

Abstract
There is a growing appreciation of the diverse roles that lipid mediators play in modulating inflammatory responses during infection. In the case of tuberculosis, virulent mycobacteria induce host production of anti-inflammatory mediators, including lipoxins, which limit the host inflammatory response and lead to necrotic cell death of infected macrophages. Recent work using the zebrafish model suggests that, while excess anti-inflammatory lipoxins are host detrimental during mycobacterial infections, excess pro-inflammatory lipids also drive host susceptibility. The balance of these inflammatory states is influenced by common human genetic variation in Asia. Fuller understanding of the mechanisms of eicosanoid-mediated inflammatory imbalance during tuberculosis infection has important implications for the development of adjunctive therapies.
Inflammatory lipid mediators are being increasingly implicated in a variety of inflammatory diseases and accordingly, there is great interest in harnessing endogenous anti-inflammatory mediators such as the lipoxins, resolvins and protectins as pharmacological agents to treat these diseases(1). While these anti-inflammatory mediators are required to stop inflammation in a timely fashion, they can also increase susceptibility to chronic infections by inhibiting the inflammatory responses required for their eradication. Indeed both bacterial and protozoal agents of chronic infection Mycobacterium tuberculosis and Mycobacterium marinum, and Toxoplasma gondii induce production of host anti-inflammatory lipoxins presumably to promote their own survival (2–5) At least two pathogens, Toxoplasma and Pseudomonas aeruginosa, encode secreted enzymes with 15-LO activity, which would directly generate these anti-inflammatory eicosanoids from host-produced intermediates (6, 7).
The model that can be derived from this dichotomy is relatively simple; that pro-inflammatory mediators evolved to protect us against acute injury and infection, that this inflammatory program has to be kept in close check by anti-inflammatory mediators, but that if their activity is too much or too soon, the resultant decrease in inflammation and immunity can make the individual more susceptible to infection. This then is the simpler of the two dichotomies presented in this review and is underscored by recent findings from our and other laboratories that lipoxin excess can increase susceptibility to TB and leprosy (Figure 1). Overlaid on the dichotomous role of the anti-inflammatory, proresolving mediators is our recent discovery of the deleterious effect of the excess of the pro-inflammatory leukotriene B (Figure 1). This surprising finding calls into question the simple model that pro-inflammatory lipid mediators have evolved to protect against infection but then promote inflammation if dysregulated. However this complex role of the leukotrienes and lipoxins can be harnessed to provide personalized TB treatment that may be life-saving in the most severe forms of TB and in all cases of the drug-resistant TB that is increasing globally (Figure 2). The layered yin and yang that we have discovered also raises evolutionary questions. This review will present these recent findings and address both the evolutionary and the therapeutic implications of these recent discoveries.
Figure 1. Cartoon diagram showing how a balance of leukotrienes and lipoxins is required for optimal protection in TB.
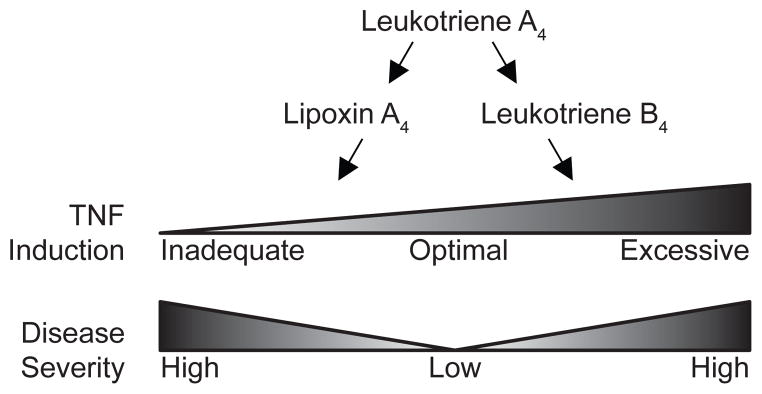
These effects are mediated through TNF expression.
Figure 2. Rationally designed therapeutic possibilities for TB in the eicosanoid pathway.
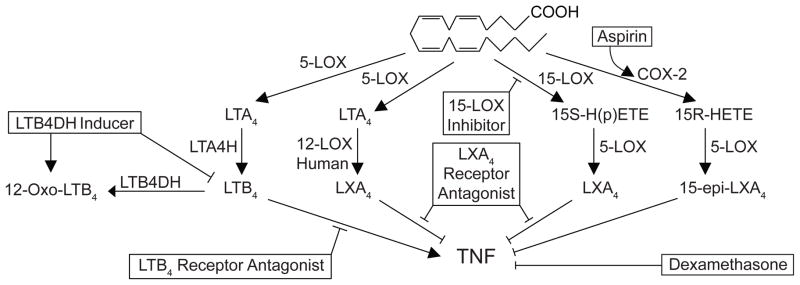
Shown are the pathways we have found to dysregulate TNF levels with possible interventional strategies to restore eicosanoid balance in boxes. The predicted effect of all interventions except the LTB4DH inducer and the LXA4 receptor antagonist have been confirmed in the zebrafish TB model.
Lipoxin Excess is Host-detrimental in TB: Mouse and Zebrafish Studies
The first evidence for the detrimental role of Lipoxin A4, in TB came from the finding that 5-lipoxygenase knockout mice were more resistant to TB this resistance was abrogated by the administration of exogenous lipoxins (2). In the meanwhile, we had developed as a valid and tractable model for TB, the zebrafish infected with M. marinum, a natural fish pathogen and a close genetic relative of the human TB bacillus (8) Not only do adult zebrafish develop TB with the necrotic granulomas characteristic of human TB, but infection in the larval fish mimics adult disease, progressing to granuloma formation (9) When we then performed a forward genetic screen to identify zebrafish with altered susceptibility to M. marinum, positional cloning of a hypersusceptible fish with higher bacterial burdens revealed leukotriene A4 hydrolase (LTA4H) as a host susceptibility factor (4). LTA4H converts the unstable epoxide LTA4 into the stable highly pro-inflammatory molecule LTB4. However, using the repertoire of tools available in the zebrafish to characterize the biology of mycobacterial infection, including pharmacological interventions and real-time, live analysis of infection, lipoxin excess rather than the absence of LTB4 was identified as driving the hypersusceptibility phenotype. In other words, the salutary effect of LTA4H is through prevention of lipoxin accumulation. Mycobacterium tuberculosis infection itself generates lipoxin production, and so the changes in the activity of this one enzyme may have large effects on a balance that is already altered by the pathogen (10). In vivo, suppression of TNF by the hypoinflammatory state was critical. Exogenous TNF was sufficient to rescue the hypersusceptibility, in contrast to exogenous LTB4 which could not. In terms of a cellular mechanism, the TNF deficiency induced by lipoxin excess reduces the microbicidal capacity of the infected macrophages, allowing exuberant intracellular bacterial growth, followed by cell lysis and release of the bacteria into the extracellular milieu where they can grow exuberantly. These in vivo findings support data from cell culture assays where lipoxins were found to induce cell necrosis (3, 10). These cell culture studies demonstrated a role for lipoxins in inhibition of PGE2 production, leading to mitochondrial damage and necrotic cell death. In a mouse TB model, lipoxins also limit cross-priming by dendritic cells, thus limiting the adaptive immune responses (11)
Human clinical studies suggest the need for “just right” LTA4H activity
In human studies, how relevant are these lipid mediators? LTB4 is detectable in the sputum of individuals with active tuberculosis (12). Using polymorphisms that were associated with functional differences in LTA4H activity, human cohorts in Vietnam and Nepal were examined for hypersusceptibility to the two most medically significant mycobacterial infections: tuberculosis and leprosy. Significant associations in susceptibility were found with host genotype, but the nature of that association was surprising. Namely, heterozygotes who carried one copy of the high-activity allele and one copy of the low-activity allele were relatively protected while homozygotes who had either two high activity alleles or two low-activity alleles had worse outcomes (4, 13) This was true both in the Vietnamese TB cohort and in the replication leprosy cohort in Nepal, where heterozygotes were least likely to have multibacillary leprosy (4). Most strikingly, when analyzing clinical data from the patients with TB meningitis in Vietnam, there was a strong association between survival and genotype. TB meningitis is one of the most lethal forms of mycobacterial disease, with mortality rates reaching 40% even with appropriate antimicrobial chemotherapy (14). Individuals heterozygous for the LTA4H activity variant were much less likely to die, with virtually all the deaths in the cohort being in the homozygotes (13).
The relevant LTA4H variant appears to be a promoter polymorphism just upstream of the transcription start site, with the high-activity genotype resulting in higher levels of LTA4H RNA and much higher protein production in lymphoblastoid cell lines from the 1000 Genomes Project. This variant is most common in Asia, with allele frequencies of the high-activity allele ranging up to 0.35, depending on population.
These findings suggested that either a hypo- or hyperinflammatory state generated through an excess of anti-inflammatory or pro-inflammatory lipid mediators, respectively, could lead to imbalances of inflammation and worse outcomes in human populations. Notably either excess state resulted in similar outcomes in terms of disease severity.
Zebrafish studies reveal mechanism of susceptibility of LTA4H excess
Insufficient inflammation makes sense as a mechanism, and corroborates the zebrafish work but how might excessive inflammation also lead to increased disease severity? The mechanism was revealed when we modeled the LTA4H-high state in the zebrafish through LTA4H RNA overexpression (13). Again, we found TNF to be the key component in mediating susceptibility, this time through its excess production. LTA4H excess increased transcriptional induction of TNF in response to infection, that was transiently beneficial by increasing intramacrophage bacterial killing (13). However, soon thereafter, the excess TNF caused lysis of the infected macrophages themselves, thus delivering the few bacteria that had escaped their enhanced killing capacity to the extracellular growth-permissive environment where they could quickly catch up in numbers to those in the LTA4H/TNF-low state (13). Further work has revealed a detailed mechanism by which TNF induces macrophage necrosis (15). TNF binding to its receptors activates the RIP1 and RIP3 kinases which then induce through a series of signaling intermediates, mitochondrial reactive oxygen species (ROS). ROS kills both bacteria and macrophage, the latter through two pathways: translocation of the redox-sensitive mitochondrial matrix protein cyclophilin D to the membrane to participate in the formation of the mitochondrial permeability transition pore complex, and the activation of lysosomal acid phosphatase with resultant ceramide overproduction that also causes cell necrosis. Indeed genetic inhibition of either pathway causes a partial reduction in LTA4H/TNF-mediated cell death, while inhibition of both together causes a near complete inhibition. The profound therapeutic implications of these findings will be discussed later but for now it becomes clear that there are many ways to generate excess inflammation. It is emerging that other pathogens may induce cell death endpoints as effective immune evasion strategies. Salmonella enterica Serovar typhimurium induces necroptosis in mouse macrophages downstream of the production of Type I interferon (16).
Pertaining to TB, all of the components of the TNF-mediated mitochondrial necrosis pathways we have described are potentially subject to genetic variations, and these could permute in many ways to alter TB susceptibility.
But there are multiple ways to generate excess inflammation, even centering around excess of pro-inflammatory lipid mediators. The fact that excess LTA4H can lead to increased infection burden suggested that excess LTB4 was sufficient to generate a hyperinflammatory state. Other variations in this pathway could similarly cause excess inflammatory states. For example, blockade of one of the enzymes responsible for inactivating LTB4 resulted in a hyperinflammatory state, with excess TNF, similar to the LTA4H overexpression (17). The excess inflammatory state due to LTA4H excess could similarly be rescued by overexpression of the LTB4-inactivating enzyme, underscoring the fine balance of pro- and anti-inflammatory cues and the ability to manipulate inflammatory state pharmacologically after understanding the underlying mechanisms and genetics of the hyperinflammatory state (17).
Heterozygous advantage and balanced inflammatory states in human populations
In humans, heterozygous genotype associates with protection from more severe disease outcome. In the TB meningitis cohort in Vietnam, heterozygosity for the LTA4H promoter polymorphism associated with improved survival relative to either homozygous state. In Nepal heterozygotes were protected from the higher burden (multibacillary) forms of leprosy (Figure 1).
Notably in a large Russian cohort, LTA4H intronic SNPs were not associated with disease susceptibility (18). However, the putative causal variant was not genotyped directly, since it had not yet been identified. Moreover, the high-activity promoter variant that is prevalent in Asia (0.35 high-activity allele frequency in Vietnam) occurs rarely among European populations, including Caucasians in Seattle and the CEU cohort from the 1000 genomes project (19). Thus it is likely that the Russian and Vietnamese populations may have different population structures, or the variant may not be sufficiently represented in the Russian population to detect an effect.
The geographic distribution of this allele is relatively unusual. The frequency of the derived, high-activity allele is high in Asia (0.35 in Vietnam, 0.29 in Japan), intermediate in Africa (0.12 allele frequency) and low in European populations (<0.04). Generally, alleles present in both Africa and Asia are also present in European populations. It is therefore possible that the allele was maintained (and perhaps selected for in Asia) while being selected against in Europe.
Although population stratification and drift may account for these differences, it is interesting to speculate that these allele frequencies may have emerged from selective pressure from infectious disease or other inflammatory conditions. Increased inflammation from the derived, high-activity allele may have conferred a selective advantage in response to infection (although it is not clear that the source of that pressure was tuberculosis per se; any infectious disease may have provided this selective pressure). Evolutionarily, the only requisite for overdominance is that the advantage that accrues to the larger group of heterozygotes outweigh the disadvantage to the homozygotes. In Europe, where this allele is largely absent, we speculate that a different historical repertoire of infectious diseases, in which increased inflammation might be host detrimental (plague might be the classic infection historically prevalent in Europe) might have selected against even the heterozygous state. In the case of systemic inflammatory response syndrome (SIRS), RIP-3 dependent necroptosis drives mortality downstream of TNF (20). A dramatic example of the deleterious effect of the excess inflammatory state in the mouse comes from cytosolic delivery of bacterial flagellin. The ensuing production of eicosanoids leads to an “eicosanoid storm” and death in less than 30 minutes(21).
Overall, these recent studies suggest that inflammatory balance can be dramatically influenced by lipid mediators, that levels of these mediators may depend on genetic variation within populations and have important functional consequences, and that host-based therapies targeting different steps of these pathways have the potential to restore inflammatory balance.
Therapeutic Implications
The findings that genetic variations leading to imbalanced production of lipid mediators can lead to TB susceptibility offers new therapeutic opportunities. A taste of such genotype-directed therapies targeting inflammatory mediators and their downstream pathways comes from our findings on treatment responsiveness in the same Vietnamese TB meningitis cohort where we examined the effect of genotype on survival. Because TB meningitis carries such a high mortality, and because inflammatory features have been associated with the disease, clinicians have used empiric anti-inflammatory glucocorticoids for many decades. The Vietnam study was done as a randomized controlled trial to see if the glucocorticoids did have a beneficial effect(5). A small but definite effect was found, so that glucocorticoids have now become standard of care as adjunctive treatment to standard antituberculous chemotherapy. Our model would predict that only the high-LTA4H expressing patients would benefit from the glucocorticoids, and indeed when we stratified patients according to LTA4H-genotype in this historical cohort, we found this to be the case (13). In fact, the low-LTA4H expressing patients appeared to derive no benefit from the steroids and may have fared worse, suggesting the need for genotype directed treatments after validating these findings in other cohorts globally. Since the LTA4H genotype affects a fundamental step in pathogenesis, we predict that its effects are not restricted to meningeal TB. In this context, glucocorticoids have been shown to have a small benefit in other forms of TB as well and it will be important to examine these. However, our work suggests additional therapies that target this pathway, many of which we have validated in the zebrafish (Figure 2). First we showed that the same glucocorticoid used in the Vietnam study had the predicted effects in the zebrafish: beneficial for the high-LTA4H genotype, while harmful to the low-LTA4H genotype. Then by rational interventions in the pathway, we were able to show that aspirin which induces the formation of a functional epimer of LXA4, was similarly beneficial to the high-LTA4H genotype while harmful to the low-LTA4H genotype. Conversely an inhibitor of 15-lipoxygenase benefitted the low-LTA4H genotype while harming the high-LTA4H genotype, presumably by inhibiting the lipoxins that provided some measure of a dampening signal to the hyperinflammatory state. One would predict that inhibiting lipoxin activity by targeting their receptors would be similarly beneficial to the low LTA4H-genotype. Interventions such as these represent treatments that must, like the glucocorticoids, be personalized to patient genotype, in this case a single base change.
However, we have also identified potential pharmacological interventions, that while helpful to only the high-LTA4H genotype, are neutral to the low-LTA4H genotype, thus obviating the need for field genotyping. Pharmacological antagonism of the LTB4 receptors represents such a therapy, further supporting our genetic work that while an excess of LTB4 is detrimental in TB, its deficiency may have no impact on this disease. Another exciting possibility would be to inhibit 5-lipoxygenase that would be predicted to be helpful in both genotypes by inhibiting both lipoxin and leukotriene; genetic knockouts of ALOX5 in mice have been shown to result in increased resistance, while human variants have been associated with susceptibility in Ghana (2, 22).
Finally, our understanding of the role of TNF as the driver of the susceptibility of LTA4H excess and the elucidation of the mechanism of programmed necrosis suggests attractive downstream therapeutic possibilities (15). RIP-1 inhibitors and ROS-scavenging drugs reverse the hypersusceptibility of LTA4H-high fish. Further downstream in the pathway, blocking macrophage necrosis also reverses hypersusceptibility. Drugs exist for both cell necrosis pathways we have identified - the cyclophilin D inhibitor alisporivir, and the tricyclic antidepressant class of drugs that enzymatically degrade acid sphingomyelinase thus preventing ceramide production. When either necrosis pathway is blocked individually, we can restore the hypersusceptible state to wildtype (corresponding to the heterozygous humans). When we block both cell death pathways, while preserving ROS production, we convert hypersusceptibility to hyperresistance. These downstream interventions also have the benefit of being neutral to the other genetic states. Moreover, by targeting downstream interventions, they can treat the hypersusceptibility coming from increased ROS production through multiple pathways: for instance TNF overproduction through routes independent of LTA4H excess.
The concept of balanced inflammation is not new, but by identifying specific molecular mechanisms underlying this balance, we are now able to rationally target and tune this balance in infectious diseases like tuberculosis. The important roles of lipid mediators in this balance has emerged from multiple animal models and is now being recognized in human cohorts as well.
Highlights.
Balanced production of inflammatory mediators is required for protection against TB
Genetic variation in lipid mediator expression can determine TB outcomes.
Lipoxin excess generates hypoinflammatory states that restrict the protective cytokine TNF.
Excessive pro-inflammatory cues from leukotriene excess are equally host-detrimental.
Host-directed therapies targeting either state have the potential to restore inflammatory balance.
Acknowledgments
The Tobin laboratory is funded through a Mallinckrodt Scholar Award, a Searle Scholar Award, the NIH Director’s New Innovator Award and the Duke University Center for AIDS Research. The Ramakrishnan laboratory is funded by the NIH, including the NIH Director’s Pioneer Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Serhan CN. The resolution of inflammation: the devil in the flask and in the details. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25:1441–8. doi: 10.1096/fj.11-0502ufm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bafica A, Scanga CA, Serhan C, Machado F, White S, Sher A, Aliberti J. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. The Journal of clinical investigation. 2005;115:1601–6. doi: 10.1172/JCI23949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, Serhan CN, Behar SM, Remold HG. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. The Journal of experimental medicine. 2008;205:2791–801. doi: 10.1084/jem.20080767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tobin DM, Vary JC, Jr, Ray JP, Walsh GS, Dunstan SJ, Bang ND, Hagge DA, Khadge S, King MC, Hawn TR, Moens CB, Ramakrishnan L. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. 2010;140:717–30. doi: 10.1016/j.cell.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thwaites GE, Nguyen DB, Nguyen HD, Hoang TQ, Do TT, Nguyen TC, Nguyen QH, Nguyen TT, Nguyen NH, Nguyen TN, Nguyen NL, Nguyen HD, Vu NT, Cao HH, Tran TH, Pham PM, Nguyen TD, Stepniewska K, White NJ, Tran TH, Farrar JJ. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N Engl J Med. 2004;351:1741–51. doi: 10.1056/NEJMoa040573. [DOI] [PubMed] [Google Scholar]
- 6.Bannenberg GL, Aliberti J, Hong S, Sher A, Serhan C. Exogenous pathogen and plant 15-lipoxygenase initiate endogenous lipoxin A4 biosynthesis. The Journal of experimental medicine. 2004;199:515–23. doi: 10.1084/jem.20031325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vance RE, Hong S, Gronert K, Serhan CN, Mekalanos JJ. The opportunistic pathogen Pseudomonas aeruginosa carries a secretable arachidonate 15-lipoxygenase. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:2135–9. doi: 10.1073/pnas.0307308101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tobin DM, Ramakrishnan L. Comparative pathogenesis of Mycobacterium marinum and Mycobacterium tuberculosis. Cellular microbiology. 2008;10:1027–39. doi: 10.1111/j.1462-5822.2008.01133.x. [DOI] [PubMed] [Google Scholar]
- 9.Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nature reviews Immunology. 2012;12:352–66. doi: 10.1038/nri3211. [DOI] [PubMed] [Google Scholar]
- 10.Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, Fortune S, Behar SM, Remold HG. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nature immunology. 2009;10:899–906. doi: 10.1038/ni.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nature immunology. 2010;11:751–8. doi: 10.1038/ni.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.el-Ahmady O, Mansour M, Zoeir H, Mansour O. Elevated concentrations of interleukins and leukotriene in response to Mycobacterium tuberculosis infection. Annals of clinical biochemistry. 1997;34 ( Pt 2):160–4. doi: 10.1177/000456329703400205. [DOI] [PubMed] [Google Scholar]
- 13**.Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, Ko DC, Zou Y, Bang ND, Chau TTH, Vary JC, Jr, Hawn TR, Dunstan SJ, Farrar JJ, Thwaites GE, King MC, Serhan CN, Ramakrishnan L. Host Genotype-Specific Therapies Can Optimize the Inflammatory Response to Mycobacterial Infections. Cell. 2012;148:434–46. doi: 10.1016/j.cell.2011.12.023. This work uses the zebrafish to model both low and high inflammatory state due to excess lipoxin or excess LTB4 and TNF. Finally, using a Vietnamese TB meningitis cohort, it finds a strong association between a common promoter variant in LTA4H and response to standard anti-inflammatory therapies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porkert MT, Sotir M, Parrott-Moore P, Blumberg HM. Tuberculous meningitis at a large inner-city medical center. The American journal of the medical sciences. 1997;313:325–31. doi: 10.1097/00000441-199706000-00002. [DOI] [PubMed] [Google Scholar]
- 15**.Roca FJ, Ramakrishnan L. TNF Dually Mediates Resistance and Susceptibility to Mycobacteria via Mitochondrial Reactive Oxygen Species. Cell. 2013;153:521–34. doi: 10.1016/j.cell.2013.03.022. Roca and Ramakrishnan delineate the cellular mechanisms by which the hyperinflammatory state due to excess LTA4H/TNF results in hypersusceptibility to mycobacterial infection. A RIP1/3 dependent mechanism leads to ROS generation, and excess ceramide production, leading to necrosis of infected macrophages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ayres JS, Trinidad NJ, Vance RE. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat Med. 2012;18:799–806. doi: 10.1038/nm.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tobin DM, Roca FJ, Ray JP, Ko DC, Ramakrishnan LR. An enzyme that inactivates the inflammatory mediator leukotriene B4 restricts mycobacterial infection. PloS one. 2013 doi: 10.1371/journal.pone.0067828. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Curtis J, Kopanitsa L, Stebbings E, Speirs A, Ignatyeva O, Balabanova Y, Nikolayevskyy V, Hoffner S, Horstmann R, Drobniewski F, Nejentsev S. Association analysis of the LTA4H gene polymorphisms and pulmonary tuberculosis in 9115 subjects. Tuberculosis. 2011;91:22–5. doi: 10.1016/j.tube.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Consortium GP. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–18. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 21*.von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K, Vance RE. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–11. doi: 10.1038/nature11351. This work links forced activation of the inflammasome via cytosolic delivery of bacterial flagellin to robust production of eicosanoids, leading to a dramatic eicosanoid storm and rapid death of animals in less than 30 minutes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herb F, Thye T, Niemann S, Browne EN, Chinbuah MA, Gyapong J, Osei I, Owusu-Dabo E, Werz O, Rusch-Gerdes S, Horstmann RD, Meyer CG. ALOX5 variants associated with susceptibility to human pulmonary tuberculosis. Hum Mol Genet. 2008;17:1052–60. doi: 10.1093/hmg/ddm378. [DOI] [PubMed] [Google Scholar]