

Abstract
Several human postnatal somatic cell types have been successfully reprogrammed to induced pluripotent stem cells (iPSCs). Blood mononuclear cells (MNCs) offer several advantages compared with other cell types. They are easily isolated from umbilical cord blood (CB) or adult peripheral blood (PB), and can be used fresh or after freezing. A short culture allows for more efficient reprogramming, with iPSC colonies forming from blood MNCs in 14 d, compared with 28 d for age-matched fibroblastic cells. The advantages of briefly cultured blood MNCs may be due to favorable epigenetic profiles and gene expression patterns. Blood cells from adults, especially nonlymphoid cells that are replenished frequently from intermittently activated blood stem cells, are short-lived in vivo and may contain less somatic mutations than skin fibroblasts, which are more exposed to environmental mutagens over time. We describe here a detailed, validated protocol for effective generation of integration-free human iPSCs from blood MNCs by plasmid vectors.
INTRODUCTION
Human iPSCs have been derived from many cell types, including blood cells, since 2007 (refs. 1–7). The original protocol using retroviral vectors expressing Oct4 (officially known as Pou5f1), Sox2, Klf4 and c-Myc (officially known as Myc) has proven to be successful in most of the reported reprogramming experiments. To make iPSCs safer for regenerative medicine and more reliable for disease modeling, efforts have been made to derive iPSCs without the integration of exogenous DNA into cellular genomes. Conceptually, the most straightforward strategies include delivery of reprogramming factors to the cells in the form of recombinant proteins or mRNAs. The feasibility of these strategies has been shown by proof-of-principle experiments8,9. However, the reprogramming efficiencies using proteins have been low, and this method requires repetitive delivery of multiple proteins and mRNAs into the fibroblast cells on a daily basis for up to 17 d, thus markedly limiting the application of these technologies in most laboratories. Although future improvement of these genetically safer technologies is required for widespread use, plasmid vectors containing the EBNA1/OriP sequences of the Epstein-Barr virus replicon have provided an effective alternative for reprogramming human somatic cells. Human iPSC lines were established by one-time transfection of neonatal foreskin fibroblasts by 2–3 plasmids encoding seven different reprogramming factors or fetal neural progenitor cells with fewer factors10,11. These studies demonstrated the feasibility of using EBNA1/OriP-containing plasmids to achieve prolonged plasmid retention and gene expression in transfected somatic cells, allowing iPSC induction of human somatic cells that require longer times (in weeks) of transgene expression. However, the efficiency using the reported episomal vectors for reprogramming postnatal human somatic cells is too low to be a routine method for most investigators.
Development of the protocol
To develop a protocol that generates integration-free iPSCs more efficiently from easily accessible cell sources, we studied the epigenetic features of various cell types, as well as reprogramming efficiencies7,12,13. We discovered that blood MNCs, either purified CD34 + hematopoietic stem/progenitor cells (HSPCs) or unfractionated cells after a brief culture, possess unique epigenetic signatures that are closer than those of the age-matched fibroblasts to embryonic stem cells (ESCs) and iPSCs7,13. A MNC expansion protocol, which preferentially enriches for erythroblast-like cells and eliminates lymphocytes, alleviates the need for isolation of CD34+ HSPCs before reprogramming. By using MNCs with favorable epigenetic profiles, we have also tested an improved EBNA1/OriP-based plasmid system that expresses five reprogramming factors (Oct4, Sox2, Klf4, c-Myc and Lin28) as a single unit linked by 2A cleavable peptide sequences. We have found that a single transfection of this single plasmid into expanded/primed umbilical CB MNCs can generate hundreds of TRA-1-60–positive iPSC colonies in 2 weeks13. Although the reprogramming efficiency with the single vector is much lower in adult blood MNCs, addition of another factor (e.g., SV40 large T antigen) by co-transfection of second episomal vector enhances the reprogramming efficiency by four- to fivefold13. Overall, this episomal vector system and the cell expansion/priming protocol make it possible to derive iPSC clones from as little as a few milliliters of postnatal blood (Fig. 1).
Figure 1.

Timeline for the MNC expansion and iPSC generation protocol. MNCs are isolated and expanded with a precise cytokine mixture over 8–9 d and transfected with episomal vectors on day 0. Human iPSC colonies derived from blood MNCs can be observed as early as 10 d after nucleofection.
We have carried out whole-genome sequencing on human iPSC lines derived using this episomal vector system14. Through rigorous sequencing analyses, we have not only further confirmed that there is no plasmid integration, but also demonstrated a low incidence of DNA sequence variation in these iPSCs compared with their somatic cell origin. Our data indicate that this episome-mediated reprogramming is not inherently mutagenic during integration-free iPSC induction. More recently, we replaced mouse feeder cells with engineered human feeder cells15. Thus, this plasmid-based protocol for derivation of integration-free human iPSCs by using blood cells and under defined culture conditions provides a powerful and safe tool for both stem cell research and future regenerative medicine.
Comparison with other methods
Several other methods have been reported using viral vectors for reprogramming of human blood cells without isolation of HSPCs16–21. The most attractive method is the use of recombinant Sendai (nonintegrating RNA) viruses to reprogram activated human T lymphocytes that can be efficiently infected21. Compared with these methods that preferentially expand and/or infect activated T lymphocytes within MNCs for reprogramming using viral vectors, the protocol described here uses plasmid DNA to generate human iPSCs that contain the intact germline genomes: they are not only free of transgene integration, but also lack V(D)J somatic rearrangements that are present in T lymphocytes. Thus, the iPSCs generated from non-lymphocytes that have intact nuclear genomes may be more suitable for therapy purposes. The absence of pre-rearranged V(D)J DNA segments in the iPSC genome may also eliminate certain safety concerns raised by the observation that mice derived from reprogrammed T cells have a high rate of T cell lymphomas22. Recently, another episomal plasmid-based protocol has also been reported to generate integration-free iPSCs from human bone marrow and CB cells without enriching for HSPCs or T lymphocytes23. The method differs from the protocol described here in the combinations of reprogramming factors and in the cell preparation conditions. The reprogramming efficiency of adult PB MNCs using the protocol described in this report is currently unclear23. Although the two methods of using episomal vectors are overall similar in derivation of integration-free iPSC lines from hematopoietic cells, they differ in the numbers of episomal vectors used and the culture conditions to prime hematopoietic cells for reprogramming. To reduce the possibility that derived IPSC lines would be from T and B lymphocytes that are present at high levels (> 50%) in CB and adult PB than in bone marrow, we cultured unfractionated MNCs for 8–14 d to enrich and expand erythroblasts (and eliminate lymphocytes) before reprogramming. Flow cytometric analysis after blood MNC expansion showed that the percentage of T or B lymphocytes present in the culture decreases to < 4%, whereas the erythroblasts increased from ~1% to a range of 11–52% (ref. 13).
Limitations of the protocol
Derivation of integration-free human iPSCs from small amounts of unfractionated human blood offers key advantages in modeling a broad range of human diseases, including those that may carry blood cell–specific somatic mutations. The intact genomes (without inserted foreign DNA sequences or V(D)J somatic recombination) of the iPSCs may be more suitable for regenerative medicine purposes. However, this protocol uses an expansion condition that preferentially expands erythroblasts, and therefore may not be suitable for reprogramming blood cells from patients who have a deficiency in erythroblast formation. In these cases, we recommend considering an alternative protocol23.
The protocol described here provides robust reprogramming of CB MNCs without a need to isolate a subpopulation. The efficiency of reprogramming adult PB MNCs is substantially (20–50×) lower than that of CB MNCs. This is a major drawback, especially when compared with the method using viral vectors with CD34 + or activated T cells21. We and others have found that adding sodium butyrate during reprogramming can enhance the efficiency of human iPSC derivation12,13,24,25, allowing sufficient numbers of iPSC colonies (≥12) to be formed after 12 d in a single experiment using 2 × 106 expanded erythroblasts initiated from 2 × 106 or fewer blood MNCs. In practice, we normally pick up to 12 candidate iPSC clones from a given donor or patient and select the two or three best clones for characterization after expansion. In case the iPSC derivation efficiency from a given patient is not high enough, one may duplicate or triplicate sample sizes by using more starting blood MNCs (abundantly available) or enhance reprogramming by other means. For example, recent work described an array of small molecules reported to enhance reprogramming efficiency. These include, but are not limited to, the inhibitors targeting the ROCK, GSK-3, TGF-β and MEK pathways26,27. Certain microRNAs (miRNAs) have also been reported to enhance the reprogramming efficiency of human fibroblast cells28,29, although their stimulatory effects have not yet been reported using nonintegrating vectors and blood cells. It is expected that certain combinations of these small molecules or miRNAs will also further boost the efficiency of the protocol described here.
Experimental design
Isolation of CB and PB MNCs
The first part of our protocol involves isolating MNCs from adult PB and umbilical CB samples. This is accomplished through the use of Ficoll-Paque Premium (P = 1.077) for density-based centrifugal separation. After a 30-min spin, MNCs stay just above the Ficoll layer, whereas red blood cells ( > 99%) traverse through and sediment at the bottom. A small amount of red blood cells left in the MNC fraction can be removed after MNC collection by treatment with red cell lysis buffer, if necessary.
Expansion and culture of blood MNCs
CB and PB MNCs are expanded over a course of 8–14 d in a serum-free medium (SFM) supplemented with a mixture of cytokines. This culture condition favors the expansion of erythroblasts and will not support lymphocyte growth. The number of viable cells will decrease over the first few days of the expansion process, but will slowly grow from day 4 onward. This expansion stage not only drives the cells into the cell cycle, but also further primes the cells to certain epigenetic states that are more easily reprogrammed13. Since the publication of the original paper13, we have found that adding more holotransferrin (up to 100 μg ml−1) to that present in the standard insulin-transferrin-selenium-X (ITS-X) solution enhances consistent erythroblast expansion. In general, CB MNCs have superior expansion capability under this condition. The reprogramming efficiency of CB MNCs is also much higher compared with that of adult PB MNCs. Therefore, we recommend using CB MNC samples for practice or as positive controls for both steps when PB MNCs are reprogrammed by this protocol.
Reprogramming of blood MNCs by episomal vector expression after transfection
The reprogramming process is initiated by a single transfection of an episomal plasmid, pEB-C5, expressing five factors (Oct4, Sox2, Klf4, c-Myc and Lin28; Fig. 2). This single plasmid has been shown to efficiently reprogram CB MNCs13. To further enhance the reprogramming efficiency of other types of cells, this vector can be supplemented with episomal plasmids expressing SV40 large T antigen (pEB-Tg) or shRNA against p53 (pEB-p53shRNA)13. Although sustained T antigen expression and p53 knockdown using integrating vectors resulted in altered genomes in derived iPSCs, transient expression of T antigen by episomal vectors during reprogramming did not have a detrimental effect on iPSC genomic integrity13,14,30. These episomal plasmids are available from Addgene (plasmid nos. 28213 [pEB-C5], 28220 [pEB-Tg] and 28222 [pEB-p53shRNA]). After a single tranfection, the cells are plated back in the expansion medium to allow recovery. Two days later, they are plated onto standard plates coated with feeder cells, with the culture medium changed to ESC medium the following day. The reprogramming procedure is similar to those that have been used for fibroblast cell reprogramming and described in detail24. In most cases, iPSC-like colonies that acquire TRA-1-60 expression on the cell surface can be observed within 2 weeks after transfection of blood MNCs.
Figure 2.
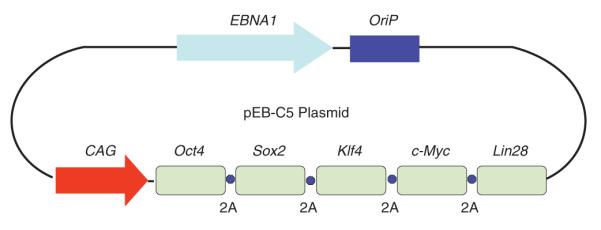
A diagram of the polycistronic episomal reprogramming plasmid pEB-C5. The EBNA1 (trans) and OriP (cis) elements derived from the Epstein-Barr virus genome enable nonintegrating plasmids to replicate and sustain in proliferating human cells. Five genes encoding reprogramming factors, Oct4, Sox2, Klf4, c-Myc and Lin28, are linked by 2A cleavable peptide sequences and are controlled by the synthetic CAG promoter. This pEB-C5 plasmid can also be used in combination with other episomal vectors expressing additional factors to further enhance reprogramming efficiency.
TRA-1-60 live staining for identifying successfully reprogrammed colonies
Along with assessing morphological differences, staining with TRA-1-60–specific antibody can be used to distinguish successfully reprogrammed colonies from other transformed non-iPSC colonies. It has been shown that the combination of morphology and TRA-1-60 live staining can reliably identify bona fide iPSC colonies12,13,24,31. After staining, individual clones can be picked and expanded for further characterization.
Materials
REAGENTS
Cord blood (CB) or peripheral blood (PB), 5–500 ml, provided in either citrate phosphate dextrose or sodium heparin preservatives ! CAUTION Informed parental and patient consent for CB and PB donation must be obtained before proceeding and must conform to institutional ethics regulations.
Blood collection tubes containing sodium heparin (BD Vacutainer, cat. no. 367871)
CB collection bags containing citrate phosphate dextrose (Pall Corporation, cat. no. 791-01)
Episomal plasmid pEB-C5 expressing five reprogramming transgenes (Addgene, plasmid no. 28213)
(Optional) episomal plasmids expressing other factors (Addgene, plasmid no. 28220 [pEB-Tg] and no. 28222 [pEB-p53shRNA])
Ficoll-Paque Premium, P = 1.077 (GE Healthcare, cat. no. 17-5442-02)
PBS without calcium and magnesium (Invitrogen, cat. no. 10010-023)
Ammonium chloride (NH4Cl; Sigma, cat. no. A0171)
Potassium bicarbonate (KHCO3; Sigma, cat. no. 431583)
EDTA disodium powder or 0.5 M solution (Invitrogen, cat. no. 15575-020) ! CAUTION EDTA disodium powder is toxic by skin contact.
Trypan blue (Invitrogen, cat. no. T10282)
Cell freezing medium (Stemgent, cat. no. 01-0013-51)
Iscove’s modified Dulbecco’s medium (IMDM; Invitrogen, cat. no. 21056023)
Ham’s F-12 (Mediatech, cat. no. 10-080-CV)
Insulin-transferrin-selenium-X supplement, 100× (ITS-X; Invitrogen, cat. no. 51500-056)
Chemically defined lipid concentrate (Invitrogen, cat. no. 11905031)
l-Glutamine (Invitrogen, cat. no. 21051024)
BSA (Sigma, A9418)
l-Ascorbic acid, 2-phosphate sesquimagnesium salt (Sigma, cat. no. A8960) ! CAUTION This salt is toxic by skin contact.
1-Thioglycerol (98%; Sigma, cat. no. M6145) ! CAUTION 1-Thioglycerol is toxic by inhalation, ingestion and skin contact.
Recombinant human stem cell factor (SCF; PeproTech, cat. no. 300-07)
Recombinant human interleukin-3 (IL-3; PeproTech, cat no. 200-03)
Recombinant human insulin-like growth factor-1 (IGF-1; PeproTech, cat. no. 100-11)
Recombinant human erythropoietin (EPO; R&D Systems, cat. no. 287-TC-500)
Dexamethasone (Sigma-Aldrich, cat. no. D2915)
Human holo-transferrin (R&D Systems, cat. no. 2914-HT-100MG)
SFM (see Reagent Setup)
Mononuclear cell (MNC) medium (see Reagent Setup)
Human CD34+ cell nucleofection kit (Lonza, cat. no. VPA-1003)
Gelatin (0.1% (wt/vol); Stem Cell Technologies, cat. no. 07903)
DMEM, high glucose (Invitrogen, cat. no. 11965-092)
FBS, defined (Hyclone, cat. no. SH3007003)
Antibiotic-antimycotic (Invitrogen, cat. no. 15240062)
l-Glutamine (Invitrogen, cat. no. 25030081)
Mouse embryonic fibroblast (MEF) medium (see Reagent Setup)
MEFs from the CF-1 strain), irradiated (GlobalStem, cat. no. GSC-6001G, or other sources)
KnockOut DMEM (Invitrogen, cat. no. 10829018)
KnockOut serum replacement (KSR; Invitrogen, cat. no. 10828028)
Modified Eagle’s medium–non-essential amino acid solution (MEM-NEAA; Invitrogen, cat. no. 11140050)
2-Mercaptoethanol (Invitrogen, cat. no. 21985023)
Fibroblast growth factor, basic (bFGF; PeproTech, cat. no. 100-18B)
(Optional) sodium butyrate (NaB; Sigma, cat. no. B5887)
Embryonic stem cell (ESC) medium (see Reagent Setup)
Conditioned medium (CM; see Reagent Setup)
TRA-1-60 mouse IgM antibody (Millipore, cat. no. MAB4360)
Alexa Fluor 555–conjugated goat anti-mouse IgM antibody (Invitrogen, cat. no. A21426)
EQUIPMENT
Class-II biological safety cabinet (SterilGARD, Baker Company)
Cell culture incubator set at 37 °C, 5% CO2 and 90% humidity (Steri-Cult by Thermo Scientific, part no. 3310)
Temperature-controlled centrifuge and swinging rotor with adapters for 15-ml and 50-ml conical tubes and microplates, capable of spinning at 2,000 r.p.m. (Allegra 6R centrifuge with GH-3.8A rotor, Beckman Coulter)
Tissue culture microscope with epifluorescence, with ×10, ×20 and ×40 objectives (TE-200U, Nikon)
Microcentrifuge (Eppendorf, model no. 5424)
Tissue culture plates, 96-, 48-, 24-, 12- and six-well plates
Tissue culture flasks, T25, T75 and T150
Conical tubes, 15 and 50 ml
Tissue culture pipettes, 2, 5, 10, 25 and 50 ml
Bottle-top filter unit with a 0.2-μm filter, 500 ml (Nalgene, cat. no. 566-020)
Countess automated cell counter (Invitrogen, cat. no. C10227)
Cell counting slides for Countess cell counter (Invitrogen, cat. no. C10312)
Cryovials (Nalgene, cat. no. 5012-0020)
Controlled-rate freezing container (Nalgene, cat. no. 5100-0001)
Liquid nitrogen storage container (Thermo Fisher, cat. no. 11-676-55)
Nucleofector II device (Amaxa/Lonza, cat. no. AAD-1001) for DNA transfection
Sterile, pulled 9-inch glass pipettes bent into ‘J’ shape, appropriate for picking colonies
Micropipettes, 10, 20, 200 and 1,000 μl
REAGENT SETUP
Red cell lysis buffer
Add 4.145 g of ammonium chloride, 0.5 g of potassium bicarbonate and 100 μl of EDTA to 400 ml of ultrapure water. Adjust the pH to the range of 7.2–7.4, and add ultrapure water to bring the volume to 500 ml. Filter-sterilize the buffer and store it at 4 °C. Use the buffer within 6 months.
SFM
To prepare 500 ml of SFM, combine 245 ml of IMDM and 245 ml of Ham’s F-12 with 5 ml of ITS-X, 5 ml of chemically defined lipid concentrate, 5 ml of l-glutamine, 0.025 g of l-ascorbic acid, 2.5 g of BSA and 9 μl of 1-thioglycerol (final concentration of 200 μM). Filter-sterilize the medium and store it at 4 °C. Use the medium within 3 weeks.
MNC medium
MNC medium contains SFM supplemented with 100 ng ml−1 SCF, 10 ng ml−1IL-3 (10 ng ml−1), 2 U ml−1 EPO, 40 ng ml−1 IGF-1, 1 μM dexamethasone and 100 μg ml−1 holo-transferrin. Prepare the medium daily immediately before use.
MEF medium
To prepare 500 ml of MEF medium, combine 450 ml of DMEM with 50 ml of FBS, 5 ml of antibiotic-antimycotic and 5 ml of l-glutamine. Filter-sterilize the medium and store it at 4 °C. Use the medium within 3 weeks.
ESC medium
To prepare 600 ml of medium, combine 470 ml of knockout DMEM with 120 ml of KSR, 6 ml of antibiotic-antimycotic, 6 ml of l-glutamine, 6 ml of MEM-NEAA, 0.6 ml of 2-mercaptoethanol and 10 ng ml−1 bFGF. Filter-sterilize the medium and store it at 4 °C. Use the medium within 3 weeks.
Gelatin (0.1% wt/vol)-coated plates
Apply 0.5 ml of 0.1% (wt/vol) gelatin to each well of a new 12-well plate. After 20 min, aspirate the gelatin and allow the plate to dry.  CRITICAL Plates must be prepared immediately before use.
CRITICAL Plates must be prepared immediately before use.
MEF-coated plates
Thaw MEFs in MEF medium and plate them on a dry, gelatin-coated 12-well plate at a density of ~100,000 cells per well. This density can be adjusted for plates of other sizes. Incubate the MEFS overnight at 37 °C, 5% CO2. Plates are best when used 1 d after MEF plating.
CM
Thaw MEFs in MEF medium and plate them on a dry, gelatin-coated 12-well plate at a density of ~100,000 cells per well. Incubate the MEFs at 37 °C, 5% CO2.The day after plating, discard the MEF medium and wash the cells once with PBS. Discard the PBS and add ESC medium (2 ml per 10 cm2 of MEFs). The next day, collect and replace the ESC medium. CM can be collected daily for 4–5 d, as long as the MEF morphology is acceptable. If the MEFs appear to be very thin, stretched, and are beginning to peel off of the plate, discontinue collection of CM. After collection, filter-sterilize and add bFGF at a concentration of 10 ng ml−1. Store the medium at 4 °C, and use it within 2 weeks.
PROCEDURE
Donor CB and PB collection
 TIMING ~10 min
TIMING ~10 min
1| Collect 5–80 ml of donor PB by venipuncture into an appropriate vessel containing sodium heparin preservative, or collect whole CB from a donor (typically 100–200 ml) into an appropriate vessel containing citrate phosphate dextrose preservative.
! CAUTION Informed consent from patients must be obtained before collection.
 PAUSE POINT CB and PB should be stored and shipped at 25 °C. Do not use samples that are older than 24 h. It is much better to freeze isolated MNCs than whole blood for later use.
PAUSE POINT CB and PB should be stored and shipped at 25 °C. Do not use samples that are older than 24 h. It is much better to freeze isolated MNCs than whole blood for later use.
Isolation of CB and PB MNCs via density gradient
 TIMING ~2.5 h
TIMING ~2.5 h
2| Dilute the CB or PB 1:2 with sterile PBS in an upright, appropriately sized sterile tissue culture flask or tube. Use a T150 flask for large volumes and a 50-ml conical tube for small volumes.
 CRITICAL STEP This and all subsequent steps should be carried out in a class II biological safety cabinet.
CRITICAL STEP This and all subsequent steps should be carried out in a class II biological safety cabinet.
3| Carefully and slowly layer 35 ml of diluted blood onto 15 ml of Ficoll-Paque Premium in a 50-ml conical tube. Repeat the process in additional conical tubes containing Ficoll-Paque Premium for the remaining diluted blood. If the last amount does not total 35 ml, add PBS.
 CRITICAL STEP Take care to minimize mixing of the layers by tilting the tube at a 45° angle and allowing the blood to run down the side of the tube onto the Ficoll layer. This will ensure a tight band of MNCs after isolation.
CRITICAL STEP Take care to minimize mixing of the layers by tilting the tube at a 45° angle and allowing the blood to run down the side of the tube onto the Ficoll layer. This will ensure a tight band of MNCs after isolation.
 CRITICAL STEP Red blood cells will settle to the bottom of the tube as the diluted blood is added. This is normal, as the red blood cells are denser than the Ficoll.
CRITICAL STEP Red blood cells will settle to the bottom of the tube as the diluted blood is added. This is normal, as the red blood cells are denser than the Ficoll.
4| Centrifuge the tubes at 750g for 30 min at 25 °C in a swinging bucket rotor.
 CRITICAL STEP This step must be performed with the centrifuge brake off. This will allow for proper separation.
CRITICAL STEP This step must be performed with the centrifuge brake off. This will allow for proper separation.
5| Aspirate the upper, yellow layer containing plasma from each tube and discard it. Carefully transfer each cloudy white interphase layer containing MNCs to a new 50-ml conical tube with a 10-ml pipette.
6| Add 30 ml of PBS to each tube containing the MNCs and centrifuge the tubes at 350g for 10 min at 4 °C. This step and subsequent steps should be performed with the centrifuge brake on.
7| Discard the supernatant, and combine and resuspend the pellets in one 50-ml conical tube with 5 ml of red cell lysis buffer. Let the cells incubate at 25 °C for 10 min. Fill the tube with PBS to dilute the lysis buffer.
8| Centrifuge the cells at 300g for 10 min at 4 °C.
9| Discard the supernatant and resuspend the pellet in 25 ml of PBS.
10| Centrifuge the cells at 300g for 10 min at 4 °C.
11| Repeat Steps 9 and 10 once.
12| Discard the supernatant and resuspend the pellet in 5 ml of PBS.
13| Count the number of viable cells (on the basis of trypan blue exclusion), after a 1:10 or greater dilution.
14| Aliquot the cells as needed for expansion.
 PAUSE POINT At this step, MNCs can be frozen for later use. Combine 5 million or more cells per milliliter of cell freezing medium. Aliquot 1 ml per vial and freeze the vials in a controlled-rate freezing container. Cells can be stored in a liquid nitrogen storage container (−150 °C) for a prolonged time. Recovery of ≥80% is expected after proper thawing. Apart from freezing, excess cells can be pelleted for future DNA and RNA extraction. Cell pellets should be stored at −80 °C.
PAUSE POINT At this step, MNCs can be frozen for later use. Combine 5 million or more cells per milliliter of cell freezing medium. Aliquot 1 ml per vial and freeze the vials in a controlled-rate freezing container. Cells can be stored in a liquid nitrogen storage container (−150 °C) for a prolonged time. Recovery of ≥80% is expected after proper thawing. Apart from freezing, excess cells can be pelleted for future DNA and RNA extraction. Cell pellets should be stored at −80 °C.
Expansion of CB and PB MNCs
 TIMING 8–14 d
TIMING 8–14 d
15| Day –8 (CB) or day –14 (PB). Plate 5–10 million viable CB or PB MNCs (fresh or after thawing) at a density of 2–5 million cells per milliliter of MNC medium in one well of a tissue culture treated six-well plate and incubate at 37 °C, 5% CO2 for 2 d.
16| Day –5 (CB) or day –11 (PB). Remove the cells and medium with a sterile pipette and place them in a sterile 15-ml conical tube.
17| Centrifuge the cells at 200g for 5 min at 25 °C. Remove and discard the supernatant and resuspend the cells in 1 ml of MNC medium.
18| Count the number of viable cells (in an aliquot).
 CRITICAL STEP At day –5 (CB) or day –11 (PB), the cell number should have decreased. This is expected.
CRITICAL STEP At day –5 (CB) or day –11 (PB), the cell number should have decreased. This is expected.
19| Plate the cells in a six-well plate at a density of 0.5–1 million per milliliter of MNC medium and incubate them for 3 d.
20| Day –2 (CB) or day –8 (PB). Repeat the cell counting and medium change as described in Steps 16–19, but incubate cells for a further 2 d (for CB) or 3–4 d (for PB).
21| Count the number of cells. For CB, count the cells after 8 d of expansion. An ~3.5-fold increase over the starting cell population should be observed. For PB, continue to change medium as described in Steps 16–19 and count the cells after 14 d of expansion. An approximately equal or greater number of cells over the starting cell population should be observed.
? TROUBLESHOOTING
Reprogramming of blood MNCs by episomal vector transfection
 TIMING ~14 d
TIMING ~14 d
22|Day 0 (day of transfection). Prepare the transfection solution provided in the Lonza human CD34+ cell nucleofector kit by mixing 0.5 ml of supplement with 2.25 ml of human CD34+ cell nucleofector solution. The solution is now ready to use and is stable for 3 months at 4 °C.
23| Count cultured CB or PB MNCs, and then transfer 2 million cells to a 15-ml conical tube containing 5 ml of PBS.
24| Centrifuge the cells at 200g for 5 min at 25 °C. Discard the supernatant completely and carefully resuspend the pellet in 100 μl of human CD34+ cell nucleofector solution.
25| Combine the 100-μl cell suspension with 10 μg of plasmid DNA (≥1 μg μl−1) of the pEB-C5 episomal vector. If you plan to use the pEB-C5 + pEB-Tg vector combination for transfection, the ratio is 8 μg of pEB-C5 to 2 μg of pEB-Tg.
26| Immediately transfer the cell/DNA mixture to a sterile cuvette (provided with the Lonza kit).
27| Place the cuvette into the cuvette holder of the Amaxa Nucleofector II device and apply program ‘T-016’.
28| When the program is complete, remove the cuvette. Carefully transfer the solution from the cuvette using a plastic pipette (provided with the Lonza kit) into 2 ml of MNC medium and plate it in one well of a 12-well plate. Incubate for 2 d at 37 °C, 5% CO2.
? TROUBLESHOOTING
29|Day 2 after transfection. Collect transfected cells with a sterile 5-ml pipette and place into a 15-ml conical tube. Centrifuge the cells at 200g for 5 min at 25 °C. Remove and discard the supernatant and resuspend the pelleted cells in 1 ml of MEF medium.
30| Count the cells and transfer them in MEF medium to a MEF-coated 12-well plate at a density of ~100,000 cells per well.
31| Spin the sealed plates at 200g for 30 min at 25 °C. When the spin is finished, incubate the plates at 37 °C, 5% CO2 overnight.
32|Day 3 after transfection. Remove the medium from the culture and transfer to 15-ml conical tubes. Add 0.5 ml of ESC medium to each well. Centrifuge the tubes at 300g for 5 min at 25 °C and aspirate the top medium without removing cell pellets at the bottom of the tubes. Resuspend the pellet with 0.5 ml of ESC medium and plate back to the MEF plate so that there is 1 ml of ESC medium per well. Incubate the cells for 48 h. If desired, NaB (0.25 mM) can be added to the cultures to enhance iPSC derivation. Continue supplementation until colonies are ready to be picked (on days 10–14 after transfection; see Step 36).
33|Day 5 after transfection. Aspirate the medium from the cultures, discard it and replace it with ESC medium at a volume of 1 ml per well. Incubate the cells for a further 48 h.
34|Day 7 after transfection. Aspirate the medium from the cultures, discard it and replace it with ESC medium at a volume of 1 ml per well. Incubate the cells for a further 48 h.
35|Day 9 after transfection. Aspirate the medium from the cultures, discard it and replace it with CM at a volume of 1 ml per well. Incubate the cells for 24 h.
 CRITICAL STEP The use of CM is necessary at this time to maintain colony growth.
CRITICAL STEP The use of CM is necessary at this time to maintain colony growth.
36|Days 10–14 after transfection. Check the cells daily, continuing to incubate and change the medium every other day. During days 10–14, large colonies should be visible and should be characterized as described in the next section before further passage.
? TROUBLESHOOTING
TRA-1-60 staining to identify fully reprogrammed colonies
 TIMING 1–2 d
TIMING 1–2 d
37| On the day before staining, prepare a 96-well MEF-coated plate as described in Reagent Setup. MEFs should be plated at a density of ~12,500 cells per well.
38| On the day of staining, dilute TRA-1-60 antibody to 1:300 and Alexa Fluor 555–conjugated secondary antibody to 1:500 in ESC medium.
39| Aspirate the medium from the colony-containing wells, discard and add 0.5 ml of the antibody-containing ESC medium to each well. Incubate the cells at 37 °C, 5% CO2 for 1 h.
40| Aspirate off the medium, discard it and wash each well with 1 ml of PBS.
41| Add 1 ml of ESC medium per well and examine the colonies with an inverted microscope properly equipped for fluorescence.
42| Cut TRA-1-60–positive colonies with sterile, pulled glass Pasteur pipettes. Remove each colony with a micropipette and place it into one well of a 96-well MEF-coated plate containing ESC medium. Repeat this process to obtain a sufficient number of clones for expansion.
43| Incubate the cells, aspirating and replacing the medium daily. Colonies should expand and be ready to passage in 4–5 d. Cultures can be passaged according to standard iPSC protocols2,24.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 1.
Table 1.
Troubleshooting table
| Step | Problem | Solution |
|---|---|---|
| 21 | Poor cell proliferation |
In our experience, the selection of blood collection tubes and the storage conditions of the blood after drawing can markedly affect the viability of blood MNCs and the subsequent expansion. In our hands, CB preserved in citrate phosphate dextrose and PB preserved in sodium heparin and sodium citrate have been successfully expanded. Blood should be transported at 25 °C and the procedure should be started within 24 h for best results. For expansion of PB cells, it may take up to 11 d to achieve sufficient cell numbers for reprogramming. In addition, the quality of the components of the SFM should be considered |
| 28 | Excessive cell death or low transfection efficiency |
It is important to have high-quality plasmid DNA. The concentration of plasmid is also important; we recommend having at least 1 μg μl−1 to minimize the volume added to the nucleofection solution |
| 36 | Poor colony growth | It is important to have high-quality feeder cells (MEFs) that are appropriate for the expansion of iPSCs. If the MEF morphology begins to look poor during cell culture for reprogramming, supplementing hESC medium with CM will help sustain the growth of iPSCs |
 TIMING
TIMING
Step 1, donor CB and PB collection: ~10 min
Steps 2–14, isolation of CB and PB MNCs using Ficoll-Paque Premium density gradient: ~2.5 h
Steps 15–21, expansion of CB and PB MNCs: 8–14 d
Steps 22–36, reprogramming of blood MNCs: ~14 d
Steps 37–43, TRA-1-60 staining: 1–2 d
ANTICIPATED RESULTS
During the first days of MNC expansion, cellular debris will be visible in the culture. This is to be expected, as only a subset of cells will expand under this culture condition. The cell number should decrease over the course of the first few days and then begin to increase. After nucleofection and culture under human ESC conditions for 10–14 d, colonies that resemble human ESC colonies in morphology should be observed. The identity of the colonies can be further confirmed by TRA-1-60 antibody live-cell staining. Approximately 200–800 TRA-1-60–positive colonies usually can be observed from 2 million input CB MNCs used for nucleofection. The efficiencies of adult MNC reprogramming are typically ~50 times lower than those of the CB MNCs.
After the TRA-1-60–positive iPSC clones are sufficiently expanded, further characterization can be performed (Fig. 3). In addition to in vitro assays, the pluripotency of these cell lines can be confirmed by teratoma formation after injection into immunodeficient mice; tissues from all three germ layers can be observed in the tumors (Fig. 3). Karyotyping by G-banding remains a reliable method to ensure the genome integrity of established iPSC lines after reprogramming and extended passaging (Fig. 3). The genome integrity of derived iPSC lines can be also assessed by high-density comparative genomic hybridization or SNP arrays as compared with their parental cells, although it cannot detect balanced translocations or single-nucleotide variations. For the latter, the increasingly affordable whole-genome sequencing may be a preferred, additional way of analyzing the genomic integrity of selected iPSC lines that will be used extensively.
Figure 3.
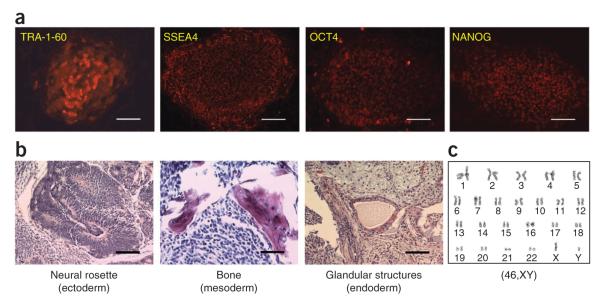
Characterization of human iPSCs derived from blood cells with episomal vectors. Details of how to perform these characterizations can be found in ref. 13. (a) Phenotypes of expanded iPSC line BC1 derived from adult bone marrow CD34+ cells by the single pEB-C5 vector. The iPSC colonies express typical pluripotency-related cell surface markers TRA-1-60 and SSEA4, as we reported previously13,24. We also detected expression of pluripotency-specific transcription factors OCT4 and NANOG24. This in vitro characterization assay can be performed as early as 2 months after transfection. (b) The pluripotency of the BC1 iPSC line was assessed by teratoma formation assay after injection into immune-deficient mice. H&E staining of the teratoma showed the presence of tissue types derived from all three embryonic germ layers. Injection for in vivo teratoma assay can be performed as early as 3 months after transfection, with teratoma excision and staining occurring 6–8 weeks after injection. (c) The BC1 iPSC line maintained a normal karyotype (46,XY) after 36 passages of expansion. Scale bars, 100 μm.
It is also important to confirm the genetic identity of iPSC lines in a laboratory that derives or uses many iPSC lines. Comparative analyses of somatic cells and iPSC lines by SNP arrays or multiple PCR using selected short tandem repeat (STR) primers provide a fair method to rule out the disparity. A standard STR-based DNA fingerprinting panel, such as the StemElite ID system by Promega, also provides a high-resolution method by which results can be shared by many laboratories using the same iPSC lines. Typical results using this panel are shown in Table 2.
Table 2.
STR profiling data for the BC1 iPSC line using the Promega StemElite ID System
| Marker | Chromosomal location (human) |
Allele 1 | Allele 2 |
|---|---|---|---|
| AMEL | Xp22.3, Yp11 | X | Y |
| CSF1PO | 5q33.3–q34 | 12 | 12 |
| D13S317 | 13q22–q31 | 11 | 11 |
| D16S539 | 16q24.1 | 9 | 10 |
| D21S11 | 21q21.1 | 28 | 28 |
| D5S818 | 5q23.2 | 11 | 11 |
| D7S820 | 7q21.1 | 8 | 10 |
| TH01 | 11p15.5 | 8 | 9 |
| TPOX | 2p25.3 | 8 | 12 |
| vWA | 12p13.31 | 15 | 20 |
| Mus | Mouse DNA | M1 | M1 |
This kit uses ten human markers plus one marker for mouse cells (Mus). As DNA was extracted from BC1 iPSCs while it was being expanded on MEF, the mouse marker (M1) was also present. Details on how to perform this characterization can be found in ref. 13.
ACKNOWLEDGMENTS
We thank members of the Cheng laboratory for discussion and sharing their experience. This work is supported by Johns Hopkins University and grants from the US National Institutes of Health (R01HL073781, U01HL107446, RC2HL101582 and T32HL007525).
Footnotes
AUTHOR CONTRIBUTIONS S.N.D., B.-K.C. and Z.Y. designed and performed experiments, analyzed data and wrote the paper; X.H. designed and performed experiments; L.C. designed experiments, analyzed data and wrote the paper.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 3.Park I, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 4.Aasen T, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008;26:1276–1284. doi: 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- 5.Mali P, et al. Improved efficiency and pace of generating induced pluripotent stem cells from human adult and fetal fibroblasts. Stem Cells. 2008;26:1998–2005. doi: 10.1634/stemcells.2008-0346. [DOI] [PubMed] [Google Scholar]
- 6.Loh Y, et al. Generation of induced pluripotent stem cells from human blood. Blood. 2009;113:5476–5479. doi: 10.1182/blood-2009-02-204800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye Z, et al. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood. 2009;114:5473–5480. doi: 10.1182/blood-2009-04-217406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim D, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warren L, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu J, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marchetto MC, et al. Transcriptional signature and memory retention of human induced pluripotent stem cells. PLoS ONE. 2009;18:e7076. doi: 10.1371/journal.pone.0007076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mali P, et al. Butyrate greatly enhances derivation of human induced pluripotent stem cells by promoting epigenetic remodeling and the expression of pluripotency-associated genes. Stem Cells. 2010;28:713–720. doi: 10.1002/stem.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chou BK, et al. Efficient human iPS cell derivation by a non-integrating plasmid from blood cells with unique epigenetic and gene expression signatures. Cell Res. 2011;21:518–529. doi: 10.1038/cr.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng L, et al. Low incidence of DNA sequence variation in human induced pluripotent stem cells generated by nonintegrating plasmid expression. Cell Stem Cell. 2012;10:337–344. doi: 10.1016/j.stem.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zou C, et al. Efficient derivation and genetic modifications of human pluripotent stem cells on engineered human feeder cell lines. Stem Cells Dev. 2012;21:2298–2311. doi: 10.1089/scd.2011.0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown ME, et al. Derivation of induced pluripotent stem cells from human peripheral blood T lymphocytes. PLoS ONE. 2010;5:e11373. doi: 10.1371/journal.pone.0011373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loh YH, et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell. 2010;7:15–19. doi: 10.1016/j.stem.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seki T, et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell. 2010;7:11–14. doi: 10.1016/j.stem.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Staerk J, et al. Reprogramming of human peripheral blood cells to induced pluripotent stem cells. Cell Stem Cell. 2010;7:20–24. doi: 10.1016/j.stem.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunisato A, et al. Direct generation of induced pluripotent stem cells from human nonmobilized blood. Stem Cells Dev. 2011;20:159–168. doi: 10.1089/scd.2010.0063. [DOI] [PubMed] [Google Scholar]
- 21.Seki T, Yuasa S, Fukuda K. Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus. Nat. Protoc. 2012;7:718–728. doi: 10.1038/nprot.2012.015. [DOI] [PubMed] [Google Scholar]
- 22.Serwold T, et al. T-cell receptor-driven lymphomagenesis in mice derived from a reprogrammed T cell. Proc. Natl. Acad. Sci. USA. 2010;107:18939–18943. doi: 10.1073/pnas.1013230107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu K, et al. Efficient generation of transgene-free induced pluripotent stem cells from normal and neoplastic bone marrow and cord blood mononuclear cells. Blood. 2011;117:e109–e119. doi: 10.1182/blood-2010-07-298331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mali P, Ye Z, Chou BK, Yen J, Cheng L. An improved method for generating and identifying human induced pluripotent stem cells. Methods Mol. Biol. 2010;636:191–205. doi: 10.1007/978-1-60761-691-7_12. [DOI] [PubMed] [Google Scholar]
- 25.Chen G, et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods. 2011;8:424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu S, et al. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell. 2010;7:651–655. doi: 10.1016/j.stem.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu J, Chau KF, Vodyanik MA, Jiang J, Jiang Y. Efficient feeder-free episomal reprogramming with small molecules. PLoS ONE. 2011;6:e17557. doi: 10.1371/journal.pone.0017557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anokye-Danso F, et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8:376–388. doi: 10.1016/j.stem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subramanyam D, et al. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 2011;29:443–448. doi: 10.1038/nbt.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gore A, et al. Somatic coding mutations in human induced pluripotent stem cells. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan EM, et al. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat. Biotechnol. 2009;27:1033–1037. doi: 10.1038/nbt.1580. [DOI] [PubMed] [Google Scholar]