

Delineating the relationships between sequence, structure, and function in biopolymers is critical to our understanding of fundamental biochemical interactions. These relationships are equally important for the design of functional biomimetic oligomers (i.e., foldamers).[1] Oligomers of N-substituted glycine, or peptoids (Figure 1a),[2] are an important class of foldamers that have been shown to possess numerous biological functions[3] and could find use in a range of fundamental and applied contexts as bio-inspired materials.[4] Peptoids are highly attractive scaffolds for such purposes, as their non-native backbones are resistant to proteolytic degradation[5] and their straightforward, modular synthesis[6] enables the ready incorporation of a range of structurally diverse amide side chains.[7] However, the design and prediction of peptoid secondary or higher-order structure a priori remains a major challenge, and few discrete structures have been characterized to date for acyclic peptoids.[8] This paucity of structure-function data limits the potential utility of peptoids, despite their many advantages.
Figure 1.

(a) The primary structure of an α-peptoid oligomer (dihedral angles omega (ω), phi (ϕ), psi (ψ), and chi (χ) labelled) and the structures of the peptoid side chains discussed in this study. Side chain abbreviations: s1npe = (S)-1-(1-naphthyl)ethyl; ph = phenyl; 2,6mph = 2,6-dimethylphenyl; 3,5mph = 3,5-dimethylphenyl; 4fph = 4-fluorophenyl. (b) Peptoid cis- and trans-amide rotamers and the isomeric preferences engendered by the two amide side chains investigated in this study in model peptoid monomer systems.
Structural studies of peptoids have been largely thwarted by the intrinsic conformational flexibility of the peptoid backbone itself. This backbone contains C-α methylene units, lacks hydrogen bond donating atoms, and, perhaps most notably, is linked by tertiary amides that can be isoenergetic between cis- and trans-amide geometries (Figure 1b). Our laboratory and others reasoned that the development of peptoid side chains capable of engendering a high degree of control over proximate main chain amide geometries could facilitate the design of well-defined peptoid structures and expand our understanding of peptoid folding.[9] We recently designed and synthesized a range of peptoid model systems to test this hypothesis, and have identified several classes of amide side chains that favor cis- or trans- main chain amides in peptoid oligomers, predominantly through steric and stereoelectronic effects. One such side chain is the α-chiral aromatic (S)-1-(1-naphthyl)ethyl (s1npe) group (Figure 1), which strongly favors cis-amide bonds (cis:trans > 6.3:1) in peptoid model systems.[9c] Moreover, Ns1npe homooligomers adopt polyproline type I (PPI)-like peptoid helices that exclusively contain cis-amides in the peptoid main chain.[8e] In turn, we found that the trans-amide peptoid rotamer is strongly enforced by N-aryl side chains (e.g., Nph, cis:trans < 0.05:1; Figure 1), and homooligomers of these residues have been calculated to give rise to extended, polyproline type II (PPII)-like peptoid helices with all trans-amides.[9b] These peptoid monomers now represent a set of building blocks with which to initiate a rational construction of new peptoid secondary structures. Herein, we report our first test of this modular design strategy for peptoids and our ensuing discovery of a new secondary structure containing an alternating sequence of aromatic residues that we designate as the “peptoid ribbon”.
In view of the strong rotameric preferences enforced in N-aryl and Ns1npe monomers, we hypothesized that linear peptoids containing an alternating sequence of these two residues would adopt a well-defined and novel secondary structure with a regular, alternating pattern of trans- and cis-main chain amides. A recent X-ray crystallographic report of an N-aryl/Ns1npe dimer by Kirshenbaum and co-workers showed that the N-aryl amide bond was trans and that the Ns1npe amide bond was cis,[10] and served to support our design hypothesis. The only known α-peptide sequences that adopt a regular, alternating pattern of trans-/cis-backbone amides are alternating L-/D-polyprolines,[11] but no formal secondary structure designation has been assigned to these peptides. We reasoned that a systematic study of short, alternating Nph/Ns1npe peptoid oligomers of increasing size would allow us to test our design premise.
We began our investigations by evaluating whether the cis- or trans-amide preferences of the Ns1npe and Nph residues in solution were affected by being adjacent to each other in a heteropeptoid sequence, or by being placed at either the N- or C-terminal positions. Two dipeptoids, 2 and 2′ (Table 1), were synthesized by standard solution phase methods (see Supp. Info. for full synthetic details),[9a, 12], purified, and evaluated by NMR spectroscopy. Each peptoid was capped at the N-terminus with an acetyl group (ac) and at the C-terminus as a dimethyl amide (dma) to (1) mimic the chemical environment of an extended peptoid chain at both termini, and (2) facilitate conformational analysis by 1H-1H nuclear Overhauser effect spectroscopy (NOESY). As expected, in CD3CN at 24 °C, the Kcis/trans values for the Ns1npe residues in 2 and 2′ were 8.7 and 10.2, respectively, while the Kcis/trans values for the Nph residues were 0.1 and 0.05, respectively. We note that the overall amide Kcis/trans for homodimers of Ns1npe and Nph in CD3CN were previously shown to be 10.8[8e] and < 0.1,[9b] respectively, suggesting that their presence in heterodimers 2 and 2′ did not impact the other residue’s rotameric preference. The cis- or trans-amide preferences of the Ns1npe and Nph residues in 2 and 2′ were also maintained in a variety of solvents (CDCl3 (Table 1); C6D6, CD3OD, DMSO-d6, and 1:1 CD3CN/D2O; see Table S-1).
Table 1.
Sequences, purities, mass spectrometry data, and overall Ns1npe Kcis/trans values for the peptoids investigated in this study.
| peptoid | Sequence [a] | purity (%) [b] | calc. mass | obs. mass (m/z; [M+Na]+) [c] | Overall Kcis/trans values for Ns1npe residues (in CDCl3) |
|---|---|---|---|---|---|
| 2 | Ac-Nph-Ns1npe-dma | >98 | 431.2 | 454.2 | 5 [d] |
| 2′ | Ac-Ns1npe-Nph-dma | >98 | 431.2 | 454.2 | 5 [d] |
| 3 | Ac-Ns1npe-Nph-Ns1npe-dma | >98 | 642.3 | 665.3 | 10 [e] |
| 4a | Ac-N2,6mph-Ns1npe-Nph-Ns1npe-dma | > 98 | 803.4 | 826.4 | 25 [e] |
| 4b | Ac-N3,5mph-Ns1npe-Nph-Ns1npe-dma | >98 | 803.4 | 826.4 | 19 [e] |
| 5 | Ac-Ns1npe-(Nph-Ns1npe)2-dma | >98 | 986.5 | 1009.5 | 47 [e] |
| 6 | Ac-(Nph-Ns1npe)3-dma | >98 | 1119.5 | 1142.5 | 30 [e] |
| 6i | Ac-N3,5mph-Ns1npe-N2,6mph-ac13C-Ns1npe-N4fph-Ns1npe-dma | >98 | 1195.6 | 1218.6 | 28 [e] |
| 7 | Ac-Ns1npe-(Nph-Ns1npe)3-dma | >98 | 1330.6 | 1353.6 | 48 [e] |
| 8 | Ac-(Nph-Ns1npe)4-dma | >98 | 1463.7 | 1486.7 | 53 [e] |
Ac = acetyl, dma = dimethyl amide; see Figure 1a for monomer abbreviations.
Determined by integration of an HPLC trace with UV detection at 220 nm.
Mass spectrometry data were acquired using high resolution ESI techniques.
Determined by integrating rotamer-related peaks in 1H-NMR spectra (500 MHz, 10 mM).
Determined by integrating rotamer-related peaks of the Ns1npe methine protons in 1H-13C HSQC spectra (700 MHz, peptoids between 4–10 mM). See Supp. Info. for full details of methods.
We next designed a trimer through octamer series of alternating peptoids 3–8 (Table 1) to determine if cis- or trans-amide preferences were maintained in longer sequences. Each was synthesized in solution and purified to homogeneity by manual column chromatography. As an initial solution-phase study of these systems, we conducted heteronuclear single-quantum correlation (HSQC) NMR experiments to determine the overall Kcis/trans value for the Ns1npe residues of each heteropeptoid. We anticipated that the N-aryl residues would adopt nearly exclusively trans-amide conformations,[9b, 9d] and thus, the overall Ns1npe-amide Kcis/trans value would correlate with the degree of overall amide-rotamer homogeneity of a given oligomer. These NMR experiments were straightforward, as the Ns1npe side chain α–methine groups that are oriented in the cis geometry are readily distinguishable from those in the trans geometry by 1H-13C HSQC.[8d] In CDCl3 (~5 mM, 24 °C), we observed an extremely high preference for cis main chain Ns1npe amides in peptoids 3–8 (Kcis/trans = 10–53; Table 1), which, in general, increased as the peptoid chain length increased. This trend correlates with that observed for homo-oligomers of Ns1npe.[8e] Also similar to Ns1npe homo-oligomers, there was a considerable temperature-dependent decrease in the overall Ns1npe amide Kcis/trans values of 8 with increasing temperatures, in both CDCl3 and CD3OD, (see Table S-2), suggesting an inherent entropic pressure for multiple rotameric conformers.
Tetramers 4a and 4b (Table 1) were specifically engineered as models for more detailed conformational analysis by NMR. Spectra for the tetramers were well dispersed, with each residue clearly distinguishable and one conformer predominating. We envisioned that upon adopting a discrete secondary structure with alternating cis- and trans-amide bonds, the i and i+3 side chains of 4a and 4b could be in close proximity, facilitating the observation of NOEs between substituents on an N-aryl side chain at the i position and protons of an Ns1npe side chain at the i+3 position. Our hypothesis proved correct, and Figure 2a depicts the NOEs that we detected between the dimethyl group on the i (N-aryl) residues and the i+3 (Ns1npe) methine and methyl protons for 4a and 4b in CDCl3 (10 mM, 24 °C; see Figures S-6 and S-7 for additional analysis).
Figure 2.

(a) The structures of peptoid tetramers 4a and 4b; side chain–side chain NOEs are indicated with blue arrows. (b) The structure of hexamer 6i. (c) An ensemble of 10 superimposed, low energy NMR-determined structures of 6i (hydrogen atoms are omitted for clarity) in CDCl3. (d) A length-wise view of the main chain atoms of one population of low energy structures of 6i. (e) An axial view down the N-terminus of 6i showing the left-handed spiral of the ribbon conformation.
The NMR data for 4a and 4b inspired our design of hexamer 6i (Figure 2b), with the goal of determining its solution-phase structure by NMR. We incorporated the 13C-labeled main chain acetyl group into the third residue of 6i for assignment purposes and the fluoro-substituted N4fph residue to provide dispersion of resonance peaks. NMR spectra of hexamer 6i were recorded in CDCl3 (10 mM, 15 °C). One major conformer was observed in all NMR experiments and the resonances were well-dispersed, enabling the unambiguous assignment of main chain methylenes, side chain s1npe methines, and all methyl groups (see Figure S-9 and Table S-3). Rotating frame Overhauser effect spectroscopy (ROESY) cross peaks were observed between i and i+3 side chains (residues 1↔4 and 2↔5), analogous to the NOEs observed in 4a and 4b. Unfortunately, ROE cross peaks between main chain methylenes were not observed. However, the HSQC data described above provided evidence that each Ns1npe amide in 6i was primarily in the cis-configuration. In total, nine distance restraints and three main chain ω torsional restraints from our experimental NMR data were included in simulated annealing molecular dynamic calculations using AMBER11 software (see Supp. Info. for full details)[13] to determine an ensemble of calculated solution-phase structures for hexamer 6i.
The alignment of the 10 lowest energy structures for 6i (out of 150 calculations) is shown in Figure 2c. These structures revealed a uniquely folded peptoid backbone of repeating turn units that resembled a ribbon-type structure. Two populations of low energy ribbon conformations were observed, with variation mainly at the C-terminus. In one population, the C-termini were extended in a continuation of the ribbon structure (Figure 2d; ensemble RMSD = 1.0 Å), while the C-termini of the second group of structures were curled inwards (ensemble RMSD = 1.1 Å; Figure S-10). Not including the C-termini, the mean ϕ and ψ values for the Ns1npe residues in 6i were −55° and 161°, respectively, and the mean ϕ and ψ values for the Naryl residues were 64° and −162°, respectively (see Table S-4 for a full listing of ensemble-averaged main chain dihedral angles). These values lie closest to the energetic minima calculated for peptoids in the αD configuration (ϕ, ψ = ± 90°, 180°), as defined by Moehle and Hofmann.[14] We also noted that the structure of 6i clearly adopts a left-handed helical twist (Figure 2e). This characteristic has been observed in α-peptide ribbon structures, which are considered to be subtypes of helical peptide conformations (β-bend → 310-helix).[15] We calculated the helical rotation between two hypothetical planes running through the two turn units in the hexamer ribbon. These planes were twisted by 34°; therefore, it would take 10.6 turn units (i.e., i–i+3 sets of residues) to complete one full helical turn of the ribbon. Presumably, the origin of the left-handed spiral in 6i is the chirality of the s1npe side chains.
Three heteropeptoids were crystalline upon slow evaporation from 1-propanol, and were further analyzed by X-ray crystallography. Solid state structures for the synthetic intermediate Br-Nph-Ns1npe-dma (representative of a peptoid dimer), trimer 3, and tetramer 4a are shown in Figure 3a, and allow one to view the progressive generation of the ribbon secondary structure as each residue is added. Tetramer 4a is the shortest peptoid that is capable of adopting one full unit of the ribbon structure, and the reverse turn of the backbone approximates the i and i+2 main chain methylene carbons of residues 1 and 3 (C•••C distance = 4.97 Å, Figure 3b). Both 3 and 4a adopted nearly identical conformations in the solid state, despite having different crystal packing interactions (see Figures S-16 and S-18), which indicates that an alternating sequence of Ns1npe and N-aryl residues can adopt a well-defined peptoid ribbon secondary structure even at short chain lengths. As expected, the amide geometries of all Ns1npe residues were cis and all N-aryl residues were trans, and the ϕ and ψ angles of these solid state structures were in close agreement with the corresponding values determined for the NMR structure of hexamer 6i (see Table S-4 for a full listing of angles). This agreement is significant, as it constitutes the first corroboration of a peptoid NMR solution structure with peptoid solid-state structures containing the same primary sequences. The similarity is well illustrated by comparison of the solid state data and NMR data for 4a. The X-ray crystal structure for 4a shows that the methine proton of the i+3 Ns1npe side chain is oriented directly towards a methyl group on the side chain of the N2,6mph (i) residue (C•••C distance = 5.05 Å, Figure 3a). Accordingly, an NOE between the protons of these same groups was observed in our analysis of 4a in CDCl3 (see above).
Figure 3.
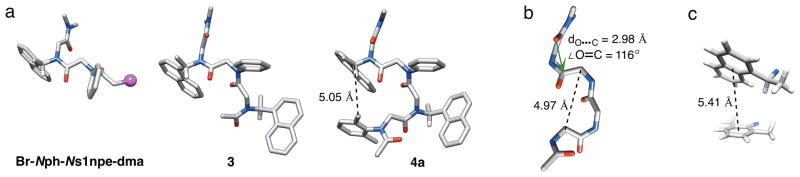
(a) X-ray crystal structures of Br-Nph-Ns1npe-dma, 3, and 4a. All hydrogen atoms except for the s1npe side chain methines have been omitted for clarity. The black dashed line indicates a side chain – side chain C ••• C distance. (b) The main chain atoms of the X-ray crystal structure of 4a illustrate the reverse turn and the green arrow depicts an n→π*C=O interaction. The black dashed line indicates a main chain Cα•••Cα distance. (c) A displaced aromatic-aromatic stacking interaction was detected between the side chains of residues 1 and 4 in the X-ray crystal structure of 4a. Only the side chain and nitrogen atoms are shown. The black dashed line indicates the centroid to centroid distance between the two aryl rings.
The solution phase and crystal structures were further analyzed for potential noncovalent interactions that may stabilize the peptoid ribbon conformation, which lacks a hydrogen bonding network. Interestingly, in all structures, every other backbone carbonyl was oriented perpendicular to one other, but the overall dipole was pointed towards the N-terminus. At the C-terminus of 4a, we detected an n→π*C=O interaction (Figure 3b).[16] Previous X-ray crystallographic studies in our laboratory have revealed that C=Oi−1•••C′i=O interactions can exist in peptoid monomers and at the N-termini of a peptoid oligomer,[9a, 9d] yet this is, to our knowledge, the first report of an C=Oi+1•••C′i=O interaction in peptoids. We analyzed all sequential backbone carbonyls in the structures of 3, 4a, and 6i for n→π*C=O interactions, and although the distances between the main chain carbonyl oxygens and the preceding or subsequent residue’s carbonyl carbon atoms were under 3.2 Å (as is typical for the n→π* interaction[16]), the angle of approach was outside of the 109° ± 10° window necessary for sufficient orbital overlap in all but the C-terminal residue of 4a.[17] We therefore surmise that n→π*C=O interactions do not play a major role in enforcing the ribbon conformation in these peptoids.
We also evaluated the structures of 4a and 6i for intramolecular aromatic–aromatic interactions between side chains by measuring the angles between the aryl planes, the distances between the aryl centroids, and the distances between nearest interresidue atoms.[18] In the X-ray crystal structure of 4a, the i and i+3 side chains are positioned in an oblique orientation of a displaced stacking interaction (angle between aromatic planes = 33.5°) with a centroid-centroid distance of 5.4 Å and nearest interresidue C–C distance of 4.6 Å (Figures 3c and S-11a). Correspondingly, the ensemble of NMR structures of 6i also revealed displaced aromatic stacking interactions between the i and i+3 residues at the beginning and end of each turn unit (see Figures S-11b–d). Elucidating the contribution, if any, of these aromatic-aromatic interactions to the thermodynamic stability of the peptoid ribbon is an important avenue for future study, and is the subject of ongoing investigations in our laboratory.
We next used circular dichroism (CD) spectroscopy to study the effects of peptoid length, solvent. and temperature on the ribbon structure. The CD spectra for all of the peptoid oligomers had nearly identical spectral features in acetonitrile, 1:1 acetonitrile/water, and methanol (negative maxima of ellipticity at 224–226 nm and broad positive peaks of ellipticity between 197–205 nm; Figures 4a–c), indicating that all of the peptoids in this series adopted similar secondary structures in both organic and polar protic solvents (at 30 μM, 20 °C). Additionally, CD data for peptoid 8 at varying concentrations (7–80 μM) in acetonitrile suggested that intermolecular interactions were not affecting the observed CD signal (see Figure S-19). Interestingly, the CD spectra of the peptoid ribbon most closely resembled data reported for poly-LD-Pro sequences,[19] which also adopt an alternating cis-/trans-amide backbone pattern. Beyond the dimer chain length, there were no length-dependent changes in the CD spectral intensities. Instead, in all solvents, we observed a difference in signal intensity between even and odd numbered chain lengths. This effect could potentially be due to the absorption properties of the s1npe side chains, which are more solvent-exposed in the ribbon as the C-terminal residue in odd numbered chain lengths.
Figure 4.

CD spectra of (a) all peptoids in acetonitrile, (b) selected peptoids in a 1:1 acetonitrile/water mixture, and (c) selected peptoids in methanol. All spectra were obtained at 30 μM and 20 °C.
The CD spectral shape for peptoids 2–8 varied slightly in the different solvents. In the acetonitrile/water mixture, shoulders were apparent in all CD traces from 227–230 nm (Figure 4b), and the shoulders were more pronounced in methanol (Figure 4c). These observations could indicate a slight change in overall conformation that is solvent-dependent, or a change in the UV-adsorption properties or orientations of the aromatic side chains in these solvents. Lastly, the thermal stability of 8 (30 μM) was examined in acetonitrile (15–75 °C), 1:1 acetonitrile/water (10–75 °C), and methanol (10–65 °C) (see Figures S-20–22). A linear decrease in signal intensity was observed with increasing temperatures; however, the spectral shape of 8 in all solvents was maintained throughout the temperature ranges investigated. This temperature destabilization can be attributed, in part, to the increase in amide bond isomerization at elevated temperatures that we observed in 1H-13C HSQC experiments for 8 (see above).
In summary, we report a new peptoid secondary structure comprised of a regular alternating sequence of Ns1npe and N-aryl residues, which we term the peptoid “ribbon”. This structure is a result of our first rational design of discretely folded peptoids using monomers with defined amide-rotamer preferences. Systematic NMR, X-ray crystallographic, and CD studies of a series of Ns1npe/Nph heteropeptiods revealed that the ribbon is stable at short chain lengths and in both protic and aprotic solvents. Perhaps most notably, we present corroborating solution-phase NMR and solid-state structures for ribbons with the same primary sequence – a level of structural characterization yet to be achieved for other peptoid secondary structures. The peptoid ribbon can be described as a succession of turn units that is similar in appearance to known LD-ribbon[15] and β-bend ribbon[20] folds in α-peptides. However, in the latter ribbon structures, all main chain amide bonds are in the trans configuration. The alternating cis-/trans- geometry of the main chain amides and the preclusion of hydrogen bonds within the peptoid backbone gives rise to a secondary structure that is, to the best of our knowledge, wholly unique to these peptoids. The steric demands of the bulky, chiral s1npe side chain undoubtedly play a dominant role in facilitating the ribbon conformation and enforcing the left-handed spiral of the peptoid ribbon. Small, model systems have proven powerful tools for understanding the processes by which foldamers and natural biopolymers fold.[1a, 1b, 21] The discovery of a peptoid ribbon secondary structure underscores the value of such model systems for peptoid design. Looking forward, we note that several classes of α-peptide ribbons are known to act as potent and selective antibiotics[22] and cell membrane-modifying agents.[23] Helical and cyclic peptoids have previously been investigated for similar functions,[24] and the peptoid ribbon could now be considered for such applications, among others.
Experimental Section
Full details of peptoid syntheses and supplemental NMR, computational, X-ray crystallographic, and CD characterization data, as well as the full citations for references 2 and 13, can be found in the Supporting Information.
Supplementary Material
Footnotes
Financial support from the NSF (CHE-0449959), ONR (N000140710255), Greater Milwaukee Foundation, and Burroughs Welcome Fund is gratefully acknowledged. Chemistry NMR facilities at UW–Madison are supported by the NIH (1 S10 RR13866-01 and 1 S10 RR08389-01) and the NSF (CHE-0342998 and CHE-9629688). The National Magnetic Resonance Facility at UW-Madison is supported in part by NIH grants P41RR02301 (BRTP/NCRR) and P41GM66326 (NIGMS). We thank Dr. Charles Fry for assistance with NMR spectroscopy, Dr. Milo Westler for assistance with AMBER calculations, Prof. Samuel Gellman for thoughtful discussions and the use of his laboratory’s CD spectrometer, and Dr. Joseph Stringer for experimental guidance.
Supporting information for this article is available on the WWW under http://www.angewandte.org
Contributor Information
Dr. Ilia A. Guzei, Department of Chemistry University of Wisconsin–Madison 1101 University Avenue Madison, WI 53706-1322, USA
Prof. Dr. Helen E. Blackwell, Email: blackwell@chem.wisc.edu, Department of Chemistry University of Wisconsin–Madison 1101 University Avenue Madison, WI 53706-1322, USA
References
- 1.a) Gellman SH. Acc Chem Res. 1998;31:173–180. [Google Scholar]; b) Goodman CM, Choi S, Shandler S, DeGrado WF. Nat Chem Biol. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Fowler SA, Blackwell HE. Org Biomol Chem. 2009;7:1508–1524. doi: 10.1039/b817980h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simon RJ, et al. Proc Natl Acad Sci U S A. 1992;89:9367–9371. doi: 10.1073/pnas.89.20.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Zuckermann RN, Kodadek T. Curr Opin Mol Ther. 2009;11:299–307. [PubMed] [Google Scholar]; b) Horne WS. Expert Opin Drug Discov. 2011;6:1247–1262. doi: 10.1517/17460441.2011.632002. [DOI] [PubMed] [Google Scholar]; c) Kesavan V, Tamilarasu N, Cao H, Rana TM. Bioconjugate Chem. 2002;13:1171–1175. doi: 10.1021/bc0255642. [DOI] [PubMed] [Google Scholar]; d) Simpson LS, Burdine L, Dutta AK, Feranchak AP, Kodadek T. J Am Chem Soc. 2009;131:5760–5762. doi: 10.1021/ja900852k. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lee J, Udugamasooriya DG, Lim HS, Kodadek T. Nat Chem Biol. 2010;6:258–260. doi: 10.1038/nchembio.333. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Reddy MM, Wilson R, Wilson J, Connell S, Gocke A, Hynan L, German D, Kodadek T. Cell. 2011;144:132–142. doi: 10.1016/j.cell.2010.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Chen X, Wu J, Luo Y, Liang X, Supnet C, Kim MW, Lotz GP, Yang G, Muchowski PJ, Kodadek T, Bezprozvanny I. Chem Biol. 2011;18:1113–1125. doi: 10.1016/j.chembiol.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Murnen HK, Rosales AM, Jaworski JN, Segalman RA, Zuckermann RN. J Am Chem Soc. 2010;132:16112–16119. doi: 10.1021/ja106340f. [DOI] [PubMed] [Google Scholar]; b) Nam KT, Shelby SA, Choi PH, Marciel AB, Chen R, Tan L, Chu TK, Mesch RA, Lee BC, Connolly MD, Kisielowski C, Zuckermann RN. Nat Mater. 2010;9:454–460. doi: 10.1038/nmat2742. [DOI] [PubMed] [Google Scholar]; c) Guo L, Zhang D. J Am Chem Soc. 2009;131:18072–18074. doi: 10.1021/ja907380d. [DOI] [PubMed] [Google Scholar]; d) Fetsch C, Grossmann A, Holz L, Nawroth JF, Luxenhofer R. Macromolecules. 2011;44:6746–6758. [Google Scholar]
- 5.a) Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Bioorg Med Chem Lett. 1994;4:2657–2662. [Google Scholar]; b) Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Drug Dev Res. 1995;35:20–32. [Google Scholar]
- 6.a) Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J Am Chem Soc. 1992;114:10646–10647. [Google Scholar]; b) Gorske BC, Jewell SA, Guerard EJ, Blackwell HE. Org Lett. 2005;7:1521–1524. doi: 10.1021/ol0502984. [DOI] [PubMed] [Google Scholar]
- 7.a) Yoo B, Kirshenbaum K. Curr Opin Chem Biol. 2008;12:714–721. doi: 10.1016/j.cbpa.2008.08.015. [DOI] [PubMed] [Google Scholar]; b) Culf AS, Ouellette RJ. Molecules. 2010;15:5282–5335. doi: 10.3390/molecules15085282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Armand P, Kirshenbaum K, Falicov A, Dunbrack RL, Jr, Dill KA, Zuckermann RN, Cohen FE. Folding Des. 1997;2:369–375. doi: 10.1016/S1359-0278(97)00051-5. [DOI] [PubMed] [Google Scholar]; b) Armand P, Kirshenbaum K, Goldsmith RA, Farr-Jones S, Barron AE, Truong KTV, Dill KA, Mierke DF, Cohen FE, Zuckermann RN, Bradley EK. Proc Natl Acad Sci U S A. 1998;95:4309–4314. doi: 10.1073/pnas.95.8.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wu CW, Kirshenbaum K, Sanborn TJ, Patch JA, Huang K, Dill KA, Zuckermann RN, Barron AE. J Am Chem Soc. 2003;125:13525–13530. doi: 10.1021/ja037540r. [DOI] [PubMed] [Google Scholar]; d) Huang K, Wu CW, Sanborn TJ, Patch JA, Kirshenbaum K, Zuckermann RN, Barron AE, Radhakrishnan I. J Am Chem Soc. 2006;128:1733–1738. doi: 10.1021/ja0574318. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Stringer JR, Crapster JA, Guzei IA, Blackwell HE. J Am Chem Soc. 2011;133:15559–15567. doi: 10.1021/ja204755p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Gorske BC, Bastian BL, Geske GD, Blackwell HE. J Am Chem Soc. 2007;129:8928–8929. doi: 10.1021/ja071310l. [DOI] [PubMed] [Google Scholar]; b) Shah NH, Butterfoss GL, Nguyen K, Yoo B, Bonneau R, Rabenstein DL, Kirshenbaum K. J Am Chem Soc. 2008;130:16622–16632. doi: 10.1021/ja804580n. [DOI] [PubMed] [Google Scholar]; c) Gorske BC, Stringer JR, Bastian BL, Fowler SA, Blackwell HE. J Am Chem Soc. 2009;131:16555–16567. doi: 10.1021/ja907184g. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Stringer JR, Crapster JA, Guzei IA, Blackwell HE. J Org Chem. 2010;75:6068–6078. doi: 10.1021/jo101075a. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Crapster JA, Stringer JR, Guzei IA, Blackwell HE. Peptide Sci. 2011;96:604–614. doi: 10.1002/bip.21599. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Jordan PA, Paul B, Butterfoss GL, Renfrew PD, Bonneau R, Kirshenbaum K. Biopolymers. 2011;96:617–626. doi: 10.1002/bip.21675. [DOI] [PubMed] [Google Scholar]; g) Caumes C, Roy O, Faure S, Taillefumier C. J Am Chem Soc. 2012;134:9553–9556. doi: 10.1021/ja302342h. [DOI] [PubMed] [Google Scholar]
- 10.Paul B, Butterfoss GL, Boswell MG, Huang ML, Bonneau R, Wolf C, Kirshenbaum K. Org Lett. 2012;14:926–929. doi: 10.1021/ol203452f. [DOI] [PubMed] [Google Scholar]
- 11.a) Benedetti E, Bavoso A, Diblasio B, Pavone V, Pedone C, Toniolo C, Bonora GM. Biopolymers. 1983;22:305–317. [Google Scholar]; b) Colapietro M, Desantis P, Palleschi A, Spagna R. Biopolymers. 1986;25:2227–2236. doi: 10.1002/bip.360251202. [DOI] [PubMed] [Google Scholar]
- 12.Hjelmgaard T, Faure S, Caumes C, De Santis E, Edwards AA, Taillefumier C. Org Lett. 2009;11:4100–4103. doi: 10.1021/ol9015767. [DOI] [PubMed] [Google Scholar]
- 13.Case DA, et al. AMBER 11. University of California; San Francisco: 2010. [Google Scholar]
- 14.Moehle K, Hofmann HJ. Biopolymers. 1996;38:781–790. doi: 10.1002/(SICI)1097-0282(199606)38:6%3C781::AID-BIP9%3E3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 15.Chandrasekaran R, Prasad BVV. Crit Rev Biochem Mol Biol. 1978;5:125–161. doi: 10.3109/10409237809177142. [DOI] [PubMed] [Google Scholar]
- 16.Choudhary A, Gandla D, Krow GR, Raines RT. J Am Chem Soc. 2009;131:7244–7246. doi: 10.1021/ja901188y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Burgi HB, Dunitz JD, Shefter E. J Am Chem Soc. 1973;95:5065–5067. [Google Scholar]; b) Burgi HB, Dunitz JD, Lehn JM, Wipff G. Tetrahedron. 1974;30:1563–1572. [Google Scholar]
- 18.a) Blundell T, Singh J, Thornton J, Burley SK, Petsko GA. Science. 1986;234:1005. [Google Scholar]; b) Brocchieri L, Karlin S. Proc Natl Acad Sci U S A. 1994;91:9297–9301. doi: 10.1073/pnas.91.20.9297. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McGaughey GB, Gagné M, Rappé AK. J Biol Chem. 1998;273:15458–15463. doi: 10.1074/jbc.273.25.15458. [DOI] [PubMed] [Google Scholar]
- 19.Mastle W, Dukor RK, Yoder G, Keiderling TA. Biopolymers. 1995;36:623–631. doi: 10.1002/bip.360360508. [DOI] [PubMed] [Google Scholar]
- 20.a) Venkatachalapathi YV, Balaram P. Biopolymers. 1981;20:1137–1145. [Google Scholar]; b) Karle IL, Flippenanderson J, Sukumar M, Balaram P. Proc Natl Acad Sci U S A. 1987;84:5087–5091. doi: 10.1073/pnas.84.15.5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dill KA. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 22.Argoudelis AD, Dietz A, Johnson LE. J Antibiot. 1974;27:321–328. doi: 10.7164/antibiotics.27.321. [DOI] [PubMed] [Google Scholar]
- 23.Desantis P, Palleschi A, Savino M, Scipioni A, Sesta B, Verdini A. Biophys Chem. 1985;21:211–215. doi: 10.1016/0301-4622(85)80008-9. [DOI] [PubMed] [Google Scholar]
- 24.a) Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. Proc Natl Acad Sci U S A. 2008;105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang ML, Shin SBY, Benson MA, Torres VJ, Kirshenbaum K. Chem Med Chem. 2012;7:114–122. doi: 10.1002/cmdc.201100358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.