

Abstract
Nemo-like kinase (NLK) is an evolutionarily conserved serine/threonine kinase and has been considered to be a suppressor of Wnt signaling in mammalian cells. Our study, however, has raised the possibility that NLK also functions as a Wnt signaling activator. In human osteosarcoma and neuroblastoma cell lines, NLK specifically enhanced β-catenin-TCF complex transcription activity. The effect required kinase activity of NLK and co-expression of the β-catenin ΔN (constitutive active mutant of β-catenin). The nuclear localization of Lymphoid enhancer factor 1 (LEF1) and β-catenin ΔN was not altered by NLK overexpression regardless of its effect on β-catenin-TCF complex activity. Reporter analysis using LEF1 mutants at known NLK target sites indicated that NLK may have different activation targets for β-catenin-TCF complex. Mutations in the potential NLK phosphorylation sites in β-catenin did not change its transcription activity either. Our results suggest that NLK positively regulates Wnt/β-catenin signaling in a cell type dependent manner through an unidentified mechanism.
Keywords: Nemo-like kinase, Wnt signaling, β-catenin, TCF, osteosarcoma, neuroblastoma
Introduction
Wnt signaling is an evolutionarily conserved developmental signal transduction pathway and is known to be involved in human carcinogenesis.1)–3) Activation of the Wnt signaling pathway results in the stabilization of cytoplasmic β-catenin which then enters the nucleus. Nuclear β-catenin binds to the T-cell factor (TCF) family member transcription factors and activates downstream target genes (http://www.stanford.edu/~rnusse/wntwindow.html). The genes encoding Wnt signaling regulators, such as APC, β-catenin and axin, are mutated in many human cancers, especially colon and liver cancers.4), 5)
Mutants in the Wnt signaling pathway are known to induce defects of the anterior-posterior cell fate determination in the Caenorhabditis elegans development.6)–8) For instance, a defect in MOM-2, a worm homolog of Frizzled, causes loss of intestinal cells and increase of pharyngeal cells, by losing posterior cell fate that induces intestinal cells at division of EMS cell.7), 8) A locus named lit-1 (loss of intestine) has been previously reported to be a critical mediator of anterior-posterior cell fate determination. 9) Genetic studies on C. elegans revealed that lit-1 encodes a protein kinase10), 11) that is a critical mediator ofWnt signaling.11) A mammalian homolog of lit-1 is Nemo-like kinase (NLK), which is related to the mitogen-activated protein kinase (MAPK).12) NLK is also the homolog of a Drosophila gene, Nmo, a mutant inducing defects in the epithelial planar polarity of the fly’s omatidia alignment.13)
A worm TCF homolog, POP-1, was initially considered to be a suppressor of Wnt signaling because a pop-1 mutant caused excessive proliferation of intestinal cells, a phenotype opposite to the one associated with Wnt defects in studies of C. elegans. 14), 15) LIT-1 phosphorylates POP-1 then mediates Wnt signaling.11), 16) Ishitani et al. correspondingly reported that NLK could suppress the transcriptional activity of the β-catenin-TCF complex through phosphorylation of TCF in human 293 cells.17) These findings led to the hypothesis that NLK may be a suppressor of TCF, and consequently, of Wnt signaling. Subsequent Drosophila studies also indicated that Nmo could be a suppressor of Wnt signaling18)–20) although Nmo mutants did not enhance the activated Armadillo phenotype, which induces apoptosis during Drosophila eye disk development.21)
Accumulation of evidence has led a different idea for function of the kinase: a TCF activator. It is noteworthy that TCF has a biphasic role in Wnt signaling; it plays a positive role when the signaling is activated and a suppressive role when the signaling is suppressed.22), 23) Korswagen et al. reported that POP-1 positively mediates Wnt signal in C. elegans.24) Siegfried et al. showed that the lit-1 gene is required for pop-1 function in the worm gonadogenesis.25) Moreover, Thorpe et al. revealed that zebrafish NLK appeared to be a positive regulator of Wnt signaling in ventrolateral mesoderm formation and patterning of the midbrain.26) Considering the evolutional conservation of Wnt signaling pathways, it is reasonable to assume that NLK may play a positive role for Wnt signaling in mammalian cells.27) Some of the reported mutant phenotypes in Drosophila Nmo18) are not compatible with the proposition that NLK is a suppressor of Wnt signaling. 26) In mice, genetic disruption of NLK caused severe lymphocyte reduction, especially of the B-cell lineage.28) Interestingly, the genetic disruption of a TCF homolog, lymphoid enhancer factor 1 (LEF1), caused severe reduction of the B220+ cell lineage in mice,29) suggesting that both NLK and LEF1 act collaboratively in the same signaling pathway concerning B-cell proliferation.
In this study, we found that NLK activates pTOPFLASH, a specific reporter plasmid of β-catenin-TCF transcription activity with repeats of TCF binding sites in the promoter sequence of the luciferase gene, in osteosarcoma (SaOS-2) and neuroblastoma (NB39-nu, and Nh12) cell lines. This increase of transcription activity of β-catenin-TCF complex was dependent on NLK kinase activity. Our results suggest that NLK can be a positive regulator of the Wnt signaling pathway in certain mammalian cell types.
Materials and methods
Plasmids
Details of the plasmids’ construction are available upon request. The plasmids pCMV-Flag-NLK, pCMV-Flag-NLK K155M were kindly provided by Dr. Kunihiro Matsumoto. The inserts of pCMV-Flag-NLK and pCMV-Flag-NLK K155M were amplified by PCR and subcloned to the Bam HI and Not I sites of pcDNA3 to construct pcDNA3-NLK-S and pcDNA3-NLK-S KD (a kinase negative mutant of NLK-S) respectively. The amplified nucleotide sequence of mouse NLK (Genbank accession number: NM 008702) was from nucleotide 488 to the stop codon. To obtain the flanking sequence of the initiation methionine for the construction of NLK-L expression vectors, we amplified mouse NLK fragment of cDNA from the mouse CG cell (kind gift from Dr. Issay Kitabayashi). The amplified cDNA was cloned into pcDNA3 to construct pcDNA3-NLK-L: the insert was from nucleotide 235 (NM 008702) to the stop codon. The kinase-negative form of the full-length NLK expression vector was constructed employing an “overlapping PCR” technique, using pcDNA3-NLK-L and pCMV-Flag-NLK K155M as templates. The insert of pGEXhLEF130) was amplified by PCR and subcloned into pcDNA3 at EcoR I and Not I sites to construct pcDNA3-HA-LEF1. pcDNA3-HA-LEF1T155A, S166A, 155/166AA, S132TSAA and TSAAS200A were produced using the Quikchange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The β-catenin expression plasmid pcDNA3-MYC-β-catenin ΔN was a kind gift from Drs. Osamu Tetsu and Frank McCormick.31) pcDNA3-MYC-β-catenin ΔNS191A, ΔNS246A, and ΔNS605A were produced using the Quikchange site-directed mutagenesis kit. pRL-tk (Promega, Madison, WI), pFOPFLASH and pTOPFLASH (Upstate, Syracuse, NY) were purchased. pCR3.1 p53, pWWP-luc, and pNF-κB-luc were described previously.32)
Cell lines, cell culture, transfection and luciferase assays
We used following cell lines: 293 cell (human kidney: epithelial), SaOS-2 (human osteosarcoma), Nh12 (human neuroblastoma), and NB39-nu (human neuroblastoma).
For luciferase assays, 293 and SaOS-2 cells were cultured with Dulbecco’s modified Eagle medium supplemented with 10% Fetal Bovine Serum (Sigma), penicillin (100 units/ml), streptomycin (0.1 mg/ml) and glutamine (0.29 mg/ml) (Penicillin-Streptomycin-Glutamine liquid, Invitrogen) in 24-well plates. Cells at 80% confluence were subjected to transfection with Lipofectamine 2000 (Invitrogen), following the manufacturer’s instructions with modifications. Neuroblastoma (Nh12 and NB39-nu) cells were cultured with RPMI1640 medium supplemented with 10% Fetal Bovine Serum, penicillin (100 units/ml), streptomycin (0.1 mg/ml) and glutamine (0.29 mg/ml), and OPI media supplement (Sigma) in 24-well plates. The neuroblastoma cell lines were transfected by the use of a fast forward method. Briefly, the DNA (0.6–1.2 μg/well) and Lipofectamine 2000 (1 μl/well) were mixed together and incubated at room temperature for 15 minutes. Cells were then trypsinized, suspended in culture medium, and placed onto a 24-well plate and the DNA-lipofectamine was added to the cell suspension. Transfected cells were incubated in a CO2 incubator for 24 to 36 hours and subjected to further analysis.
Luciferase activity was analyzed using the dual-luciferase assay system (Promega). 50 ng of pRL-tk, 0.1 μg of luciferase reporter plasmids, and effector plasmids were mixed with Lipofectamine 2000 and placed in 24-well plates. The total amount of plasmid DNA per well was adjusted by adding pcDNA3. After 24 hours of transfection, the cells were lyzed with a passive lysis buffer (Promega), following the manufacturer’s instructions. Each experiment was performed at least twice in triplicate. Luminescence was assayed with a VERITAS luminomator (Turner bioscience, USA).
Antibodies and immunofluorescence analysis
Antibodies to the Hemagglutinin (HA) epitope tag (mouse: 12CA5, rat: 3F10, F. Hoffmann-La Roche, Basel, Switzerland) and Myc (9E10: Santa-Cruz Biotechnology, Santa-Cruz, CA) were used.
Cultured cells were grown on 25 mm diameter collagen-coated glass coverslips (Iwaki, Tokyo, Japan) placed on 6-well plate wells. Cells on coverslips were washed and fixed with 4% paraformaldehyde in PBS on ice for 10 minutes, followed by permeabilization with 100% methanol at −20 °C for 10 minutes. To perform indirect immunofluorescence, cells were incubated with primary antibodies in 50 mM Tris-Cl (pH 7.4), 150 mM sodium chloride, 4% (weight/volume) bovine serum albumin (F. Hoffmann-La Roche) overnight at 4 °C, followed by secondary antibodies (Alexa flore-488 and 655, Molecular Probes, Eugene, OR). Nuclei were visualized with DAPI in Vectashield (Vector laboratory, Burlingame, CA). The fluorescence microscopy system (Olympus, Tokyo, Japan) was used for generation of fluorescent images.
Results
NLK enhances the transcription activity of the β-catenin-TCF complex in osteosarcoma and neuroblastoma cell lines
It has been previously suggested that mouse NLK may have a shorter isoform, translated from an alternative initiation codon at the 72nd amino acid from the N-terminal end.33) We generated mammalian NLK expression vectors to examine whether the shorter NLK isoform can have different function from full-length protein. Figure 1 shows a schematic diagram of the expression vectors used in this study. We designed to express Flag-NLK, the full-length NLK without a tag sequence (named NLK-L), and the shorter NLK isoform (named NLK-S). The native NLK mRNA sequences flanking each potential initiation codon were used as leader sequences for translation in mammalian cells (see Materials and methods).
Fig. 1.
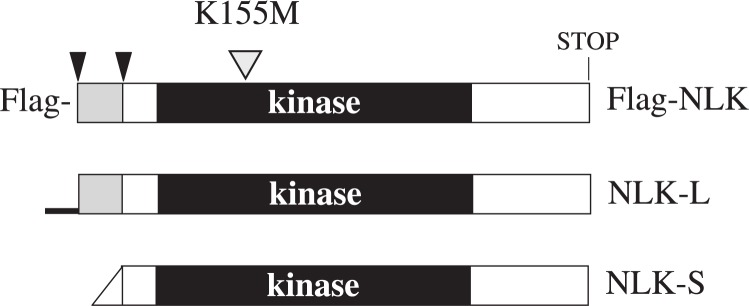
N-terminal structure of NLK expression vectors.
Schematic diagram of NLK constructs used in this study. NLK-L is the full-length protein without N-terminal tag sequences. NLK-S is the shorter NLK isoform. The rectangles indicate the open reading frame while the protein kinase domains are shown as black boxes. The tapered end on the left side of NLK-S indicates the leader sequence for the potential initiation codon (see Materials and methods). The thick line at the N-terminal of NLK-L indicates 5’ untranslated leader sequences of mouse NLK. The histidine rich regions are shown as hatched rectangles. The arrowheads indicate the methionine codons at the N-terminal regions. The mutation site of the kinase negative NLK is indicated with a hatched triangle above Flag-NLK.
We tested whether NLK isoforms can suppress the transcription activity of β-catenin ΔN (constitutive active mutant β-catenin31)) by the use of pTOPFLASH, a specific reporter for Wnt/β-catenin signaling, in 293 and SaOS-2 cells. In 293 cells, Flag-NLK suppressed reporter gene expression, a finding in agreement with previous reports.17), 34) NLK-S also suppressed the reporter gene expression but to a lesser degree (Fig. 2A). Unexpectedly, NLK showed apparent induction of the reporter gene expression in SaOS-2 cells, suggesting that NLK enhances β-catenin-TCF transcription (Fig. 2A).
Fig. 2.

NLK activates the pTOPFLASH transcription in osteosarcoma and neuroblastoma cells.
The vertical axis shows the relative values of normalized fire fly luciferase activity in the cell lysates. The luciferase activity of each cell lysate was further normalized with that of the lysate transfected with pcDNA3-MYC-β-catenin ΔN only as 1 otherwise specified. The error bars indicate the standard error of measurement (SEM). A. NLK can activate the pTOPFLASH transcription in SaOS-2 (osteosarcoma) cells but not in 293 (epithelial) cells. 0.3 μg/well and 0.1 μg/well of MYC-β-catenin ΔN and NLK expression vectors were used respectively. B. Specific enhancement of pTOPFLASH transcription by NLK depends on its kinase activity and co-expression of β-catenin ΔN. The amount of MYC-β-catenin ΔN and NLK expression vectors used was 0.1 μg/well. In the case of leftmost panel, which indicates the lack of enhancement of β-catenin-TCF activity by NLK without β-catenin ΔN, luciferase activity of each cell lysate was further normalized with that of the lysate transfected with pTOPFLASH only as 1. C. NLK can activate pTOPFLASH reporter with β-catenin ΔN in neuroblastoma cells. The amount of MYC-β-catenin ΔN and NLK expression vectors used was 0.3 μg/well.
We then examined whether NLK can activate the pTOPFLASH reporter gene expression without β-catenin ΔN, in which case we would hypothesize that NLK may be able to activate endogenous β-catenin or may activate pTOPFLASH through another pathway. Both Flag-NLK and NLK-S failed to activate pTOPFLASH without β-catenin ΔN in SaOS-2 cells, indicating that NLK activates Wnt/β-catenin signaling through the β-catenin-TCF complex (Fig. 2B, left panel). The kinase-negative NLK mutants did not enhance the reporter gene expression (Fig. 2B, middle panel), thus showing that the enhancement of expression by NLK is kinase-dependent. In addition, NLK-L could also induce pTOPFLASH expression in SaOS-2 cells in kinase-dependent manner (Fig. 2B, middle panel). Therefore, all NLK isoforms, regardless of their N-terminal sequences, enhanced β-catenin-TCF complex activity in SaOS-2 cells (Fig. 2A and B). Reporter gene analysis with pFOPFLASH, a negative control reporter for pTOPFLASH with mutant TCF/LEF binding sites, showed no activation, also indicating that NLK activates β-catenin-TCF transcription (Fig. 2B, right panel). Similarly, overexpression of NLK caused activation of the pTOPFLASH in Nh12 and NB39-nu neuroblastoma cell lines in a kinase-dependent manner (Fig. 2C).
Enhancement of transcription activity by NLK was specific to the β-catenin-TCF complex in osteosarcoma cells
We subsequently sought to examine whether the NLK short isoform could activate transcription factors other than the β-catenin-TCF complex. For this, we tested the endogenous activity of NF-κB and co-expressed p53 (expression vector: pCR3.1 p53) with overexpressed NLK-S and its kinase-negative mutants in SaOS-2 cells, using the pNF-κB-luc and pWWP-luc reporter genes respectively. NLK-S slightly reduced the expression of both reporters (Fig. 3). This moderate suppression of transcription factors has also been observed previously for Flag-NLK.32) Taken together, these results suggest that NLK is a specific activator of β-catenin-TCF complex in osteosarcoma and neuroblastoma cells.
Fig. 3.

NLK-S does not activate the reporter gene expression for NF-κB and p53 in SaOS-2 cells.
The amount of NLK expression vectors is 0.1 μg/well.
The amount of co-transfected p53 expression vector was 0.1 μg/well for the analysis of pWWP-luc reporter.
NLK does not change the nuclear localization of LEF1 in either 293 cells or SaOS-2 cells
It is known that the transcription activity of the worm TCF homolog POP-1 is regulated through its subcellular localization.14), 15), 35), 36) When the Wnt signaling is activated, the nuclear concentration of POP-1 is reduced and remaining nuclear POP-1 binds to β-catenin homologs and mediates the signal.35), 36) If the same mechanism in C. elegans occurred in some mammalian cells such as SaOS-2, NLK would reduce the nuclear localization of the TCF/LEF transcription factors for activation of Wnt signaling. It is an interesting question if NLK could differently regulate the localization of TCF between 293 cells and SaOS-2 cells. On the other hand, β-catenin ΔN usually accumulates in the nucleus to activate target genes. To examine whether subcellular localization of β-catenin ΔN is differently regulated by NLK depending on the cell type, Hemagglutinin (HA) tagged LEF1 (a member of the TCF transcription factor family) and β-catenin ΔN were co-expressed with NLK in either 293 or SaOS-2 cells. Co-expression with NLK did not alter the nuclear localization of either LEF1 or β-catenin ΔN in either 293 or SaOS-2 cells (Fig. 4A and B, respectively).
Fig. 4.

Overexpression of NLK-S did not change the nuclear localization of LEF1 and stabilized β-catenin in 293 cells (A) or SaOS-2 cells (B). Immunofluorescence analysis of LEF1 and β-catenin without (upper row) and with (lower row) NLK-S. For both cell lines, 1×105 of cells were transfected with 1 μg each of pcDNA3-HA-LEF1, pcDNA3-MYC-β-catenin ΔN with or without 1 μg pcDNA3-NLK-S in 6-well plate wells.
Activation of β-catenin-TCF by NLK is not mediated by phosphorylation of the conserved Threonine 155 and Serine 166 sites in LEF1
It has been shown that TCF is the target molecule of phosphorylation by NLK for the suppression of β-catenin-TCF complex activity. Moreover, it was shown that the phosphorylation sites were evolutionarily conserved between worm POP-1 and its mammalian homolog TCF/LEF.17), 34) These sites are matched to the consensus target phosphorylation sites of the MAPK superfamily (-X-S/T-P-X-). 37) It is an open question whether NLK phosphorylates the same LEF1 sites in order to activate and suppress its transcription activity. To examine this issue, we constructed HA-LEF1 T155A, S166A, and 155/166AA mutants and used a luciferase assay with pTOPFLASH as a reporter. Since preliminary experiments revealed that HA-LEF1, co-transfected with β-catenin ΔN, enhanced pTOPFLASH reporter gene expression in NB39-nu neuroblastoma cells but not in other cells (data not shown), we used this cell line to examine the effect of LEF1 mutants. Overexpression of the LEF1 mutants, T155A, S166A and 155/166AA, could induce pTOPFLASH reporter gene expression (Fig. 5A).
Fig. 5.

Activation of pTOPFLASH by NLK-S is not mediated by phosphorylation of the conserved Threonine 155 and Serine 166 sites in LEF1.
A and B. The dual luciferase assays of pTOPFLASH and pFOPFLASH with β-catenin ΔN in NB39-nu cells are shown. The luciferase activity of each cell lysate was further normalized with that of the lysate transfected with pcDNA3-MYC-β-catenin ΔN only as 1. The pcDNA-HA-LEF1 mutants used in the assay are indicated at the bottom of the graph (AA indicate the pcDNA-HA-LEF1-155/166AA). The amount of MYC-β-catenin ΔN, HA-LEF1, and NLK expression vectors used was 0.3 μg/well.
The activation of β-catenin-LEF1 complex by NLK was specific to pTOPFLASH, as the pFOPFLASH reporter was not activated (Fig. 5A). The transcription activity of the 155/166AA double mutant was very similar to that of wild-type LEF1 even when NLK was co-expressed (Fig. 5A). Furthermore, additional mutations in other potential phosphorylation sites (S132A/T155A/S166A triple mutant as S132TSAA and T155A/S166A/S200A triple mutant as TSAAS200A) also did not affect pTOPFLASH activity with NLK and β-catenin ΔN co-expression (Fig. 5B). The NLK kinase-negative mutant failed to activate the pTOPFLASH reporter gene expression when co-expressed with β-catenin ΔN and LEF1 mutants (Fig. 5B). These results indicated that the conserved phosphorylation sites in TCF/LEF would not be the target for transcription activation by NLK.
β-catenin phosphorylation site mutations do not affect its transcription activation by NLK
In C. elegans, LIT-1, an NLK homolog, can phosphorylate not only a TCF homolog, POP-1, but also a β-catenin homolog, WRM-1.11) It is therefore possible that NLK phosphorylates β-catenin to activate β-catenin-TCF transcription. To address this question, we constructed expression vectors for β-catenin ΔN mutated at potential phosphorylation sites by NLK (ΔNS191A, ΔNS246A, and ΔNS605A). Figure 6 shows that mutational status of β-catenin ΔN at the phosphorylation sites did not alter its transcription activity by NLK overexpression in SaOS-2 cells.
Fig. 6.

Mutations of phosphorylation sites in β-catenin would not affect the transcription activation by NLK-S.
The dual luciferase assays of pTOPFLASH with stabilized β-catenin in SaOS-2 cells were shown. The amount of MYC-β-catenin ΔN mutants and NLK-S expression vectors used was 0.1 μg/well.
Discussion
The present study revealed a new function of NLK in Wnt signaling: activation of β-catenin-TCF complex transcription activity. NLK enhanced pTOPFLASH transcription activity in osteosarcoma and neuroblastoma cells. This enhancement of transcription activity by NLK is dependent of co-expression of β-catenin. We also found that the NLK kinase activity is essential for the enhancement of the β-catenin-TCF complex transcription activity. Nuclear localization of the LEF1 and β-catenin ΔN was not altered by the co-expression of NLK in either epithelial (293) or osteosarcoma cells. Finally, we showed that the mutational status of certain potential phosphorylation sites of β-catenin and LEF1 did not alter their transcription activity by NLK overexpression.
Our finding that NLK activates pTOPFLASH reporter with β-catenin ΔN may appear to contradict previous studies, including our own,17), 30), 34), 38) which proposed NLK as a suppressor of β-catenin-TCF transcription activity. Even in the present study, however, NLK isoforms suppressed pTOPFLASH reporter to some degree in 293 cells. Thus, the effect of mammalian NLK on Wnt signaling seems to depend on the cell type; NLK plays some suppressive role in Wnt signaling in 29317) and DLD-1 cell lines30) and a positive role in SaOS-2, NB39-nu and Nh12 cell lines (the present study). This cell type specificity may explain the complexity of the phenotypes in genetically disrupted model organisms.18), 26), 28) It is unclear, however, whether NLK plays any positive role for β-catenin-TCF transcription activity in normal mammalian development. The molecular mechanism of this differential activity of NLK among cell lines is also an open question. One important aspect in this issue is that the cell types in which NLK plays a suppressive role are epithelial origin (293 and DLD-1). Some factors specific to the epithelial differentiation may induce suppressive outcome by NLK in Wnt/β-catenin signaling in these cell lines. It may also need to consider the possibility that NLK need other factors for activation of β-catenin-TCF complex and 293 cells lacks such factors (see following text).
How does NLK enhance the transcription activity of β-catenin-TCF transcription activity in SaOS-2 and neuroblastoma cell lines? The activation mechanism of β-catenin-TCF complex by NLK may be different from that of POP-1 by LIT-1 in C. elegans since NLK overexpression did not alter the nuclear localization of β-catenin or LEF1. Our results clearly indicate that activation of the β-catenin-TCF transcription activity by NLK depends on its kinase activity. Moreover, activation of the reporter gene expression by NLK is specific to the β-catenin-TCF complex in these cell lines (see Figs. 2 and 3). It is reasonable to assume that either β-catenin or TCF or both should be the NLK substrates. Ishitani et al. suggested that DNA binding of LEF1 decreased with its phosphorylation at threonine 155 and serine 166 by overexpression of NLK.17), 34) In C. elegans, the phosphorylation sites in POP-1 by LIT-1 were identified and these sites did not correspond to the target sites in LEF1 by NLK.16) It is quite possible that the NLK target sites for activation in the β-catenin-TCF complex are not conventional MAP kinase target sites. Alternatively, NLK may be an upstream kinase for yet unidentified kinases that phosphorylate the β-catenin-TCF complex at different sites. Identification of the NLK targets for activation of β-catenin-TCF complex will provide an important clue to understanding the regulatory mechanism of cell growth and differentiation.
Acknowledgments
We thank Drs. Osamu Tetsu and Frank McCormick for providing pcDNA3-Myc-β-catenin ΔN, our colleagues at the Molecular Oncology Division of National Cancer Center Research Institute for providing essential reagents, and Dr. Yuji Owada for his critical comments. This work was supported in part by a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1).Bienz, M. and Clevers, H. (2000) Cell 103, 311–320 [DOI] [PubMed] [Google Scholar]
- 2).Fearnhead, N.S., Britton, M.P. and Bodmer, W.F. (2001) Hum. Mol. Genet. 10, 721–733 [DOI] [PubMed] [Google Scholar]
- 3).Reya, T. and Clevers, H. (2005) Nature 434, 843–850 [DOI] [PubMed] [Google Scholar]
- 4).Polakis, P. (1999) Curr. Opin. Genet. Dev. 9, 15–21 [DOI] [PubMed] [Google Scholar]
- 5).Peifer, M. and Polakis, P. (2000) Science 287, 1606–1609 [DOI] [PubMed] [Google Scholar]
- 6).Sawa, H., Lobel, L. and Horvitz, H.R. (1996) Genes. Dev. 10, 2189–2197 [DOI] [PubMed] [Google Scholar]
- 7).Rocheleau, C.E., Downs, W.D., Lin, R., Wittmann, C., Bei, Y., Cha, Y.H., Ali, M., Priess, J.R. and Mello, C.C. (1997) Cell 90, 707–716 [DOI] [PubMed] [Google Scholar]
- 8).Thorpe, C.J., Schlesinger, A., Carter, J.C. and Bowerman, B. (1997) Cell 90, 695–705 [DOI] [PubMed] [Google Scholar]
- 9).Kaletta, T., Schnabel, H. and Schnabel, R. (1997) Nature 390, 294–298 [DOI] [PubMed] [Google Scholar]
- 10).Meneghini, M.D., Ishitani, T., Carter, J.C., Hisamoto, N., Ninomiya-Tsuji, J., Thorpe, C.J., Hamill, D.R., Matsumoto, K. and Bowerman, B. (1999) Nature 399, 793–797 [DOI] [PubMed] [Google Scholar]
- 11).Rocheleau, C.E., Yasuda, J., Shin, T.H., Lin, R., Sawa, H., Okano, H., Priess, J.R., Davis, R.J. and Mello, C.C. (1999) Cell 97, 717–726 [DOI] [PubMed] [Google Scholar]
- 12).Brott, B.K., Pinsky, B.A. and Erikson, R.L. (1998) Proc. Natl. Acad. Sci. USA 95, 963–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Choi, K.W. and Benzer, S. (1994) Cell 78, 125–136 [DOI] [PubMed] [Google Scholar]
- 14).Lin, R., Thompson, S. and Priess, J.R. (1995) Cell 83, 599–609 [DOI] [PubMed] [Google Scholar]
- 15).Lin, R., Hill, R.J. and Priess, J.R. (1998) Cell 92, 229–239 [DOI] [PubMed] [Google Scholar]
- 16).Lo, M.C., Gay, F., Odom, R., Shi, Y. and Lin, R. (2004) Cell 117, 95–106 [DOI] [PubMed] [Google Scholar]
- 17).Ishitani, T., Ninomiya-Tsuji, J., Nagai, S., Nishita, M., Meneghini, M., Barker, N., Waterman, M., Bowerman, B., Clevers, H., Shibuya, H. and Matsumoto, K. (1999) Nature 399, 798–802 [DOI] [PubMed] [Google Scholar]
- 18).Verheyen, E.M., Mirkovic, I., MacLean, S.J., Langmann, C., Andrews, B.C. and MacKinnon, C. (2001) Mech. Dev. 101, 119–132 [DOI] [PubMed] [Google Scholar]
- 19).Mirkovic, I., Charish, K., Gorski, S.M., McKnight, K. and Verheyen, E.M. (2002) Mech. Dev. 119, 9–20 [DOI] [PubMed] [Google Scholar]
- 20).Zeng, Y.A. and Verheyen, E.M. (2004) Development 131, 2911–2920 [DOI] [PubMed] [Google Scholar]
- 21).Freeman, M. and Bienz, M. (2001) EMBO Rep. 2, 157–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Tutter, A.V., Fryer, C.J. and Jones, K.A. (2001) Genes Dev. 15, 3342–3354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Waterman, M.L. (2004) Cancer Metastasis Rev. 23, 41–52 [DOI] [PubMed] [Google Scholar]
- 24).Korswagen, H.C., Herman, M.A. and Clevers, H.C. (2000) Nature 406, 527–532 [DOI] [PubMed] [Google Scholar]
- 25).Siegfried, K.R. and Kimble, J. (2002) Development 129, 443–453 [DOI] [PubMed] [Google Scholar]
- 26).Thorpe, C.J. and Moon, R.T. (2004) Development 131, 2899–2909 [DOI] [PubMed] [Google Scholar]
- 27).Bowerman, B. (2005) Cell 121, 662–664 [DOI] [PubMed] [Google Scholar]
- 28).Kortenjann, M., Nehls, M., Smith, A.J., Carsetti, R., Schuler, J., Kohler, G. and Boehm, T. (2001) Eur. J. Immunol. 31, 3580–3587 [DOI] [PubMed] [Google Scholar]
- 29).Reya, T., O’Riordan, M., Okamura, R., Devaney, E., Willert, K., Nusse, R. and Grosschedl, R. (2000) Immunity 13, 15–24 [DOI] [PubMed] [Google Scholar]
- 30).Yasuda, J., Tsuchiya, A., Yamada, T., Sakamoto, M., Sekiya, T. and Hirohashi, S. (2003) Biochem. Biophys. Res. Commun. 308, 227–233 [DOI] [PubMed] [Google Scholar]
- 31).Tetsu, O. and McCormick, F. (1999) Nature 398, 422–426 [DOI] [PubMed] [Google Scholar]
- 32).Yasuda, J., Yokoo, H., Yamada, T., Kitabayashi, I., Sekiya, T. and Ichikawa, H. (2004) Cancer Sci. 95, 52–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Kortenjann, M., Wehrle, C., Nehls, M.C. and Boehm, T. (2001) Gene 278, 161–165 [DOI] [PubMed] [Google Scholar]
- 34).Ishitani, T., Ninomiya-Tsuji, J. and Matsumoto, K. (2003) Mol. Cell. Biol. 23, 1379–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Kidd, A.R., 3rd, Miskowski, J.A., Siegfried, K.R., Sawa, H. and Kimble, J. (2005) Cell 121, 761–772 [DOI] [PubMed] [Google Scholar]
- 36).Maduro, M.F., Kasmir, J.J., Zhu, J. and Rothman, J.H. (2005) Dev. Biol. 285, 510–523 [DOI] [PubMed] [Google Scholar]
- 37).Davis, R.J. (1993) J. Biol. Chem. 268, 14553–14556 [PubMed] [Google Scholar]
- 38).Ishitani, T., Kishida, S., Hyodo-Miura, J., Ueno, N., Yasuda, J., Waterman, M., Shibuya, H., Moon, R.T., Ninomiya-Tsuji, J. and Matsumoto, K. (2003) Mol. Cell. Biol. 23, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]