

Abstract
Retinal degeneration leads to progressive photoreceptor cell death, resulting in vision loss. Subsequently, inner retinal neurons develop aberrant synaptic activity, compounding visual impairment. In retinal ganglion cells, light responses driven by surviving photoreceptors are obscured by elevated levels of aberrant spiking activity. Here, we demonstrate in rd10 mice that targeting disruptive neuronal circuitry with a gap junction antagonist can significantly reduce excessive spiking. This treatment increases the sensitivity of the degenerated retina to light stimuli driven by residual photoreceptors. Additionally, this enhances signal transmission from inner retinal neurons to ganglion cells, potentially allowing the retinal network to preserve the fidelity of signals either from prosthetic electronic devices, or from cells optogenetically modified to transduce light. Thus, targeting maladaptive changes to the retina allows for treatments to use existing neuronal tissue to restore light sensitivity, and to augment existing strategies to replace lost photoreceptors.
Introduction
Retinal degeneration (RD) is a family of blinding diseases that stem from a variety of mutations in photoreceptor cells and the retinal pigment epithelium (Hartong et al., 2006). Visual impairment from RD occurs as a result of progressive photoreceptor degeneration, compounded by the concurrent structural remodeling of inner retinal neurons, which includes changes in glutamate receptor expression, and synaptic disorganization (for review, see Jones et al., 2012). Shortly after disease onset, alterations to neuronal physiology occur, and oscillatory activity develops in bipolar, amacrine, and ganglion cells (Margolis et al., 2008; Borowska et al., 2011). This aberrant neuronal activity can exacerbate visual impairment, as it obscures signaling from bipolar cells to ganglion cells (Yee et al., 2012). Thus, the loss of photoreceptors is compounded by maladaptive interactions within the inner retinal network, as neuronal oscillations interfere with signal transmission from the retina to the brain. This suggests that treatment strategies may extend beyond replacing photoreceptor function to address inner retinal dysfunction.
Recent studies have investigated the source of this aberrant activity. A possible mechanism is the disruption of a network of gap junction-linked amacrine and bipolar cells, which has been shown to oscillate in wild-type (wt) retina when metabotropic glutamate receptors (mGluRs) are blocked, inducing conditions similar to the loss of photoreceptors (Trenholm et al., 2012). It has been found that blocking gap junction transmission significantly reduces aberrant oscillations (Borowska et al., 2011; Menzler and Zeck, 2011; Yee et al., 2012).
We hypothesized that reducing or abolishing oscillatory activity will increase the fidelity of signals transmitted through degenerated retina. To test this, we recorded spiking activity from retinal ganglion cells (RGCs) of rd10 mice, which carry a mutation in the Pde6b gene, a frequent cause of retinitis pigmentosa in humans (Gargini et al., 2007). As in other forms of RD that primarily affect rod photoreceptors, cones in rd10 retinas remain present and functional for an extended period after rod death, maintaining residual light sensitivity (Thyagarajan et al., 2010; Stasheff et al., 2011). However, light responses originating from surviving cone photoreceptors are obscured by aberrant spiking activity. Our data indicate that reducing or eliminating aberrant oscillatory activity with the gap junction blocker meclofenamic acid (MFA) increases sensitivity to light stimuli in degenerated retinas, by increasing signal fidelity between RGCs and inner retinal cells. Thus, reducing synaptic noise is crucial for enhancing signal transmission in degenerated retina, which not only rescues surviving cone function but can also augment the efficacy of interventions that replace lost photoreceptor function.
Materials and Methods
Animals.
In all experimental procedures, animals were treated according to the regulations in the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, in accordance with protocols approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College, and with the National Institutes of Health Guide for the care and use of laboratory animals. Mice of either sex were obtained from The Jackson Laboratory.
Preparation of retinal whole mounts.
Methods for whole-mount tissue preparation have been described in detail previously (Toychiev et al., 2013). After the animal was killed, its eyes were enucleated and placed in bicarbonate-buffered Ames solution, constantly equilibrated with 95% O2 and 5% CO2. The cornea, iris, and lens were removed. The retina was dissected into four equal quadrants. Quadrants were attached photoreceptor surface down to a modified Millicell filter (Millipore). This preparation was transferred to a recording chamber on the stage of an upright Nikon FN1 microscope equipped with Hoffman modulation contrast optics (Modulation Optics). The recording chamber was constantly bathed (1 ml/min) with bicarbonate-buffered Ames solution, constantly equilibrated with 95% O2 and 5% CO2. Pharmacological agents were applied using an eight-channel superfusion system (Warner Instruments). All experiments were performed at a temperature of 32°C. All reagents were obtained from Sigma.
Electrophysiology.
To compare RGCs of similar classes, α-like RGCs were targeted based on large soma size and number of primary dendritic processes. In some instances, to increase the number of matched RGC classes across control and rd10 groups, mice homozygous for the Thy1-YFP allele (B6.Cg-Tg(thy1-YFP)/J) with genetically prelabeled RGCs were used as control. Extracellular spiking activity was obtained from RGCs in a loose-patch mode using the electrode filled with HEPES-buffered extracellular Ringer's solution, containing the following (in mm): 137 NaCl, 2.5 KCl, 2.5 CaCl2, 1.0 MgCl2, 10 Na-HEPES, 28 glucose, pH 7.4. Electrodes were pulled from borosilicate glass (1B150F-4; WPI) with a P-97 Flaming/Brown puller (Sutter Instruments) and had a measured resistance of ∼4–7 MΩ. All recordings were made with a MultiClamp 700B patch-clamp amplifier (Molecular Devices) using Signal (CED). Data were filtered at 5 kHz with a four-pole Bessel filter and were sampled at 15 kHz. After recordings, the cellular membrane under the pipette was ruptured to allow dye filling of the cell for morphological phenotyping.
Cell identification.
The pipette solution was supplemented with 0.05% sulforhodamine B or AlexaFluor-568 hydrazide to visualize nonprelabeled cells. Contrast and fluorescent images of the cell were documented with a modified Nikon D5000 DSLR attached to the microscope. The preparation was immediately placed into a glass-bottom culture dish (MatTek) and transferred to the stage of a Nikon C-1 confocal microscope. A z-stack of 160 images was acquired at 0.5 μm steps at a resolution of 1024 × 1024 pixels. Dendritic stratification was measured relative to the proximal (0%) to the distal margins (100%) of the inner plexiform layer. In general, ON cells were defined as stratifying at <60%, with OFF cells stratifying at >60% of inner plexiform layer depth. The precise depth of RGC dendritic stratification was confirmed by immunostaining against choline acetyl transferase (goat anti-ChAT, 1:2000; Millipore Bioscience Research Reagents).
Retinal stimulation and analysis.
Light stimulation and electrical stimulation were both performed in whole-mount retina. The microscope's illuminator was used to deliver a 200 μm spot of light centered on the RGC. An aperture, a series of neutral density filters, the FN-C LWD condenser (Nikon), and a Uniblitz shutter (Vincent Associates) were used to control the size, intensity, focal plane, and duration of the stimulus, respectively. Stimulation routines were controlled by Signal 2 software (CED). The tissue was light adapted at 30 cd/m2. To generate an intensity-response function, RGCs were presented with a 1 s stimulus separated by 60 s blank trials while maintaining the background light level throughout. The intensity of the light stimulus was changed in steps of either 0.25 or 0.5 log units, ranging from 10 to 10,000 Rh*/rod/s above adapting level, covering the range of cone-mediated responses (Deans et al., 2002). For electrical stimulation, a positive current pulse (2 ms, 0.003–1.0 mA; Grass Technologies) was applied to the bipolar cells in outer plexiform layer using a patch pipette filled with extracellular solution (see Figure 3A, top). The stimulating electrode was first inserted into the bipolar cell layer. Once the stimulating electrode was stable and the retinal tissue had settled (2–3 min), the RGC above was targeted for recordings. Figure 3A (bottom) displays a fluorescent image from an actual experiment, showing the positioning of both the stimulating and recording electrodes. To measure how aberrant synaptic inputs interfere with responses, signal-to-noise ratio (SNR) was calculated from spiking responses by dividing instantaneous AP firing frequency during the response (signal) by the firing frequency of unstimulated activity (noise) over an equivalent 1 s epoch. The data points were fitted with the Hill equation as follows:
 |
where Rmax is the maximum response, I is the stimulus intensity, b is the slope factor, and c is the stimulus intensity resulting in the half-maximal response. The c value was used to determine sensitivity of the response (Sampath and Rieke, 2004). Statistics were performed using Sigmaplot (Systat Software).
Figure 3.

Blockade of aberrant activity improves electrically evoked responses in RD. A, Paradigm for recording electrically evoked responses in situ. For illustration purposes, a cross-sectional view is shown. In the whole-mount retina, bipolar cells were activated with focal electrical stimuli (0.1–100 μA; 2 ms) delivered by an extracellular electrode placed in the inner nuclear layer. Fluorescence image shows a patch electrode (left) attached to an RGC cell body and a stimulation electrode (right) inserted into bipolar cell layer. In this case, both electrodes are filled with solution containing sulforhodamine. B, Electrically evoked spiking activity recorded in loose-patch mode from rd10 RGCs before and after application of MFA. Red traces represent stimulus timing. Dashed vertical lines indicate response onset. C, Responses fit by the Hill equation. D, MFA increases sensitivity to electrical stimuli (n = 6; p < 0.001, paired t test). Error bars indicate SEM. *p < 0.01.
Results
The following data demonstrate that, in rd10 retina: (1) residual cone-driven light responses are obscured by elevated RGC spiking activity; (2) blocking gap junction transmission reduces spiking and increases light sensitivity; and (3) the fidelity of responses to direct electrical stimulation of the inner retina is increased after the block of gap junctions.
Light responses are masked by aberrant bursting in RD retina
First, to establish the effect of elevated resting activity on evoked activity, we recorded light responses from RGCs in rd10 retinas aged between postnatal day 37 and postnatal day 46, an age where rod photoreceptors have mostly degenerated but cone photoreceptors are present and functional, enabling the retina to respond to light (Gargini et al., 2007; Stasheff et al., 2011). To isolate the cone contribution to light responses, we recorded spiking activity from RGCs in light-adapted whole-mount retinas. “Bursting” activity was defined as repetitive spiking activity in which groups of ≥3 spikes recur at a frequency of 3–12 Hz. The underlying synaptic currents in these cells are referred to as oscillatory currents. When using a spot of light producing 250 rhodopsin photoisomerizations per second (Rh*/s), responses in rd10 RGCs were indistinguishable from elevated resting spiking activity, or “bursting,” in rd10 RGCs (Fig. 1A; compare with wt in Fig. 2D). The resting action potential frequency (AP/s) of rd10 RGCs is several times greater than that of age-matched wt controls (16.10 ± 0.71, n = 26 vs 1.16 ± 0.13, n = 10, p < 0.01, ANOVA), masking the light-evoked activity. Although most RGCs exhibited bursting, the amount of bursting varied between cells, with a subset of quiescent cells that did not burst (Fig. 1A, right). It is notable that bursting in rd10 RGCs is distinguished from the sustained spiking activity observed in certain wt RGCs. Although these wt RGCs have an elevated resting spike rate, spikes are not temporally grouped as seen in rd10 (Fig. 1B, left). This is reflected in autocorrelograms of the respective spiking activity, in which rd10 activity has both a peak at ∼0 s and frequency harmonics, in contrast to wt activity having a trough at ∼0 s and no harmonics (Fig. 1B, right).
Figure 1.
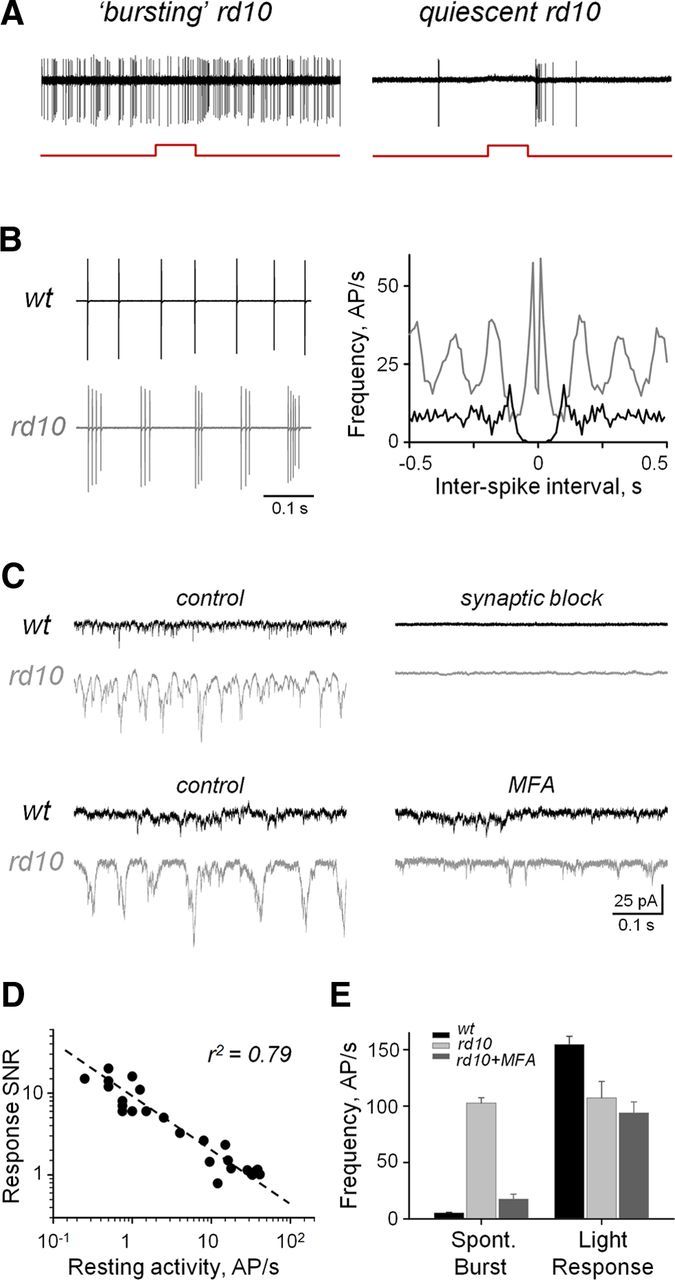
Aberrant bursting activity masks light responses in RD. A, Resting activities and light responses from example bursting and quiescent rd10 Off RGCs. Red steps under spiking traces indicate timing of the stimulus. B, Resting spiking activity (left) and autocorrelograms of spiking activity in wt and rd10 RGCs (right). C, Spontaneous EPSCs under conditions of synaptic block and after application of MFA (50 μm). sEPSCs were recorded in whole-cell patch-clamp mode at a holding potential of −60 mV, a reversal for Cl− currents. Synaptic block used a mixture of strychnine (5 μm), SR-95531 (10 μm), TPMPA (100 μm), CNQX (10 μm), and d-AP5 (50 μm), selective inhibitors of glycine, GABA, AMPA, and NMDA receptors, respectively. D, Response SNR as a function of resting spiking activity (n = 26 RGCs, of which 12 On and 14 Off). E, Average instantaneous frequency rates within individual spontaneous bursts (left group) and light-evoked response (right group) in wt (n = 33; 13 On, 20 Off), and bursting rd10 RGCs before (n = 152; 72 On, 80 Off) and after (n = 38; 18 On, 20 Off) treatment with MFA (50 μm). AP/s, Action potentials per second. Error bars indicate SEM.
Figure 2.

Blockade of aberrant activity restores light responses in RD retina. Maximum intensity projection fluorescence images of representative On (A) and Off (C) rd10 RGCs. Top views, Inverted contrast images of RGCs filled with Alexa-568. Side views, RGC dendritic stratifications (red) proximal and distal to cholinergic amacrine cell On and Off ChAT bands (green), respectively. B, D, Series of light-evoked responses from corresponding On (B, P39) and Off (D, P46) rd10 RGCs before (middle) and after application of MFA (right, 50 μm), and age-matched wt controls (left). Spiking responses were obtained to a 200 μm spot of light. Red steps indicate stimulus timing. Dashed vertical lines highlight the onset of the response. E, Intensity-response function measurements, fitted with the Hill equation. F, Same plots from E with expanded y-axis illustrating weak responses in untreated rd10 RGCs. G, Light sensitivity profiles of wt (n = 5) and rd10 (n = 8) RGCs in control and treated conditions. Sensitivity values were obtained from the Hill equation (see Materials and Methods). MFA treatment increases light sensitivity of rd10 RGCs (n = 8; p = 0.009, paired t test). AP/s, Action potentials per second. Error bars represent SEM. *p < 0.01.
Next, we demonstrated that aberrant activity could be blocked. It has been established that the source of this activity is synaptic input to RGCs, rather than an intrinsic property (Margolis et al., 2008). As expected, blocking synaptic inputs to wt and rd10 RGCs eliminates spontaneous excitatory currents (Fig. 1C, top). Recent studies have identified a key role for gap junctions in mediating spontaneous bursting activity in RD retina (Borowska et al., 2011; Menzler and Zeck, 2011; Yee et al., 2012). Therefore, we applied MFA (50 μm), a potent gap junction blocker (Pan et al., 2007), to target aberrant activity. Application of MFA eliminated oscillatory currents but preserved excitatory input to rd10 RGCs (Fig. 1C, bottom). This treatment had no effect on wt or quiescent rd10 RGCs.
To quantify the level at which resting activity affected the fidelity of the light response of the cell, an SNR was calculated (see Materials and Methods). Across RGCs, a linear correlation was revealed between the resting spike rate and the SNR (Fig. 1D), demonstrating that elevated spontaneous activity reduces signal transmission. Moreover, the resting activity in RD retina is conflated with light responses. Spontaneous bursts of spiking activity cannot be discriminated from evoked responses in rd10 RGCs, as the instantaneous spike rates of individual spontaneous bursts are similar to the spike rates of light responses (Fig. 1E, light gray bars).
Light sensitivity is restored by blocking aberrant bursting
Next, we tested whether blocking aberrant bursting would increase the overall light sensitivity of RGCs, by recording responses to light stimuli over a range of intensities. After MFA treatment, the spontaneous bursting frequency of rd10 RGCs decreased significantly (Fig. 1E, dark gray bars). At lower light intensities (<250 Rh*/s), light responses in untreated rd10 RGCs were indistinguishable from resting activity (SNR ∼ 1). MFA reduced bursting, and light responses were clearly distinct from background activity over a broader range of light intensities, particularly at lower intensities. This was the case for both On and Off cells (Fig. 2B,D).
Sensitivity was calculated by fitting the Hill equation to a plot of SNR as a function of stimulus intensity, then taking the c value, the stimulus intensity eliciting the half-maximum response (Fig. 2E; see Materials and Methods). Treatment with MFA significantly increased the sensitivity of rd10 RGCs compared to untreated conditions (0.83 ± 0.05, n = 5 vs 0.34 ± 0.12, n = 5, p < 0.01). This was particularly true for lower light intensities (Fig. 2F). Sensitivity in wt cells was not affected by MFA (p = 0.25, n = 4; paired t test), indicating that this treatment is not detrimental to cone-mediated responses, consistent with MFA primarily affecting the rod pathway (Veruki and Hartveit, 2009). MFA-treated rd10 RGCs were still less sensitive than untreated wt RGCs (1.46 ± 0.01, n = 4, p < 0.01), consistent with photoreceptor loss after disease onset (Fig. 2G). These data establish that reducing gap junction-mediated aberrant activity in RD retina restores sensitivity to light stimuli.
Blocking aberrant bursting improves signal transmission through RD retina
Retinal degeneration is a progressive disease that, at late stages, leads to the loss of functional photoreceptor cells. Surviving cones lose their outer segments, affecting their ability to respond to light (Lin et al., 2009; Busskamp et al., 2010). At this stage, treatment strategies aim to replace the function of lost photoreceptors either with light-sensitive prosthetic devices or by using optogenetic tools, such as halorhodopsin or channelrhodopsin-2, to confer light sensitivity to surviving neurons (Freeman et al., 2011; Sahel and Roska, 2013). However, even with an auxiliary mode of phototransduction, a dysfunctional retinal network can still impair signal transmission. Under these circumstances, is reducing aberrant activity still a valid approach? To answer this, we electrically stimulated cells presynaptic to RGCs, bypassing photoreceptors (Fig. 3A). This approach simulates the activation of surviving retinal circuitry that is achieved with either prosthetic devices or optogenetic tools, which are independent of the function of photoreceptor outer segments.
As with light stimuli, responses to electrical stimulation were masked by aberrant bursting, which was reduced with MFA treatment (Fig. 3B). MFA significantly increased the peak SNR of rd10 RGCs (3.35 ± 0.87 to 6.73 ± 0.64, n = 6; p < 0.001, paired t test), as well as sensitivity (0.81 ± 0.28 to 1.98 ± 0.86, n = 6; p < 0.001, paired t test) (Fig. 3C,D). A stimulus of 1.06 ± 0.46 μA was sufficient to evoke a half-maximum response in RGCs, in contrast to 15.45 ± 5.41 μA before treatment. This is a marked improvement, particularly considering that the area of focal electrical activation is a fraction of the dendritic field of the recorded RGC; that is, cells transmitting the electrically evoked signal to the RGC are outnumbered by cells transmitting aberrant noise. These data demonstrate that the physiological changes induced by RD compromise photoreceptor-independent signal transmission through the inner retina.
Discussion
Our data have demonstrated that increased resting spiking activity has a disruptive effect on the ability to encode light responses in rd10 RGCs. Light sensitivity can be restored by selectively blocking gap junctions to reduce excessive spiking, which also improves the signal transmission within the inner retina.
Photoreceptor degeneration is the primary effect of retinal degenerative diseases, such as retinitis pigmentosa. As the disease progresses, the retina undergoes a phased process of remodeling that results in soma hypertrophy and migration, the retraction of bipolar cell dendrites, and the sprouting of neurites from all cell classes, which fasciculate and form microneuromas (Jones et al., 2012). However, aberrant neuronal activity develops soon after disease onset and is present in mouse models of RD before dendritic retraction occurs and while cone photoreceptors remain (Gargini et al., 2007; Stasheff et al., 2011).
The source of aberrant activity in degenerated retina remains unclear, although it is established that it is not the result of intrinsic RGC activity (Margolis et al., 2008). Several sources of these oscillations have been proposed. Disrupted inhibitory inputs from the amacrine cell network have a critical role in mediating dystrophic activity (Yee et al., 2012). This activity may also involve intrinsic bipolar cell oscillations, which can be elicited in wt retina by either blocking either inhibitory transmission (Borowska et al., 2011; Yee et al., 2012), or blocking mGluRs, essentially mimicking the loss of glutamatergic input to bipolar cells from photoreceptors (Trenholm et al., 2012). Our data demonstrates that blocking gap junctions significantly reduces oscillations and restores light sensitivity. Why is this? Because a large diversity of gap junction interactions exist in the retina, between numerous cell classes (Bloomfield and Volgyi, 2009), it is difficult to ascertain which play a role in generating or propagating this activity. A possible mechanism is that rod degeneration induces aberrant activity in the rod pathway, whereas the still functional cone pathway receives aberrant input from the rod pathway. Gap junctions are essential in transmitting rod pathway signals to RGCs via the cone pathway (Deans et al., 2002). This interaction may introduce noise into the cone pathway at the level of bipolar cells, reducing light sensitivity at the level of RGCs. Another possibility is that gap junctions mediate input from an unidentified class of dystrophic oscillating amacrine cells (Yee et al., 2012). Blocking gap junctions may prevent these disruptions of the cone pathway, enhancing the fidelity of light-driven signals from residual cones.
Preserving cone-mediated signal transmission is important, as central vision in humans relies on cones. Rod degeneration induces peripheral scotomas (Hartong et al., 2006), but rescuing peripheral cone function could preserve some degree of peripheral vision. In humans, retinal degeneration can be clinically detected before a significant loss of vision, which allows interventions to take advantage of a large population of surviving photoreceptors. To optimize the function of these cells, treatment must compensate for aberrant neuronal activity, as this can degrade image processing at and beyond the retina. We have demonstrated one approach to this treatment strategy, by blocking gap junctions. The limitation of this approach is that gap junctions are crucial to crossover interactions between visual pathways of the retina, such as between On and Off circuits, and between mechanisms for dark and light adaptation (Bloomfield and Volgyi, 2009), which may thus introduce new complications to visual processing. On the other hand, blocking gap junctions either pharmacologically (Fig. 2) or genetically (Deans et al., 2002; Bloomfield and Volgyi, 2009) does not significantly affect cone-mediated light sensitivity. Additionally, alternative approaches to blocking aberrant activity may be even more problematic. For instance, although blockade of inhibitory transmission has been shown to eliminate high-frequency oscillations in degenerated retina, it increases low-frequency oscillations from bipolar cells (Yee et al., 2012). Targeting spiking amacrine cells with tetrodotoxin can also reduce oscillations (Trenholm et al., 2012). However, this intervention would also block the spiking of RGCs, which would prevent retinal output to the brain. Ideally, a genetic approach to silence aberrant bursting cells, with small hairpin RNA, could be used to provide a more permanent solution. However, until the specific genes can be identified and targeted, a pharmacological approach is a viable substitute. Our approach has the additional benefit of using MFA (Meclomen), an FDA-approved NSAID. Together, this provides a safe and selective way of silencing aberrant activity while preserving cone-mediated light responses.
In conclusion, we have shown that targeting aberrant neuronal circuitry can significantly improve visually evoked responses from surviving cones in RD. This strategy can use the time window wherein functional surviving photoreceptors are present. Additionally, reducing aberrant activity could be beneficial in conjunction with therapies that restore photoreceptor function, using optogenetics and/or retinal prosthetics (Freeman et al., 2011; Sahel and Roska, 2013). MFA and related NSAIDs are promising candidates for further testing. Together, these data demonstrate that targeting aberrant activity, either alone or in combination with methods to restore phototransduction, can be an effective strategy for treating RD.
Footnotes
This work was supported by National Institutes of Health Grant R01-EY020535 to B.T.S.
The authors declare no competing financial interests.
References
- Bloomfield SA, Völgyi B. The diverse functional roles and regulation of neuronal gap junctions in the retina. Nat Rev Neurosci. 2009;10:495–506. doi: 10.1038/nrn2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowska J, Trenholm S, Awatramani GB. An intrinsic neural oscillator in the degenerating mouse retina. J Neurosci. 2011;31:5000–5012. doi: 10.1523/JNEUROSCI.5800-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busskamp V, Duebel J, Balya D, Fradot M, Viney TJ, Siegert S, Groner AC, Cabuy E, Forster V, Seeliger M, Biel M, Humphries P, Paques M, Mohand-Said S, Trono D, Deisseroth K, Sahel JA, Picaud S, Roska B. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science. 2010;329:413–417. doi: 10.1126/science.1190897. [DOI] [PubMed] [Google Scholar]
- Deans MR, Volgyi B, Goodenough DA, Bloomfield SA, Paul DL. Connexin36 is essential for transmission of rod-mediated visual signals in the mammalian retina. Neuron. 2002;36:703–712. doi: 10.1016/S0896-6273(02)01046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman DK, Rizzo JF, 3rd, Fried SI. Encoding visual information in retinal ganglion cells with prosthetic stimulation. J Neural Eng. 2011;8 doi: 10.1088/1741-2560/8/3/035005. 035005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargini C, Terzibasi E, Mazzoni F, Strettoi E. Retinal organization in the retinal degeneration 10 (rd10) mutant mouse: a morphological and ERG study. J Comp Neurol. 2007;500:222–238. doi: 10.1002/cne.21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- Jones BW, Kondo M, Terasaki H, Lin Y, McCall M, Marc RE. Retinal remodeling. Jpn J Ophthalmol. 2012;56:289–306. doi: 10.1007/s10384-012-0147-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin B, Masland RH, Strettoi E. Remodeling of cone photoreceptor cells after rod degeneration in rd mice. Exp Eye Res. 2009;88:589–599. doi: 10.1016/j.exer.2008.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DJ, Newkirk G, Euler T, Detwiler PB. Functional stability of retinal ganglion cells after degeneration-induced changes in synaptic input. J Neurosci. 2008;28:6526–6536. doi: 10.1523/JNEUROSCI.1533-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzler J, Zeck G. Network oscillations in rod-degenerated mouse retinas. J Neurosci. 2011;31:2280–2291. doi: 10.1523/JNEUROSCI.4238-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan F, Mills SL, Massey SC. Screening of gap junction antagonists on dye coupling in the rabbit retina. Vis Neurosci. 2007;24:609–618. doi: 10.1017/S0952523807070472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahel JA, Roska B. Gene therapy for blindness. Annu Rev Neurosci. 2013;36:467–488. doi: 10.1146/annurev-neuro-062012-170304. [DOI] [PubMed] [Google Scholar]
- Sampath AP, Rieke F. Selective transmission of single photon responses by saturation at the rod-to-rod bipolar synapse. Neuron. 2004;41:431–443. doi: 10.1016/S0896-6273(04)00005-4. [DOI] [PubMed] [Google Scholar]
- Stasheff SF, Shankar M, Andrews MP. Developmental time course distinguishes changes in spontaneous and light-evoked retinal ganglion cell activity in rd1 and rd10 mice. J Neurophysiol. 2011;105:3002–3009. doi: 10.1152/jn.00704.2010. [DOI] [PubMed] [Google Scholar]
- Thyagarajan S, van Wyk M, Lehmann K, Löwel S, Feng G, Wässle H. Visual function in mice with photoreceptor degeneration and transgenic expression of channelrhodopsin 2 in ganglion cells. J Neurosci. 2010;30:8745–8758. doi: 10.1523/JNEUROSCI.4417-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toychiev AH, Sagdullaev B, Yee CW, Ivanova E, Sagdullaev BT. A time and cost efficient approach to functional and structural assessment of living neuronal tissue. J Neurosci Methods. 2013;214:105–112. doi: 10.1016/j.jneumeth.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenholm S, Borowska J, Zhang J, Hoggarth A, Johnson K, Barnes S, Lewis TJ, Awatramani GB. Intrinsic oscillatory activity arising within the electrically coupled AII amacrine-ON cone bipolar cell network is driven by voltage-gated Na+ channels. J Physiol. 2012;590:2501–2517. doi: 10.1113/jphysiol.2011.225060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veruki ML, Hartveit E. Meclofenamic acid blocks electrical synapses of retinal AII amacrine and on-cone bipolar cells. J Neurophysiol. 2009;101:2339–2347. doi: 10.1152/jn.00112.2009. [DOI] [PubMed] [Google Scholar]
- Yee CW, Toychiev AH, Sagdullaev BT. Network deficiency exacerbates impairment in a mouse model of retinal degeneration. Front Syst Neurosci. 2012;6:8. doi: 10.3389/fnsys.2012.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]