

Abstract
Intracellular protein transport and localization to subcellular regions are processes necessary for normal protein function. Fluorescent proteins can be fused to proteins of interest to track movement and determine localization within a cell. Currently, fluorescence microscopy combined with image processing is most often used to study protein movement and subcellular localization. In this contribution we evaluate a high-throughput time-resolved flow cytometry approach to correlate intracellular localization of human LC3 protein with the fluorescence lifetime of enhanced green fluorescent protein (EGFP). Subcellular LC3 localization to autophagosomes is a marker of the cellular process called autophagy. In breast cancer cells expressing native EGFP and EGFP-LC3 fusion proteins, we measured the fluorescence intensity and lifetime of (i) diffuse EGFP (ii) punctate EGFP-LC3 and (iii) diffuse EGFP-ΔLC3 after amino acid starvation to induce autophagy-dependent LC3 localization. We verify EGFP-LC3 localization with low-throughput confocal microscopy and compare to fluorescence intensity measured by standard flow cytometry. Our results demonstrate that time-resolved flow cytometry can be correlated to subcellular localization of EGFP fusion proteins by measuring changes in fluorescence lifetime.
OCIS codes: (260.2510) Fluorescence; (170.6920) Time-resolved imaging; (140.3518) Lasers, frequency modulated; (170.1530) Cell analysis
1. Introduction
The localization of proteins to subcellular regions is closely related to protein function, and aberrant protein localization is a hallmark of many disease states such as cancer [1]. In fact, locating the subcellular regions where proteins assemble is common practice among the massive field of fluorescence microscopy, immunofluorescence imaging, and optical spectroscopy in studies aimed at understanding protein function and cell regulation.
With fluorescence microscopy, the subcellular localization of a fluorescently labeled or fused target protein can be determined (i.e. association and disassociation with other proteins, presence in specific organelles, etc.). Using such methods has led to a more complete understanding with regard to the relationship between protein localization and function. Therefore many optical platforms that can detect subcellular localization of proteins (mainly microscopy) are essential for diagnostics, and basic research with cells and tissues. One drawback to microscopy-based platforms is the inability to rapidly evaluate the location of proteins, cell-to-cell, for extremely large populations (>10,000) of cells while retaining high-resolution and high-contrast appropriate for subcellular quantitation. Confocal and multi-photon approaches can provide moderate throughput by slight microfluidic design modifications and image processing automation [2–5]. Alternatively, imaging flow cytometry systems have higher throughput but suffer from lower contrast and resolution [6–8].
In this report, we demonstrate a way in which the localization of enhanced green fluorescent protein (EGFP)-LC3 fusion protein can be correlated to changes in fluorescence lifetime at the throughput of a flow cytometer while remaining independent of protein concentration. That is, the fluorescence lifetime of EGFP is the cytometric parameter measured and correlated to changes in the subcellular localization of EGFP-LC3 fusion protein to the autophagosome during autophagy.
The fluorescence lifetime (denoted most often by the Greek symbol tau, τ) of a fluorophore is recognized as the average time that a molecule spends in its excited state before radiatively decaying back to its ground state whereby photons of a lower energy are emitted. The rate of fluorescence decay is unique among fluorophores but also depends on the biochemical environment surrounding the fluorescent species. For example, the subcellular location of a fluorophore, the pH surrounding it, fluctuations in temperature, and the presence of other bulky species can alter fluorescence decay kinetics (see Reference [9] for a comprehensive overview).
Fluorescence lifetime has been developed and introduced in flow cytometry using different platforms [10–13], with more recent advancement in digital frequency-domain techniques [14]. Frequency-domain theory involves modulating the laser excitation source at a continuous radio frequency followed by a measurement of fluorescence lifetime by capturing a time-delayed characteristic of the emission signal. That is, the emitted light is modulated at the same frequency, however owing to the fluorescence decay kinetics is attenuated and shifted in time relative to the excitation signal. The shift is captured by the difference in phase angles of the modulated emission and excitation signals (in this case, light scatter). Using a flow cytometer, the fluorescence lifetime can be quantified in real time by exploiting a homodyne mixing approach with analog devices or by purely measuring digitally modulated waveforms and performing Fourier analysis on the signals digitally. The relationship between the phase shift, ϕ, and the fluorescence lifetime, τ, is shown in Eq. (1) where ω is the angular modulation frequency. This equation represents the average fluorescence lifetime when assuming single exponential decay kinetics. Under this assumption the average times over which a fluorophore will become excited and decay (as it is a probabilistic function) is equivalent to the fluorescence lifetime. In some cases other functions have emerged showing that the probability of fluorescence decay for a given molecule could follow other forms (i.e. power-law, multi-exponential, stretched exponential). Other forms and analysis methods such as lifetime moment analysis (LiMA) can predict different fluorescence lifetimes as opposed to simply one average value [15–17]. Owing to the single-frequency methodology with the flow cytometry system (described later) we report average fluorescence lifetimes dependent on the phase angle.
| (1) |
In this study we find that the fluorescence lifetime of EGFP when fused to LC3 is altered by changes in subcellular localization of the protein. This is measured using a single modulation frequency time-resolved flow cytometer. By corroborating with confocal microscopy, our results demonstrate that fluorescence lifetime can be a sensor of protein localization.
2. Background
Autophagy is a normal cellular process that is associated with various cell stresses. This process can be induced by cell starvation, hypoxic conditions, or DNA damage. The LC3 protein is a critical component of the process of autophagy and the localization of LC3 to autophagosomes, specific organelles in a cell that form during autophagy, is a standard biomarker in autophagic cells. Under normal conditions, a basal level of autophagy occurs in cells, but autophagy induction by amino acid starvation converts the soluble form of LC3 to an autophagosome-associated, membrane-bound LC3 [18]. The formation of autophagosomes consists of several steps, and LC3 plays a structural role in the final step of the process. Autophagosomes enwrap parts of the cytosol and various organelles such as mitochondria, endoplasmic reticulum and peroxisomes. Additionally, during the maturation process, lysosomes bind to autophagosomes in order to form an autolysosome that can degrade old and damaged organelles by hydrolyses. Thus, autophagy acts as a source of energy during starvation conditions [19].
Current methods to quantify autophagy rely on digital imaging and fluorescent microscopy to measure the accumulation of LC3 in autophagosomes using EGFP-LC3 fusion proteins [20, 21]. In addition to microscopy-based methods, flow cytometric assays for measuring autophagy are possible with the EGFP-LC3 fusion protein but rely on a decrease in fluorescence intensity values as an indicator of autophagy induction. Yet both approaches present challenges because it is difficult to discriminate between autophagy-associated EGFP-LC3 punctae and background EGFP-LC3 aggregates. Moreover, the EGFP-LC3 fusion protein can be partially degraded after autolysosome formation, resulting in the accumulation of free EGFP, and this may complicate measurements [22, 23]. Ultimately, overall autophagic activity and autophagic flux are difficult to assess by measuring fluorescence intensity alone.
We hypothesized that the fluorescence lifetime of EGFP-LC3 localized diffusely in a cell or autophagosome-associated punctate distribution could be resolved using a flow cytometer capable of time-resolved measurements of fluorescence lifetime.
3. Materials and methods
3.1 Cell culture, transfection, and autophagy induction
MCF-7 human breast adenocarcinoma cells were cultured in Dulbecco’s modified Eagle medium (Life Technologies Corp., Carlsbad, CA) supplemented with 10% FBS in 5% CO2 at 37°C in a humidified incubator. Three plasmid constructs were transfected into MCF-7 cells at 95% confluence using Lipofectamine 2000 reagent (Life Technologies Corp., Carlsbad, CA) according to the manufacturer’s protocol. The plasmid constructs used in our experiments were obtained from Addgene (Cambridge, MA). pcDNA-EGFP, native EGFP; EGFP-LC3 (pEX-EGFP-hLC3B), EGFP fused to the human LC3 protein; and EGFP-ΔLC3 (pEX-EGFP-mhLC3B G120A, a mutant form of LC3 that does not localize to autophagosomes when cells are starved) [24, 25]. Autophagy was induced in transfected MCF-7 cells by 3 hour amino acid starvation using Hank’s Balanced Salt Solution (HBSS) (Life Technologies Corp., Carlsbad, CA).
3.2 Microscopy
MCF-7 cells were prepared for confocal microscopy by culturing on 35 mm non-coated cover slips (Fluorodish Cell Culture Dish, World Precision Instruments, Sarasota, FL) followed by fixation with 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MO) in PBS at room temperature for 15 min followed by washing two times. Cells were stained with DAPI (4',6-Diamidino-2-phenylindole) dihydrochloride (300 nM in PBS) (Life Technologies Corp., Carlsbad, CA) to detect the DNA at 37°C for 5 minutes and used for verifying the presence of cells in the microscope field. Confocal images (Leica TCS SP5 laser scanning confocal microscope, 1024 × 1024 pixel imaging, Leica Microsystems) were obtained using a 63X oil immersion objective and samples were excited with 405 nm or 488 nm lasers. Fluorescence emission was collected at 440-490 nm to detect DAPI staining and at 500-560 nm to detect the presence of EGFP. The photomultiplier tube (PMT) voltage in the detection channel was fixed (400 V), ensuring consistent sensitivity, no saturation, and verification that pixel intensities were in the linear range of the detector.
3.3 Flow cytometry
All samples were analyzed using (1) a standard BD Accuri C6 Flow Cytometer (Becton, Dickinson and Company); and (2) a laboratory constructed time-resolved flow cytometer (TRFC). For analysis, cells were harvested from culture and centrifuged at 300 x g for 5 min then re-suspended in PBS followed by fixation with 4% paraformaldehyde at room temperature for 15 min. The BD Accuri C6 Flow Cytometer was used for standard cytometric measurements using 488 nm laser for excitation and measuring emission at 530/30 nm to verify the presence of EGFP in the cells.
The TRFC is illustrated schematically in Fig. 1 ; this approach adds a single average fluorescence lifetime parameter to complement the other intensity-based list mode data that a standard cytometer provides. It is based on a homodyne mixing approach that enables real-time extraction of the phase-shifted fluorescence signal, and is discussed in greater detail elsewhere [11, 13, 26].
Fig. 1.

Schematic of the time-resolved flow cytometry (TRFC) system. A modulated laser excites individual cells that are flowing in single file (direction is out of the page in the image). Fluorescence is collected (side scatter also, but not illustrated for brevity) whereby the PMT measures the phase-shifted frequency modulated signal. For fluorescence lifetime acquisition analog mixing hardware permit the measurement of the tangent of the phase-shift (proportional to lifetime), which is collected by a high-speed data acquisition system.
Briefly, a 488 nm solid-state laser diode (Vortran® Stradus Sacramento, CA) was digitally modulated at 6 MHz via direct digital modulation with an arbitrary function generator (Tektronix® AFG-3102, Beaverton, OR). The laser beam was elliptically shaped by two crossed cylindrical lenses (focal lengths: 30 and 5.4 cm) and then focused onto an inverted flow chamber (cross section 200 μm2: Beckman-Coulter, Inc., Biosense model). Cells were introduced by syringe pump (New Era Pump Systems Inc., model NE-1000, Farmindale, NY) into the flow chamber, hydrodynamically focused, and passed through the laser beam at typical flow rate of 10 μL/min. Cytometric pulses from the cytometer were read at 90° to the excitation direction via side scatter (SSC) and fluorescence (FL) photomultiplier tube (PMT; Hamamatsu, Inc., model R636-10) channels. The signals from the PMTs were amplified using high bandwidth preamplifiers. The SSC channel was filtered with a band-pass filter (488/10 nm) at the laser line to remove the fluorescence contributions. The FL channel was filtered with a long pass filter, with a wavelength cut dependent on the fluorescence emission (490 nm sharp cut off long pass filter). The final processed signals were then routed directly into a digital data acquisition system [27] with a clock speed of 50 MSPS (mega samples per second).
In order to accomplish homodyning, two general mixing steps are initiated downstream of the photomultiplier tube signal output. That is, the modulated fluorescence signal is mixed with two phase-shifted reference signals, where the reference signal is phase-locked to the signal driving the laser modulation. Equation (2) is the mathematical representation of the total fluorescence signal, F(t), which contains a Gaussian contribution (owing to the laser profile) and an RF “square wave” modulated component, which is the summation of multiple odd-harmonics (owing to the nature digitally modulating a solid-state laser). F(t) is then, a function of time, t, where m is the modulation, ϕ is the shift in phase related to the excited state kinetic processes of fluorescence emission, ω is the angular frequency of modulation, and b and t0 are parameters of the Gaussian response.
| (2) |
To complete the homodyne process the signal represented by Eq. (2) is mixed with a −90° phase-shifted reference sine wave and a 0° phase-shifted reference sine wave. The resulting two signals after low-pass frequency filtering become dependent on the sine of ϕ and the cosine of ϕ, respectively, where ϕ is the phase shift between the excitation signal and the emission signal. Finally, a ratio module is used to divide sinϕ by cosϕ and effectively achieve a value of tanϕ (recall Eq. (1) for the relationship between the fluorescence lifetime, τ, and tanϕ).
For calibration the fluorescence lifetime of non-fluorescent samples (i.e. τ = 0) were acquired, and the fluorescence lifetime of a lifetime standard (Flow-Check fluorospheres, Beckman Coulter, τ~7 ns) were also measured. Using the analog homodyne approach three parameters are measured: (1) the low-pass frequency filtered FL emission pulse height; (2) the low-pass frequency filtered SSC pulse height; and (3) the tanϕ, which is the value proportional to the fluorescence lifetime [recall Eq. (1)]. For all seven samples, approximately 1000 cells were measured and data are expressed as the means ± S.E.M of at least three independent experiments.
4. Results
4.1 Distribution of the EGFP-LC3 protein in MCF-7 cells after autophagy induction
Fluorescence microscopy was used to determine EGFP-LC3 localization in amino acid-starved MCF-7 cells prior to measuring fluorescence intensity and lifetime by flow cytometry to ensure the presence of EGFP-LC3 punctae after starvation. Displayed are the transiently transfected, paraformaldehyde-fixed MCF-7 cells imaged after 3-hour amino acid starvation to induce autophagy. Two columns of images in Fig. 2 include (1) unstarved and starved MCF-7 cells expressing the indicated native EGFP, EGFP-LC3, and EGFP-ΔLC3 using the green fluorescence channel to validate the expression of EGFP in the cells and to identify autophagy-associated punctae. Fluorescence images of MCF-7 cells transiently expressing native EGFP (not fused to LC3 protein) with and without amino acid starvation for 3 hours, Figs. 2(a) and 2(d), demonstrate the absence of punctae formation. Next, MCF-7 cells transiently expressing the EGFP-LC3 protein with amino acid starvation for 3 hours (Fig. 2(e)) have distinct punctae distribution consistent with autophagosomes formation [24]. Note the presence of some irregularly shaped high intensity fluorescence spots (EGFP protein aggregates) in the MCF-7 cells transiently expressing EGFP-LC3 without amino acid starvation (Fig. 2(b)). This observation is consistent with previously published results showing that EGFP-LC3 protein aggregation can be induced when transiently over-expressed in cells regardless of autophagy induction [28]. Image processing with ImageJ (http://rsb.info.nih.gov/ij/) resulted in an average of 32 ( ± 1.8) punctae for the starved and 15 ( ± 1.2) aggregates for unstarved MCF-7 cells quantified for 20 cells. Due to presence of EGFPaggregates in the The area between 0.07 and 0.65 µm2 were counted as autophagosomes [29]. Lastly, images of MCF-7 cells transiently expressing with EGFP fused to mutant LC3 (EGFP-ΔLC3) with and without amino acid starvation for 3 hours demonstrated no punctate distribution consistent with the inability of ΔLC3 to localize to autophagosomes, Figs. 2(c) and 2(f).
Fig. 2.
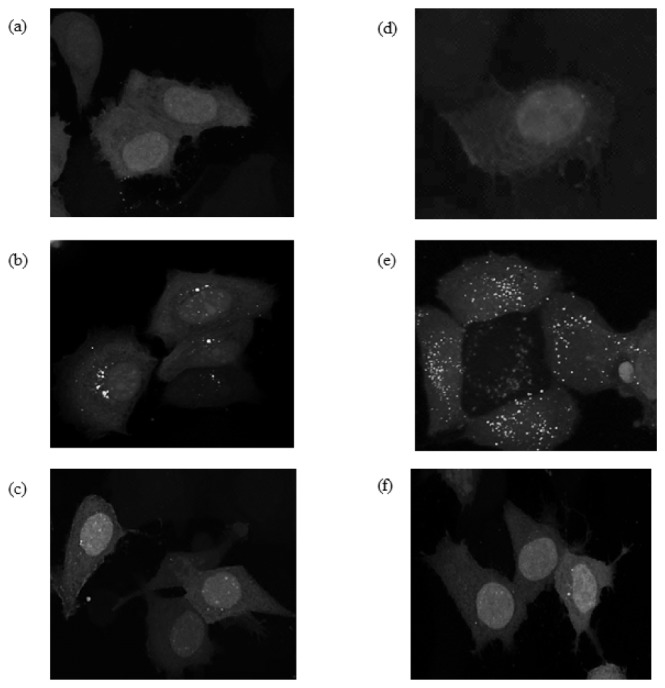
Detection of EGFP in MCF-7 breast cancer cells expressing EGFP, EGFP-LC3, or EGFP-ΔLC3 under non-starved conditions (a)-(c), and after autophagy induction by amino acid starvation for 3 hours (d)-(f). MCF-7 cells expressing EGFP (a) and (d), EGFP-LC3 (b) and (e), or EGFP-ΔLC3 (c) and (f) were imaged with confocal microscopy to show fluorescence intensity detected between 500 and 560 nm. These data are representative of at least 3 independent experiments.
4.2 Flow cytometry measurements
The displayed histograms represent one out of several repeated measurements of the fluorescence intensity and fluorescence lifetime for MCF-7 cells expressing EGFP, EGFP LC3, or EGFP-ΔLC3 after amino acid starvation for 3 hours to induce autophagy. The two columns of histogram statistics are for one experiment and include (1) the average fluorescence intensity, Figs. 3(a) -3(c) of MCF-7 cells transiently transfected with the indicated plasmid using a BD Accuri C6 Flow Cytometer, and (2) the average fluorescence lifetimes of MCF-7 cells transiently transfected with the indicated plasmids, Figs. 3(d)-3(f) measured with the TRFC. Markers with upper and lower bounds fixed for all data are provided to exemplify differences in the fluorescence intensity and fluorescence lifetimes.
Fig. 3.
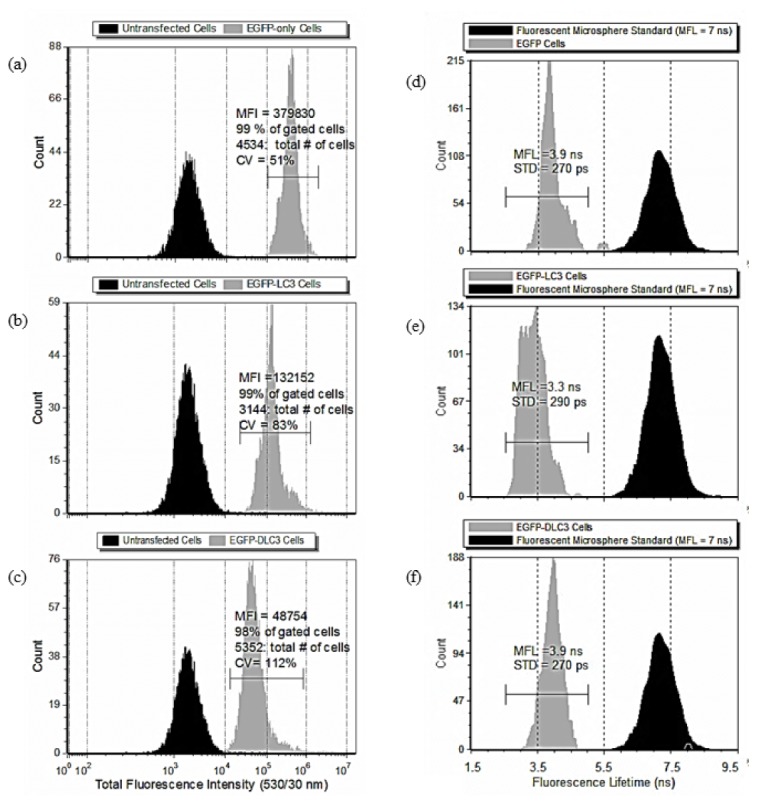
Decreased mean fluorescence lifetime (MFL) of EGFP upon subcellular localization to autophagosomes. Histograms indicating the mean fluorescence intensity (MFI) of amino acid-starved MCF-7 cells expressing EGFP (a), EGFP-LC3 (b), or EGFP-DLC3 (D = Δ) (c). MFI is shown for MCF-7 cells not transfected with EGFP as a negative control. Histograms indicating MFL in amino acid-starved MCF-7 cells expressing EGFP (d), EGFP-LC3 (e), or EGFP-DLC3 (D = Δ) (f). MFI data were obtained by 488 nm excitation and 530/30 nm emission using the BD Accuri C6 Flow Cytometer and the MFL data were obtained by 488 nm excitation and > 490-nm emission on the TRFC instrument described previously. These data are representative of 5 independent experiments.
In Fig. 3(a), the histogram represents data from approximately 5,000 MCF-7 cells transfected with EGFP (grey population). The histogram is an example of results obtained for these cells and the range in total fluorescence intensity measured for each cell. Also plotted are data from MCF-7 cells that were not transfected (black population) to provide a relative measurement of the autofluorescence background level. The mean fluorescence intensity (MFI) and statistics on the fluorescence characteristics of the population of transiently transfected cells are provided. Figures 3(b) and 3(c) show data for approximately 5,000 cells expressing EGFP-LC3 and EGFP-ΔLC3, respectively. The histograms indicate the average fluorescence intensity change for each cell population. When measured by standard flow cytometry, changes in the mean fluorescence intensity integrated across cells change simply owing to the differences in transient transfection and expression of native or fused EGFP. Overall, the MFI was consistent for at least 3 independent experiments and differences in MFI between the native EGFP and fused EGFP was reproducible.
The changes in fluorescence intensity were also unrelated to the presence of punctae. The ratio of the average MFI for EGFP-LC3 and EGFP-ΔLC3 relative to native EGFP was 0.45 and 0.2. Thus, cell populations expressing EGFP fusion proteins, were less fluorescent than cells with native EGFP.
To evaluate the fluorescence lifetime of EGFP, at least 3 independent fluorescence lifetime experiments were performed. That is, separate populations of starved cells were measured and the mean fluorescence lifetime (MFL) values for each population were averaged together. For repeated experiments (n ≥ 3) the MCF-7 cells expressing native EGFP (i.e. not fused to the LC3 or ΔLC3 protein) had an average MFL of 4 ns (300 ps standard error). The average MFL for MCF-7 cells expressing EGFP-LC3 fusion protein was reduced to 3.6 ns (200 ps standard error), and MCF-7 cells that expressed EGFP-ΔLC3 had fluorescence lifetimes that averaged to a MFL of 4 ns (200 ps standard error). In addition to the starved (autophagy induced) state, the MFL values were averaged for cells under unstarved conditions. The results for EGFP, EGFP-LC3, and EGFP-ΔLC3 are 4.2 ns (840 ps standard error), 3.8 ns (230 ps standard error) and 3.8 ns (240 ps standard error), respectively. Histograms of fluorescence intensity and lifetime for unstarved EGFP expressing cells were not used to quantify changes in fluorescence lifetime associated with autophagosome localization due to the presence of aggregates in unstarved cells (Fig. 2.) that could affect fluorescence lifetime measurements independent of localization to the autophagosome. Also, there is a possibility that changes in the biochemical milieu of the cell caused by amino-acid deprivation could affect the fluorescence lifetime of EGFP.
5. Discussion
The flow cytometry measurements obtained in this study in combination with fluorescence microscopy work suggest that the fluorescence lifetime of EGFP is sensitive to the formation of punctate EGFP distributions. Fluorescence lifetime values were altered during the process of autophagy and were correlated with the subcellular relocation of EGFP when bound to the LC3 protein during autophagy. Our study adds to works by others in which EGFP fluorescence lifetimes (ranging from 1 to 4 ns) are altered depending on the immediate microenvironment of the protein, the location within a cell, the attachment to other proteins, and cellular changes such as apoptosis and cell cycle stage [15, 16, 30–38].
Most lifetime work has been investigated with fluorescence lifetime imaging microscopy (FLIM) platforms, and although these measurements were conducted in a well-characterized lifetime cytometer [11–14, 39–41] FLIM measurements are indeed helpful to evaluate lifetime gradients across a cell and pixel-by-pixel. Yet flow cytometry has several advantages in that it can help hunt for rare events and reveal trends in population owing to dynamic changes with living cells. With time-resolved flow cytometry, a new parameter that is independent of fluorescence intensity can help to reveal changes in fluorescent proteins that are not as easily revealed with intensity-only systems. With the cytometry system described herein, a single, average fluorescence lifetime is collected, using a single modulation frequency concept. Unique fluorescence decays such as stretched and bi-exponential decay have been reported for fluorescent proteins, typically during resonance energy transfer scenarios. Therefore future work in our lab will exploit both phase and modulation lifetimes using square wave modulation where multi-frequency data can be extracted. With heterogeneous fluorescence lifetime measurements by TRFC we foresee a new high-throughput method for protein localization studies that can resolve multiple decays from single cytometric events.
Through cell growth treatment conditions (amino acid starvation), we were able to control the localization of EGFP to punctae by fusing this fluorescent protein to LC3. This allowed us to measure fluorescence lifetime shifts and compare those to overall fluorescence intensity changes. Presently, there are no ideal cytometry assays for autophagy. One approach involves staining the autolysosomes with acridine orange. This method is not optimal since acridine orange staining is not autolysosome-specific and stains non-autophagy-associated lysosomes [22, 42]. Another method is GFP-LC3 based, and quantifies the autophagic flux through the sensitivity of GFP-LC3 fusion protein to the acidic environment of autolysosome that leads to gradual attenuation of the GFP fluorescence intensity during the autophagy process [43, 44]. However our results indicate the attenuation of EGFP-ΔLC3 fluorescence during amino-acid starvation (data not shown). Thus, the attenuation of fluorescence is not dependent on GFP being localized to the autophagosome formation.
In general, this work prefaces a way in which compound screening can be performed rapidly to test for effects on protein localization. This is significant because protein localization is often indicative of various cellular processes and can also be used as a biomarker for environmental stresses on cells. Aberrant protein localization has been associated with the development of many disease states such as cancer. For example, the function of the cell-cycle inhibitor p27kip1 is dependent on cytoplasmic or nuclear localization. In the nucleus, p27kip1 function to inhibit cell proliferation while in the cytoplasm this protein functions to promote carcinogenesis [45, 46]. Current methods to detect protein localization involve immunofluorescence and are not considered high-throughput due to dependence on microscopy-based imaging and time-consuming image processing steps. If autophagy can quantitatively be detected, then TRFC could screen thousands of compounds for those that modulate autophagy. Such a tool would allow for the identification of novel cellular mechanisms that regulate autophagy. Recognizing autophagy at early stages has some benefits in the context of cancer. That is, the formation of autophagosomes provides energy for cells to survive, which implies that cancer cells could leverage this state for continued energy and growth. It is worth noting here that the role of autophagy remains controversial in that it could be favorable or antagonistic during carcinogenesis [47]. Additionally, autophagy is an important part of maintaining cells’ genomic stability by removing old and damaged organelles, and has been found to play a role in preventing the formation of protein aggregation-related diseases such as Alzheimer’s, Huntington’s and Parkinson’s disease [48]. Therefore, new and improved methods for carefully observing and evaluating autophagy at the large scale are essential.
Overall the study described herein shows that changes in the fluorescence lifetime of EGFP can be used as a sensor of LC3 protein localization induced by autophagy as verified by confocal microscopy. Ultimately this foreshadows such use for high-throughput quantification of protein localization to enhance both basic cell research and potentially lead to better diagnostic tools for certain disease states.
Acknowledgment
This work is supported by NIH Grant: R15 EB012013-01 and a New Mexico State University Vice President for Research Office Interdisciplinary Research Grant. We thank Dr. Peter Cooke (Electron Microscopy Facility, NMSU) for his assistance with confocal microscopy.
References and links
- 1.Hung M. C., Link W., “Protein localization in disease and therapy,” J. Cell Sci. 124(20), 3381–3392 (2011). 10.1242/jcs.089110 [DOI] [PubMed] [Google Scholar]
- 2.Tkaczyk E. R., Tkaczyk A. H., “Multiphoton flow cytometry strategies and applications,” Cytometry A 79(10), 775–788 (2011). 10.1002/cyto.a.21110 [DOI] [PubMed] [Google Scholar]
- 3.Swedlow J. R., “Innovation in biological microscopy: current status and future directions,” Bioessays 34(5), 333–340 (2012). 10.1002/bies.201100168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao M., Schiro P. G., Kuo J. S., Koehler K. M., Sabath D. E., Popov V., Feng Q., Chiu D. T., “An automated high-throughput counting method for screening circulating tumor cells in peripheral blood,” Anal. Chem. 85(4), 2465–2471 (2013). 10.1021/ac400193b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoo H. J., Park J., Yoon T. H., “High throughput cell cycle analysis using microfluidic image cytometry (μFIC),” Cytometry A 83(4), 356–362 (2013). 10.1002/cyto.a.22261 [DOI] [PubMed] [Google Scholar]
- 6.Murphy R. F., “Communicating subcellular distributions,” Cytometry A 77(7), 686–692 (2010). 10.1002/cyto.a.20933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barteneva N. S., Fasler-Kan E., Vorobjev I. A., “Imaging flow cytometry: coping with heterogeneity in biological systems,” J. Histochem. Cytochem. 60(10), 723–733 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elliott G. S., “Moving pictures: imaging flow cytometry for drug development,” Comb. Chem. High Throughput Screen. 12(9), 849–859 (2009). 10.2174/138620709789383204 [DOI] [PubMed] [Google Scholar]
- 9.J. R. Lakowicz, Principles of Fluorescence Spectroscopy (Springer, New York, 2006). [Google Scholar]
- 10.Pinsky B. G., Ladasky J. J., Lakowicz J. R., Berndt K., Hoffman R. A., “Phase-resolved fluorescence lifetime measurements for flow cytometry,” Cytometry 14(2), 123–135 (1993). 10.1002/cyto.990140204 [DOI] [PubMed] [Google Scholar]
- 11.Steinkamp J. A., Crissman H. A., “Resolution of fluorescence signals from cells labeled with fluorochromes having different lifetimes by phase-sensitive flow cytometry,” Cytometry 14(2), 210–216 (1993). 10.1002/cyto.990140214 [DOI] [PubMed] [Google Scholar]
- 12.Steinkamp J. A., Keij J. F., “Fluorescence intensity and lifetime measurement of free and particle-bound fluorophore in a sample stream by phase-sensitive flow cytometry,” Rev. Sci. Instrum. 70(12), 4682–4688 (1999). 10.1063/1.1150143 [DOI] [Google Scholar]
- 13.Houston J. P., Naivar M. A., Jenkins P., Freyer J. P., “Digital Analysis and Sorting of Fluorescence Lifetime by Flow Cytometry,” Cytometry A 77(9), 861–872 (2010). 10.1002/cyto.a.20930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houston J. P., Naivar M. A., Freyer J. P., “Capture of fluorescence decay times by flow cytometry,” Curr. Protoc. Cytom. (2012). 10.1002/0471142956.cy0125s59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esposito A., Gerritsen H. C., Wouters F. S., “Fluorescence lifetime heterogeneity resolution in the frequency domain by lifetime moments analysis,” Biophys. J. 89(6), 4286–4299 (2005). 10.1529/biophysj.104.053397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calleja V., Ameer-Beg S. M., Vojnovic B., Woscholski R., Downward J., Larijani B., “Monitoring conformational changes of proteins in cells by fluorescence lifetime imaging microscopy,” Biochem. J. 372(1), 33–40 (2003). 10.1042/BJ20030358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seefeldt B., Kasper R., Seidel T., Tinnefeld P., Dietz K. J., Heilemann M., Sauer M., “Fluorescent proteins for single-molecule fluorescence applications,” J Biophotonics 1(1), 74–82 (2008). 10.1002/jbio.200710024 [DOI] [PubMed] [Google Scholar]
- 18.Munafó D. B., Colombo M. I., “A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation,” J. Cell Sci. 114(Pt 20), 3619–3629 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Corcelle E. A., Puustinen P., Jäättelä M., “Apoptosis and autophagy: Targeting autophagy signalling in cancer cells -‘trick or treats’?” FEBS J. 276(21), 6084–6096 (2009). 10.1111/j.1742-4658.2009.07332.x [DOI] [PubMed] [Google Scholar]
- 20.Kimura S., Noda T., Yoshimori T., “Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3,” Autophagy 3(5), 452–460 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Mizushima N., Yoshimori T., Levine B., “Methods in Mammalian Autophagy Research,” Cell 140(3), 313–326 (2010). 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bampton E. T. W., Goemans C. G., Niranjan D., Mizushima N., Tolkovsky A. M., “The dynamics of autophagy visualized in live cells - From autophagosome formation to fusion with endo/lysosomes,” Autophagy 1(1), 23–37 (2005). 10.4161/auto.1.1.1495 [DOI] [PubMed] [Google Scholar]
- 23.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T., “LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing,” EMBO J. 19(21), 5720–5728 (2000). 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanida I., Yamaji T., Ueno T., Ishiura S., Kominami E., Hanada K., “Consideration about negative controls for LC3 and expression vectors for four colored fluorescent protein-LC3 negative controls,” Autophagy 4(1), 131–134 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Tanida I., Ueno T., Kominami E., “Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met(121) to expose Gly120 for lipidation and targeting to autophagosomal membranes,” J. Biol. Chem. 279(46), 47704–47710 (2004). 10.1074/jbc.M407016200 [DOI] [PubMed] [Google Scholar]
- 26.P. Jenkins, M. A. Naivar, J. P. Freyer, A. Arteaga, and J. P. Houston, “Flow Cytometric Separation of Spectrally Overlapping Fluorophores Using Multifrequency Fluorescence Lifetime Analysis,” (International Society for Optics and Photonics, San Francisco, CA, 2011). [Google Scholar]
- 27.Naivar M. A., Parson J. D., Wilder M. E., Habbersett R. C., Edwards B. S., Sklar L., Nolan J. P., Graves S. W., Martin J. C., Jett J. H., Freyer J. P., “Open, reconfigurable cytometric acquisition system: ORCAS,” Cytometry A 71(11), 915–924 (2007). 10.1002/cyto.a.20445 [DOI] [PubMed] [Google Scholar]
- 28.Kuma A., Matsui M., Mizushima N., “LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization,” Autophagy 3(4), 323–328 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Kristensen A. R., Schandorff S., Høyer-Hansen M., Nielsen M. O., Jäättelä M., Dengjel J., Andersen J. S., “Ordered organelle degradation during starvation-induced autophagy,” Mol. Cell. Proteomics 7(12), 2419–2428 (2008). 10.1074/mcp.M800184-MCP200 [DOI] [PubMed] [Google Scholar]
- 30.Suhling K., Siegel J., Phillips D., French P. M., Lévêque-Fort S., Webb S. E., Davis D. M., “Imaging the environment of green fluorescent protein,” Biophys. J. 83(6), 3589–3595 (2002). 10.1016/S0006-3495(02)75359-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Treanor B., Lanigan P. M., Suhling K., Schreiber T., Munro I., Neil M. A., Phillips D., Davis D. M., French P. M., “Imaging fluorescence lifetime heterogeneity applied to GFP-tagged MHC protein at an immunological synapse,” J. Microsc. 217(1), 36–43 (2005). 10.1111/j.0022-2720.2005.01430.x [DOI] [PubMed] [Google Scholar]
- 32.Clayton A. H., Hanley Q. S., Verveer P. J., “Graphical representation and multicomponent analysis of single-frequency fluorescence lifetime imaging microscopy data,” J. Microsc. 213(1), 1–5 (2004). 10.1111/j.1365-2818.2004.01265.x [DOI] [PubMed] [Google Scholar]
- 33.Pliss A., Zhao L., Ohulchanskyy T. Y., Qu J., Prasad P. N., “Fluorescence lifetime of fluorescent proteins as an intracellular environment probe sensing the cell cycle progression,” ACS Chem. Biol. 7(8), 1385–1392 (2012). 10.1021/cb300065w [DOI] [PubMed] [Google Scholar]
- 34.Ito T., Oshita S., Nakabayashi T., Sun F., Kinjo M., Ohta N., “Fluorescence lifetime images of green fluorescent protein in HeLa cells during TNF-alpha induced apoptosis,” Photochem. Photobiol. Sci. 8(6), 763–767 (2009). 10.1039/b902341k [DOI] [PubMed] [Google Scholar]
- 35.Nakabayashi T., Wang H. P., Kinjo M., Ohta N., “Application of fluorescence lifetime imaging of enhanced green fluorescent protein to intracellular pH measurements,” Photochem. Photobiol. Sci. 7(6), 668–670 (2008). 10.1039/b800391b [DOI] [PubMed] [Google Scholar]
- 36.Nakabayashi T., Nagao I., Kinjo M., Aoki Y., Tanaka M., Ohta N., “Stress-induced environmental changes in a single cell as revealed by fluorescence lifetime imaging,” Photochem. Photobiol. Sci. 7(6), 671–674 (2008). 10.1039/b805032e [DOI] [PubMed] [Google Scholar]
- 37.Elgass K., Caesar K., Schleifenbaum F., Stierhof Y. D., Meixner A. J., Harter K., “Novel application of fluorescence lifetime and fluorescence microscopy enables quantitative access to subcellular dynamics in plant cells,” PLoS ONE 4(5), e5716 (2009). 10.1371/journal.pone.0005716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pepperkok R., Squire A., Geley S., Bastiaens P. I., “Simultaneous detection of multiple green fluorescent proteins in live cells by fluorescence lifetime imaging microscopy,” Curr. Biol. 9(5), 269–274 (1999). 10.1016/S0960-9822(99)80117-1 [DOI] [PubMed] [Google Scholar]
- 39.Cui H. H., Valdez J. G., Steinkamp J. A., Crissman H. A., “Fluorescence lifetime-based discrimination and quantification of cellular DNA and RNA with phase-sensitive flow cytometry,” Cytometry A 52(1), 46–55 (2003). 10.1002/cyto.a.10022 [DOI] [PubMed] [Google Scholar]
- 40.Steinkamp J. A., “Phase-sensitive detection methods for resolving fluorescence emission signals and directly quantifying lifetime,” Methods Cell Biol. 42(Pt B), 627–640 (1994). 10.1016/S0091-679X(08)61099-7 [DOI] [PubMed] [Google Scholar]
- 41.Cao R., Pankayatselvan V., Houston J. P., “Cytometric sorting based on the fluorescence lifetime of spectrally overlapping signals,” Opt. Express 21(12), 14816–14831 (2013). 10.1364/OE.21.014816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mizushima N., “Methods for monitoring autophagy,” Int. J. Biochem. Cell Biol. 36(12), 2491–2502 (2004). 10.1016/j.biocel.2004.02.005 [DOI] [PubMed] [Google Scholar]
- 43.Shvets E., Elazar Z., “Flow cytometric analysis of autophagy in living mammalian cells,” Methods Enzymol. 452, 131–141 (2009). 10.1016/S0076-6879(08)03609-4 [DOI] [PubMed] [Google Scholar]
- 44.Hundeshagen P., Hamacher-Brady A., Eils R., Brady N. R., “Concurrent detection of autolysosome formation and lysosomal degradation by flow cytometry in a high-content screen for inducers of autophagy,” BMC Biol. 9(1), 38 (2011). 10.1186/1741-7007-9-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liang J., Zubovitz J., Petrocelli T., Kotchetkov R., Connor M. K., Han K., Lee J. H., Ciarallo S., Catzavelos C., Beniston R., Franssen E., Slingerland J. M., “PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest,” Nat. Med. 8(10), 1153–1160 (2002). 10.1038/nm761 [DOI] [PubMed] [Google Scholar]
- 46.Chu I. M., Hengst L., Slingerland J. M., “The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy,” Nat. Rev. Cancer 8(4), 253–267 (2008). 10.1038/nrc2347 [DOI] [PubMed] [Google Scholar]
- 47.Mathew R., White E., “Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night,” Curr. Opin. Genet. Dev. 21(1), 113–119 (2011). 10.1016/j.gde.2010.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cecconi F., Levine B., “The role of autophagy in mammalian development: cell makeover rather than cell death,” Dev. Cell 15(3), 344–357 (2008). 10.1016/j.devcel.2008.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]