

Abstract
The synaptic toxicity of soluble amyloid-β (Aβ) oligomers plays a critical role in the pathophysiology of Alzheimer's disease (AD). Here we report that overexpressed α1-takusan, which we previously identified as a protein that enhances synaptic activity via interaction with PSD-95, mitigates oligomeric Aβ-induced synaptic loss. In contrast, takusan knockdown results in enhanced synaptic damage. α1-Takusan interacts with tau either directly or indirectly, and prevents Aβ-induced tau hyperphosphorylation and mitochondrial fragmentation. Deletion analysis identified the second domain (D2) within the takusan protein that is required for PSD-95 clustering and synaptic protection from Aβ. A 51 aa sequence linking D2 to the PDZ-binding C terminus was found to be as effective as full-length takusan in protecting synapses from Aβ-induced damage. Moreover, a sequence containing the D2 from the human protein discs large homolog 5, when linked to a C-terminal PDZ-binding motif, can also increase the clustering of PSD-95 in cortical dendrites. In summary, α1-takusan protects synapses from Aβ-induced insult via interaction with PSD-95 and tau. Thus, takusan-based protein sequences from either mouse or human may be of potential therapeutic benefit in AD.
Introduction
Alzheimer's disease (AD) is the most common form of dementia in the elderly. Emerging evidence suggests that amyloid-β (Aβ)-induced synaptic damage is dependent on the aberrant processing of the microtubule-associated protein tau (Ballatore et al., 2007; Morris et al., 2011). Although previously known as an axonal protein, tau is also expressed in dendrites and postsynaptic densities (PSDs), albeit at much lower levels (Ittner et al., 2010). In AD transgenic mice expressing mutant human amyloid precursor protein (APP), AD-like defects are mitigated by the reduction of total or dendritic tau expression (Roberson et al., 2007, 2011; Ittner et al., 2010). In cultured neurons, oligomeric Aβ application induces tau hyperphosphorylation and dendritic disruption, which can be rescued by antibodies against Aβ or by tau reduction with RNAi (De Felice et al., 2008; Zempel et al., 2010; Jin et al., 2011). Tau hyperphosphorylation has also been observed in cultured human neurons differentiated from AD patient-derived IPSCs (Israel et al., 2012). Collectively, these findings suggest that dendritic/synaptic tau and its hyperphosphorylation play an important role in Aβ-induced synaptic loss.
PSD-95 is a scaffold protein that plays a critical role in synaptic development and plasticity by anchoring a variety of postsynaptic proteins to the postsynaptic density [including NMDA-type glutamate receptors (NMDARs) and AMPA-type glutamate receptors (AMPARs)] (Sheng and Hoogenraad, 2007). Aβ oligomers may directly interact with PSD-95 as well as with NMDARs and AMPARs at postsynaptic sites (Lacor et al., 2007; Pham et al., 2010; Zhao et al., 2010). Moreover, the expression of PSD-95 proteins is reduced in AD brains (Gylys et al., 2004; Love et al., 2006), and the degree of reduction correlates with both the level of Aβ oligomers and the severity of dementia (Pham et al., 2010; Proctor et al., 2010; Sultana et al., 2010). AD transgenic neurons and WT neurons exposed to Aβ oligomers contain less PSD-95, concomitant with spine loss and reduction in AMPAR expression (Almeida et al., 2005; Roselli et al., 2005; Spires et al., 2005). It is thus possible that PSD-95 disruption is an important step in Aβ-induced synaptic injury.
α1-Takusan (Tksn) belongs to the takusan gene family, characterized by the takusan domain (formerly called DUF622) that is shared by the discs large homolog 5 (DLG5) protein in humans (Tu et al., 2007). We previously reported that cultured neurons overexpressing α1-takusan showed increased PSD-95 clustering, dendritic spine density, and AMPA- and NMDA-mediated glutamate receptor activity (Tu et al., 2007). We thus reasoned that α1-takusan might protect synapses from Aβ-induced deterioration. In the current study, we induced synaptic dysfunction in cultured neurons through exposure to naturally secreted Aβ oligomers from 7PA2 cells (Podlisny et al., 1995) and found that synaptic damage was reduced by overexpressed α1-takusan, but was enhanced by takusan knockdown. We further show that overexpressed α1-takusan coimmunoprecipitates with tau and reduces Aβ-induced tau hyperphosphorylation at postsynaptic sites, preventing excessive mitochondrial fragmentation. Finally, we performed deletion studies and identified the D2 responsible for synaptic protection. Together, this work identifies potential mechanisms of synaptic protection by α1-takusan and paves the way for development of takusan-based therapeutics for AD.
Materials and Methods
Cell culture.
Low- or high-density (5 × 103 or 4 × 105 cells per 35 mm dish, respectively) primary mouse hippocampal or rat cerebrocortical cultures were prepared from E17 mice or rats of either sex as previously described (Tu et al., 2007; Nakanishi et al., 2009). In brief, cerebral cortices or hippocampi were enzymatically dissociated (papain; Collaborative Research), mechanically dispersed into a single-cell suspension, and plated onto glass coverslips coated with 0.1 mg/ml poly-l-lysine (Sigma). The cells were maintained in neural basal medium supplied with B27, 0.5 mm glutamine, and 1× Pen/Strep (Invitrogen) until use.
Preparation of naturally secreted Aβ-containing conditioned medium.
7PA2 cells [Chinese hamster ovary (CHO) cells expressing mutant (V717F) human APP] secrete SDS-stable, low-n Aβ oligomers (Podlisny et al., 1995). Conditioned medium (CM) was collected from 7PA2 cells as described in detail previously (Podlisny et al., 1995). In brief, the medium of 100% confluent 7PA2 cells [or regular CHO cells as control (Ctrl)] was replaced with serum-free Neural Basal Medium (Invitrogen) overnight. The collected CM was then spun briefly to remove cell debris. The aliquots were stored at −80°C until used. The existence of naturally secreted Aβ oligomers in 7PA2 CM was confirmed by immunoblotting using anti-β-amyloid (monoclonal, clone 6E10, Covance).
Immunocytochemistry.
Cultured neurons were infected overnight with Semliki Forest Virus (SFV; Invitrogen) encoding EGFP alone or EGFP-α1-takusan, as described previously (Tu et al., 2007), and then exposed to control CM or 7PA2 CM for 8–12 h. The cells were fixed with 4% PFA and permeabilized with 0.1% Triton X-100 in PBS. After blocking with 10% goat serum in PBS for 30–60 min, cells were incubated with anti-PSD-95 mouse monoclonal antibody (NeuroMap) and anti-synapsin I rabbit polyclonal antibody (Millipore) overnight at 4°C, followed by Alexa Fluor-conjugated secondary antibodies (Invitrogen). Images were captured by deconvolution microscopy and analyzed with SlideBook 4.0 software (Intelligent Imaging Innovations). In these images, we counted clusters stained with anti-PSD-95 along primary and secondary dendrites. For each neuron, we counted PSD-95 clusters, typically along a dendritic length of at least 30 μm. At least eight neurons were analyzed for each condition. We then determined the density of these clusters per 10 μm for each neuron and performed a statistical analysis on this result. In addition, the number of PSD-95 clusters juxtaposed to synapsin I signal was also quantified. Statistical significance was determined by one-way ANOVA and post hoc testing (for multiple comparisons) or Student's t test (for two-way comparisons).
Mitochondria were visualized using MitoTracker Red (Life Technologies), as suggested by the manufacturer. Mitochondria normally exhibit elongated tubular structures. However, under pathological conditions, they undergo fragmentation into small, round dots, as verified under deconvolution microscopy with 3D reconstruction. The 3D rendering of acquired z-stacks and the quantification of mitochondrial length and number were performed using Volocity software (PerkinElmer), as described previously (Cho et al., 2009).
Quantification of dendritic spines.
To visualize dendritic spines, cells were infected for 5–7 d with lentiviral vectors carrying EGFP. The cells were then infected overnight with SFV encoding mCherry or mCherry-α1-takusan fusion protein, followed by exposure to control or 7PA2 CM for 8–12 h. The cells were fixed with 4% PFA. We counted the number of dendritic spines along primary and secondary dendrites on projected deconvolved images, typically along a 30 μm distance. At least 10 neurons were analyzed for each condition. The density of dendritic spines was calculated as the number of spines per 10 μm. Statistical significance was determined by one-way ANOVA (for multiple comparisons) or Student's t test (for two-way comparisons).
Electrophysiological recordings.
Cultured neurons were infected with SFV encoding EGFP alone or EGFP-α1-takusan and then exposed overnight to control CM or 7PA2 CM. AMPAR-mediated spontaneous miniature EPSCs (mEPSCs) were recorded, as previously described (Tu et al., 2007). Briefly, d-AP5 (50 μm) and picrotoxin (50 μm) were added to the extracellular solution to block NMDA and GABA receptor-mediated spontaneous postsynaptic currents, respectively. In addition, TTX (1 μm) was added and allowed to equilibrate for at least 20 min to block Na+ channels and resulting action currents. All recordings were made at room temperature at a holding potential of −70 mV. The currents were digitally sampled at 10 kHz and filtered at 2–5 kHz with a Bessel characteristic. Solution changes were made with fast-flow, gravity-fed tubes gated by valves. Data acquisition and analysis were made with pClamp 10.2 (Molecular Devices) or a synaptic “mini” analysis program (Synaptosoft). Cumulative distribution curves were generated, and statistical significance was determined by the Kolmogorov–Smirnov test.
Immunoprecipitation and immunoblotting.
High-density primary cerebrocortical cells were used for immunoblotting and immunoprecipitation (IP) experiments. The procedures were modified from methods previously described in detail (Tu et al., 2007). In brief, cells for IP were infected with SFV expressing EGFP or EGFP-α1-takusan overnight and then lysed at 4°C overnight in 1× PBS/EDTA containing 1% Triton X-100, 0.1% SDS, and a protease inhibitor cocktail. On the second day, the supernatant was collected and incubated with anti-EGFP antibody (rabbit, 1:100; Invitrogen) and Dynabeads protein G (Invitrogen). After overnight rotation at 4°C, the beads were washed and the proteins binding to the antibodies on the beads were dissociated using 2× SDS loading buffer containing β-mercaptoethanol. The samples were then ready for immunoblotting. After brief sonication and boiling, the samples were fractionated by SDS-PAGE and transferred to nitrocellulose or PVDF membranes. The membranes were then probed with various primary antibodies, as follows: PSD-95 (mouse monoclonal, NeuroMap); phosphorylated tau (p-tau) AT8 (mouse monoclonal, ThermoFisher); tau (mouse monoclonal 5E2, Millipore); tau H-150 (rabbit polyclonal, Santa Cruz Biotechnology); tubulin (mouse monoclonal, Sigma); EGFP (mouse monoclonal, Santa Cruz Biotechnology; rabbit polyclonal, Agilent); Flag (M2, Sigma); and appropriate secondary antibodies. The immunosignals were captured on Kodak x-ray film and quantified using ImageJ version 1.37c. Statistical significance was determined by ANOVA or Student's t test.
Proximity ligation assays.
Proximity ligation assays (PLAs) were performed using the Duolink kit (OLINK Bioscience) to detect close interaction between two proteins within a distance of <40 nm. Cultured neurons were fixed, permeabilized, and blocked as described in the Immunocytochemistry section. Following the application of rabbit anti-EGFP (Agilent) and mouse anti-tau 1 (Millipore) antibodies overnight at 4°C, the cells were incubated with secondary antibodies (against rabbit and mouse) from the Duolink kit according to the manufacturer's protocol. These antibodies are coupled with PLA “plus” and “minus” oligonucleotide probes, respectively. When two PLA probes are in close proximity (<40 nm), the DNA strands can interact through the subsequent addition of two other circle-forming DNA oligonucleotides. After the two oligonucleotides are joined by enzymatic ligation, they are amplified via rolling circle amplification using a polymerase. After several hundredfold replication of the circular DNA template, amplified DNA strands were hybridized by fluorescence-labeled complimentary oligonucleotides.
Results
Overexpressed α1-takusan protects neurons from Aβ-induced synaptic loss
We previously showed that forced expression of α1-takusan in cultured hippocampal neurons enhanced their synaptic activity based on the observation that such manipulation increased dendritic spine density, PSD-95 clustering, and mEPSCs (Tu et al., 2007). In contrast, exposure of cultured neurons to Aβ results in reduction of dendritic spine and PSD-95 cluster density, which are signs of synaptic injury (Sheng et al., 2012). We thus postulated that overexpressed α1-takusan might counteract the effects of Aβ toxicity in neurons. To test this hypothesis, we established a system in which cultured neurons were challenged by soluble Aβ oligomers, which has been suggested to be the most neurotoxic form of Aβ (Selkoe, 2008). We chose to use the CM of 7PA2 CHO cells as the source of soluble Aβ oligomers (Podlisny et al., 1995) as this CM contains naturally secreted Aβ oligomers that are thought to be more pathophysiologically relevant than synthetic oligomers (Selkoe, 2008). We confirmed through immunoblotting that the CM contained single-digit nanomolar levels of low-n Aβ oligomers (Fig. 1A). We incubated cortical neurons at 21–28 d in vitro (DIV) with Ctrl CHO (i.e., non-Aβ-containing CM) CM or Aβ oligomer-containing CM from 7PA2 cells, and quantified the level of PSD-95 protein in whole-cell lysates (Fig. 1B,C, −Tksn). As expected, Aβ incubation resulted in the reduction of PSD-95 protein levels at 1, 2, and 6 h of exposure. We next transduced cortical neurons with SFV encoding the EGFP fusion protein EGFP-α1-takusan (+Tksn). The following day, the cells were challenged with control CM or Aβ-containing 7PA2 CM, and PSD-95 protein level was measured in total lysates. After a 6 or 12 h exposure to Aβ, the level of PSD-95 was significantly higher than that in nontransfected neurons (Fig. 1B,C, +Tksn). Finally, in the absence of Aβ, overexpressed α1-takusan did not alter the total level of PSD-95 in whole-cell lysates (Fig. 1D).
Figure 1.

Overexpression of α1-takusan alleviates Aβ-induced loss of PSD-95 and PSD-95/Syn clusters in cultured neurons. A, SDS-stable Aβ oligomers are enriched in the CM of 7PA2 cell cultures. One milliliter of 7PA2 CM (lane 3) or CM from control CHO cells (lane 2) was precipitated with a conventional TCA method or with anti-Aβ antibodies and immunoblotted with monoclonal antibody 6E10. Lane 1 was loaded with 5 ng (or 1.1 pmol, which is equivalent to 1.1 nm when diluted in 1 ml of medium) of synthetic monomeric Aβ1–42 peptide as a control. mo, Monomeric; di, dimeric; tri, trimeric Aβ. Relative molecular masses (in kilodaltons) are shown. B, Cultured cerebrocortical neurons, uninfected (−Tksn) or takusan infected (+Tksn), were exposed to control CM (top) or Aβ-containing CM (bottom) for the period of time indicated. Cell lysates were prepared and subjected to immunoblotting with antibodies against PSD-95 and tubulin. C, Level of PSD-95 from three (−Tksn) or four (+Tksn) experiments was normalized to tubulin and expressed as a percentage of control (determined at 0 min; right). The level of PSD-95 was significantly decreased in −Tksn cells at 1 h (73.5 ± 2.6%), 2 h (61.0 ± 5.4%), and 6 h (55.6 ± 9.2%) after Aβ application, but not in +Tksn cells at 6 h (92.8 ± 2.2%) or 12 h (103.6 ± 7.4%). D, Takusan overexpression does not increase the total PSD-95 in cultured cortical cells. Neurons were infected with SFV encoding EGFP or EGFP-α1-takusan overnight and then subjected to immunoblotting using anti-PSD-95 and anti-tubulin (top). The level of PSD-95 protein was normalized to tubulin and expressed as a percentage of control/EGFP (n = 4, bottom). E, Experimental scheme, Cultured neurons (21–28 DIV) were infected overnight with SFV encoding EGFP or EGFP-α1-Tksn and then exposed to Ctrl or Aβ CM for an additional 8–12 h. F, ICC using antibodies against PSD-95 (red) and synapsin I (Syn; blue) in the dendrites of cultured neurons under the following four conditions: i, EGFP/Ctrl CM; ii, EGFP/Aβ CM; iii, Tksn/Ctrl CM; or iv, Tksn/Aβ CM. Scale bar, 5 μm. G, Quantification of density of PSD-95 clusters (top) and PSD-95/Syn coclusters (bottom). Forced expression of Tksn significantly increased the density of both clusters in the absence of Aβ (compare i, iii in both graphs). Aβ exposure significantly reduced the density of both clusters in EGFP-expressing cells but not in Tksn-expressing cells (n = 8). H, Quantification of Aβ-induced reduction (percentage of control) in the density of PSD-95 clusters (top) and PSD-95/Syn coclusters (bottom). Aβ-induced loss of both clusters was significantly reduced in Tksn-expressing cells (PSD-95, 22.4 ± 3.8%; PSD-95/Syn clusters, 26.4 ± 4.9%) compared with EGFP-expressing cells (54.9 ± 4.6% and 54.7 ± 6.8%, respectively). I, Quantification of dendritic PSD-95 immunosignal. Exposure to Aβ resulted in decreased intensity of dendritic PSD-95 (by 41.37 ± 6.64%) in cells expressing EGFP. In contrast, cells expressing takusan did not exhibit a significant reduction in PSD-95 ICC signal after Aβ exposure (n = 8). All values are mean ± SEM. n.s., Not significant. *p < 0.05, **p < 0.01, by one-way ANOVA (C, G, I) or Student's t test (D, H).
We next examined whether forced expression of α1-takusan in cultured neurons would alleviate Aβ-induced synaptic injury. In the first set of experiments (Fig. 1E), cultured hippocampal neurons were infected overnight with SFV encoding EGFP or EGFP fusion protein EGFP-α1-Tksn. The transduced cells were then exposed to control CM or 7PA2 (Aβ) CM for 8–12 h. The cells were subsequently subjected to immunocytochemistry (ICC) for the evaluation of synaptic integrity. In Figure 1, F and G, we present density measurements for PSD-95 clusters as well as for PSD-95 clusters that are juxtaposed with presynaptic synapsin I signal (PSD-95/Syn coclusters). Consistent with our previous report (Tu et al., 2007), overexpressed α1-takusan significantly increased the density of both types of clusters in cultured neurons in the absence of Aβ oligomers (Fig. 1G, top and bottom, compare columns i, iii). After Aβ exposure, the density of both clusters was significantly decreased in cells expressing EGFP alone (Fig. 1G, compare columns i, ii) but not in cells expressing α1-takusan (Fig. 1G, compare columns iii, iv). We measured Aβ-induced damage by calculating percentage reduction in cluster density and found a significantly smaller reduction in α1-takusan-expressing cells than in EGFP-expressing cells (Fig. 1H). We also found that the total PSD-95 immunosignal in dendrites (i.e., clustered and nonclustered signals) was significantly decreased in EGFP-expressing cells, but not in takusan-expressing cells (Fig. 1I).
To determine the effects of takusan overexpression on Aβ-induced dendritic spine loss, we first transduced cultured hippocampal neurons with a lentivirus encoding EGFP to visualize dendritic spines. We then infected these cells overnight with SFV encoding mCherry or mCherry-α1-Tksn and exposed them to control CM or 7PA2 (Aβ) CM for an additional 8–12 h. Aβ exposure of transduced cells led to a significant loss in dendritic spine density in both mCherry-expressing cells (Fig. 2A,B, compare columns i, ii) and takusan-expressing cells (Fig. 2A,B, compare columns iii, iv). However, as shown in Figure 2C, the percentage loss in spine density following Aβ exposure was significantly lower in takusan-expressing cells than in EGFP-expressing cells. In fact, considering both PSD-95 clusters and dendritic spines as measures of synaptic integrity, α1-takusan reversed or compensated for the effect of Aβ insult to near-normal levels. For example, synaptic integrity was protected to the extent that neurons receiving the α1-takusan expression vector plus Aβ CM exhibited a similar number of synapses as neurons receiving control vector and Ctrl CM (Figs. 1G, 2B, compare columns i, iv).
Figure 2.

Forced expression of α1-takusan alleviates Aβ-induced loss of dendritic spines in cultured neurons. A, Cultured hippocampal neurons were infected with SFV encoding mCherry (i, ii) or mCherry-α1-Tksn (iii, iv) and then exposed to Ctrl (i, iii) or Aβ CM (ii, iv). Dendritic spines were visualized by EGFP expressed from a lentiviral vector. Scale bar, 5 μm. B, The density of dendritic spines was significantly decreased by Aβ-application in both mCherry-expressing cells (ii vs i) and Tksn-expressing cells (iv vs iii), and was increased by Tksn overexpression (iii vs i, and iv vs ii). Values (clusters per 10 μm) for the four conditions are as follows; i, 2.70 ± 0.16; ii, 1.28 ± 0.17; iii, 4.96 ± 0.37; iv, 3.40 ± 0.35. C, Aβ-induced synaptic loss in the density of dendritic spines (percentage control) is significantly less in the cells overexpressing mCherry-α1-takusan than in the cells overexpressing mCherry (31 vs 52%, p < 0.05). Values are mean ± SEM. n = 10. *p < 0.05, **p < 0.01, one-way ANOVA or Student's t test.
We next performed gene knock-down experiments using an α-takusan shRNA (NN002) that targets the most conserved sequence in the α-takusan subfamily, thus knocking down the expression levels of the majority of the gene family by ∼50%, as previously demonstrated (Tu et al., 2007). In contrast to the overexpression experiments, knocking down α-takusan genes resulted in increased synaptic damage (Fig. 3). This result suggests that endogenous α1-takusan functions to protect against Aβ-mediated synaptic impairment.
Figure 3.

Knocking down endogenous α-takusan variants exacerbates Aβ-induced synaptic loss in cultured hippocampal cells. Cultured hippocampal neurons infected with a lentiviral vector encoding EGFP and control shRNA (Ctrl shRNA; i, ii) or EGFP and takusan shRNA NN002 (Tksn shRNA; iii, iv) were exposed for 6–8 h to Ctrl (−; i, iii) or Aβ CM (+; ii, iv). The infected cells were visualized by coexpressed EGFP. A, Immunohistochemistry using antibodies against PSD-95 (red) and synapsin I (Syn; blue) on dendrites of neurons. Scale bar, 5 μm. B, C, Quantification of the density of PSD-95 clusters (B) and juxtaposed PSD-95/Syn coclusters (C). Values are mean ± SEM. n = 10. *p < 0.05, **p < 0.01, by one-way ANOVA.
Additionally, we studied the electrophysiological properties of neurons to determine whether overexpressed α1-takusan functionally mitigates Aβ-induced synaptic damage. Cultured cortical neurons (22 DIV) were infected for at least 6 h with SFV encoding EGFP or EGFP-α1- Tksn and then exposed overnight to control CM or 7PA2 (Aβ) CM. AMPAR-mediated mEPSCs were then recorded in these neurons (Fig. 4A). Consistent with our previous report (Tu et al., 2007), Tksn overexpression in the absence of Aβ resulted in an increase in mEPSC amplitude (Fig. 4B, inset) and a rightward shift in the amplitude distribution plot (Fig. 4B, compare i, iii). Overnight exposure to Aβ significantly decreased mEPSC amplitude in EGFP-expressing cells, but not in Tksn-expression cells (Fig. 4B, compare i and ii, i and iv), suggesting that Tksn overexpression normalizes synaptic activity in the presence of Aβ. In addition, Aβ exposure decreased mEPSC frequency, as demonstrated by the increased interevent interval (Fig. 4C, inset), and the rightward shift in the interevent interval distribution plot (Fig. 4C, compare i, iii) in EGFP-expressing cells. Tksn overexpression in the absence of Aβ did not change the mean interspike interval (Fig. 4C, inset, compare i, iii). However, Tksn overexpression prevented the Aβ-induced decrease in mEPSC frequency (and hence the increase in interevent interval; Fig. 4C, inset, compare ii, iv). A decrease in mEPSC frequency can signify either a presynaptic defect in neurotransmission or a frank loss of synapses. Thus, together the immunocytochemistry and electrophysiology results are consistent with the notion that α1-takusan overexpression mitigates Aβ-induced synaptic dysfunction and loss.
Figure 4.

Forced expression of α1-takusan alleviates Aβ-induced dysfunction in AMPAR-mediated mEPSCs in cultured neurons. A, Representative traces recorded from cultured neurons infected with SFV encoding EGFP or EGFP-α1-Tksn and then exposed to control or Aβ-containing CM. B, Cumulative plot and mean value (inset) of mEPSC amplitude. In EGFP-expressing cells, Aβ exposure caused a significant (p < 0.01 by pairwise Kolmogorov–Smirnov test) leftward shift in the cumulative plot and decreased the mean amplitude of mEPSCs (inset, compare i, ii). In the absence of Aβ, Tksn-expressing cells manifested an increase in mEPSC amplitude compared with EGFP-expressing control cells (compare i, iii). Aβ exposure had no apparent effect on Tksn-expressing cells (compare iii, iv). C, Cumulative plot and mean value (inset) of mEPSC interevent intervals, reflecting mEPSC frequency. Tksn expression in the absence of Aβ did not alter the mEPSC interevent interval (inset, compare i, iii). Aβ exposure resulted in increased mEPSC interevent interval (or decreased frequency) in neurons expressing EGFP (compare i, ii), but not in neurons expressing Tksn (compare iii, iv). Values are mean ± SEM. n = 7–9 in each group. **p < 0.01 by one-way ANOVA.
To determine whether α1-takusan expression can repair Aβ-induced synaptic injury that has already occurred, we reversed the order of Aβ application and forced expression of α1-takusan (Fig. 5A). We first applied Ctrl or Aβ CM to cultured cortical neurons (21–28 DIV) overnight. The next day, we transduced the cells with SFV encoding EGFP or EGFP-α1-takusan. The transduced cells were kept in the medium containing Ctrl or Aβ CM for an additional 24 h, and immunohistochemistry was subsequently performed to evaluate synaptic integrity. Cells expressed high levels of EGFP and EGFP-α1-takusan fusion proteins within 24 h of transduction (Fig. 5B, left). We then quantified the density of PSD-95/Syn coclusters (Fig. 5B, right). As summarized in Figure 5C, the cluster density in Tksn-expressing cells exposed to Aβ CM (column iv) was significantly higher than that in EGFP-expressing cells exposed to Aβ CM (column ii), and was comparable to that in EGFP-expressing cells exposed to Ctrl CM (column i). We also found that Aβ-induced loss of cluster density was significantly reduced by forced expression of α1-takusan (Fig. 5D). Collectively, these data suggest that overexpressed α1-takusan can counteract and even reverse synaptic damage induced by soluble Aβ oligomers.
Figure 5.
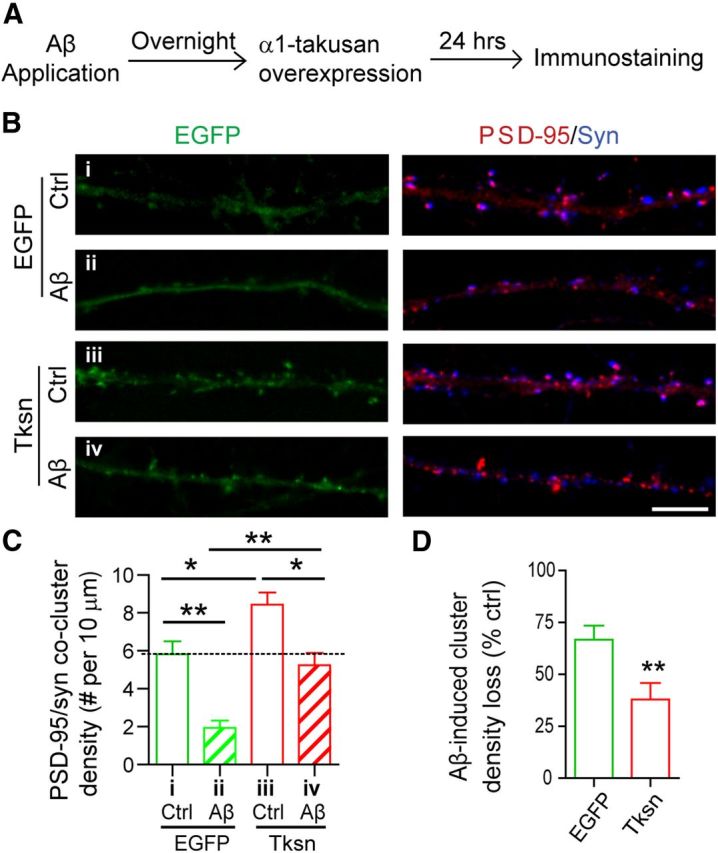
Forced expression of α1-takusan following Aβ application in cultured cortical neurons alleviates Aβ-induced loss of PSD-95/Syn coclusters. A, Experimental scheme showing that the order of takusan transduction and Aβ application was reversed from the experiments in Figure 1. Cultured neurons were exposed overnight to Ctrl or Aβ CM. The cells were then transduced with SFV encoding EGFP or EGFP-α1-Tksn and maintained in Ctrl or Aβ CM for an additional 24 h. B, Images of EGFP signals (left) and immunocytochemistry (right) using antibodies against PSD-95 (red) and synapsin I (Syn; blue) in the dendrites of cultured neurons. Scale bar, 5 μm. C, Quantification of PSD-95/Syn cocluster density in neuronal dendrites. Forced expression of Tksn following Aβ application reversed synaptic injury to the extent that the density of PSD-95/Syn coclusters in cells exposed to Aβ CM and transduced with Tksn was similar to that in cells exposed to Ctrl CM and transduced with EGFP. D, Quantification of Aβ-induced reduction (percentage control) in the density of PSD-95/Syn coclusters. Aβ-induced cluster loss in Tksn-expressing cells (38.0 ± 7.8%) was significantly less than that in EGFP-expressing cells (66.8 ± 6.7%). All values are mean ± SEM. n = 8. *p < 0.05, **p < 0.01, one-way ANOVA (C) or Student's t test (D).
We then tested whether α1-takusan protects cultured neurons from synaptic injury caused by NMDA application, since NMDAR activation is implicated at least in part in Aβ-induced toxicity (Kamenetz et al., 2003; Shankar et al., 2007). Cultured cortical neurons infected with SFV encoding mCherry or mCherry-α1-takusan were exposed for 5 min to 50 μm NMDA, a sublethal concentration determined by our previous studies (Nakanishi et al., 2009). We visualized dendritic spines using EGFP encoded by a lentiviral vector. As reported previously (Halpain et al., 1998), NMDA exposure resulted in a significant (p < 0.05) reduction in the density of dendritic spines in cells expressing mCherry alone (Fig. 6A,B, compare second column, first column) 24 h after NMDA application. This reduction was blocked by pretreatment with 10 μm memantine, an NMDAR antagonist, before the addition of NMDA (data not shown), indicating that NMDA toxicity is mediated by NMDA receptor activation. As shown previously (Tu et al., 2007; Fig. 2), overexpressed α1-takusan significantly increased spine density in cultured neurons in the absence of NMDA (Fig. 6B, compare third column, first column). Importantly, spine density in Tksn-expressing cells exposed to NMDA was not significantly changed relative to mCherry-expressing cells exposed to vehicle (Fig. 6B, compare fourth column, first column), but was significantly higher than in mCherry-expressing cells exposed to NMDA (Fig. 6B, compare fourth column, second column). We also found that NMDA-induced dendritic varicosity (Hasbani et al., 2001) was significantly reduced by takusan overexpression (Fig. 6, arrowheads). Eighty percent of mCherry-expressing neurons displayed dendritic varicosity following NMDA exposure, while in takusan-expressing neurons this proportion was reduced to 10% (p < 0.01, Fisher exact test). Together, our findings establish a potential role for α1-takusan as a therapeutic agent that effectively mitigates or reverses synaptic damage induced by oligomeric Aβ application or other excitotoxic insults (Talantova et al., 2013).
Figure 6.

Forced expression of α1-takusan alleviates NMDA-induced loss of dendritic spines in cultured cortical neurons. Cultured neurons transfected with plasmid vector encoding mCherry or mCherry-α1-takusan (Tksn) were exposed to vehicle (Veh) or 50 μm NMDA. A, Dendritic spines were visualized by cotransfected EGFP. Arrowheads indicate the sites of NMDA-induced dendritic varicosity. Scale bar, 5 μm. B, Quantification of dendritic spine density. NMDA exposure produced a significant (p < 0.05) reduction in the density of dendritic spines in mCherry-expressing cells, but not in Tksn-expressing cells. In addition, forced expression of Tksn drastically reduced the number of neurons with dendritic varicosity (8 of 10 in mCherry-expressing cells vs 1 of 10 in Tksn-expressing cells; p < 0.01 by Fisher's exact test). Values are mean ± SEM. n = 10. n.s., Not significant. *p < 0.05, one-way ANOVA or Student's t test.
Overexpressed α1-takusan inhibits Aβ-induced tau hyperphosphorylation
A primary pathology in AD is Aβ-induced tau hyperphosphorylation, which leads to the dissociation of tau from the microtubule cytoskeleton, aggregate formation of phosphorylated tau, and neuronal death (De Felice et al., 2008; Zempel et al., 2010; Jin et al., 2011). To gain mechanistic insights into the protective effects of α1-takusan, we examined the level of tau phosphorylation in neurons exposed to Aβ. We transduced cultured cortical neurons with SFV encoding EGFP or EGFP-α1-takusan. After overnight transduction, we exposed the cells to Ctrl or Aβ CM for an additional 8–12 h. We fractionated PSD lysates from neurons and performed Western blot analysis to determine the expression level of p-tau and total tau (Fig. 7A). We then calculated the level of tau phosphorylation as the ratio of p-tau to total tau. As shown in Figure 7A, Aβ exposure resulted in a significant increase in tau phosphorylation in EGFP-transduced cells (p < 0.05), but not in α1-takusan-transduced cells. Interestingly, in the absence of Aβ, overexpressed α1-takusan had no significant effect on tau phosphorylation. It is therefore likely that takusan mitigates Aβ-induced synaptic damage through the blockade of tau hyperphosphorylation at the PSD. These results also suggest that the biochemical mechanism underlying the protection of synaptic PSD-95 clusters by takusan overexpression following Aβ application is distinct from that underlying the increase in the density of PSD-95 clusters before Aβ application.
Figure 7.
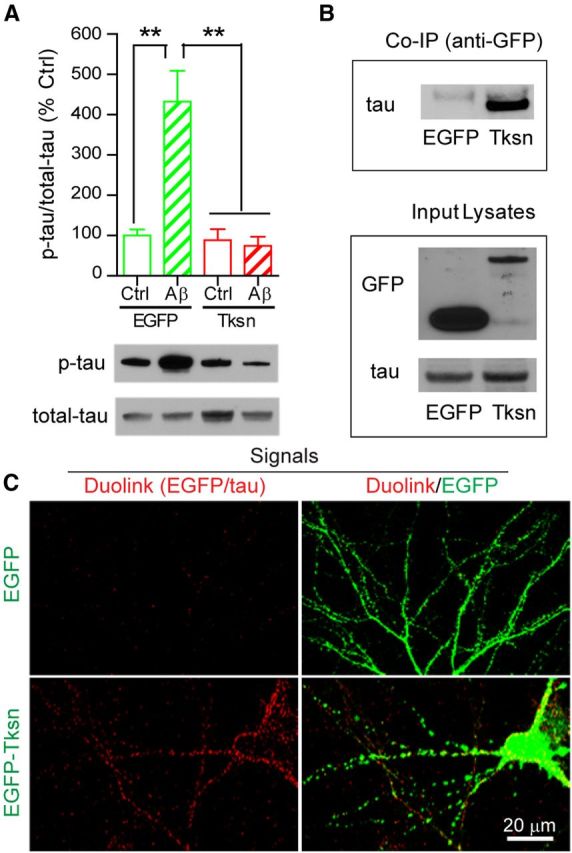
Overexpressed α1-takusan interacts with tau and inhibits Aβ oligomer-induced tau hyperphosphorylation at postsynaptic sites of cultured cortical neurons. A, Overexpressed α1-takusan inhibits Aβ-induced tau hyperphosphorylation. Immunoblotting of PSD-enriched fractions prepared from cultured cortical neurons transduced overnight with SFV encoding EGFP or EGFP-α1-takusan (Tksn) and followed by application of Ctrl or Aβ CM for an additional 8–12 h. The graph at the top shows the level of tau phosphorylation, which was calculated as the ratio of p-tau to total-tau and then normalized to the control (EGFP/Ctrl CM). Representative gel images are shown at the bottom. Aβ application significantly increased the p-tau/total tau ratio in cells transduced with EGFP (432.3 ± 76.7% of control), but not in the cells transduced with Tksn (74.2 ± 22.7% of control). Values are mean ± SEM. n = 4. **p < 0.01, one-way ANOVA. B, Co-IP experiments showing that overexpressed EGFP-α1-takusan interacts with tau in cultured cortical neurons. Cell lysates were prepared from cultured cortical neurons transduced with SFV encoding EGFP or EGFP-α1-takusan. Lysates were precipitated with anti-GFP antibody and then subjected to immunoblotting using anti-tau antibody. The top panel shows that endogenous tau proteins were coprecipitated with EGFP-α1-takusan, but not with EGFP. The bottom panel shows the presence of EGFP, EGFP-α1-takusan, and tau in the lysates. C, Duolink (in-cell Co-IP) experiments showing the interaction between EGFP-α1-takusan and endogenous tau in cultured neurons. Cultured cortical neurons were transduced with SFV encoding EGFP or EGFP-α1-takusan. The Duolink procedure using antibodies against GFP and tau produced robust red signals, which indicate that the two antibodies are localized in close proximity (<40 nm) in cells expressing EGFP-α1-takusan, but not in cells expressing EGFP (left panels). The overlay images (right panels) show that the majority of red signals are colocalized with EGFP-α1-takusan on dendrites, suggesting that tau and EGFP-α1-takusan interact with each other predominantly at postsynaptic sites.
α1-Takusan may function to prevent Aβ-induced tau hyperphosphorylation through an interaction, possibly direct or indirect, with tau. To test this idea, we performed co-IP experiments using cultured cortical neurons in which α1-takusan was overexpressed. Specifically, we transduced cortical neurons overnight with SFV encoding EGFP or EGFP-α1-takusan and then performed co-IP experiments using anti-GFP antibodies. The co-IP products were then subjected to Western blot analysis using anti-tau antibodies, as illustrated in Figure 7B. Tau protein coimmunoprecipitated with EGFP-α1-takusan, but not with EGFP, suggesting that α1-takusan and tau are present in the same protein complex in cultured cortical neurons.
To further verify the interaction identified by co-IP, we performed PLAs (Söderberg et al., 2006) using the Duolink in-cell interaction system (OLINK Bioscience). PLAs generate an immunoreactive signal (red) only if two antigens (target proteins) are localized within 40 nm of each other (Fredriksson et al., 2002). Using antibodies against GFP and tau, we performed PLAs in cultured cortical neurons infected with SFV encoding EGFP or EGFP-α1-takusan. Compared with EGFP-expressing cells, takusan-expressing cells showed robust red fluorescent Duolink signals (Fig. 7C, left), indicating that tau and α1-takusan proteins are in close proximity and likely interact with each other, either directly or indirectly via additional proteins such as PSD-95, which can be coimmunoprecipitated with tau in the PSD (Ittner et al., 2010). Indeed, the Duolink signals form clusters in dendrites rather than in cell bodies coincident with EGFP-α1-takusan (green) subcellular localization (Fig. 7C, right). Thus, these results suggest that takusan proteins interact with tau at postsynaptic sites.
Overexpressed α1-takusan prevents Aβ-induced mitochondrial fragmentation
Abnormal mitochondrial dynamics have been associated with AD pathogenesis (Knott et al., 2008; Reddy and Beal, 2008; Wang et al., 2009). Indeed, a recent study found that p-tau interacts directly with the mitochondrial fission protein Drp1 and plays an important role in Aβ-induced mitochondrial fragmentation and synaptic deficiencies (Manczak and Reddy, 2012). We therefore postulated that forced expression of α1-takusan may reduce Aβ-induced mitochondrial fragmentation by inhibiting tau hyperphosphorylation. To test this idea, we transduced cultured hippocampal neurons with SFV encoding EGFP or EGFP-α1-takusan. After overnight transduction, the cells were exposed to Ctrl or Aβ CM for an additional 8–12 h (Fig. 8A). We monitored Aβ-induced morphological changes in mitochondria by 3D deconvolution fluorescence microscopy and determined mitochondrial fragmentation (fission) by using the 3D rendering of acquired z-stack images, as described previously (Cho et al., 2009). We found that Aβ application caused a significant decrease in the size and a significant increase in the number of mitochondria, indicating excessive mitochondrial fragmentation in the neurons overexpressing EGFP, but not in the neurons overexpressing EGFP-α1-takusan (Fig. 8B,C). However, Aβ exposure did not cause a significant change in the total mitochondrial mass as determined by the measurement of the total mitochondrial volume (Fig. 8D). These findings suggest that Aβ-induced mitochondrial fragmentation is blocked by overexpressed α1-takusan.
Figure 8.

Overexpressed α1-takusan prevents Aβ-induced excessive mitochondrial fragmentation in cultured hippocampal neurons. Cultured neurons were infected overnight with SFV encoding EGFP or EGFP-α1-takusan (Tksn) followed by exposure to Ctrl or Aβ CM for an additional 8–12 h. A, Images of mitochondria in the dendrites of neurons in the following four conditions: i, EGFP/Ctrl CM; ii, EGFP/Aβ CM; iii, Tksn/Ctrl CM; iv, Tksn/Aβ CM. B–D, The size (B), number (C), and volume (D) of mitochondria were quantified in these neurons. Decreased size and increased number suggest elevated mitochondrial fragmentation in the cells overexpressing EGFP (ii), but not in the cells overexpressing Tksn (iv). Values are mean ± SEM. n = 20. **p < 0.01, one-way ANOVA.
α1-Takusan requires the D2 for self-clustering and synaptic clustering of PSD-95
We next sought to identify protein domains of α1-takusan responsible for synaptic enhancement and protection. Based on predicted secondary structures and exon/intron organization, we tentatively divided the α1-takusan sequence into four modular domains and the C-terminal PDZ-binding motif (Fig. 9A). Beginning with the N terminus, D1 and D3 are predicted to form coiled-coil domains according to the COILS program (www.ch.embnet.org/software/COILS_form.html). D2, which is located between D1 and D3, is encoded mostly by exon 5 and is extremely well conserved among α-takusan family variants. In contrast, some α-takusan variants lack D4 (Tu et al., 2007). We generated α1-takusan mutants by deleting each of the four domains (designated as ΔD1–ΔD4; Fig. 9A) and expressed them in human embryonic kidney 293T (HEK293T) cells (Fig. 9B,C). While EGFP proteins are diffuse in the cytoplasm, EGFP-α1-takusan proteins form clusters (Fig. 9B), presumably representing self-multimers of takusan proteins as we have previously reported (Tu et al., 2007). Interestingly, deletion mutants ΔD1, ΔD3, and ΔD4 all formed clusters in HEK293T cells, but ΔD2 did not (Fig. 9B,D), suggesting that the D2 is critical for takusan self-clustering. Consistent with this finding, co-IP experiments showed that EGFP-ΔD2 cannot bind to Flag-α1-takusan, while EGFP-tagged takusan as well as takusan ΔD1, ΔD3, and ΔD4 mutants can (Fig. 9E).
Figure 9.

Identification of the domains required for α1-takusan clustering. A, A schematic diagram of the full-length α1-takusan (Tksn) and its deletion mutants ΔD1–ΔD4, each of which lacks one of the four domains (D1–D4). The number of amino acids in each domain is indicated. The deleted sequence in each of the four mutants is represented by a dashed line. B, Fluorescent images of EGFP, EGFP-Tksn, and EGFP-Tksn deletion mutants expressed in HEK293T cells. In these cells, EGFP-Tksn forms clusters, while EGFP shows dispersed localization. Among deletion mutants, ΔD1, ΔD3, and ΔD4 show cluster formation similar to Tksn, while ΔD2 shows dispersed localization similar to EGFP. C, Western blot using anti-GFP antibodies shows the expression of transfected EGFP, EGFP-Tksn, and EGFP-Tksn deletion mutants in HEK293T cells. D, Evaluation of cluster formation by EGFP, EGFP-Tksn, and deletion mutants in HEK293T cells. The sizes of fluorescence signals were quantified in cells expressing EGFP, EGFP-Tksn, or deletion mutants and were expressed as the percentage of EGFP control. The fluorescence signals observed in cells expressing Tksn, ΔD1, ΔD3, or ΔD4 proteins are significantly smaller in size than those seen in cells expressing EGFP or ΔD2. Values are mean ± SEM. n = 4. **p < 0.01, one-way ANOVA. E, Co-IP experiments showing that D2 is required and sufficient for the self-clustering of takusan. Co-IP experiments used anti-EGFP antibodies for immunoprecipitation and anti-Flag antibody for immunoblotting on protein lysates prepared from HEK293T cells expressing Flag-α1-takusan and EGFP, EGFP-α1-takusan (E-Tksn), or one of the EGFP (E)-tagged takusan mutants. These data show that EGFP-Tksn, ΔD1, ΔD3, and ΔD4 proteins bind to Flag-Tksn, while ΔD2 and EGFP controls do not. The results of co-IP experiments are schematically summarized at the bottom.
We subsequently assessed the impact of these domains on PSD-95 clustering. We coexpressed mCherry-PSD-95 together with each of the takusan deletion constructs in HEK293T cells. mCherry-PSD-95 proteins form clusters when they are coexpressed with EGFP-Tksn, ΔD1, ΔD3, or ΔD4, but not with EGFP control or EGFP-ΔD2 (Fig. 10A–C). These results suggest that the self-clustering of α1-takusan and the clustering of mCherry-PSD-95 in HEK293T cells requires the D2. We then expressed mCherry-PSD-95 together with EGFP, EGFP-Tksn or EGFP-ΔD2 in cultured cortical neurons (Fig. 10D). Compared with the EGFP control, EGFP-Tksn increased the density of mCherry-PSD-95 clusters (Fig. 10D,E), consistent with the observed effects of α1-takusan on endogenous PSD-95 (Fig. 1). In contrast, coexpression with ΔD2 had no effect on the cluster density of mCherry-PSD-95 (Fig. 10D,E). Collectively, these results indicate that the D2 of α1-takusan is necessary for enhanced PSD-95 clustering in cultured cortical neurons.
Figure 10.
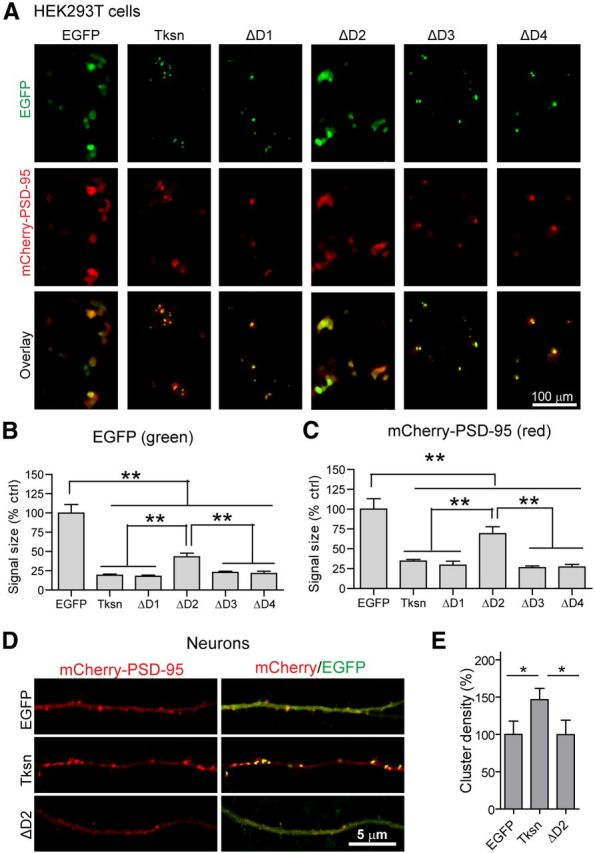
D2 is required for PSD-95 clustering by α1-takusan. A, Images showing overexpressed mCherry-PSD-95 (red, middle) and EGFP or one of the following EGFP fusion proteins (green, top): EGFP-Tksn, EGFP-ΔD1, EGFP-ΔD2, EGFP-ΔD3, or EGFP-ΔD4 in HEK293T cells. Overlay images of mCherry and EGFP are shown at the bottom. Scale bar, 100 μm. B, The size of green fluorescent signals was used as a measure of protein clustering. Similar to the results shown in Figure 9, B and D, EGFP-Tksn, ΔD1, ΔD3, and ΔD4 fusion proteins form clusters of much smaller sizes, while EGFP and EGFP-ΔD2 proteins are dispersed. n = 4. C, The clustering of mCherry-PSD-95 proteins was also analyzed in the same manner. The fluorescence signals in cells expressing Tksn, ΔD1, ΔD3, or ΔD4 are significantly smaller in size than in cells expressing EGFP or ΔD2, suggesting increased clustering. D, Images showing the level of overexpressed mCherry-PSD-95 (red) and EGFP, EGFP-α1-takusan (Tksn), or EGFP-ΔD2 (green) in the dendrites of cultured cortical neurons. The left panels show red signals from mCherry, and the right panels are the overlay of mCherry and EGFP. E, Quantification of mCherry-PSD-95 clusters in the dendrites. The density of mCherry-PSD-95 clusters was significantly increased by overexpressed Tksn (146.6 ± 15.0% of control), but not by ΔD2 overexpression (99.6 ± 19.2% control). n = 10. Values are normalized to EGFP controls and are mean ± SEM. **p < 0.01, *p < 0.05, one-way ANOVA.
D2 recapitulates synaptic protection of α1-takusan against Aβ
To address whether the D2 combined with the C-terminal PDZ-binding motif is sufficient for synaptic protection, we generated the following three fusion proteins: D2a, D2b, and D2c. In each instance, a 45 aa peptide within the D2 was directly fused to a 6 aa peptide from the C terminus of α1-takusan, the C terminus of α5-takusan, or the plasmid vector, respectively (Fig. 11A). We previously reported that α5-takusan binds to PSD-95 with a higher affinity than α1-takusan in HEK293T cells (Tu et al., 2007), while the vector-derived 6 aa sequence is not predicted to bind to PDZ (according to Scansite prediction, http://scansite.mit.edu/). In HEK293T cells, we expressed mCherry-PSD-95 together with EGFP, EGFP-Tksn, EGFP-D2b, or EGFP-D2c (Fig. 11B). Like EGFP-Tksn, both EGFP-D2b and EGFP-D2c proteins form self-clusters (Fig. 11C). mCherry-PSD-95 proteins, however, form clusters with EGFP-D2b but not with EGFP-D2c, suggesting that the PDZ-binding motif is required for clustering PSD-95 (Fig. 11D). In cultured cortical neurons, we overexpressed mCherry-PSD-95 together with EGFP, EGFP-Tksn, EGFP-D2a, or EGFP-D2b (Fig. 11E). Similar to full-length α1-takusan, both D2a and D2b increased the cluster density of coexpressed mCherry-PSD-95 (Fig. 11F). These results suggest that the D2, when linked with the PDZ-binding C terminus, is sufficient to cluster PSD-95 in cortical dendrites.
Figure 11.

D2 is sufficient to enhance PSD-95 clustering. A, A schematic diagram depicting the structures of full-length α1-takusan (Tksn) and three takusan-derived proteins, D2a, D2b, and D2c. Each takusan derivative contains a D2 of α1-takusan and a 6 aa sequence, which is the PDZ-binding motif of α1-takusan (D2a), the PDZ-binding motif of α5-takusan (D2b), or the non-PDZ-binding vector sequence (D2c). B, Images of EGFP (green, left), mCherry-PSD-95 (red, middle), and overlay (right) in HEK293T cells cotransfected with mCherry-PSD-95, and either EGFP, EGFP-Tksn, EGFP-D2b, or EGFP-D2c. C, The size of green fluorescent signals was used as a measure of protein clustering. EGFP-Tksn, D2b, and D2c fusion proteins form clusters of much smaller sizes, while EGFP proteins are dispersed. D, The clustering of mCherry-PSD-95 proteins was analyzed in the same manner. The fluorescence signals in cells expressing Tksn or D2b, but not D2c, are significantly smaller in size than in EGFP control cells. E, Images (overlay) showing the expression of mCherry-PSD-95 (red) and cotransfected EGFP, EGFP-Tksn, EGFP-D2a, or EGFP-D2b (green) in cultured cortical neurons. F, Quantification of the density of mCherry-PSD-95 clusters (cluster number per 10 μm length) on the dendrites of neurons cotransduced with EGFP, EGFP-Tksn, EGFP-D2a, or EGFP-D2b. The density of mCherry-PSD-95 clusters was significantly increased in neurons overexpressing Tksn (5.52 ± 0.57), D2a (6.47 ± 1.00), or D2b (6.54 ± 0.42) when compared with EGFP control (3.36 ± 0.46). Values are mean ± SEM. n = 8–18. **p < 0.01.
We next aimed to determine whether the D2 recapitulates the ability of takusan to protect synapses from Aβ oligomer-induced damage. To this end, cultured cortical neurons were transfected overnight with vectors encoding mCherry, mCherry-ΔD2, mCherry-Tksn, or mCherry-D2b before being exposed to Ctrl or Aβ CM for 8–12 h (Fig. 12). Dendritic spines were visualized using EGFP-encoding lentiviral vectors (Fig. 12A), subsequently quantified (Fig. 12B), and then expressed as the percentage loss in spine density following Aβ exposure (Fig. 12C). These experiments show that while overexpressed ΔD2 exhibits no apparent synaptic effect relative to the mCherry control, the effect of the D2b mutant is indistinguishable from that of the full-length α1-takusan. Namely, forced expression of D2b not only increased dendritic spine density in the absence of Aβ application, but also significantly mitigated Aβ-induced dendritic spine loss. Together, we have identified a protein sequence that is required for synaptic clustering of PSD-95 and is sufficient, when linked with the PDZ-binding motif, to protect neurons from Aβ oligomer-induced synaptic injury.
Figure 12.
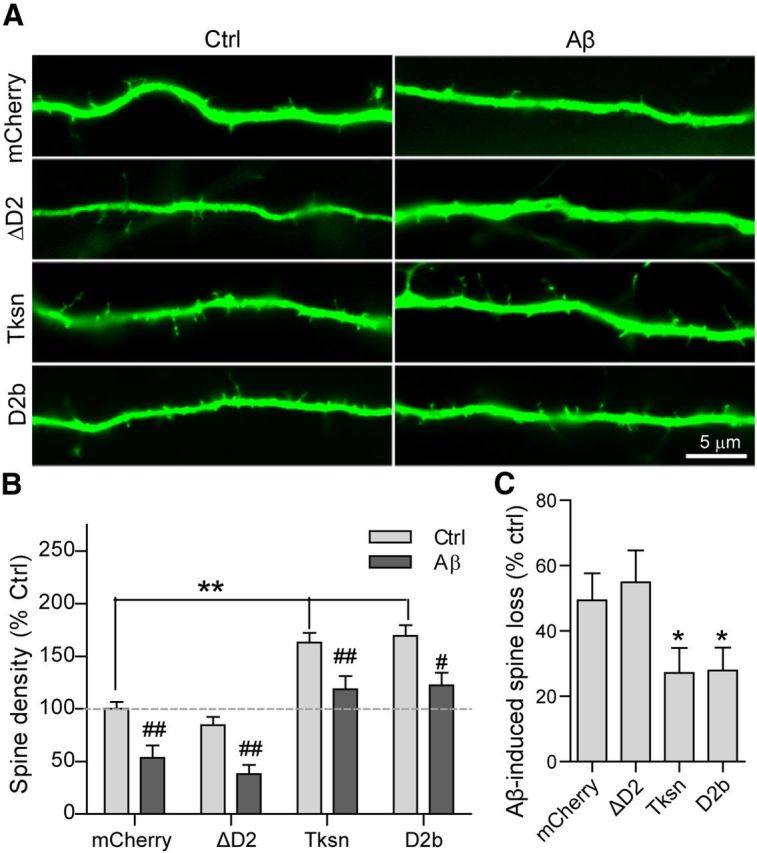
D2 is required and sufficient for mitigation of Aβ oligomer-induced loss of dendritic spines. A, Cultured neurons were infected with SFV encoding mCherry, mCherry-ΔD2, mCherry-Tksn, or mCherry-D2b, and then exposed to Ctrl or Aβ CM for an additional 8–12 h. Dendritic spines were visualized by EGFP in a lentiviral vector. B, Quantification of dendritic spine density normalized to the control (mCherry/Ctrl CM). In the absence of Aβ (light gray bars), overexpressed Tksn or D2b, but not ΔD2, caused a significant increase in the density of dendritic spines (**p < 0.01, one-way ANOVA). Aβ application caused a significant decrease in the density of spines in all types of cells (dark gray bar vs light gray bar: #p < 0.05, ##p < 0.01, one-way ANOVA). However, the level of spine density upon Aβ exposure in the cells expressing either Tksn or D2b, but not ΔD2, is comparable to that in the control cells (dark gray bars, Tksn, D2b, or ΔD2; light gray bars, mCherry). C, Quantification of Aβ-induced loss (percentage of control) in the density of dendritic spines in cultured neurons overexpressing mCherry (49 ± 8%), ΔD2 (55 ± 10%), Tksn (27 ± 8%), or D2b (28 ± 7%). Overexpressed Tksn or D2b, but not ΔD2, significantly reduced Aβ-induced spine loss compared with mCherry control. There was no difference between the effects of Tksn and D2b. Values are mean ± SEM. n = 10. *p < 0.05, one-way ANOVA.
Interestingly, the protein sequence in the D2 of human DLG5 protein shows high homology with mouse α1-takusan (Fig. 13A). We thus tested whether the D2 of human DLG5 has the ability to form self-clusters and to enhance PSD-95 clustering when linked to a PDZ-binding motif. We generated a fusion protein, hD2b, which contains human DLG5 D2 and the same C-terminal PDZ-binding motif as D2b (Fig. 13A). In HEK293T cells, mCherry-PSD-95 was cotransfected with EGFP, EGFP-D2b, or EGFP-hD2b (Fig. 13B). Similar to EGFP-D2b, EGFP-hD2b formed self-clusters in the cytoplasm, while EGFP was located diffusely (Fig. 13C). Additionally, mCherry-PSD-95 formed clusters with both D2b and hD2b, but not with EGFP, suggesting that hD2b like D2b has the ability to cluster PSD-95 in cultured HEK293T cells (Fig. 13D). We also cotransfected mCherry-PSD-95 together with EGFP, EGFP-D2b, or EGFP-hD2b in cultured cortical neurons (Fig. 13E). Similar to D2b, hD2b increased the cluster density of dendritic PSD-95 (Fig. 13F). These results suggest that the human D2, when linked with a C-terminal PDZ-binding motif, is sufficient to cluster PSD-95 in cortical dendrites, and can thus be explored further for its therapeutic potential in the treatment of AD.
Figure 13.

Human D2 mediates self-clustering and enhances PSD-95 clustering. A, Protein sequence alignment between two takusan derivatives, D2b and hD2b. Each of the derivatives contains the D2 of mouse α1-takusan (top, D2b) or human DLG5 (bottom, hD2b), and the same C-terminal PDZ-binding motif taken from mouse α5-takusan. The residues that are identical (*), strongly similar in properties (:), or weakly similar (.) between the two D2 sequences are indicated. B, Images of EGFP (green, left), mCherry-PSD-95 (red, middle), and overlay (right) in HEK293T cells cotransfected with mCherry-PSD-95 and either EGFP, EGFP-D2b, or EGFP-hD2b. C, The magnitude of EGFP signal was used as a quantitative measure of protein clustering. EGFP-D2b and hD2b fusion proteins form clusters of much smaller size, while EGFP proteins appear to be dispersed in the cytosol. D, Clustering of mCherry-PSD-95 proteins analyzed as above. Fluorescent signal in cells expressing D2b or hD2b was significantly smaller in magnitude than in EGFP control cells. E, Overlaid images showing expression of mCherry-PSD-95 (red) and cotransfected EGFP, EGFP-D2b, or EGFP-hD2b (green) in cultured cortical neurons. Boxed areas magnified in bottom panels. F, Quantification of mCherry-PSD-95 cluster density (cluster number per 10 μm length) on the dendrites of neurons cotransduced with EGFP, EGFP-D2b, or EGFP-hD2b. Values are mean ± SEM. n = 4 (C, D) or 8 (F). **p < 0.01.
Discussion
Dendritic spine structures are dynamically regulated by synaptic activity, and the loss of dendritic spines and functional synapses are early hallmarks in the pathophysiology of AD (Sheng et al., 2012). In Tg2576 and PDAPP AD transgenic mice, spine loss is detected long before the appearance of amyloid plaques (Lanz et al., 2003; Jacobsen et al., 2006), suggesting that soluble Aβ species have a primary role in this pathology. In the current study, we have evaluated the integrity of synapses by measuring the density of dendritic spines, PSD-95 clusters, and PSD-95/Syn coclusters. We have found that Aβ-induced loss of PSD-95 clusters (Figs. 1H, 5D) and dendritic spines (Fig. 2C) is reduced in takusan-expressing cells relative to EGFP-expressing cells. However, two-way ANOVA analysis did not demonstrate a significant statistical interaction between takusan and Aβ. Since takusan overexpression can enhance the basal level of synaptic integrity, it is possible that this amelioration is, in part, a result of the ability of takusan to boost the density of PSD-95 clusters and dendritic spines back to a “pseudonormal” level in an Aβ-independent manner. However, the observation that takusan decreases the percentage of (as opposed to net) Aβ-induced loss of both spine density and PSD-95 clustering indicates that takusan-induced spines and PSD-95 clusters are more resistant to Aβ toxicity than native spines and PSD-95 clusters. Given the ability of α1-takusan to self-cluster and interact with PSD-95, α1-takusan likely ameliorates Aβ-induced synaptic damage by facilitating PSD-95 clustering (Figs. 9, 10, 11; Tu et al., 2007). Consequently, the synapses containing takusan-PSD-95 clusters may be more resistant to Aβ-induced synaptic damage. Finally, we found that takusan increased dendritic expression as well as synaptic clustering of PSD-95 (by ICC; Fig. 1F–I) without affecting the total level of PSD-95 protein (determined by Western blotting of whole-cell lysates; Fig. 1D). These results are consistent with the notion that takusan overexpression protects synapses by recruiting PSD-95 into dendrites from other subcellular localizations.
The knock-down experiments (Fig. 3) suggest that endogenous α-takusan may also protect against Aβ-induced synaptic loss. However, these experiments produced a relatively modest effect on synaptic damage. It should be noted that the shRNA sequence used in these experiments targets only the α-takusan subfamily. However, non-α-takusan members may also contain a D2 homologous (but not identical) to the D2 of α-takusan (Tu et al., 2007) and thus may partly compensate for the loss of α-takusan in these knock-down experiments.
We observed the protective effect of takusan not only when takusan was overexpressed before Aβ application (Figs. 1, 2), but also when it was overexpressed after Aβ had caused synaptic damage (Fig. 5). These data suggest that takusan, or molecules mimicking its action, may be exploited as therapeutics to prevent or even reverse synaptic degeneration in AD. Furthermore, we found that forced expression of α1-takusan reversed or compensated for the effect of NMDA-induced spine loss (Fig. 6). This suggests that takusan or peptide mimetics may be useful as therapeutics not only against Aβ-induced synaptic damage but also other excitotoxic insults.
Tau was first identified as a protein essential for microtubule assembly (Weingarten et al., 1975) and for the stabilization of the microtubule cytoskeleton (Lee and Rook, 1992). It has since been recognized that during the course of AD, tau becomes hyperphosphorylated and deposits as neurofibrillary tangles in affected brain regions (Ballatore et al., 2007). Hyperphosphorylated tau accumulates and colocalizes with Aβ oligomers at synapses in both human AD brains (Fein et al., 2008; Sokolow et al., 2012) and mouse AD brains (Takahashi et al., 2010). It has further been determined that the level of synaptic phosphorylated tau correlates inversely with the number of existing synapses. Indeed, multiple studies in animal models of AD have shown that Aβ-induced synaptic injury/neuronal loss can be prevented by either reducing tau expression or preventing tau phosphorylation (Roberson et al., 2007; Ittner et al., 2010; Vossel et al., 2010; Jin et al., 2011; Roberson et al., 2011). In this study, we find that overexpressed α1-takusan coimmunoprecipitates with tau in cultured neurons, inhibiting Aβ-induced tau hyperphosphorylation in the PSD (Fig. 7) and mitigating Aβ-induced synaptic loss. Our findings strongly support the idea that tau hyperphosphorylation plays an important role in the process of AD-associated synaptic degeneration. In support of this finding, others have shown that synaptic dysfunction in rodents is dependent on tau phosphorylation as transgenic mice expressing pseudo-hyperphosphorylated tau display synaptic pathology, while tau phosphorylation-deficient mice do not (Hoover et al., 2010). We thus propose that the synaptic protection by overexpressed α1-takusan is mediated, at least in part, by its ability to interact with tau either directly or indirectly and to prevent tau hyperphosphorylation at postsynaptic sites.
A growing body of evidence suggests that tau pathologies are strongly associated with mitochondrial impairment, which leads to synaptic starvation and dysfunction, and ultimately to neuronal damage in AD (Mandelkow et al., 2003; Vossel et al., 2010; Zempel et al., 2010; Kopeikina et al., 2011; Manczak and Reddy, 2012; Shahpasand et al., 2012). Interestingly, a recent study has shown that hyperphosphorylated tau proteins interact directly with the mitochondrial fission protein Drp1 in postmortem AD brains (Manczak and Reddy, 2012). In the same brain specimens, elevated levels of Drp1 and excessive mitochondrial fragmentation were reported (Manczak et al., 2011). These studies suggest that hyperphosphorylated tau, through its interaction with Drp1 may play important roles in Aβ-induced mitochondrial dysfunction. If this is the case, then Aβ-induced mitochondrial fragmentation should be inhibited by inhibiting tau hyperphosphorylation. In support of this notion, we found that overexpressed α1-takusan prevents Aβ-induced mitochondrial fragmentation in cultured neurons (Fig. 8).
Forced expression of PSD-95 in cultured neurons is sufficient to increase the density of dendritic spines (El-Husseini et al., 2000). Also, the level of PSD-95 is decreased in the cortex and hippocampus of human postmortem AD brains, and this reduction correlates with disease pathology (Pham et al., 2010; Proctor et al., 2010; Sultana et al., 2010). Evidence further suggests that Aβ oligomers specifically target excitatory synapses and interact with PSD-95 at postsynaptic sites (Lacor et al., 2004; Pham et al., 2010). Consistent with this idea, the level of PSD-95 is decreased significantly after a ≥1 h application of Aβ in cultured neurons (Roselli et al., 2005; Fig. 1). These findings suggest that PSD-95 plays an important role in the synaptic pathogenesis of AD. Since overexpressed α1-takusan interacts with PSD-95 in cultured neurons increasing PSD-95 synaptic clustering, this interaction may be mechanistically critical to the synaptic protection conveyed by takusan. It is possible that the association between takusan and PSD-95 leads to a more stabilized PSD-95 protein complex, rendering clusters less susceptible to Aβ-induced damage. In addition, we identified the D2 responsible for this interaction. The D2 of α1-takusan is critical not only for clustering of itself (Fig. 9) and PSD-95 (Fig. 10), but also in mitigating Aβ-induced synaptic loss (Fig. 12). A possible mechanism by which takusan stabilizes PSD-95-containing clusters could involve (1) multimerization of takusan via its D2 and (2) recruitment of PSD-95 to takusan multimers. Moreover, this process may require both tau and the microtubule network, since tau interacts with PSD-95 at postsynaptic sites (Ittner et al., 2010). It has also been shown that PSD-95 interacts with the microtubule end-binding protein EB3 and dynamically regulates the dendritic microtubule cytoskeleton (Sweet et al., 2011). Thus, takusan, PSD-95, and tau proteins may work in concert to stabilize postsynaptic protein complexes and to protect neuronal synapses under pathological conditions.
Finally, based on our deletion studies, we have generated two takusan-derived proteins (D2a and D2b, each of which contains a portion of D2 and the C-terminal PDZ-binding motif) with therapeutic potential. These truncated takusan derivatives are as effective as full-length α1-takusan in protecting synapses from Aβ-induced damage (Fig. 12). Of note, the D2 of α1-takusan is shared by DLG5, a protein that is conserved throughout evolution and is also expressed in humans. Moreover, the fusion protein hD2b, which consists of the human DLG5 D2 and a PDZ-binding motif, can also enhance PSD-95 clustering in cultured neurons (Fig. 13). Interestingly, the PDZ-binding motif of hD2b is identical to the C terminus of the human protein rho guanine nucleotide exchange factor 33 (NCBI Reference Sequence, NP_001138923.2). Therefore, we believe that these protein sequences hold promise not only for mechanistic investigation of synaptic activity and protection against Aβ-induced synaptic injury, but also for developing therapeutic peptides against AD and possibly other neurological diseases involving synaptic degeneration.
Footnotes
This work was supported in part by National Institutes of Health Grants R01 NS054880 and R01 AG031893 (to F.-F.L.); R01 AG021173, R01 NS046673, R01 AG030197, R01 AG038710 and R03 AG034366 (to H.X.); and R01 EY05477, P01 HD29587 and P01 ES016738 (to S.A.L.). Additional support was provided by grants from the Alzheimer's Association (to H.X.) and National Institute of Neurological Disorders and Stroke Neuroscience Core Grant P30 NS076411 (to S.A.L.).
The authors declare no competing financial interests.
References
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, Chen-Dodson E, Kinney GG, Klein WL. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. doi: 10.1126/science.290.5495.1364. [DOI] [PubMed] [Google Scholar]
- Fein JA, Sokolow S, Miller CA, Vinters HV, Yang F, Cole GM, Gylys KH. Co-localization of amyloid beta and tau pathology in Alzheimer's disease synaptosomes. Am J Pathol. 2008;172:1683–1692. doi: 10.2353/ajpath.2008.070829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gústafsdóttir SM, Ostman A, Landegren U. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol. 2002;20:473–477. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in alzheimer's disease: increased amyloid-β and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–1817. doi: 10.1016/S0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpain S, Hipolito A, Saffer L. Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J Neurosci. 1998;18:9835–9844. doi: 10.1523/JNEUROSCI.18-23-09835.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbani MJ, Schlief ML, Fisher DA, Goldberg MP. Dendritic spines lost during glutamate receptor activation reemerge at original sites of synaptic contact. J Neurosci. 2001;21:2393–2403. doi: 10.1523/JNEUROSCI.21-07-02393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LS. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-[beta] toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/S0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, Koffie RM, Frosch MP, Hyman BT, Spires-Jones TL. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer's disease brain. Am J Pathol. 2011;179:2071–2082. doi: 10.1016/j.ajpath.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid β oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz TA, Carter DB, Merchant KM. Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol Dis. 2003;13:246–253. doi: 10.1016/S0969-9961(03)00079-2. [DOI] [PubMed] [Google Scholar]
- Lee G, Rook SL. Expression of tau protein in non-neuronal cells: microtubule binding and stabilization. J Cell Sci. 1992;102:227–237. doi: 10.1242/jcs.102.2.227. [DOI] [PubMed] [Google Scholar]
- Love S, Siew LK, Dawbarn D, Wilcock GK, Ben-Shlomo Y, Allen SJ. Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol Aging. 2006;27:797–803. doi: 10.1016/j.neurobiolaging.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Manczak M, Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer's disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet. 2012;21:2538–2547. doi: 10.1093/hmg/dds072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet. 2011;20:2495–2509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003;24:1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410–426. doi: 10.1016/j.neuron.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi N, Tu S, Shin Y, Cui J, Kurokawa T, Zhang D, Chen HS, Tong G, Lipton SA. Neuroprotection by the NR3A subunit of the NMDA receptor. J Neurosci. 2009;29:5260–5265. doi: 10.1523/JNEUROSCI.1067-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A, Salmon D, Galasko D, Michael S, Savas JN, Yates JR, Glabe C, Masliah E. Progressive accumulation of amyloid-β oligomers in Alzheimer's disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 2010;277:3051–3067. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid β-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- Proctor DT, Coulson EJ, Dodd PR. Reduction in post-synaptic scaffolding PSD-95 and SAP-102 protein levels in the Alzheimer inferior temporal cortex is correlated with disease pathology. J Alzheimers Dis. 2010;21:795–811. doi: 10.3233/JAD-2010-100090. [DOI] [PubMed] [Google Scholar]
- Reddy PH, Beal MF. Amyloid β, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer's disease. Trends Mol Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu GQ, Palop JJ, Noebels JL, Mucke L. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roselli F, Tirard M, Lu J, Hutzler P, Lamberti P, Livrea P, Morabito M, Almeida OF. Soluble β-amyloid1–40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J Neurosci. 2005;25:11061–11070. doi: 10.1523/JNEUROSCI.3034-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, Toyoshima Y, Hasegawa M, Hisanaga S. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. J Neurosci. 2012;32:2430–2441. doi: 10.1523/JNEUROSCI.5927-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Sheng M, Sabatini BL, Südhof TC. Synapses and Alzheimer's disease. Cold Spring Harb Perspect Biol. 2012;4:a005777. doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Sokolow S, Henkins KM, Bilousova T, Miller CA, Vinters HV, Poon W, Cole GM, Gylys KH. AD synapses contain abundant Aβ monomer and multiple soluble oligomers, including a 56-kDa assembly. Neurobiol Aging. 2012;33:1545–1555. doi: 10.1016/j.neurobiolaging.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: insights into their potential roles for loss of synapses and memory, accumulation of Aβ, and neurodegeneration in a prodromal stage of Alzheimer's disease. J Neurosci Res. 2010;88:469–477. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet ES, Previtera ML, Fernández JR, Charych EI, Tseng CY, Kwon M, Starovoytov V, Zheng JQ, Firestein BL. PSD-95 Alters microtubule dynamics via an association with EB3. J Neurosci. 2011;31:1038–1047. doi: 10.1523/JNEUROSCI.1205-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK. Co-occurrence of Alzheimer's disease β-amyloid and tau pathologies at synapses. Neurobiol Aging. 2010;31:1145–1152. doi: 10.1016/j.neurobiolaging.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, Dziewczapolski G, Nakamura T, Cao G, Pratt AE, Kang YJ, Tu S, Molokanova E, McKercher SR, Hires SA, Sason H, Stouffer DG, Buczynski MW, Solomon JP, Michael S, et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. 2013;110:E2518–E2527. doi: 10.1073/pnas.1306832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu S, Shin Y, Zago WM, States BA, Eroshkin A, Lipton SA, Tong GG, Nakanishi N. Takusan: a large gene family that regulates synaptic activity. Neuron. 2007;55:69–85. doi: 10.1016/j.neuron.2007.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Aβ-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Neurochem. 2009;109:153–159. doi: 10.1111/j.1471-4159.2009.05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM. Aβ oligomers cause localized Ca2+ elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WQ, Santini F, Breese R, Ross D, Zhang XD, Stone DJ, Ferrer M, Townsend M, Wolfe AL, Seager MA, Kinney GG, Shughrue PJ, Ray WJ. Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption. J Biol Chem. 2010;285:7619–7632. doi: 10.1074/jbc.M109.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]