours (A) and 24 hours (B).
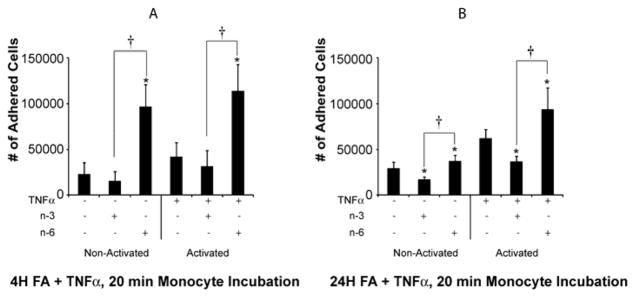
To assess the effects of FA on endothelial cells’ ability to bind monocytes, ECs were grown to confluency and treated with n-3 and n-6 FA (5ug/ml) with or without TNF-α (100ng/ml) for 4 and 24 hours. At 4 hours, in the absence or presence of TNF-α, adhesion of monocytes to the endothelial monolayer is higher in the n-6 FA treated cells than in the non-treated or n-3 FA treated cells. This remains true at 24 hours. Means ± SD (n=8). *= P ≤ 0.05 for n-3 or n-6 treatment compared to the control without FA. † ≤ P ≤ 0.001 between n-3 and n-6 FA treatment groups.
Interestingly, longer treatment periods of ECs with FAs (24 hours) also led to significant changes in adhesion of monocytes to endothelial cells, both in the non-activated (p<0.0001 for the overall group effect) and activated conditions (p<0.0001 for the overall group effect) (Figure 1b). In the non-activated state, there was a significant increase in the adhesion of monocytes to the EC monolayer in the presence of n-6 FA (p=0.03) and a significant decrease in monocyte adhesion in the presence of n-3 FA (p=0.001). The response between n-3 and n-6 FA was significantly different (p<0.001). In the activated state, n-3 FAs led to a significant decrease (p=0.02) and n-6 FA led to a significant increase (p=0.003) in monocyte adhesion to ECs, compared with TNF-α alone. The response between n-3 and n-6-treated FA was also significantly different in the activated state (p<0.001).
While the direct effects of n-3 FA on monocyte adhesion (decrease in adhesion) are more pronounced with a longer treatment period, our results suggest that n-6 FAs lead to an increase in monocyte adhesion to the endothelial monolayer in a manner that is less dependent on time of exposure. This increased binding of the monocytes to ECs with n-6 FA was also observed with fluorescence microscopy (Figure 2).
Figure 2. Monocyte Adhesion to Endothelial Cells at Fluorescence Microscopy after Treatment with n-6 Fatty Acids.
Confluent ECs were incubated with 5 μg/ml AA (n-6 FA) or vehicle alone for 5 hr prior to a 30-minute incubation with calcein-AM labeled U937 cells. The coverslips were gently dipped in PBS to remove unbound U937 cells then imaged by fluorescence microscopy. ECs were labeled with Hoescht stain (Blue) and U937 cells were labeled with Calcein-Am (Green). Fluorescence microscopy demonstrates an increase in monocytes binding to ECs after treatment with n-6 FA.
The effects of n-6 FA on endothelial cells are mediated through the cyclooxygenase pathway
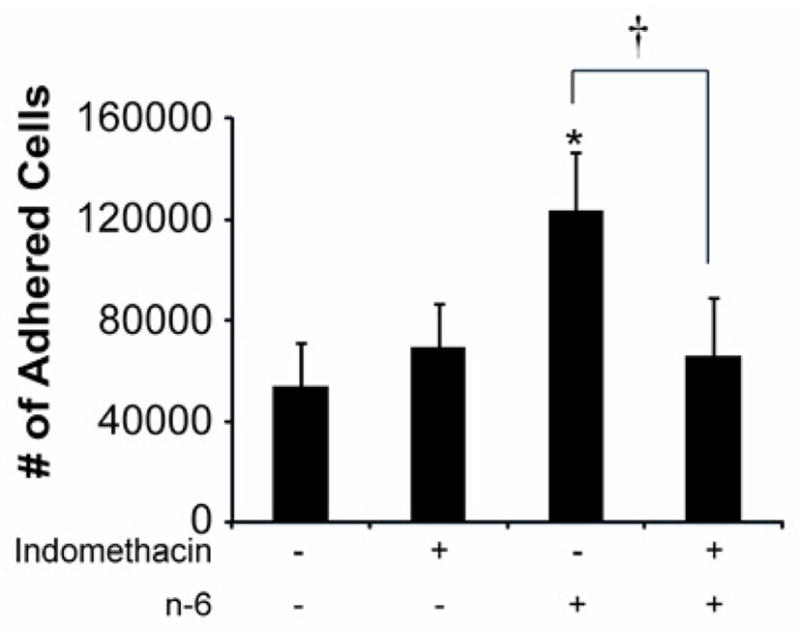
In order to assess if the cellular effects of n-6 FAs is mediated through the cyclooxygenase (COX) pathway, ECs were exposed to indomethacin, a COX-2 inhibitor, in the presence and absence of the n-6 FA AA. Pre-treatment with indomethacin at the dose of 50 uM significantly decreased the binding of monocytes to the endothelial monolayer in the presence of AA (p=0.001) (Figure 3). Indomethacin alone did not have a significant impact on the adhesion of monocytes to ECs (p=1.00)
Figure 3. Effect of Pre-Treatment of Endothelial cells with Indomethacin, a COX-2 inhibitor on Monocyte Adhesion.

To evaluate if the cellular effects of n-6 FA are mediated through the cyclooxygenase (COX) pathway, ECs were exposed to indomethacin, a COX-2 inhibitor, at the dose of 50 uM in the presence and absence of the n-6 FA AA. Indomethacin decreased the adhesion of monocytes to the n-6 treated endothelial monolayer. Indomethacin alone did not affect adhesion. Means ± SD (n=8). *=P ≤ 0.001 n-6 treated endothelial monolayer compared to no-treatment. † = P = 0.001 for n-6 treated endothelial monolayer compared to n-6 treated endothelial monolayer following pre-treatment with indomethacin.
An increase in inflammatory and adhesion molecules gene expression could mediate the effects of n-6 on endothelial cells
In order to investigate molecular mechanisms involved in the effects of PUFAs, gene expression in ECs was investigated, in the presence or absence of PUFAs, and with and without activation with TNF-α (Figure 4). The primers used are described in Table 1.
Figure 4. Gene Expression in Endothelial Cells After Treatment of n-3 and n-6 Fatty Acids using qRTPCR analysis.

To assess molecular mechanisms involved in the alterations in monocyte binding to treatment with FAs (4 hours), qRTPCR analysis of gene expression was done. Treatment of endothelial cells with FA in both the activated (with TNFα) and non-activated states shows a marked increase in expression of adhesion molecules and inflammatory mediators in n-6 compared to n-3 FA. Means ± SD (n=4). *= P ≤ 0.05 n-3 or n-6 treatment compared to the control without FA. †= P ≤ 0.05 between n-3 and n-6 FA treatment groups.
Treatment with FAs led to significant changes in gene activation (Figure 4). At baseline, in the absence of TNF-α, there was a significant increase in ICAM-1, VCAM, E-Selectin as well as IL-6 and TNF-α with n-6 FAs. The gene activation response after treatment with PUFAs (in the absence of TNF-α) differed between n-3 and n-6 FA for ICAM-1, VCAM, E-Selectin, IL-6 and TNF-α.
In the presence of the activator TNF-α, only n-6 FAs led to significant changes in gene activation. This was characterized by an increase in ICAM and VCAM, as well as an increase in IL-6. Similarly, the pattern of gene activation was significantly different between n-3 and n-6 FA in the presence of TNF-α.
Overall, this suggests that n-6 FAs lead to an activation of ECs, characterized by an increase in gene expression of inflammatory mediators, i.e. IL-6 and TNF-α and an increase in adhesion molecules. These responses differ from the response to n-3 FAs.
DISCUSSION
Monocyte-endothelial interactions and inflammation are central to the initiation of atherosclerosis. Our data demonstrate that treatment of ECs with the n-6 FA AA leads to an increase in monocyte adhesion to ECs, likely mediated by an increase in ICAM-1, VCAM and E-Selectin. n-6 FAs also appear to increase inflammatory markers in the ECs. With regards to this finding, to our knowledge, this is the first study demonstrating a direct increase in IL-6 and TNF-α gene expression with the n-6 fatty acid AA in endothelial-type cells. Furthermore, the COX pathway appears central to these interactions. Conversely, n-3 FA appears to decrease adhesion of monocytes to ECs but this effect is more modest. These findings have important clinical implications, particularly in resolving the controversy concerning the association of specific PUFAs in the diet (i.e., n-6 FAs and n-3 FAs) on atherosclerotic syndromes7,18.
Effects of n-6 FAs on monocyte adhesion to the endothelial monolayer and on inflammation
In this experiment, we have consistently demonstrated an increase in monocyte-endothelial cell adhesion after treatment with n-6 FAs. This effect appears to be mediated by an increase in ICAM, VCAM and E-Selectin. In previous literature, these interactions were more controversial. Reissig et al reported that linoleic acid (LA), a precursor of AA, led to a decrease in monocyte-endothelial adhesion19. Others have reported no changes20. Consistent with our results, some authors have reported a stimulatory effect of n-6 FA on adhesion molecule expression and adhesion of monocytes to endothelial cells21–23. Dichtl et al demonstrated that LA led to an increase in VCAM-1 protein and mRNA, which was mediated through NF-KB signaling21. Findings of Toborek et al also support this with an increase in NF-KB, VCAM-1 and ICAM-1 after stimulation of ECs with LA22.
AA is the precursor for an extensive array of eicosanoids (20-carbon FA metabolites), including all the 2-series prostaglandins, thromboxane A2, prostacyclin (PGI2), the 4- series leukotrienes, and a variety of cytochrome P-450 metabolites. These compounds are bioactive and several mediate inflammatory responses, stimulate platelet aggregation, and produce vasoconstriction. Although these are all essential metabolic functions, in excess and unopposed, they can promote atherosclerotic disease and thrombus formation. Our findings provide evidence that AA directly impacts production of inflammatory mediators in the ECs with an increase in IL-6 and TNF-α.
Enzymatic involvement: COX-2
In the present study, we demonstrated that pre-treatment with indomethacin led to less monocyte adhesion to ECs in the presence of AA, pointing towards the importance of COX-2 in the mechanisms of action of AA. Indomethacin sits in the catalytic pocket of COX-2, thus inhibiting the conversion of the n-6 FA, AA, to its by-products prostaglandins and lipoxygenases and other inflammatory mediators. The inability of AA to make its downstream products in the presence of indomethacin is most likely the cause of its consequent inability to influence adhesion. Although indomethacin led to a decrease in monocyte adhesion to ECs in the presence of n-6 FAs, the clinical relevance of these findings need not to be overstated as several large studies have demonstrated adverse effects of COX-2 inhibition in cardiovascular disease (reviewed in 24). No recommendation can be made based on our findings with regards to COX-2 inhibition clinically, however our findings support the mechanism of action of AA on monocyte-endothelial adhesion through the cyclooxygenase pathway.
Some of the biological effects of PUFA are believed to be related to their incorporation into cell membrane phospholipids. There is evidence that in the presence of n-3 FA, PGD3 replaces PGD225, leading to competitive inhibition at the COX-2 level. This replaces AA with the prostanoid derivatives of EPA, which are potentially less pro- thrombotic and vasoconstrictive than AA derivatives26. The present study provides further evidence that effects of PUFA on monocyte-endothelial interactions are mediated by the COX-2 enzyme. The effects of PUFA could lead to alterations in the NF-KB27–29 system and PPARα30. Future investigations will include studies of the AA-induced expression of chemokines/cytokines and downstream products that are changed by the presence of NSAIDS.
Effects of n-3 FA on monocyte adhesion to the endothelial monolayer
Our study confirms prior work demonstrating that treatment with EPA leads to a decrease in adhesion of monocytes to ECs 31–35. Our study further elucidates the effects of PUFA on adhesion molecule expression. We demonstrated that treatment of n-3 or n-6 FA had relatively differential effects on adhesion molecules, providing further evidence that the actions of PUFA are related to changes in adhesion molecule expression (ICAM-1, VCAM-1 and E-Selectin) as well as changes in cytokine expression (i.e., IL-6 and TNF-α).
De Caterina and Libby demonstrated that a decrease in monocyte adhesion to ECs with n- 3 FA was related to a reduction in VCAM-1, E-Selectin, ICAM-1, IL-6 and IL-832. Wang et al, in human aortic endothelial cells treated with EPA and DHA, demonstrated a decrease in VCAM-1 by cell-surface enzyme-linked immunoabsorbent assay, which was confirmed by protein analysis of the cell lysates36. Although other pieces of evidence support alterations in VCAM-1, E-Selectin, and ICAM-1 with PUFA treatment29,33,37, some conflicting evidence negates such changes in adhesion molecule gene expression38,39. Taken together, our findings support the majority of the data presently available, supporting a beneficial effect of n-3 FA on reduction in leukocyte adhesion to the endothelium, steps that are critical in early atherogenesis. This parallels findings from Thies et al demonstrating that n-3 FAs modulate the cellular and structural composition of the atherosclerotic plaque, in a manner to reduce rupture or ulceration40.
Clinical implications of the present findings
Our findings add to existing knowledge suggesting a protective effect of n-3 FAs at the cellular level as relates to monocyte-endothelial interactions. They support the recent recommendations from the American Heart Association (AHA) promoting a diet rich in n-3 FAs. The AHA suggests that the general population should aim to consume fish, especially oily fish, at least twice a week41. For patients with documented CAD, recommended consumption is of ~1gm of EPA+DHA per day, preferably from fish high in EPA and DHA, as this is associated with a reduced risk of both sudden death and death from CAD42,43. Hence, our findings provide further evidence for the importance of nutrition in addition to optimal medical therapy in patients suffering from cardiovascular disease.
Despite the evidence of beneficial effects of n-3 FAs, definitive evidence that consumption of n-6 FAs for primary and secondary prevention of atherosclerosis is less clear. The AHA has recently recommended that consumption of n-6 FA be increased to 5–10% of energy intake, in order to reduce CAD risk that may potentially be related to lower intake44. Inconsistencies in the evidence base have rendered these recommendations controversial7,45–47. Our findings suggest a cautionary approach n-6 dietary advice, in view of a possible increase in inflammation and monocyte binding at the level of the endothelial monolayer. Further evidence from translational studies also support this. For example, Ambring et al studied n-6:n-3 PUFA ratios in healthy subjects while on a Swedish diet or a Mediterranean diet high in fish and flaxseed oil48. The Mediterranean diet led to a lower n-6:n-3 ratio as well as a lower total number of circulating leukocyte and platelets. Ferrucci et al. assessed the relationship between plasma PUFAs to circulating inflammatory markers in a community-based sample, and found that the total n-3 PUFAs were independently associated with lower levels of pro-inflammatory markers and higher anti-inflammatory markers independent of confounders49. A study by Dwyer et al demonstrated that dietary AA significantly enhances the apparent atherogenic effect of genotype, whereas increased dietary intake of n-3 FAs blunted this effect50. Furthermore, the large cardiovascular trials have demonstrated clinical benefits to increasing the dose of n-3 PUFAs in the diet1–3. Based on the concentration of PUFAs in human serum51, we would hypothesize that the levels used in this study should be looked at as a pharmacological effect of a physiological dose, which correlates with evidence from the literature.
Limitations
The cell lines used in this study are not primary human endothelial cell lines, which could lead to different responses compared with primary human endothelial cell lines. Additionally, the adhesion experiments were performed in static conditions, which presents another level of complexity as to how our results could be interpreted for human vasculature. Dose-response curves for n-6 and n-3 FAs have been performed by the authors in previous work for osteoblasts, epithelial cells, endothelial cells, prostate cancer, and colorectal cancer cells, and were not repeated here. The best concentration for cellular response has been in the 5μg/ml range for both types of FAs for all cell types. Furthermore, this concentration remains within human physiological range, estimated to be on average 16.1 mg/100ml in Japanese male and 21.1 mg/100ml in Dutch males sera for AA, and 3.7 mg/100ml and 1.0 mg/100ml for EPA in Japanese and Dutch sera, respectively51. These factors, as well as general limitations when comparing in vitro to in vivo studies restrict the direct applicability to clinical settings. Nonetheless, the findings provide a clearer understanding of the effects of PUFAs on the monocyte-endothelial interactions and inflammation.
CONCLUSIONS
We conclude that AA increases binding of monocytes to the ECs through a mechanism that is related to an increased expression of ICAM-1, VCAM and E-Selectin, while EPA likely decreases the binding of monocytes to ECs. Equally important is the direct effect of AA in inducing inflammation through an increase in IL-6 and TNF-α, a novel finding. Taken together, these data indicate that PUFAs have the ability to alter monocyte adhesion to ECs, strengthening the evidence of the importance of dietary PUFAs on early events in atherosclerosis.
Acknowledgments
The authors would like to thank Ms. Amy Markowitz (UCSF) for her editorial assistance on the manuscript.
Sources of Funding:
This research was funded by Dr. Grenon’s start-up funds from the University of California San Francisco and the Northern California Institute for Research and Education. The project described was supported by Award Number KL2RR024130 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
SMG, JPH, MSC, MHF designed research; SMG, JPH, JAZ conducted research; CO provided essential materials; SMG, JAZ, JPH, MHF performed data analysis; SMG, MHF, MSC and JAZ wrote the manuscript; CDO and JPH critically reviewed the manuscript; SMG had the primary responsibility for the final content. All authors read and approved the final manuscript. None of the authors have any conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burr ML, Fehily AM, Gilbert JF, et al. Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: diet and reinfarction trial (DART) Lancet. 1989;2:757–61. doi: 10.1016/s0140-6736(89)90828-3. [DOI] [PubMed] [Google Scholar]
- 2.Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet. 1999;354:447–55. [PubMed] [Google Scholar]
- 3.Yokoyama M, Origasa H, Matsuzaki M, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. 2007;369:1090–8. doi: 10.1016/S0140-6736(07)60527-3. [DOI] [PubMed] [Google Scholar]
- 4.Grenon SM, Hughes-Fulford M, Rapp J, Conte MS. Polyunsaturated fatty acids and peripheral artery disease. Vasc Med. 2012;17:51–63. doi: 10.1177/1358863X11429175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marik PE, Varon J. Omega-3 dietary supplements and the risk of cardiovascular events: a systematic review. Clin Cardiol. 2009;32:365–72. doi: 10.1002/clc.20604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Das UN. Essential fatty acids and their metabolites could function as endogenous HMG-CoA reductase and ACE enzyme inhibitors, anti-arrhythmic, anti-hypertensive, anti-atherosclerotic, anti-inflammatory, cytoprotective, and cardioprotective molecules. Lipids Health Dis. 2008;7:37. doi: 10.1186/1476-511X-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramsden CE, Hibbeln JR, Majchrzak SF, Davis JM. n-6 fatty acid-specific and mixed polyunsaturate dietary interventions have different effects on CHD risk: a meta-analysis of randomised controlled trials. Br J Nutr. 2010;104:1586–600. doi: 10.1017/S0007114510004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 9.Hansson GK, Libby P, Schonbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res. 2002;91:281–91. doi: 10.1161/01.res.0000029784.15893.10. [DOI] [PubMed] [Google Scholar]
- 10.Osterud B, Bjorklid E. Role of monocytes in atherogenesis. Physiol Rev. 2003;83:1069–112. doi: 10.1152/physrev.00005.2003. [DOI] [PubMed] [Google Scholar]
- 11.Ringseis R, Eder K. Fatty acids and signalling in endothelial cells. Prostaglandins Leukot Essent Fatty Acids. 2010;82:189–98. doi: 10.1016/j.plefa.2010.02.022. [DOI] [PubMed] [Google Scholar]
- 12.Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci U S A. 1983;80:3734–7. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sundstrom C, Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937) Int J Cancer. 1976;17:565–77. doi: 10.1002/ijc.2910170504. [DOI] [PubMed] [Google Scholar]
- 14.Braut-Boucher F, Pichon J, Rat P, Adolphe M, Aubery M, Font J. A non-isotopic, highly sensitive, fluorimetric, cell-cell adhesion microplate assay using calcein AM-labeled lymphocytes. J Immunol Methods. 1995;178:41–51. doi: 10.1016/0022-1759(94)00239-s. [DOI] [PubMed] [Google Scholar]
- 15.Hatton JP, Pooran M, Li CF, Luzzio C, Hughes-Fulford M. A short pulse of mechanical force induces gene expression and growth in MC3T3-E1 osteoblasts via an ERK 1/2 pathway. J Bone Miner Res. 2003;18:58–66. doi: 10.1359/jbmr.2003.18.1.58. [DOI] [PubMed] [Google Scholar]
- 16.Boonyaratanakornkit JB, Cogoli A, Li CF, et al. Key gravity-sensitive signaling pathways drive T cell activation. Faseb J. 2005;19:2020–2. doi: 10.1096/fj.05-3778fje. [DOI] [PubMed] [Google Scholar]
- 17.Hughes-Fulford M, Sugano E, Schopper T, Li CF, Boonyaratanakornkit JB, Cogoli A. Early immune response and regulation of IL-2 receptor subunits. Cell Signal. 2005;17:1111–24. doi: 10.1016/j.cellsig.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 18.Grenon SMH-FM, Rapp J, Conte MS. Polyunsaturated Fatty Acids and Peripheral Arterial Disease. Vascular Medicine. doi: 10.1177/1358863X11429175. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reissig D, Rassoul F, Salvetter J, Wagner O, Richter V. Effect of fatty acids on expression of endothelial leukocyte adhesion molecules. Eur J Nutr. 2003;42:224–7. doi: 10.1007/s00394-003-0408-4. [DOI] [PubMed] [Google Scholar]
- 20.Holthe MR, Andersson Y, Lyberg T. Lack of proinflammatory effects of free fatty acids on human umbilical cord vein endothelial cells and leukocytes. Acta Obstet Gynecol Scand. 2005;84:672–8. doi: 10.1111/j.0001-6349.2005.00799.x. [DOI] [PubMed] [Google Scholar]
- 21.Dichtl W, Ares MP, Jonson AN, et al. Linoleic acid-stimulated vascular adhesion molecule-1 expression in endothelial cells depends on nuclear factor-kappaB activation. Metabolism. 2002;51:327–33. doi: 10.1053/meta.2002.29963. [DOI] [PubMed] [Google Scholar]
- 22.Toborek M, Lee YW, Garrido R, Kaiser S, Hennig B. Unsaturated fatty acids selectively induce an inflammatory environment in human endothelial cells. Am J Clin Nutr. 2002;75:119–25. doi: 10.1093/ajcn/75.1.119. [DOI] [PubMed] [Google Scholar]
- 23.Reiterer G, Toborek M, Hennig B. Quercetin protects against linoleic acid-induced porcine endothelial cell dysfunction. J Nutr. 2004;134:771–5. doi: 10.1093/jn/134.4.771. [DOI] [PubMed] [Google Scholar]
- 24.Amer M, Bead VR, Bathon J, Blumenthal RS, Edwards DN. Use of nonsteroidal anti-inflammatory drugs in patients with cardiovascular disease: a cautionary tale. Cardiol Rev. 2010;18:204–12. doi: 10.1097/CRD.0b013e3181ce1521. [DOI] [PubMed] [Google Scholar]
- 25.Tull SP, Yates CM, Maskrey BH, et al. Omega-3 Fatty acids and inflammation: novel interactions reveal a new step in neutrophil recruitment. PLoS Biol. 2009;7:e1000177. doi: 10.1371/journal.pbio.1000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dyerberg J, Bang HO, Stoffersen E, Moncada S, Vane JR. Eicosapentaenoic acid and prevention of thrombosis and atherosclerosis? Lancet. 1978;2:117–9. doi: 10.1016/s0140-6736(78)91505-2. [DOI] [PubMed] [Google Scholar]
- 27.Wang L, Lim EJ, Toborek M, Hennig B. The role of fatty acids and caveolin-1 in tumor necrosis factor alpha-induced endothelial cell activation. Metabolism. 2008;57:1328–39. doi: 10.1016/j.metabol.2008.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen W, Esselman WJ, Jump DB, Busik JV. Anti-inflammatory effect of docosahexaenoic acid on cytokine-induced adhesion molecule expression in human retinal vascular endothelial cells. Invest Ophthalmol Vis Sci. 2005;46:4342–7. doi: 10.1167/iovs.05-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goua M, Mulgrew S, Frank J, Rees D, Sneddon AA, Wahle KW. Regulation of adhesion molecule expression in human endothelial and smooth muscle cells by omega-3 fatty acids and conjugated linoleic acids: involvement of the transcription factor NF-kappaB? Prostaglandins Leukot Essent Fatty Acids. 2008;78:33–43. doi: 10.1016/j.plefa.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Sethi S, Ziouzenkova O, Ni H, Wagner DD, Plutzky J, Mayadas TN. Oxidized omega-3 fatty acids in fish oil inhibit leukocyte-endothelial interactions through activation of PPAR alpha. Blood. 2002;100:1340–6. doi: 10.1182/blood-2002-01-0316. [DOI] [PubMed] [Google Scholar]
- 31.Weber C, Erl W, Pietsch A, Danesch U, Weber PC. Docosahexaenoic acid selectively attenuates induction of vascular cell adhesion molecule-1 and subsequent monocytic cell adhesion to human endothelial cells stimulated by tumor necrosis factor-alpha. Arterioscler Thromb Vasc Biol. 1995;15:622–8. doi: 10.1161/01.atv.15.5.622. [DOI] [PubMed] [Google Scholar]
- 32.De Caterina R, Libby P. Control of endothelial leukocyte adhesion molecules by fatty acids. Lipids. 1996;31 (Suppl):S57–63. doi: 10.1007/BF02637052. [DOI] [PubMed] [Google Scholar]
- 33.De Caterina R, Cybulsky MI, Clinton SK, Gimbrone MA, Jr, Libby P. The omega-3 fatty acid docosahexaenoate reduces cytokine-induced expression of proatherogenic and proinflammatory proteins in human endothelial cells. Arterioscler Thromb. 1994;14:1829–36. doi: 10.1161/01.atv.14.11.1829. [DOI] [PubMed] [Google Scholar]
- 34.De Caterina R, Massaro M. Omega-3 fatty acids and the regulation of expression of endothelial pro-atherogenic and pro-inflammatory genes. J Membr Biol. 2005;206:103–16. doi: 10.1007/s00232-005-0783-2. [DOI] [PubMed] [Google Scholar]
- 35.Yamada H, Yoshida M, Nakano Y, et al. In vivo and in vitro inhibition of monocyte adhesion to endothelial cells and endothelial adhesion molecules by eicosapentaenoic acid. Arterioscler Thromb Vasc Biol. 2008;28:2173–9. doi: 10.1161/ATVBAHA.108.171736. [DOI] [PubMed] [Google Scholar]
- 36.Wang TM, Chen CJ, Lee TS, et al. Docosahexaenoic acid attenuates VCAM-1 expression and NF-kappaB activation in TNF-alpha-treated human aortic endothelial cells. J Nutr Biochem. 2011;22:187–94. doi: 10.1016/j.jnutbio.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 37.De Caterina R, Cybulsky MA, Clinton SK, Gimbrone MA, Jr, Libby P. Omega-3 fatty acids and endothelial leukocyte adhesion molecules. Prostaglandins Leukot Essent Fatty Acids. 1995;52:191–5. doi: 10.1016/0952-3278(95)90021-7. [DOI] [PubMed] [Google Scholar]
- 38.Mayer K, Merfels M, Muhly-Reinholz M, et al. Omega-3 fatty acids suppress monocyte adhesion to human endothelial cells: role of endothelial PAF generation. Am J Physiol Heart Circ Physiol. 2002;283:H811–8. doi: 10.1152/ajpheart.00235.2002. [DOI] [PubMed] [Google Scholar]
- 39.Schaefer MB, Wenzel A, Fischer T, et al. Fatty acids differentially influence phosphatidylinositol 3-kinase signal transduction in endothelial cells: impact on adhesion and apoptosis. Atherosclerosis. 2008;197:630–7. doi: 10.1016/j.atherosclerosis.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 40.Thies F, Garry JM, Yaqoob P, et al. Association of n-3 polyunsaturated fatty acids with stability of atherosclerotic plaques: a randomised controlled trial. Lancet. 2003;361:477–85. doi: 10.1016/S0140-6736(03)12468-3. [DOI] [PubMed] [Google Scholar]
- 41.Lichtenstein AH, Appel LJ, Brands M, et al. Diet and lifestyle recommendations revision 2006: a scientific statement from the American Heart Association Nutrition Committee. Circulation. 2006;114:82–96. doi: 10.1161/CIRCULATIONAHA.106.176158. [DOI] [PubMed] [Google Scholar]
- 42.Kris-Etherton PM, Harris WS, Appel LJ. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation. 2002;106:2747–57. doi: 10.1161/01.cir.0000038493.65177.94. [DOI] [PubMed] [Google Scholar]
- 43.Wang C, Harris WS, Chung M, et al. n-3 Fatty acids from fish or fish-oil supplements, but not alpha-linolenic acid, benefit cardiovascular disease outcomes in primary- and secondary-prevention studies: a systematic review. Am J Clin Nutr. 2006;84:5–17. doi: 10.1093/ajcn/84.1.5. [DOI] [PubMed] [Google Scholar]
- 44.Harris WS, Mozaffarian D, Rimm E, et al. Omega-6 fatty acids and risk for cardiovascular disease: a science advisory from the American Heart Association Nutrition Subcommittee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Cardiovascular Nursing; and Council on Epidemiology and Prevention. Circulation. 2009;119:902–7. doi: 10.1161/CIRCULATIONAHA.108.191627. [DOI] [PubMed] [Google Scholar]
- 45.Fritsche KL. Too much linoleic acid promotes inflammation-doesn’t it? Prostaglandins Leukot Essent Fatty Acids. 2008;79:173–5. doi: 10.1016/j.plefa.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 46.Simopoulos AP. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med (Maywood) 2008;233:674–88. doi: 10.3181/0711-MR-311. [DOI] [PubMed] [Google Scholar]
- 47.Kris-Etherton P, Fleming J, Harris WS. The debate about n-6 polyunsaturated fatty acid recommendations for cardiovascular health. J Am Diet Assoc. 2010;110:201–4. doi: 10.1016/j.jada.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 48.Ambring A, Johansson M, Axelsen M, Gan L, Strandvik B, Friberg P. Mediterranean-inspired diet lowers the ratio of serum phospholipid n-6 to n-3 fatty acids, the number of leukocytes and platelets, and vascular endothelial growth factor in healthy subjects. Am J Clin Nutr. 2006;83:575–81. doi: 10.1093/ajcn.83.3.575. [DOI] [PubMed] [Google Scholar]
- 49.Ferrucci L, Cherubini A, Bandinelli S, et al. Relationship of plasma polyunsaturated fatty acids to circulating inflammatory markers. J Clin Endocrinol Metab. 2006;91:439–46. doi: 10.1210/jc.2005-1303. [DOI] [PubMed] [Google Scholar]
- 50.Dwyer JH, Allayee H, Dwyer KM, et al. Arachidonate 5-lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis. N Engl J Med. 2004;350:29–37. doi: 10.1056/NEJMoa025079. [DOI] [PubMed] [Google Scholar]
- 51.Hirai K, Horiuchi R, Ohno Y, Higuchi H, Asano Y. Lower eicosapentaenoic acid and higher arachidonic acid levels in Sera of young adults in the Netherlands than in Japan. Environ Health Prev Med. 2000;5:60–5. doi: 10.1007/BF02932005. [DOI] [PMC free article] [PubMed] [Google Scholar]