

Abstract
Metalloenzymes efficiently catalyze some of the most important and difficult reactions in nature. For many years, coordination chemists have effectively used small molecule models to understand these systems. More recently, protein design has been shown to be an effective approach for mimicking metal coordination environments. Since the first designed proteins were reported, much success has been seen for incorporating metal sites into proteins and attaining the desired coordination environment but until recently, this has been with a lack of significant catalytic activity. Now there are examples of designed metalloproteins that, although not yet reaching the activity of native enzymes, are considerably closer. In this review, we highlight work leading up to the design of a small metalloprotein containing two metal sites, one for structural stability (HgS3) and the other a separate catalytic zinc site to mimic carbonic anhydrase activity (ZnN3O). The first section will describe previous studies that allowed for a high affinity thiolate site that binds heavy metals in a way that stabilizes three-stranded coiled coils. The second section will examine ways of preparing histidine rich environments that lead to metal based hydrolytic catalysts. We will also discuss other recent examples of the design of structural metal sites and functional metalloenzymes. Our work demonstrates that attaining the proper first coordination geometry of a metal site can lead to a significant fraction of catalytic activity, apparently independent of the type of secondary structure of the surrounding protein environment. We are now in a position to begin to meet the challenge of building a metalloenzyme systematically from the bottom-up by engineering and analyzing interactions directly around the metal site and beyond.
Keywords: de novo metalloprotein design, Zn hydrolase, Hg thiolates
1. Introduction
Protein design is a powerful approach for understanding metal sites in biology through reproduction of coordination environments and active site structures within a peptidic scaffold. Metal sites can efficiently catalyze many of the most complex and essential reactions in nature [1,2]. The ability to reproduce these reactivities in a designed system is a true test of our knowledge of protein folding and how structure and function are related in metalloproteins. Relative to protein design in general, designing metal sites into proteins can be challenging due to the variability of metal ions in their oxidation states, ligand donor sets, and preferred geometries. However, many metal sites can also serve as spectroscopic tags to assist in characterization and assessment of the success of the design, thus speeding up the design process. Additionally, the variability and sheer catalytic power inherent in metal ions increases the variety of functions that can be achieved, which may allow for a wider range of novel metalloenzymes for commercial use. Since the first reports of designed proteins and metalloproteins began appearing over 30 years ago, significant progress has been made in reproducing many native metal sites for a large variety of metals and metal cofactors [3]. However, designed metalloproteins have often operated with a significant lack of catalytic efficiency relative to their designed counterparts [4–6]. Now there are several examples of functional metalloproteins with increasing enzymatic activities, suggesting that rivaling native metalloenzymes is a more accessible goal than ever. Reaching this point, however, has required many advances in metalloprotein (and non-metalloprotein) folding and design. Specifically, one must be able to design structural sites where a metal can impart thermodynamic stability, direct protein folding and/or stabilize non-preferred metal binding geometries. These advances in understanding the balance between the protein scaffold stability and metal ion coordination preferences often naturally lead to the design of catalytically active metal sites using a variety of approaches. Our ultimate goal is to generate efficient metalloenzymes through a bottom up (de novo or “from scratch”) approach in which a primary sequence is chosen that folds into the predicted structure (requiring a deep understanding of protein folding, structure, and function). Other strategies, such as the redesign of native protein scaffolds, also provide new insight for metal-protein interactions. Through these complementary approaches, one might expect to be able to apply the insights gained throughout these studies to achieve one of the larger goals of the field: the design of novel metalloproteins with improved properties (stability, efficiency) and/or new catalytic activities. Such constructs, which are not found in nature, may be useful for a range of applications, including industrial processes where they could represent cheaper, more stable, and environmentally friendly alternatives to the current catalysts.
Metals in proteins have a wide range of functions that can be broadly classified into a few types: structural, catalytic, and electron transfer. Understanding how the protein matrix can fine-tune the properties of metal ions to affect such a variety of functions is an important question being actively addressed by protein design. Here, we will focus on designed metalloproteins as they relate to structural and catalytic metal functions. Readers interested in the design of metalloproteins with electron transfer capabilities are referred to other review papers [7,8]. After a discussion of general protein design approaches and scaffolds, we will review the design of structural metal sites (Section 3), highlighting our work on the de novo design of metal-binding three-stranded coiled coils (3SCCs) in Section 3.2, and discuss examples of functional de novo designed metalloenzymes (Section 4), with an emphasis on those reported recently, including our combined structural and hydrolytic dual-site metalloprotein (Section 4.1).
2. Metalloprotein design approaches and scaffold choices
Based on selection of the starting scaffold, metalloprotein design approaches can be categorized broadly into protein redesign or de novo (“from scratch” in its purest form) protein design. Metalloprotein redesign encompasses a variety of approaches including redesign using non-metalloprotein scaffolds as a starting point and using existing metal sites and redesigning them into new metal sites with altered properties. De novo metalloprotein design, on the other hand, requires both design of a unique protein scaffold and incorporation of the metal site(s). Computational and/or empirical approaches to the design can be taken. Computational design of metalloproteins is currently limited due to the inherent variability in metal sites and a lack of suitable parameters to describe them (especially for the design of metal sites with non-preferred geometries), but nonetheless there are successes in this area [9–12]. Empirical design can be less limiting but requires more knowledge about the templates being used and the design target. A combination of the two is often used. Further, proteins and metalloproteins can be designed combinatorially or rationally. Combinatorial design is typically used when little is known about the design goal and involves preparing a library of designs to sample, although it can be difficult to sample enough sequence variety [12]. Rational design is the ideal approach and allows more control over the outcome but also requires more in-depth knowledge of the structure and function. This approach has seen much success with the design of heme proteins [13]. Often, a combination of rational and combinatorial approaches can be a very effective strategy. This review will focus on rationally designed metalloproteins.
2.1. Protein redesign
One can take a redesign approach by beginning with a native protein scaffold and incorporating a novel metal site through introduction of the necessary ligands into a non-metalloprotein (or into a new location of an existing metalloprotein), redesign of an existing metal site, or incorporation of a metal cofactor or complex. Evolved proteins tend to be fairly stable and can handle the multiple mutations required to engineer a metal site. If chosen correctly, they can be easily obtained in high yield and are already well characterized. It can even be argued that nature uses this approach. Although there are a large number of characterized sequences and structures available, there are only ~1000 unique protein folds known and often, many different types of proteins with diverse functions use a single protein fold (an important example of this is the Greek key β-barrel fold) [12]. It is possible that nature may only use a small number of thermodynamically stable scaffolds to design repeatedly many proteins with diverse active site structures and functions. Relative to de novo design, this approach can currently offer a wider variety of potential scaffolds, as there are relatively few robust de novo designed scaffolds (Section 2.3). On the other hand, this could also prove to be a drawback as it may be difficult to divulge hidden structural features in complex native proteins. Nevertheless, there is much that has been and will continue to be learned from the redesign of existing metalloproteins. Although this review will focus on de novo designed proteins, there are several recent and impressive reports of redesigned functional metalloproteins [10,14–23] and many others that have been described in previous reviews [9,12,13,24–29].
2.2. De novo protein design
The de novo, or in its purest form, “from scratch”, approach to metalloprotein design is to build a well-folded protein structure from a primary sequence not directly related to that of any natural protein and subsequently engineer a metal binding site [30]. The basis of de novo design usually involves a minimization or simplification of protein structure [31,32]. One of the goals is to attain a minimal specific structure that removes much of the excess of a native protein but retains sufficient complexity for protein-like functions. The main advantage of this approach is that it eliminates most of the convoluted behaviors that make characterizing many native metalloproteins difficult. It is likely that this simplified approach to characterizing metal binding and function may uncover otherwise hidden subtleties in structure-function relationships. With a deeper understanding of the necessary features, increasing amounts of complexity can then be incorporated, as deemed necessary, into these minimal designs. Ultimately, careful analyses of de novo designed metalloproteins, combined with the information learned from redesign endeavors, should lead to the ability to prepare de novo designed metal-based biocatalysts for a variety of desired functions not necessarily found in nature and for a variety of applications.
2.3. Scaffolds for designing metal binding sites
Design of a metal binding site, regardless of the approach, first requires a stable protein scaffold into which mutations can be made to incorporate ligands for metal (or metal cofactor) binding. For protein redesign, the scaffold is typically chosen based on the goal of the design and the properties of the native scaffold and nature provides many existing scaffolds to choose from [12]. A variety of approaches can be taken including engineering a metal or metal cofactor binding site into a non-metalloprotein [9,10,13–15,25–27], redesigning an existing metal site [17,33–35], removing and inserting entire loop sequences containing the desired metal binding site [12,18,24], and generating multimeric protein structures with metal-mediated protein interfaces [16,36–39].
Far fewer de novo designed scaffold choices have been prepared to date, which limits the range of metalloprotein environments that may be examined. Ultimately, one goal of de novo design would be to expand this repertoire to include as least as many, if not more, scaffolds than those provided by nature. Initial attempts at de novo protein design lead to structures that formed molten globules, but now there are a number of successful examples of small uniquely folded proteins. Hydrophobicity provides a strong driving force for folding but many more subtle features also need to be incorporated for both stability and specificity such as hydrogen bonding, electrostatics, side-chain packing, and conformational preferences. Most de novo designed proteins consist of α-helices and are either coiled coil structures made up of self-assembling peptides or α-helical bundles made up of helix-loop-helix motifs [6,30,40]. The fact that α-helices have intrastrand interactions with residues that are close in the primary sequence to stabilize their structures is one aspect (as opposed to interstrand hydrogen bonding found in β-sheets) that made them an attractive initial target for design.
Hodges and coworkers pioneered the de novo design of α-helical coiled coils [30,41–44]. The α-helical coiled coil is made up of a bundle of parallel or antiparallel amphipathic α-helices with a left-handed superhelical twist [45–48]. The primary sequence contains a seven residue repeating sequence (the heptad repeat), with positions designated a–g. The residues in the a and d positions are directed towards the interior of the structure and tend to be hydrophobic while hydrophilic residues in the remaining positions are involved in electrostatic interactions. The association of hydrophobic a and d residues provides part of the driving force for assembly of the coiled coil. Helix-inducing residues, such as Ala, may be placed in the c position. Figure 1 illustrates these interactions for parallel two-, three-, and four-stranded coiled coils (XSCCs where X = 2, 3, or 4). Specific oligomers of α-helices can be attained through the rational design of core packing interactions (typically by varying the size of the hydrophobic residues) and surface electrostatics (which also contribute to stabilization of parallel or antiparallel configurations of coiled coils) [32,42,49–52]. The heptad repeat approach is a highly versatile method that has yielded many successful designed metalloproteins (Figure 2a, b) [9,53–70]. In particular, our group has designed the TRI family of peptides, which are α-helical coiled coils that fold into specific aggregates depending on pH, peptide length, and sequence composition (these will be described in detail in Section 3.2) [3,53,63,71].
Figure 1.
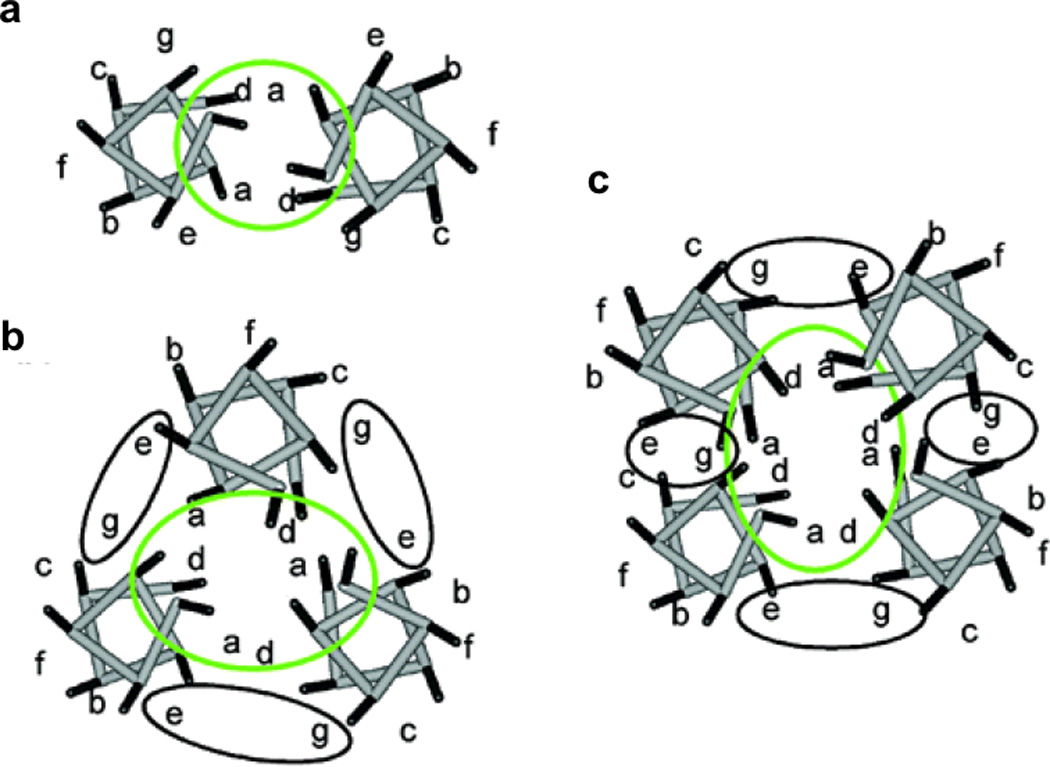
Helical wheel diagrams for parallel (a) two-, (b) three-, and (c) four-stranded coiled coils. Figure was reproduced with permission from ref. [53]. Copyright 2004 American Chemical Society.
Figure 2.
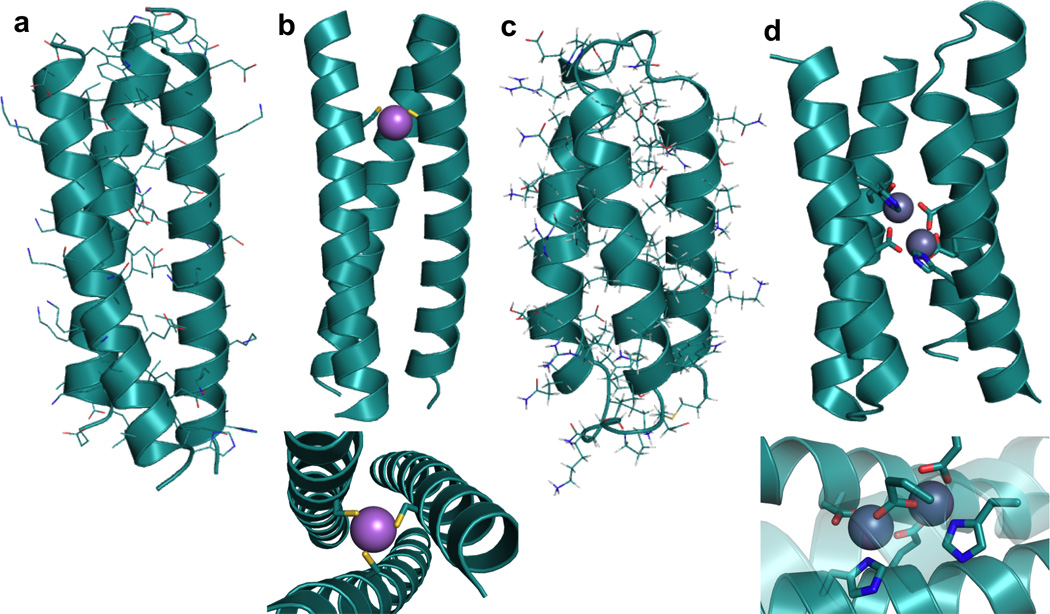
Structures of representative de novo designed α-helical coiled coils and bundles. (a) X-ray structure of a three-stranded coiled coil, CoilSer generated from PDB 1COS [50], (b) X-ray structure of a metal-bound three-stranded coiled coil, [AsIII]S(CSL9C)3 generated from PDB 2JGO [68] with side-on view (top) and top-down view (bottom); As-S metal-ligand bond distances all 2.3 Å, (c) NMR structure of a three-helix α-helical bundle protein generated from PDB 2A3D [73], (d) X-ray structure of a dinuclear metal site in a designed four-helix bundle protein (diZnII-DF1) generated from PDB 1EC5 [11] with side-on full view (top) and close-up view of metal site (bottom); All metal-ligand bond distances are in the range 1.8–2.1 Å.
Another structure type extensively explored in protein design is the α-helical bundle. Although often designed based on coiled coils, α-helical bundles can be structurally different in that the helices gradually diverge from a point near one end of the bundle [72,73]. The most common are the four-helix bundles and one of the earliest examples of a designed four-helix bundle is α4 [31,74]. This small protein was prepared by taking short helical sequences that could self-associate into a tetramer, adding hairpin loops to make a dimer of helix-loop-helix motifs, and finally incorporating a third loop to create a single primary sequence. Although this bundle adopted a folded, globular conformation in aqueous solution, the core was too conformationally flexible (it contained only Leu residues) leading it to exhibit some molten globule-like characteristics rather than assuming a single native state. Insertion of metal binding residues to create a ZnHis3 site did help induce a more native-like state into α4 [49,75]. Now, there are many examples of well-folded α-helical bundles that adopt a single native state due to the incorporation of a variety of more specific interactions both in the hydrophobic core and on the hydrophilic surface [30,73,76]. There are also examples where metal sites are engineered into α-helical bundles [11,75,77–79]. Our group has recently reported the incorporation of a heavymetal binding Cys3 site into a three-helix α-helical bundle (α3D, Figure 2c) that was originally designed based on CoilSer (CS, a de novo designed 3SCC, Table 1, Figure 2a, b) [73,78]. DeGrado and coworkers have reported an extensive amount of work on the dimetal-containing four-helix bundle Due Ferri (DF, Figure 2d, to be discussed in detail in Section 4.3) [4,11,80– 83].
Table 1.
CoilSer and TRI peptide family sequences used in these studies.
| Peptide | a b c d e f g | a b c d e f g | a b c d e f g | a b c d e f g | ||
|---|---|---|---|---|---|---|
| CoilSer (CS) | Ac-E | WEALEKK | LAALESK | LQALEKK | LEALEHG | -NH2 |
| CSL9C | Ac-E | WEALEKK | CAALESK | LQALEKK | LEALEHG | -NH2 |
| CSL19C | Ac-E | WEALEKK | LAALESK | LQACEKK | LEALEHG | -NH2 |
| CSL9PenL23H | Ac-E | WEALEKK | PenAALESK | LQALEKK | HEALEHG | -NH2 |
| TRI | Ac-G | LKALEEK | LKALEEK | LKALEEK | LKALEEK | G-NH2 |
| TRIL9C | Ac-G | LKALEEK | CKALEEK | LKALEEK | LKALEEK | G-NH2 |
| TRIL12C | Ac-G | LKALEEK | LKACEEK | LKALEEK | LKALEEK | G-NH2 |
| TRIL16C | Ac-G | LKALEEK | LKALEEK | CKALEEK | LKALEEK | G-NH2 |
| TRIL12CL16C | Ac-G | LKALEEK | LKACEEK | CKALEEK | LKALEEK | G-NH2 |
| TRIL2WL9C | Ac-G | WKALEEK | CKALEEK | LKALEEK | LKALEEK | G-NH2 |
| TRIL2WL23C | Ac-G | WKALEEK | LKALEEK | LKALEEK | CKALEEK | G-NH2 |
| TRIL23H | Ac-G | LKALEEK | LKALEEK | LKALEEK | HKALEEK | G-NH2 |
| TRIL2WL23H | Ac-G | WKALEEK | LKALEEK | LKALEEK | HKALEEK | G-NH2 |
| TRIL9CL23H | Ac-G | LKALEEK | CKALEEK | LKALEEK | HKALEEK | G-NH2 |
| Baby | Ac-G | LKALEEK | LKALEEK | LKALEEK | G-NH2 | |
| BabyL9C | Ac-G | LKALEEK | CKALEEK | LKALEEK | G-NH2 | |
| BabyL12C | Ac-G | LKALEEK | LKACEEK | LKALEEK | G-NH2 | |
| Grand | Ac-G | LKALEEK | LKALEEK | LKALEEK | (LKALEEK)2 | G-NH2 |
C- and N-termini are capped by Ac and NH2 groups, respectively.
Although the de novo design of proteins has been dominated by preparation of α-helical coiled coils and bundles, there has also been substantial effort directed towards the formation of β-sheet and related structures. This is relatively challenging, not only because of the need to create more interstrand interactions between residues well separated in sequence, but because there are issues with aggregation, overall stability, folding cooperativity, and challenges in designing turn sequences. Residues that are bulky and hydrophobic, such as Tyr, Phe, Thr, Ile, Val, and Trp, seem to have a higher propensity towards the formation of β-sheet structures. Despite these challenges, there are several successes in the design of β-sheet structures [84–89] and related scaffolds including mixed α/β structures [90–93], γ turns [94], β-hairpin peptides [95–98], and small peptides with loop region metal clusters [99,100]. There is also limited success in the de novo design of β-structured scaffolds for functional metalloproteins; the design of a redox-active rubredoxin mimic is an example of one of these rare β-sheet metalloproteins [101].
3. Designing structural metal sites
Many metal ions play a structural role in their protein environments. They can act to simply stabilize a structure by inducing or assisting folding or as switches for converting one conformation into another (for example, to regulate activity). These are most commonly represented by zinc-thiolate sites (as in zinc fingers which are typically comprised of ZnCys2His2 coordination environments) and calcium sites (calmodulin), although others exist [102–104]. There are examples of designed metal binding sites in which the metal plays a structural role [53,64][70]. In addition to describing our work involving heavy metal binding, particularly with HgII to thiol sites in α-helical coiled coils in Section 3.2, we will highlight other examples and recent approaches to the design of structural metal binding sites in proteins.
3.1. Metal-assisted and metal-induced folding in α-helices and α-helical coiled coils
In the field of metalloprotein design, there are many examples in which metal-ion assisted folding and self-assembly of α-helical coiled coils generates well-defined secondary and tertiary structures [64,69,105–110]. Much of this work has been reviewed, but some of what is relevant to the most recent studies is described in the context of this paper. Figure 3 provides models of some of the important constructs described below.
Figure 3.
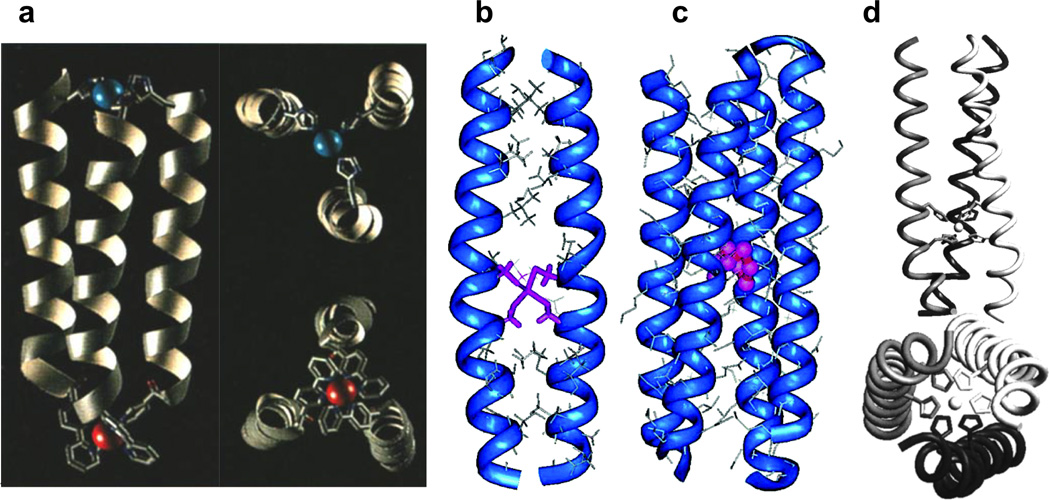
Examples of metal-stabilized α-helical structures described in this section. (a) Computer-generated model of the parallel three-helix bundle CuIIRuII metalloprotein with full side-on view (left) and close-up views of each metal site (right). Figure was reproduced with permission from ref. [113]. Copyright WileyVCH Verlag GmbH & Co. KGaA. (b) Energy-minimized computer model of the CdII-bridged C16C19 peptide dimer (2SCC) with CdII bound in a tetrahedral geometry. Figure was reproduced with permission from ref. [7]. Copyright 2006 American Chemical Society. (c) Computer-generated model of the tetrameric CuI-C16C19-GGY metalloprotein (4SCC). Figure was reproduced with permission from ref. [7]. Copyright 2006 American Chemical Society. (d) Model of the NiII-His6 complex of IZ-3adH (3SCC) with full side-on view (top) and a bottom-up view from the C-termini (bottom). Figure was reproduced with permission from ref. [109]. Copyright 1998 American Chemical Society.
Ghadiri et al. were among the first to exploit the design of metal binding sites in peptides to promote the formation of α-helices [111,112] and coiled coils [106,113]. They designed a 17 mer peptide (Ac-AEAAAKEAAAKX1AAAX2A-CONH2 where X1 = His and X2 = His or Cys) which could bind CdII, CuII, NiII, and ZnII, all with an enhancement in α-helicity from 54% to ~90%. Using the His/His version of this peptide, binding of an exchange inert metal complex, [Ru(NH3)5(OH2)]2+, leads to the formation of a macrocyclic complex cis-[Ru(NH3)4(His)2peptide]3+ with 80% α-helical content. The group has also designed a 15-mer amphiphilic peptide with a 2,2’-bipyridine moiety at the N-terminus which, upon binding NiII, CoII, or RuII, self-assembles to form a 3SCC with > 70% α-helical content [106]. This construct was also used as a template for incorporation a CuII(His)3 site to form a heterodinuclear three-helix construct (Figure 3a) [113]. Further, a stable parallel four-helix bundle metalloprotein is obtained by incorporating another 15 residue amphiphilic peptide with a pyridyl group at the N-terminus and binding RuII [114].
Along similar lines, the Ogawa group has also reported the use of metal binding sites to generate α-helical bundles. Kharenko et al. designed a 32 mer peptide C16C19-GGY (Ac-K(IEALEGK)2(CEACEGK)(IEALEGK)GGY-CONH2), which has Cys residues in adjacent a and d positions of one of the heptads, resembling the Cys-X-X-Cys motif found in rubredoxin. Upon binding CdII, the apo-peptide went from a random coil to a 2SCC containing a single CdII ion (Figure 3b) [7,54]. Interestingly, when CuI is bound to the same peptide, a four-helix bundle metalloprotein containing a tetranuclear Cu4S4 cofactor is generated (Figure 3c) [115]. Further, upon binding Pt(en)(NO3)2 to the 30-residue peptide AQ-Pal14Pal21 (which contains two 4-pyridylalanine, or Pal, binding residues on its surface and exists as a 2SCC in its apo form) results in a significant conformational change to a metal-bridged four-helix bundle [107].
Tanaka and coworkers have used a heptad repeat to design a parallel 3SCC peptide (in the presence of bound metal ions) with the sequence YGG(IEKKIEA)4 called IZ. The peptide IZ-3aH was generated by substitution of an isoleucine residue on the third heptad with a His and IZ-3adH could be generated with two His substitutions [109,116]. While this sequence showed only random coil-like secondary structure in solution, it was induced to fold into a 3SCC in the presence of metal ions such as CuII, ZnII, CoII, and NiII (Figure 3d). Metal binding could be followed by monitoring the folding with circular dichroism (CD) as a function of the metal concentration, yielding dissociation constants in the low µM range. Substituting Cys residues into the IZ peptides also yields metal binding sites for CdII and HgII [110]. Recently, the group reported a study in which they fused the IZ-3adH peptide to another functional protein (a derivative of a naturally occurring DNA-binding domain) to create a metal-ion-controlled DNA binding protein which could modulate hydrolysis of the DNA site and inhibit RNA transcription by T7 RNA polymerase [117].
As will be discussed in Section 3.2, the Pecoraro group has shown that heavy-metal thiolate binding can also be used to induce and mediate the folding of 2- and 3SCCs.
Further, as will be discussed in Section 4.4, the Ball group has exploited the metal-induced α-helicity of 2SCCs and the inherent properties of coiled coil structures to design dirhodium-based catalysts for a variety of roles relevant to enzymatic function.
3.2. Binding of HgII to the TRI family of α-helical coiled coils
Our group’s work is based on the TRI family of de novo designed peptides [3,71] for which the parent sequence, Ac-G(LKALEEK)nG-NH2, is based on that of CoilSer (CS) [50,118] and consists of four heptad repeats. This heptad repeat sequence results in amphipathic α-helices with all Leu residues in the a and d positions on the hydrophobic face, Ala in the c position, and Lys and Glu residues in most of the remaining positions on the hydrophilic face. In aqueous solution, the hydrophobic residues aggregate at low pH to form a 2SCC and as the pH is raised above 5.5, salt bridge interactions between Lys and Glu at the interfaces between helices stabilize a parallel 3SCC. The a and d residues point towards the interior of the coiled coil to form the hydrophobic core. Substitution of these Leu residues with residues such as Cys or His yields metal-binding sites. Figures 4 and 5 display the crystal structures of apo-CSL9C (a site) and apo-CSL19C (d site), respectively [67]. Peptide sequences are given in Table 1. These structures demonstrate the differences between these types of sites in the absence of metal. For the a site, the Cys side chains show alternate conformations with the predominant form preorganized for metal binding. Comparison of this apo-peptide with the corresponding metal-bound version ([AsIII]S(CSL9C)3 [68], where we use [AsIII]S to indicate that AsIII is bound at the Cys3 site) suggests only minor changes in the Cys residue geometries where they are slightly more constrained with metal binding. The d site residues, on the other hand, exist in a single conformation different than those observed in CSL9C, resulting in a larger cavity which can accommodate metals with larger ionic radii, such as PbII.
Figure 4.
Ribbon diagrams of the X-ray crystal structure of apo(CSL9C)3 showing the orientation of the Cys ligands. The Cys side chains are shown as red sticks with thiol groups colored yellow. Top-down views from the N-termini show (a) the orientation of the major conformer with all Cys side chains pointing towards the interior of the trimer and (b) the minor conformer with Cys side chains pointing towards the helical interface. Side views further demonstrate the flexibility of this site. (c) In the major conformer, the thiol groups point towards the N-termini and (d) in the minor conformer, the thiol groups point towards the C-termini. Figure was reproduced with permission from ref. [67]. Copyright 2010 American Chemical Society.
Figure 5.
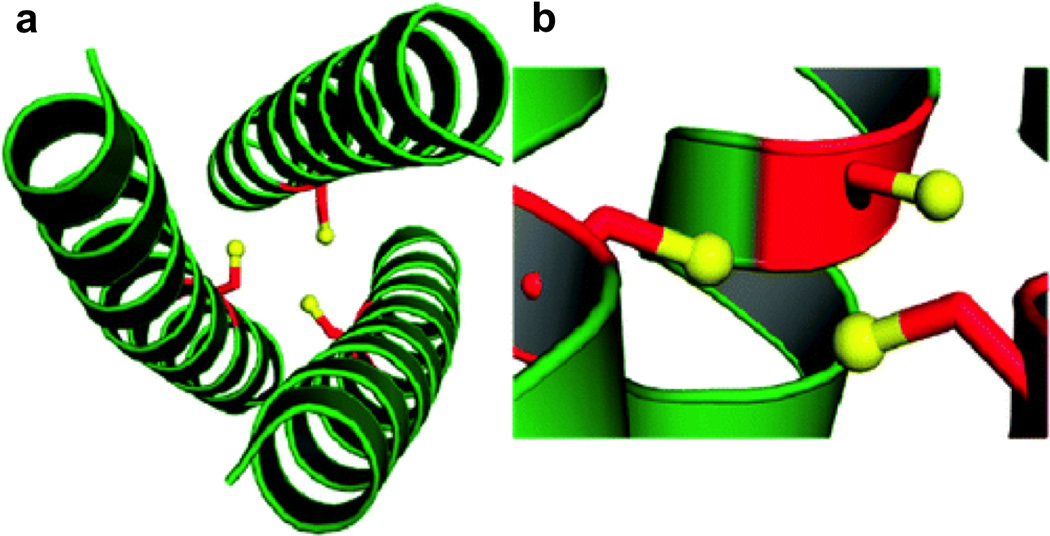
Ribbon diagrams of the X-ray crystal structure of apo(CSL19C)3 showing the orientation of the Cys ligands. The Cys side chains are shown as red sticks with thiol groups colored yellow. The top-down view from the N-termini shows (a) that two of the Cys ligands point towards the interior of the coiled coil and the third toward the helical interface. The side view demonstrates that two thiol groups point towards the C-termini while the third is almost perpendicular to the helical axis. Figure was reproduced with permission from ref. [67]. Copyright 2010 American Chemical Society.
We have also shown that non-natural amino acids can be used for metal recognition. Specifically, Penicillamine (Pen) can be used to increase steric bulk directly around the metal binding pocket, thus enforcing lower coordination geometries than may otherwise be observed [66,119,120]. Further, alternate chirality amino acids create smaller metal binding pockets when placed in the Leu layer above the metal binding site [121]. Moreover, placement of a d-amino acid ligand in the metal binding Leu layer alters the ligand environment [122]. Specifically, we have reported the crystal structures of apo-CSL16Pen and apo-CSL16d-Pen, which demonstrate that incorporation of a d-amino acid ligand results in a metal binding site that is not preorganized for metal binding (Figure 6). This work provides a structural explanation for the relatively poor binding of CdII to d-Pen as compared to l-Pen.
Figure 6.
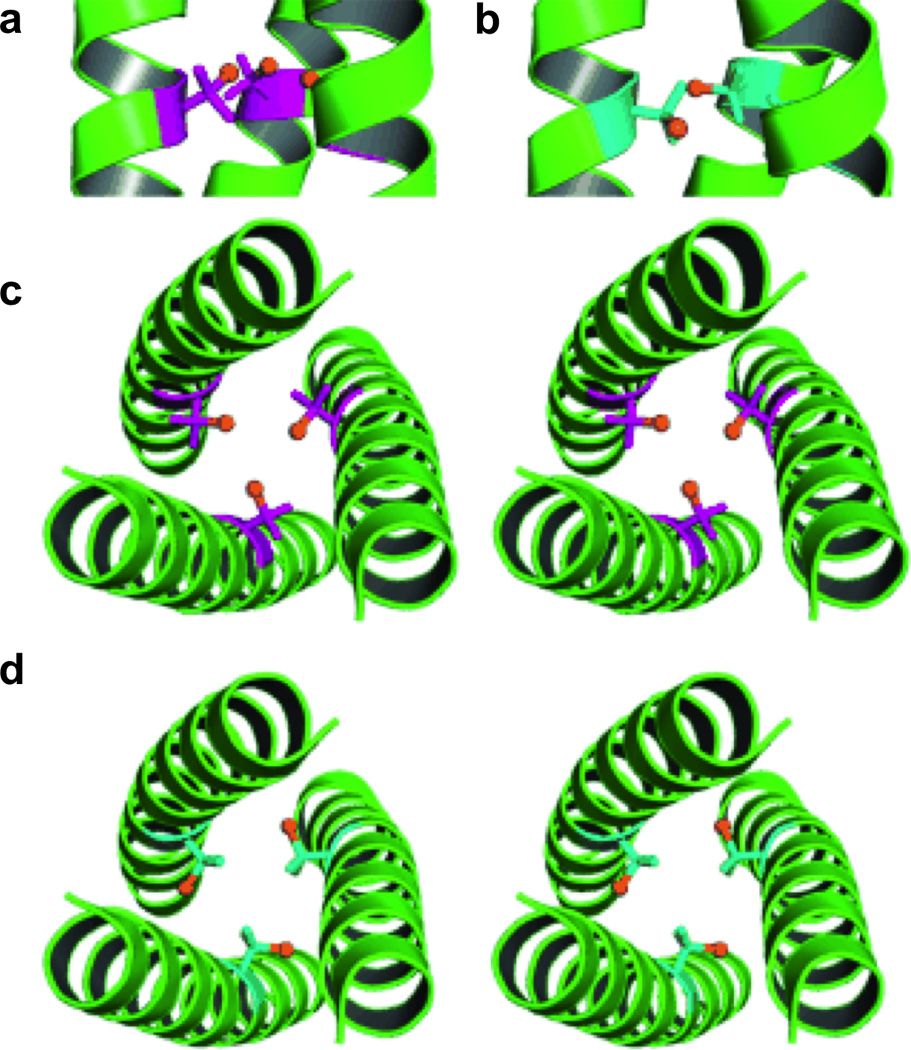
Ribbon diagrams of the X-ray crystal structures of apo(CSL16Pen)3 and apo(CSL16d-Pen)3 displaying the orientation of the Pen side chain. The l-(purple) and d-Pen (blue) side chains are shown in stick form with the thiol group colored orange. Side views show the (a) l-Pen residues oriented toward the N-termini and the (b) d-Pen residues oriented toward the C-termini. Top down stereo views from the N-termini are shown and display (c) l-Pen residues oriented toward the interior of the coiled coil and (b) d-Pen residues oriented toward the helical interface. Figure was reproduced with permission from ref. [122]. Copyright © 2009 WILEYVCH Verlag GmbH & Co. KGaA, Weinheim.
In theory, the three-fold symmetric nature of the coiled coils can allow for the preparation of most metal geometries within its scaffold (Figure 7). With substitution of a single Leu layer, trigonal planar or trigonal pyramidal structures can be achieved depending on the stereochemical preference of the metal. A tetrahedral geometry may be observed with the inclusion of a solvent molecule and with two solvent molecules, a trigonal bipyramidal metal site might be observed. Substituting two adjacent Leu layers and inserting the appropriate metal may yield an octahedral site (Suzuki et al. have reported an α-helical trimer with two adjacent His3 sites which can bind NiII, ZnII, and CoII in octahedral geometries [109]). Further, a linear metal-binding site may be attained by either lowering the pH to stabilize the 2SCC, incorporating the appropriate metal and metal:peptide ratios, as will be described for HgII, or at lower pH’s in the presence of a 3SCC (where the third protein ligand present is not bound), as demonstrated in the structure of [HgII]S[ZnII(OH2/OH−)]N(CSL9PenL23H)3n+, which will be discussed in detail in Section 4.1 (the nomenclature of this complex now accounts for a coordinated solvent molecule to the ZnII ion: if the pH conditions are such that the solvent may occur as both H2O and OH− then OH2/OH− will be used). Therefore, using a three-fold symmetric coiled coil as a scaffold should allow for an extensive range of different metal binding structures for a variety of metals and amino acid ligands within the same relative protein environment.
Figure 7.
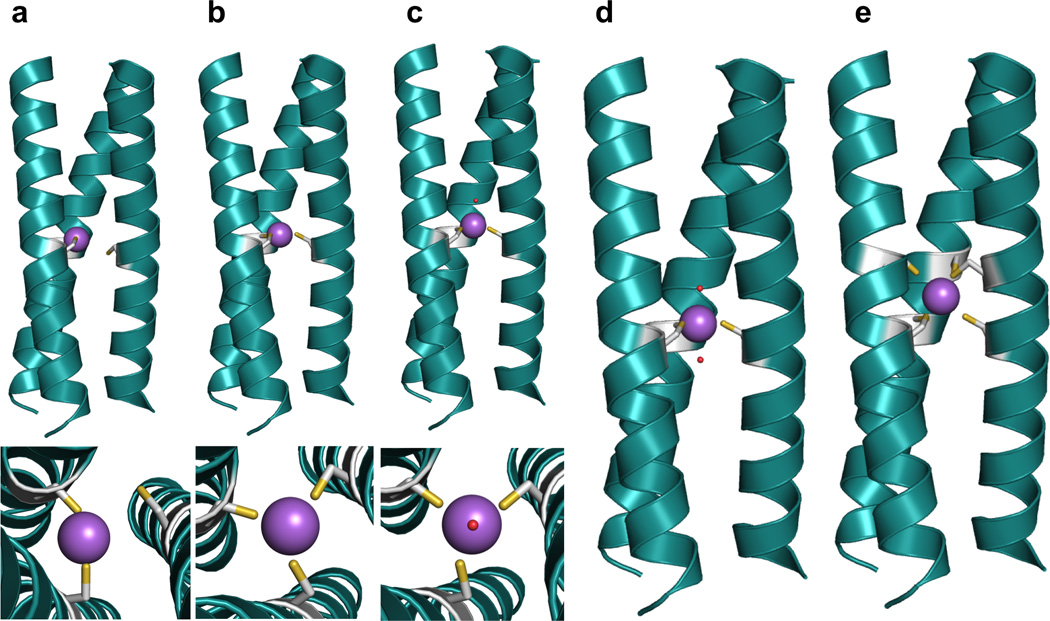
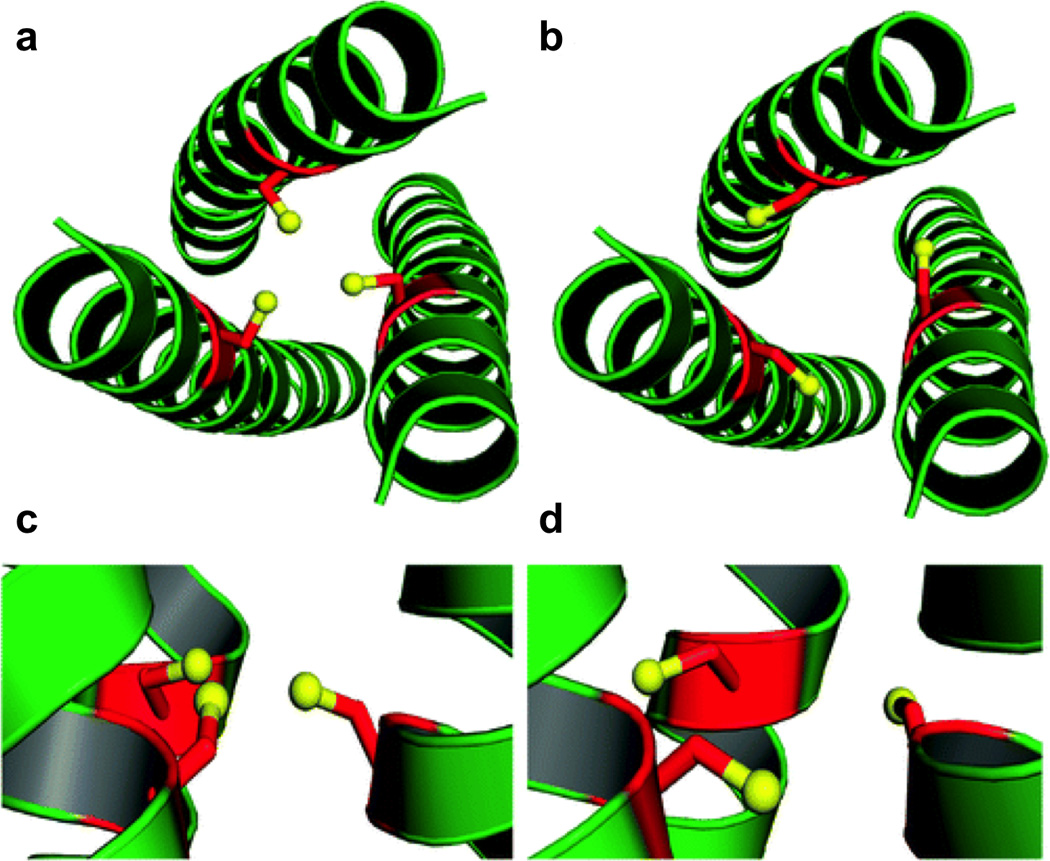
Pymol models of possible metal-binding geometries in a 3SCC. The metal-binding ligands are Cys residues and the metal is colored purple. (a) side-on view (top) and top-down view (bottom) of two-coordinate or linear geometry, (b) side-on view (top) and top-down view (bottom) of three-coordinate trigonal planar geometry, c) side-on view (top) and top-down view (bottom) of four-coordinate tetrahedral geometry, (d) side-on view of five-coordinate trigonal bipyramidal geometry, and (e) side-on view of six-coordinate octahedral metal geometry. Models are generated using the crystal structure of [AsIII]S(CSL9C)3 PDB 2JGO [68].
Until recently, most of our work on this system has focused on the preparation of thiolato coordination environments for metals such as HgII, CdII, PbII, AsIII and BiIII with the goal of understanding how heavy metals are coordinated in biological environments [3,53,59–62,67,68,71,119,120,123–128]. Although the toxic effects of these metals are well known, the causes are often not well understood due to a lack of suitable models, especially in the case of HgII [63]. By preparing thiophilic metal binding sites in the TRI family of proteins, we have not only been able to gain a better understanding of how heavy metals are coordinated in native metalloproteins, but also have learned how to selectively control these coordination environments. Previous reviews have discussed HgII and CdII binding in some detail [53,59,63,65], so here we will focus on describing HgII binding as a means to achieve a structurally stabilizing metal binding site in a 3SCC.
Our studies began by examining HgII binding to peptides such as TRIL16C and TRIL12C, where Leu has been substituted for Cys in an a site and a d site, respectively (sequences of the peptide to be discussed can be found in Table 1). As we have shown with the crystal structures of apo(CSL9C)3 and apo(CSL19C)3 (Figures 4 and 5) and will describe in terms of solution studies below, this seemingly slight shift in sequence position is significant for metal binding. To describe the binding of HgII to these peptides, a variety of conditions must be considered (Figure 8). At low pH (< 5.5), 2SCCs containing a linearly-bound HgII are formed. It is well known that this is the preferred geometry of HgII [129,130]. More interestingly, three distinct behaviors are observed at higher pH. When the peptide monomer (single α-helix) to HgII ratio is 2:1, 2SCCs with two-coordinate HgII are formed for both peptides, indicating that the HgII binding preference dominates the preferred bundle aggregation state. When this ratio is increased to 3:1, and in the pH range of 5.5–7, both peptides contain two-coordinate HgII within a 3SCC, simply suggesting that the third Cys remains protonated and unbound. Finally, for TRIL16C above pH 8.6 and TRIL12C above pH 9.5, fully three-coordinate trigonal planar HgII is obtained. Based on an equilibrium between two- and three-coordinate HgII within the 3SCC, the pKa for deprotonation of the third Cys can be determined and is 7.8 for the a site and 8.5 for the d site, indicating that metal binding to Cys residues is dependent on the a vs. d substitution pattern. Importantly, this represents the first peptidic system to bind HgII as a trigonal thiolato complex in aqueous solution and is an excellent model for the regulatory protein, mercury resistance regulator (MerR) [3,71,131]. We have also reported the crystal structure of the complex, [HgII]S[ZnII(OH2/OH−)]N(CSL9PenL23H)3n+ (at pH 8.5), which contains a trigonal HgS3 site in one trimer of the asymmetric unit (ASU) and a T-shaped HgII site in the other [70], crystallographically confirming our spectroscopic data. This structure will be discussed in more detail in Section 4.1. Through preparation of dual cysteine peptides (in which two adjacent layers of Leu are substituted), we were able to generate exclusively four-coordinate HgS4 with the sequence TRIL12CL16C, representing a model of HgII-substituted rubredoxin [132,133]. Extensive spectroscopic analysis of our system using numerous techniques for probing the metal coordination over a broad range of timescales (including UV-visible characterization, 199Hg NMR, 199mHg perturbed angular correlation (PAC, where 199mHg is a metastable isotope of 199Hg which emits two successive γ rays upon its decay) and X-ray absorption (XAS) spectroscopies) provides a summary of the properties expected for homoleptic HgII thiolates in biological systems (Table 2) [63].
Figure 8.

Species present at different TRIL9C/HgII ratios and pH values. Figure was reproduced from ref. [199] with permission of the copyright holders. Figure 9. Linear-free energy correlation between folding preferences of the peptides in the absence of metal to the binding of a third strand of peptide to a divalent HgIIS2 species. Figure was reproduced with permission from ref. [200]. Copyright © 2007 WILEYVCH Verlag GmbH & Co. KGaA, Weinheim.
Table 2.
Spectroscopic values for [HgII]S(TRILXC)X complexes.
| Hg coordination mode |
λ (Δε)a | pKa | RHgS (Å)b | δ (199Hg ppm) |
PAC (νQ, η)c |
|---|---|---|---|---|---|
| Linear 2-coordinate | 240 (2700)d | - | 2.32d | −844e | 1.53, 0.13e |
| Trigonal 3-coordinate a site | 247 (19200)f 265 (11900)f 295 (5800)f |
7.6 ± 0.2g | 2.44f, 2.23l | −185e | 1.16, 0.25e |
| Trigonal 3-coordinate d site | 230 (21300)h 247 (15000)h 297 (5500)h |
8.5 ± 0.2h | 2.44h | −316i | - |
| Linear 2-coordinate within a 3SCC | 247 (2000)j | - | 2.29, 2.13, 3.06m | −908e | 1.56, 0.23e |
| Tetrahedral 4-coordinate | 230 (8100)k 289 (7100)k |
- | - | −500k | - |
. λ given in nm and Δε given as M−1 cm−1;
. Average Hg-S EXAFS bond lengths;
. νQ given in GHz and η is a unitless quantity;
. Data for TRIL16C from [71];
. Data for TRIL9C from [199];
. Data for TRIL16C from [126];
. Data for TRIL9C from [124];
. Data for TRIL12C from [126];
. Data for TRIL19C from [199];
. Data for TRIL12C from [201];
. Data for TRIL12CL16C from [133];
. Data from the crystal structure of [HgII]S[ZnII(OH2/OH−)]N(CSL9PenL23H)3n+ for the trigonal HgS3 site. Average of the three Hg-S bonds to Pen, 2.21, 2.07, and 2.41 Å [70];
. Data from the crystal structure of [HgII]S[ZnII(OH2/OH−)]N(CSL9PenL23H)3n+ for the T-shaped Hg site [70].
A deeper analysis of the relationship between the peptide self-association affinity and HgII (and CdII) binding with a series of peptides demonstrated a linear free energy correlation with metal binding (Figure 9). In the case of HgII, this helped confirm the notion that trigonal HgII complexation is a direct result of the designed peptide recognition. Further, attempts to form heterotrimeric complexes between TRIL2WL9C and TRIL2WL23C and HgII did not succeed, demonstrating that control over orientation of the peptides is retained in the face of metal binding. The self-association affinity as it relates to HgII binding can be examined more deeply by considering the peptides used in the analysis. This affinity can be increased by increasing the hydrophobic contacts in the coiled coil by incorporating an extra heptad (this sequence is called Grand instead of TRI) and can likewise be decreased by either removing a heptad (to generate a sequence named Baby) or substituting less hydrophobic residues for Leu around the metal site (such as Val, Ala, and Gly). At the conditions under which these peptides are typically studied (low µM concentrations), Baby exists as random coils in solution, TRI is folded into an α- helical coiled coil, and Grand forms an even tighter coiled coil complex. Typically, the α-helical character correlates with association of the coiled coils (amphipathic α-helices will not be well-folded in aqueous solution if their hydrophobic faces are exposed), therefore, by monitoring the signal at 222 nm using CD, we can determine how well-folded a given peptide is. Fitting a plot of the signal at 222 nm vs. increasing concentration of guanidine hydrochloride (GuHCl) denaturant can yield the free energy of folding for the complex. As expected, when Leu residues are replaced with smaller and less hydrophobic residues, the peptide association becomes weaker (~1–2 kcal/mol per residue per peptide). This effect is directly correlated with the affinity of HgII (and CdII) for each of the peptides, demonstrating that it is not the metal’s geometric preference controlling the association of the coiled coils, but the free energy of folding of the peptides. This conclusion supports the theory that in biological systems, the surrounding protein matrix mediates folding and stabilization of unusual metal sites.
Figure 9.

Linear-free energy correlation between folding preferences of the peptides in the absence of metal to the binding of a third strand of peptide to a divalent HgIIS2 species. Figure was reproduced with permission from ref. [200]. Copyright 2005 American Chemical Society.
Trigonal planar HgII complexes could be obtained in all of these designs, but the kinetics of metal binding is different and can again be related to the self-association of the peptides. Above we described how a heptad could be added or removed from TRI to produce the Grand and Baby sequences, respectively. While TRI is well-folded at a 10 µM concentration, BabyL9C has only 20% helicity (% helicity is the measured molar ellipticity as a fraction of the maximum ellipticity for a fully α-helical complex of this size). However, in the presence of HgII, BabyL9C could still form a stabilized 3SCC. Initially, this challenged our notion that a preorganized site is necessary to stabilize alternate coordination geometries around metals in designed systems. To this end, we derived a thermodynamic model to explain how small, unstructured peptides could ultimately stabillize a [HgII(SR)3]− site [53,125]. First, a 2SCC is nucleated by the addition of HgII to a solution of peptide because two-coordinate linear HgII binding to thiolates is a highly favored process. When a third equivalent of peptide monomer is added, peptide-peptide interactions overwhelm the coordination preference of the metal to give a 3SCC final structure. Kinetic studies examining BabyL9C and HgII binding showed that the initial binding and formation of a [HgII]S(BabyL9C)2 site in a 2SCC is very fast while formation of [HgII]S(BabyL9C)2(H-BabyL9C) with addition of more peptide is rate-limiting. This step may be described by the stepwise aggregation-deprotonation (StepAD) model in which the third helix binds to the 2SCC in either a parallel (productive complex) or antiparallel fashion (unproductive) and every time antiparallel binding occurs, the helix must first dissociate then reassociate in a parallel manner (Figure 10). This is followed by deprotonation of the third Cys residue at the appropriate pH to form a fully trigonal [HgII]S(BabyL9C)3− complex. The pKa for deprotonation depends on the location of Cys in the sequence, but is independent of the length of the sequence (this pKa is 7.8 for both a site peptides, TRIL9C and BabyL9C while for TRIL12C and BabyL12C, both d site peptides, it is 8.4).
Figure 10.
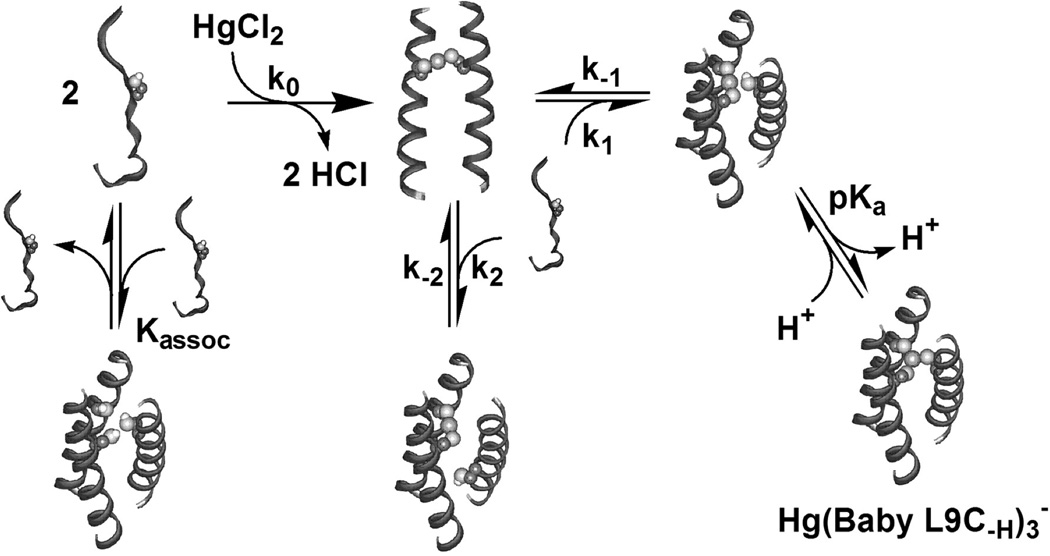
Stepwise aggregation-deprotonation (StepAD) mechanism for the encapsulation of HgII by the 3SCC, (BabyL9C)3. HgII reacts in a fast step to form [HgII]S(BabyL9C)2. Formation of [HgII]S(BabyL9C)2(H-BabyL9C) is the rate-limiting association and, depending on pH, rapidly converts to [HgII]S(BabyL9C)3−. Figure was reproduced from ref. [125] with permission of the copyright holders.
The case of HgII binding to the Baby peptide is particularly informative as to how metals can stabilize protein folds. Since Baby exists at µM concentrations as unassociated and unfolded individual peptides, addition of metal is essential for proper folding. Stopped-flow kinetic analysis revealed that under all of these conditions, HgII complexation to two thiolates first generates the [HgII]S(Baby)2 species which then acts as a nucleating agent for the third strand of the bundle to associate [125]. Thus, we can conclude that the presence of the metal site stabilizes the preferred fold (3SCC) by initiating the assembly of the bundle through the non-preferred 2SCC. Once the system begins to fold by forming the 2SCC, it subsequently adopts the preprogramed 3SCC structure. This example illustrates that a metal site may induce a desired protein fold by stabilizing an early folding intermediate even though the final, desired fold leads to an undesired coordination environment for the metal. This work clearly demonstrates the intricate balance and interplay between metal binding preferences and protein folding.
The difference in the observed pKa values for HgII binding to BabyL9C and BabyL12C prompts a further examination of the difference between a and d sites. A comparison between HgII-induced folding in BabyL9C and BabyL12C revealed not only different pKas but also a lower extinction coefficient (for the ligand-to-metal charge transfer transition representing HgII-thiolate bond formation) and HgII binding affinity and a lower free energy change for HgII complex formation with BabyL12C. A concentration dependence study of coiled coil association revealed a higher self-association affinity for the a site peptide. Kinetic studies revealed a faster rate for HgII binding to BabyL12C, likely due to this lower self-association preference (more monomers in solution can lead to more rapid nucleation). Modeling studies demonstrated the Cys conformation found in d site peptides is less favorable than that in a sites because the d site amino acid must twist from its normal position in the apo-bundle and orient more towards the core, whereas the a site residue resembles a more preorganized metal binding site. As discussed earlier in this section, these modeling studies have now been confirmed crystallographically with apo structures of CSL9C and CSL19C [67].
Further kinetic studies performed on TRIL9C and TRIL12C showed the same a vs. d site pattern where HgII insertion into the d site was faster than the a site. However, the mechanism of metal insertion into these prefolded peptides must be different than that derived for the unfolded peptides. There are two possibilities (Figure 11); one is the breathing mechanism in which the 3SCC may open up and allow the metal ion to insert and the other is the dissociative mechanism which is similar to the StepAD mechanism in which the third helix must first dissociate and then reassociate in the proper orientation to result in a productive 3SCC. The observed biphasic behavior of HgII encapsulation can be most easily rationalized with the dissociative mechanism; if some are misfolded then the step where a peptide must be lost from the 3SCC and then reassociated in the correct manner would represent a slower second phase. However, the breathing mechanism cannot be ruled out since the same pattern as was observed for Baby is observed for TRI, where the kinetics for d site insertion is faster. More experiments are needed to rationalize these mechanisms; however, some general conclusions for the field of protein design can be drawn from this work. One important observation is that a highly stable apo-protein may not be kinetically optimal for metal binding (another example where this is the case is described in Section 4.3). It is likely that nature makes compromises between rapid kinetic transfer and high metal ion affinity. Further, highly stable scaffolds are not necessarily required to enforce a non-preferred metal coordination environment (often required for functional metalloproteins) and may actually be detrimental to the kinetics of metal ion insertion.
Figure 11.

Possible mechanisms for insertion of HgII into the folded peptides. Figure was reproduced with permission from ref. [53]. Copyright 2004 American Chemical Society.
4. Designing functional metalloproteins
The ultimate goal of metalloprotein design is to be able to prepare metalloproteins that can efficiently catalyze virtually any reaction as native enzymes can do for biological reactions in mild, aqueous environments with as-yet unrivalled speed and specificity. Attempts at generating catalytic proteins and metalloproteins came soon after the first protein design papers, but until recently most have demonstrated rather poor efficiencies (if they have any reactivity at all). Many of the attempts at designed enzyme design have been reviewed fairly recently [6,9] so here we will focus on some of the most recent examples of functional de novo designed metalloproteins, specifically those which are hydrolytic and redox active.
4.1 A dual-site combined structural and functional metalloprotein for hydrolytic catalysis
Two-site metalloproteins in which there are two different metals in two separate designed sites are rather rare in the protein design literature [113,134], and no corresponding reactivity has been reported. However, many complex multi-site metalloproteins are found in nature and these can catalyze some of the most challenging transformations. Therefore, an important advance in metalloprotein design is gaining the understanding of how to prepare structures that selectively bind different metals in different sites for different purposes. To begin to meet this challenge, we have reported an example of a de novo designed 3SCC with two metal sites, the first for structural stabilization and the second with hydrolytic activity within just a few hundred-fold of a native enzyme (Figure 12) [70].
Figure 12.
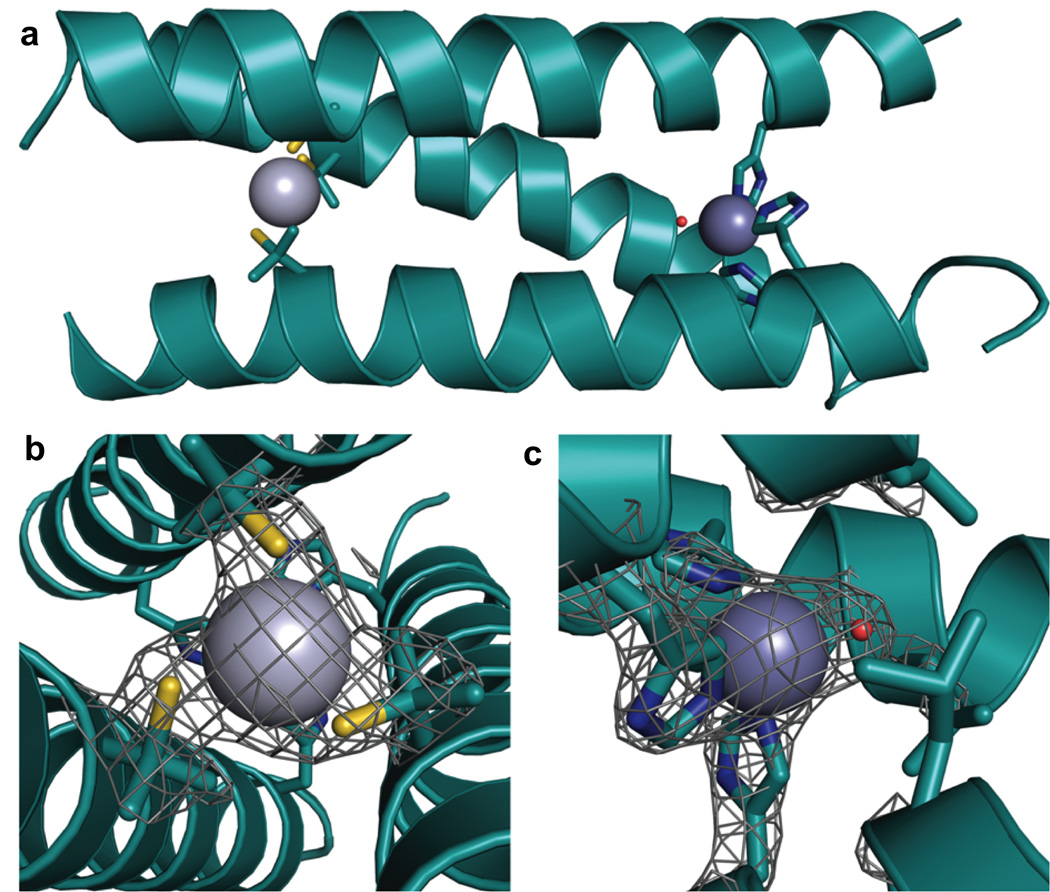
Ribbon diagrams of the X-ray crystal structure of [HgII]S[ZnII(OH2/OH−)]N(CSL9PenL23H)3n+ at pH 8.5. The Pen and His side chains are shown in stick form (sulfur = yellow; nitrogen = blue; oxygen = red). (a) One of two trimers found in the asymmetric unit of the crystal structure. (b) Top-down view of the structural trigonal thiolate site, HgIIS3, confirming the proposed structure of HgII in Cys-containing TRI. (c) Side view of the tetrahedral catalytic site, ZnIIN3O, which closely mimics carbonic anhydrase and matrix metalloproteinase active sites. Figure was reproduced from ref. [70] with permission of the copyright holders.
Our strategy to distinguish the sites quantitatively was to use an ion with high affinity for thiolate ligation but low affinity for His ligands (HgII) while exploiting the high stability of the HgII(thiolate) ligation to allow discrimination of ZnII solely to the His position. Thus, the structural site is made up of HgII bound to three thiolates, which stabilizes the pre-folded structure. In Section 3.2, we discussed how Cys-substituted TRI peptides can be used to enforce a trigonal coordination environment on HgII and also how HgII can be used to induce the folding of a random coil structure with the Baby peptides. We aimed to exploit this work in order to generate a peptide in which the presence of HgII in a pre-folded structure would lead to a more thermodynamically stable complex in the face of the presence of a second, separate metal binding site made up of His residues. The aim was to incorporate His residues in place of one layer of Leu residues in the interior of TRI for generation of an active site similar to that in carbonic anhydrase (ZnIIN3O), which catalyzes the reversible hydration of carbon dioxide (CO2) and is one of the fastest known zinc metalloenzymes [135]. Although these sequences can withstand multiple thiol substitutions [127], it was not originally known whether or not the incorporation of His residues would lead to a stable construct. The sequence chosen for these studies was TRIL9CL23H, with a His substitution in the 23rd position near the C-terminal end of the trimer and Cys in the 9th position for the HgII-binding site near the N-terminus (Table 1). Based on previous CS crystal structures containing solely thiol substitutions, the C-terminal end of the 3SCC tends to be more ‘frayed’ and, therefore, it was hypothesized that this location could better accommodate bulkier residues. After ensuring that this peptide bound HgII in a trigonal fashion under appropriate conditions according to previous studies [63], CD denaturation titrations were performed to monitor the stability of three constructs, [HgII]S(TRIL9CL23H)3−, apo(TRIL9CL23H)33− (no bound HgII), and apo(TRIL23H)3 (no thiol site). A plot of the molar ellipticity versus concentration of GuHCl is quite striking in its representation of how the presence of HgII confers significant stability on the designed peptide (Figure 13). The complexes (TRIL23H)3, (TRIL9CL23H)33− and [HgII]S(TRIL9CL23H)3− are well-folded α-helical coiled coils at pH 8.5, albeit with a bit lower α-helical content (~70–80%) than any reported peptides containing a single Cys substitution (usually above 90%) due to fraying of the coiled coil at the C-terminal end below the His site (Figure 14). Denaturation of the complexes revealed that in the absence of metal, the doubly-substituted peptide unfolded at the lowest concentration of GuHCl, (TRIL23H)3 was more resistant to denaturation, and when HgII was present there was a dramatic shift in the midpoint of unfolding, demonstrating a significant increase in stability of the complex. Unfortunately, none of these unfolding curves fit a two-state model and so quantitative free energy values could not be reported. Importantly, we were able to use this stabilization to our benefit to obtain X-ray quality crystals of an analogous dual metal site complex [HgII]S[ZnII(OH2/OH−)]N(CSL9PenL23H)3n+ (the comparable CS sequence has similar solution properties to TRI), which illuminates how HgII and ZnII bind to the structural and catalytic sites, respectively (Figure 12). The ASU of this structure contains two independent, well-folded 3SCCs. In one, HgII is bound with trigonal planar geometry to the sulfur atoms of Pen and ZnII is bound four-coordinate to three His residues and a chloride ion. In the second 3SCC, HgII is bound in a T-shaped fashion, where one of the HgII-S distances is significantly longer than the others (2.29, 2.13, and 3.06 Å) and ZnII is again four-coordinate and bound to three His residues and a solvent molecule (water/hydroxide). The HgII sites in this crystal represent the first structural characterization of HgII-binding in this family of peptides, support the models proposed based on spectroscopic methods (Section 3.2), and the trigonal site provides the first crystallographic model for tris(cysteinato)HgII in a protein environment, hence serving as a model for mercury complexation by MerR. Furthermore, the ZnIIHis3X site closely resembles the first coordination sphere of the active site in carbonic anhydrase (CA) (Figure 15) and related ZnIIHis3X sites found in matrix metalloproteinases [136]. Much of the protein matrix beyond the first coordination sphere is very different between the model and CA (α-helices vs. β-sheets being the most noticeable), but there are also differences in hydrogen bonding residues, water channels and even the identities of the coordinating nitrogen atoms from the imidazole rings. An important objective of metalloprotein design is to assess whether a specific fold (e.g., β-barrel) is essential for the formation of a particular coordination environment and/or catalytic activity. Our model may aid in addressing what the minimal unit required for catalytic activity is (which is one important question of de novo design). In other words, is it possible to remove an active site from a native protein, embed it in a minimized and completely different protein fold, obtain the same metallocenter, and retain activity? Clearly, the answer to the structural question is clear, one can have dramatically different folds (α-helices versus β-sheets) and still prepare an essentially identical first coordination sphere for the metal. Addressing the activity question should allow one to assess which structural features are necessary for an efficient metalloenzyme through building up secondary interactions that would certainly be required to achieve optimal active site properties. To this end, we first tested if the construct could hydrolyze the activated ester, p-nitrophenyl acetate (pNPA), a non-physiological substrate used extensively for analyzing the reactivity of CA and its mutants and often with small molecule synthetic enzyme models. In fact, [HgII]S[ZnII(OH2/OH−)]N(TRIL9CL23H)3n+ exhibits enzyme-like saturation kinetics with kcat/KM up to 23.3 M−1 s−1 at pH 9.5, which is ~550-fold more efficient than small molecule model complexes [137–139]. Further, it is within ~100-fold of that of CAII at pH 8 (its maximum efficiency), the fastest isozyme of the α-CA enzymes [140,141]. The reaction is pH-dependent, analogous to that for CA, with increasing activity correlated to increasing pH, but the pKa is 2 units higher (8.8) than that for CAII (6.8) (which has evolved to operate most efficiently under neutral pH conditions) [135].
Figure 13.
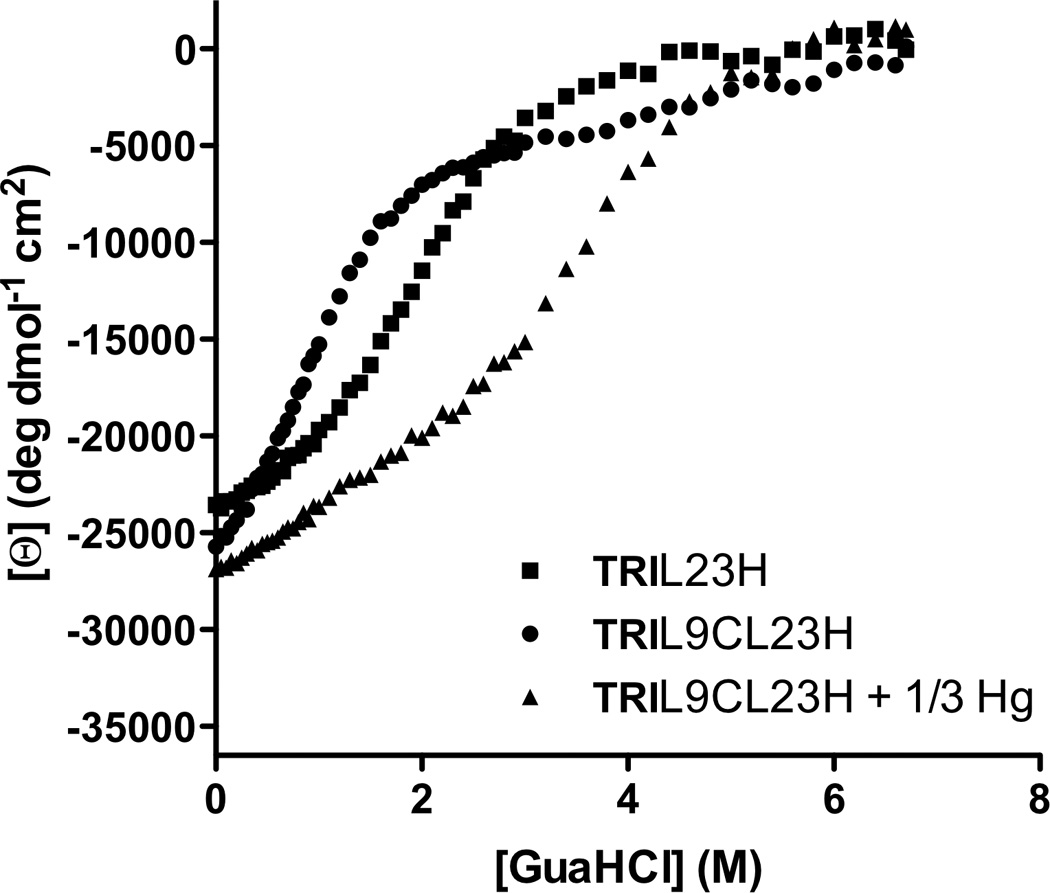
Guanidine hydrochloride denaturation titrations represented by the molar ellipticity values [Θ] at 222 nm versus denaturant concentration for TRIL23H (■), TRIL9CL23H (●), and TRIL9CL23H + 1/3 HgII (▲). Figure was reproduced from ref. [70] with permission of the copyright holders.
Figure 14.

Comparison of the size of the active site cavities of (a) modeled His3 site using the structure of [AsIII]S(CSL9C)3 (PDB 2JGO)[68] and (b) the actual structure containing the His3 site, [HgII]S[ZnII(OH2/OH−)]N(CSL9PenL23H)3 n+ (PDB 3PBJ) [70]. (c) Overlay of the two sites with the model in gray and the actual structure in cyan.
Figure 15.
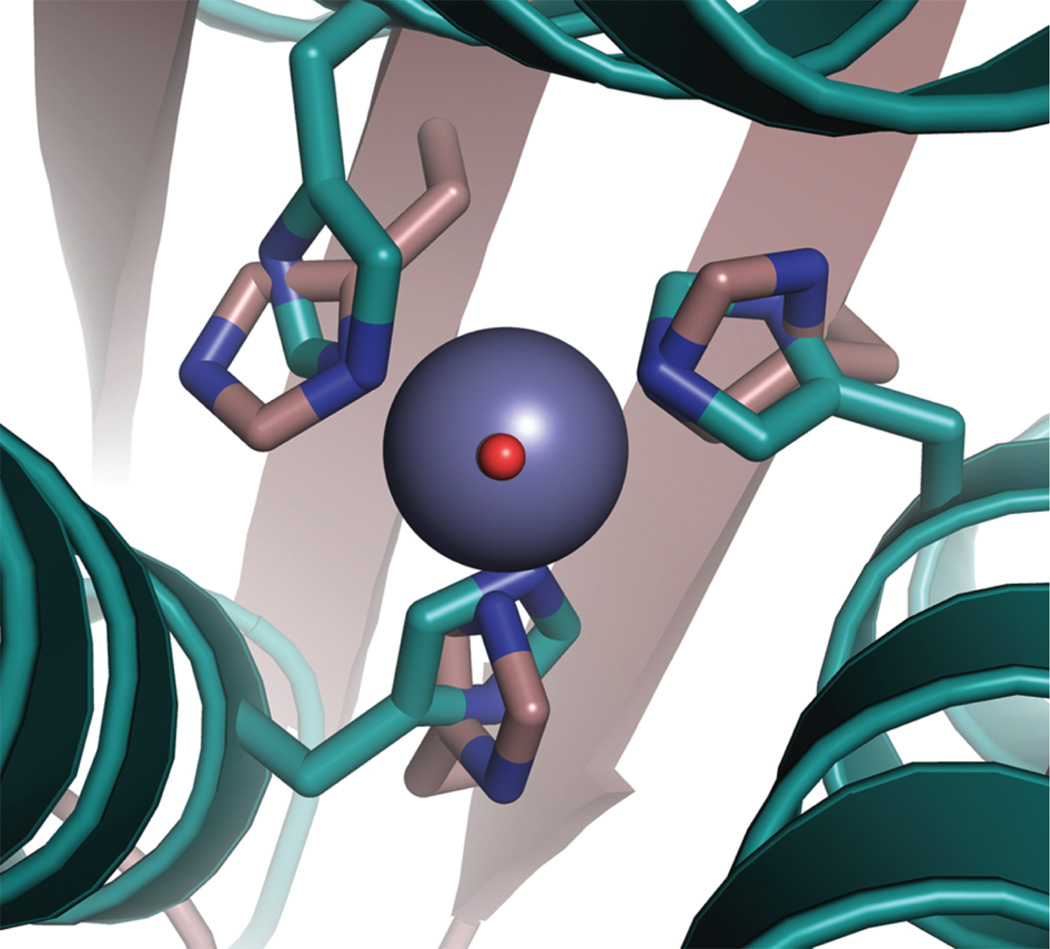
Overlay of the ZnIIN3O site in [HgII]S[ZnII(OH2/OH−)]N(CSL9PenL23H)3 n+ (cyan, PDB 3BPJ) with the active site of human CAII (tan, PDB 2CBA). The solvent molecule of [HgII]S[ZnII(OH2/OH−)]N(CSL9PenL23H)3 n+ is shown in red and that for CA lies directly underneath. Figure was reproduced from ref. [70] with permission of the copyright holders.
Although the focus of this review is meant to be on de novo designed metalloenzymes there are other examples of hydrolytic zinc enzymes which do not strictly fit in this category but have “de novo” zinc sites in preexisting protein scaffolds (there are no other completely de novo designed hydrolytic zinc metalloenzymes reported, although there was a report of a non-metalloprotein hydrolytic designed enzyme which was later retracted [142,143]). The Baker group has reported a redesigned zinc-containing mouse adenosine deaminase for organophosphate hydrolysis [17]. The approach the authors used to develop this enzyme harnesses de novo enzyme computational design methods to identify scaffolds with backbones that can support active sites for the target reaction, therefore, addressing solely the geometric compatibility of the site with the transition state and not the reactivity of wild-type functional groups. After directed evolution to optimize the original active site (where only surrounding residues were altered and not the first coordination sphere around ZnII, which is His3Asp in a trigonal bipyramidal geometry with one open coordination site), the enzyme could catalyze diethyl 7-hydroxycoumarinyl (DECP) hydrolysis with a catalytic efficiency of 9750 M−1 s−1. Only seven mutations, of which four were shown to be the minimal required set, resulted in a 2500-fold improvement in the hydrolytic activity. Given that this design addresses reactivity to a different substrate than we have studied, and with a different first coordination sphere around the ZnII center, it cannot be directly compared to our system. On the other hand, Der et al. have recently reported the design of a ZnHis3 site at the interface of two copies of the Rab4-binding domain of rabenosyn which catalyzes the efficient hydrolysis of pNPA and p-nitrophenyl phosphate (pNPP) [16]. In this work, the authors used computational methods to introduce a metal binding site on the surface of a monomeric protein in order to direct the formation of a dimer and development of a protein interface. The Tezcan group has reported extensive studies on the development of multimeric protein complexes in a similar fashion, whereby the relatively strong interactions formed between metals and protein side chains are used to direct their formation and can be followed by subsequent optimization of protein-protein interface interactions (weaker hydrogen bonds and Van der Waals forces) [36–38]. Along these lines, Der and coworkers, through incorporation of two His residues on the monomer surface (based on computation), developed a ZnHis3 site (the intended site was ZnHis4, two from each monomer, but crystallographic analysis showed that the fourth residue did not bind) [39]. After determining that this metal center contained an open coordination site, filled by tartrate in the crystal structure (Figure 16), the authors decided to test it for esterase activity towards pNPA and pNPP. MID1-Zn effectively catalyzes pNPA hydrolysis with more than 50 turnovers and a rate of 0.22 s−1 and KM = 0.47 mM at pH 8.5. The reaction is pH-dependent (increasing efficiency with increasing pH) and has a kinetic pKa of 8.2 ± 0.1 and a maximal efficiency of 630 ± 90 M−1 s−1, which is ~27-fold more efficient than our model at pH 9.5. The authors discuss the importance of the binding cleft in achieving this efficiency. Synthetic models are relatively ineffective given that they have no binding cleft, although this can be improved somewhat in apolar solvents which can simulate the apolarity of an active site cleft [144]. Although pNPP is intrinsically less reactive, MID1-Zn still effectively catalyzes its hydrolysis with an efficiency of 14 M−1 s−1 at pH 8.5. The KM for this reaction, 12 µM, is 40× lower than that for pNPA because of favorable electrostatic interactions between the negatively charged phosphate group and positively charged ZnII active site.
Figure 16.
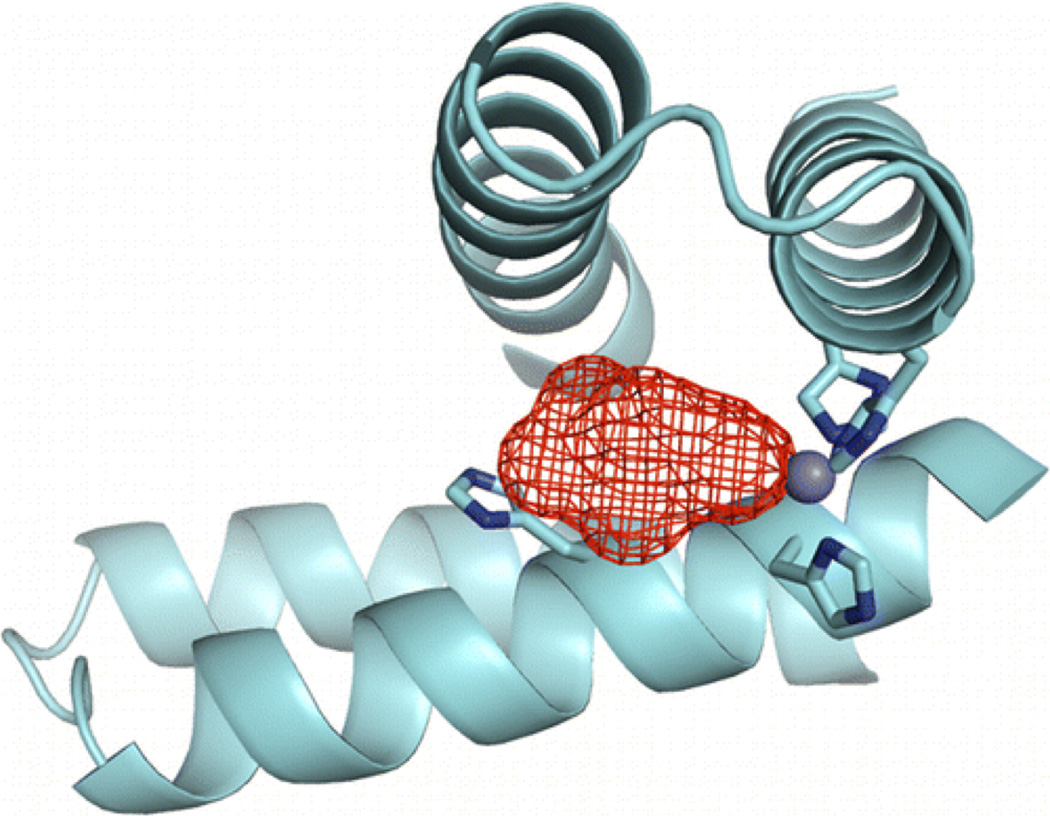
X-ray crystal structure of MID1-zinc, a designed metal-mediated protein interface. The red mesh represents the active site cleft above the open coordination site of the ZnHis3 metal site. Figure was reproduced with permission from ref. [16]. Copyright 2012 American Chemical Society.
Evaluation of the native reaction catalyzed by CA, CO2 hydration, yields an efficiency at pH 9.5 for [HgII]S[ZnII(OH2/OH−)]N(TRIL9CL23H)3n+ of ~1.8 × 105 M−1 s−1, which is within ~500-fold of that of CA (Table 3). This efficiency is also superior to that reported for any small molecule model [145–152]. Although MID1-Zn with a ZnHis3 site discussed above is 27-fold more efficient than [HgII]S[ZnII(OH2/OH−)]N(TRIL9CL23H)3n+ at pNPA hydrolysis, no activity has been reported for CO2 hydration with the designed protein dimer and so no comparisons can be made [16]. It is expected that this design would also catalyze CO2 hydration but it would be interesting to observe how the CO2 hydration activity may correlate in order to identify more fully any further effects that may be imparted by the surrounding protein scaffolds on each model (such as steric effects). Regardless, it is important to point out that, for our model, the scaffold does not begin to incorporate secondary interactions around the metal site, yet can gain an efficiency for an evolved reaction within ~500-fold of one of nature’s fastest metalloenzymes. Upon evaluation of the effects of these secondary interactions on CA (numerous mutagenesis studies have been reported [135]), the hydrogen bonding interaction of ZnII-bound solvent with Thr199 seems to be one of the most important [153,154]. When this residue is mutated to Ala, ~100-fold efficiency is lost for both hydrolytic reactions and the kinetic pKa increases by nearly two pH units. The resultant properties of the CA mutant are strikingly similar to those of our model for pNPA hydrolysis. This study serves to highlight the importance of secondary interactions in fine-tuning an enzyme, although a significant amount of activity can apparently be gained by simply mimicking the first coordination sphere of a native zinc metalloenzyme. As compared to small molecule models, a substantial amount of catalytic power can be gained by embedding a metal site into a protein scaffold.
Table 3.
Comparisons of the kinetics for CO2 hydration by selected catalysts.
| Catalyst | kcat (s−1) | kcat/KM (M−1 s−1) | k1 (M−1 s−1) | k2 (s−1) |
|---|---|---|---|---|
| [HgII]S[ZnII(OH2/OH−)]N(TRIL9CL23H)3a | 1.8 (± 0.4) × 103 | 1.8 (± 0.3) × 105 | ||
| CAIIb | 8.2 × 105 | 9.2 × 107 | ||
| H2Oc | 6.6 (± 0.4) × 10−4 | 37 (± 2) × 10−3 | ||
| OH−(d) | 12.1 (± 0.4) × 103 | |||
| Zn(imidazole)n•(OH)+(e) | 6.0 | |||
| Zn[tris(4,5-di-n-propyl-2-imidazolyl phosphine)f | 2480 | |||
| Zn[([12]aneN3)(OH)]+(g) | 581 ± 64 |
. Taken from reference [70], pH 9.5 and at 25° C, the complex can perform;
. Taken from reference [202], pH-independent values;
. Taken from reference [203] according to the equation: CO2 (aq) + H2O ⇋ H2CO3 where k1 represents the second-order reaction rate for the forward reaction (water as one reactant) and k2 represents the pseudo-first-order reaction, where the water concentration is assumed constant at 55.6 M;
. Taken from reference [203] according to the equation: CO2 (aq) + OH− ⇋ HCO3− where k1 represents the second-order reaction rate for the forward reaction (hydroxide as one reactant);
. Taken from reference [204], pH 6.63, ZnSO4 in imidazole buffer;
. Taken from reference [149], pH 6.55 in 80% ethanol at 25° C;
Further, the role of a separate stabilizing structural site can be evaluated. Although the complex is pre-folded in its absence, its presence demonstrates that the structure remains intact throughout the analysis. Analysis of the analogous complex in the absence of the HgS3 site indicates that there is no loss in activity due to the extra stabilization. This is especially important for developing multi-site metalloproteins where one may want to incorporate multiple sites for a variety of functions (for example, one for electron transfer and another for catalysis) without detrimentally affecting the folding or function of the protein. Incorporation of additional mutations to promote favorable secondary interactions may even be offset by the presence of a structural site. This study clearly demonstrates the utility of α-helical bundles and coiled coils towards designing functional multi-metal site proteins [70].
4.2. De novo design of a redox-active copper metalloprotein
Another very important goal of de novo metalloprotein design is to generate systems capable of performing redox chemistry. As compared to the development of hydrolytic catalysts, there is another level of complexity in preparing redox catalysts; one must optimize the metal binding site to accommodate both the oxidized and reduced versions of the enzyme. Copper centers play an important role in many biological systems, including denitrification. In particular, copper nitrite reductase (NiR) carries out the dissimilatory reduction of nitrite to nitric oxide (NO2− + e− + 2H+ → NO + H2O) [155–157]. This enzyme contains two distinct copper sites, a Type 1 center which provides the electron necessary for reduction (originally provided by pseudoazurin) and a Type 2 (T2) center where the catalytic conversion occurs. The T2 center has a copper ion coordinated to three His residues and a water molecule in a distorted tetrahedral environment. This coordination environment is very similar to that of the ZnHis3O site in [HgII]S[ZnII(OH2/OH−)]N(CSL9PenL23H)3n+ (discussed earlier in Section 4.1), and an overlay of the two sites is shown in Figure 17. Given that CuI and ZnII are both d10 metal ions, we thought it reasonable that copper could bind to the His3 site in our 3SCCs. There are several previous reports for copper binding in α-helical coiled coils; however, none of these have been extensively characterized for each of the oxidation states of copper [7,55,56,58,77,113,117,134,158–161]. We have now reported this characterization for CuI/II binding in our TRIL23H and TRIL2WL23H peptides along with nitrite reductase reactivity (Table 1) [162]. 1H NMR studies, in which the chemical shifts for the imidazole protons are measured in the presence and absence of CuI, indicate that at a 1:3 metal:peptide ratio and pH greater than 4.45, a [CuI]N(TRIL23H)3+ complex is formed, where all three His imidazoles are coordinated to CuI. X-ray absorption near-edge spectroscopy (XANES) data for [CuI]N(TRIL2WL23H)3+ confirms the presence of three-coordinate Cu at pH 5.9 and 7.4. Extended X-ray absorption fine structure (EXAFS) data further verifies this assignment (the three Cu-N distances are 1.93 Å, consistent with a three-coordinate Cu structure) and indicates that the trigonal planar CuN3 site is distorted. CuII binding was characterized using UV-visible and electron paramagnetic resonance (EPR) spectroscopies. A broad absorption band at 640 nm, with extinction coefficients of 133–138 M−1 cm−1 (pH 5.8–7.4) is assigned to d-d transitions and consistent with a CuIIHis3 coordination environment including one or two exogenous water ligands. EPR spectra display features typical of T2 copper centers (pH 5.19–7.80). The g values (g// = 2.27 for both [CuII]N(TRIL23H)32+ and [CuII]N(TRIL2WL23H)32+) are somewhat smaller and the hyperfine splitting (A// = 186 G and 188 G for [CuII]N(TRIL23H)32+ and [CuII]N(TRIL2WL23H)32+, respectively) is somewhat larger than those observed for NiR (which has a CuII(His)3(OH2) environment with g// = 2.34 ± 0.03 and A// = 125 ± 35 G for Rhodobacter sphaeroides [163]). The larger hyperfine splitting we observe for the designed complex is more consistent with a five-coordinate structure. The metal center in the TRI system is likely Cu(His)3(OH2)2 with CuII in a distorted square pyramidal geometry containing three quasi inplane imidazole rings. When 27 equivalents of nitrite are added to [CuII]N(TRIL23H)3, a 9 G decrease in AII is observed with no further changes up to 212 equivalents. A similar 13 G decrease has been observed for R. sphaeroides NiR, representing addition and binding of nitrite [163]. This supports direct binding of nitrite to the Cu center in [CuII]N(TRIL23H)3.
Figure 17.
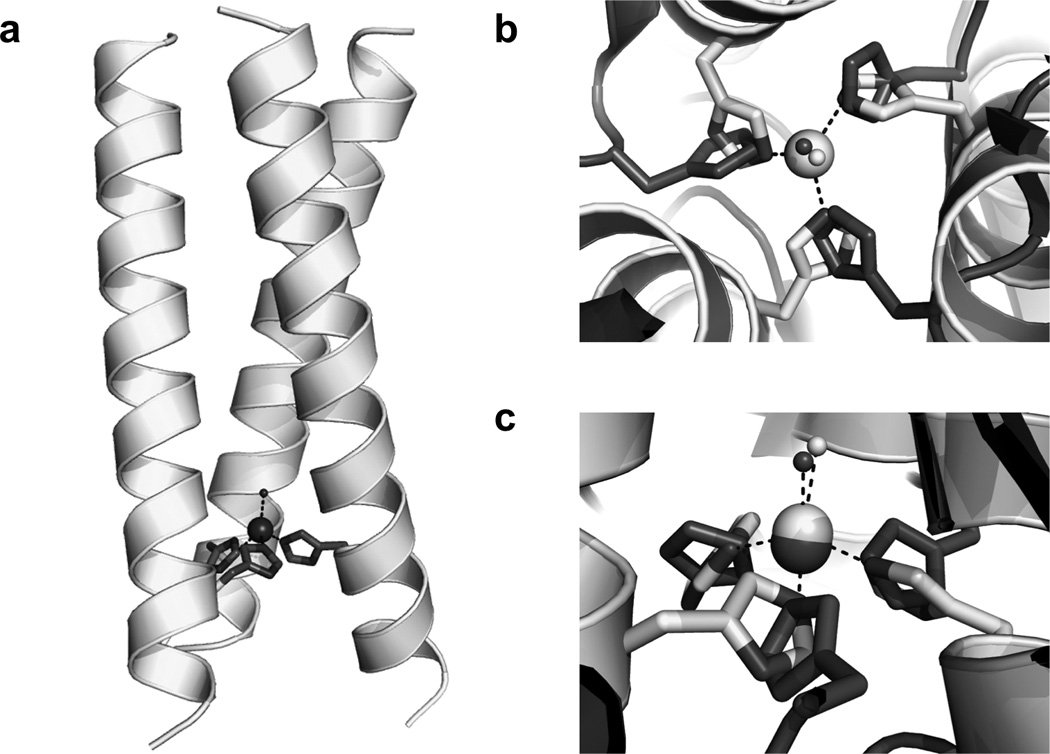
Model of [CuI/II]N(TRIL23H)3+/2+ based on the structure of [HgII]S[ZnII(OH2/OH−)]N(CSL9PenL23H)3n+ (PDB 3PBJ). (a) side view of the model, (b) Top-down view from the N-termini of the metal sites with the ZnII(H2O)(His)3 site in light grey superimposed on the Type 2 CuII(OH2)(His)3 site in R. sphaeroides NiR (PDB 2DY2) in dark grey. (c) side view of the superimposed metal sites, as in b. Figure was reproduced from ref. [162] with permission of the copyright holders.
Given that the goal of the work was to obtain a redox-active model, it is important to estimate the reduction potential of the copper center. To do this the binding affinities for CuI and CuII were separately determined at pH 5.9 and 7.4. The dissociation constants for CuII are 40 ± 8 nM and 8.7 ± 1.1 nM at pH 5.9 and 7.4, respectively, and those for CuI represent even stronger binding, 3.1 ± 0.7 pM and 0.20 ± 0.06 pM at pH 5.9 and 7.4, respectively. Using these values and the Nernst equation, the calculated reduction potentials are 400 ± 30 mV at pH 5.9 and 430 ± 30 mV at pH 7.4. This reduction potential is higher than those for T2 copper centers in NiR (218 mV at pH 6, 137 mV at pH 8.4 for R. sphaeroides [164] and 220–310 mV at pH 7.0 for A. cycloclastes and A. xylosoxidans [165]) likely because CuI in the (TRIL2WL23H)3 peptide complex is three-coordinate with a distorted trigonal planar environment (based on XAS data described above). This type of structure should stabilize the CuI oxidation level and may explain the high positive potential. Regardless, these potentials suggested that there may be some nitrite reductase activity and the EPR data suggested also that nitrite could bind to copper, so we decided to investigate the redox reactivity of the complex.
First, we found that stoichiometric ascorbate could be used to reduce [CuII]N(TRIL23H)3, as confirmed by loss of the absorbance from the 640 nm d-d band. Excess nitrite was then added to a solution of [CuI]N(TRIL23H)3, resulting in an increase of absorbance at 640 nm. Full recovery of absorbance was never observed but three more successive additions of ascorbate all lead to recoveries over 65% and, importantly, demonstrate that the copper center can be continuously cycled between oxidation states. This is important for developing catalytic redox metal sites. Next, the complex was shown to produce the product, NO, by reaction with one equivalent of nitrite at pH 5.8 and trapping the gas as the colored [Fe(NO)(EDTA)]2− complex. The activity of the complex was then monitored under catalytic conditions (an excess of both nitrite and sacrificial electron donor). For this study, the ascorbate oxidation (representing reduction of the Cu center and correspondingly, nitrite) could be monitored through a decrease in its absorbance at 265 nm. After background correction, it was concluded that the complex can perform five turnovers of the reaction over the course of 3.7 hours (with turnovers expressed as moles of electrons per mole of [CuII]N(TRIL23H)32+). Notably, the ascorbate oxidation rate has a linear dependence on the concentration of [CuII]N(TRIL23H)32+, demonstrating that the observed activity is corelated with the designed metalloprotein. Both the rate of ascorbate oxidation and the reoxidation of [CuI]N(TRIL23H)3+ were measured as a function of pH. Reoxidation of [CuI]N(TRIL23H)3+ was measured up to pH 7.4, where no significant increase in absorbance is observed over two hours. The rate of ascorbate oxidation was measured from pH 5.3–6.5. Pseudo-first-order rate constants can be obtained in both cases and are given in Table 4. The pseudo-first-order rate constant at pH 5.9 for [CuII]N(TRIL23H)32+, k1st,Asc, is 4.40×10−4 s−1 (five turnovers in 3.7 hours). This is much slower than the nitrite reductase activity of the NiR enzymes (1500 s−1 at pH 5.8 for Alcaligenes faecalis NiR, Table 4) [166]. However, our de novo designed metallopeptide is the only example of a stable and functional CuHis3 site in aqueous solution. Many model complexes have examined NiR reactivity in non-aqeuous solutions, which do not accurately represent the native system, although these rates are higher than those observed in aqueous solutions and some model the His3 environment fairly well [167–170]. Those which are functional in aqueous solution have far lower rates and some produce N2O rather than NO [171–176]. Those systems which produce multiple turnovers are mostly heterogeneous systems, where the catalyst is linked to an electrode surface [171–173,176]. Another example uses photoactivated [RuII(bpy)3]2+ as the electron donor and can do at least four turnovers in 15 min, but a significant decrease in NO generation is observed after two turnovers [174]. We have demonstrated that [CuI/II]N(TRIL23H)3+/2+ does not show any evidence of catalyst decomposition over the 175 minute reaction and that the product of our reaction is predominantly NO, with no N2O detected. This work further illustrates the utility of de novo designed metallopeptides in developing functional models, specifically those of native redox metalloenzymes, which provide a number of benefits over traditional synthetic model complexes, including stability and solubility in water, enforcement of lower metal coordination number (including inhibition of dimerization reactions), and presentation of a hydrophobic environment with a single metal cofactor. Therefore, not only is this complex the only example of a stable, functional CuHis3 site in aqueous solution, it represents a simplified functional CuT2 center in the absence of the CuT1 electron transfer center found in the native enzyme. This can allow for future study of the catalysis independent of the Cu-Cu electron transfer process and characterization of the CuT2 spectroscopic features that are often hidden by those of the CuT1 center. One may imagine that the combination of an aqueous model within a protein framework in the absence of complications of the native enzyme may lead to a deeper understanding of the function and other properties of CuNiRs.
Table 4.
Pseudo-first-order rate constants for the oxidation of [CuI]N(TRIL23H)3+ (kfirst,Cu) and of oxidation of ascorbate (kfirst,Asc) as a function of pH and compared to kcat for Cu nitrite reductases and a representative model complex.
| pH | kfirst,Cu × 10−4 (s−1)a | kfirst,Asc × 10−4 (s−1)a |
AfNiR (kcat, s−1) |
AxNiR (kcat, s−1)d |
CuMe2bpaCl2 (kcat, s−1)e |
|---|---|---|---|---|---|
| 5.3 | - | 12 ± 3 | |||
| 5.5 | - | 9 ± 1 | 0.063 | ||
| 5.8 | 5.2 ± 0.3 | 4.6 ± 0.3 | 1500b | ||
| 5.9 | - | 4.40 ± 0.16 | |||
| 6.0 | 2.8 ± 0.3 | 2.4 ± 0.2 | |||
| 6.5 | 0.65 ± 0.03 | 0.68 ± 0.03 | 620 ± 10c | ||
| 7.0 | 0.22 ± 0.08 | - | 89 ± 3 |
. Taken from reference [162]. Note that five turnovers are measured at pH 5.9 in 3.7 hours at 25° C;
. Taken from reference [166], measured at 25° C;
. Taken from reference [205], measured at 25° C with Pseudoazurin as the electron donor;
. Taken from reference [206], measured at 4° C;
. Bis(6-methyl-2-pyridylmethyl)amine (Me2bpa). Taken from reference [171], electrochemical catalysis (25° C, in collagen matrix on electrode surface) with significant decrease in activity after two turnovers.
4.3. De novo design of a functional diiron protein
Another level of complexity can be added to the de novo design of metalloprotein systems with the development of binuclear metal centers. DeGrado and coworkers have developed the Due Ferri (two-iron; DF) family of artificial metalloproteins as models for diiron and dimanganese enzymes, a diverse set of enzymes capable of catalyzing both hydrolytic and oxygen dependent reactions [82,83]. The binuclear metal site in these proteins is bridged by a combination of oxo, hydroxo, or carboxylate donors and although the protein folds of this diverse family of metalloproteins are complex, a simple four-helix bundle houses the active sites (Figure 2d) [11]. DeGrado et al. have designed four-helix bundle proteins to aid in understanding how polarity, solvent accessibility, and electrostatic environment influence the properties of these proteins, with a focus on diiron sites. The work demonstrates that minimal protein models can be used to track structural changes as small as a methyl group and connect them to the enzyme’s activity. Their success lies in iterative design and rigorous characterization of each model. Retrostructural analysis of diiron proteins was used to design the parent model, DF1, which is an antiparallel dimer of 48 residue helix-loop-helix (α-2) motifs that binds a di-metal cofactor near its center [11]. The helical bundle has a slight left-handed tilt as in classical four-helix bundles and antiparallel 4SCCs and its geometries may be discussed in terms of the heptad nomenclature described in Section 2.3 (Figure 18). Each helix has a single Glu residue in an a position which projects towards the center of the bundle. Two carboxylates bridge both metals, while the other two each interact with a single metal in a mono or bidentate binding configuration. Two His residues are located at d positions, three residues away from the bridging Glu side chains and coordinate each metal in a monodentate fashion to form a EXXH binding motif. Small side chains occur at the analogous d positions opposite the His residues and control oxygen and substrate access. Residues in the e and b positions line the two sides of the active site (e/b interface) and the other two sides are lined by c and g residues (c/g interface). Those residues along the b/e interface play primarily a structural role and are tightly packed. Those residues at the c/g interface form hydrogen bonds to the ligand His residues on one side and on the other side define entry to the active site. The a and d positions above and below the ligand residues form the top and bottom of the active site. These are often hydrophobic and their association provides part of the driving force for association of the α-helical bundle.
Figure 18.
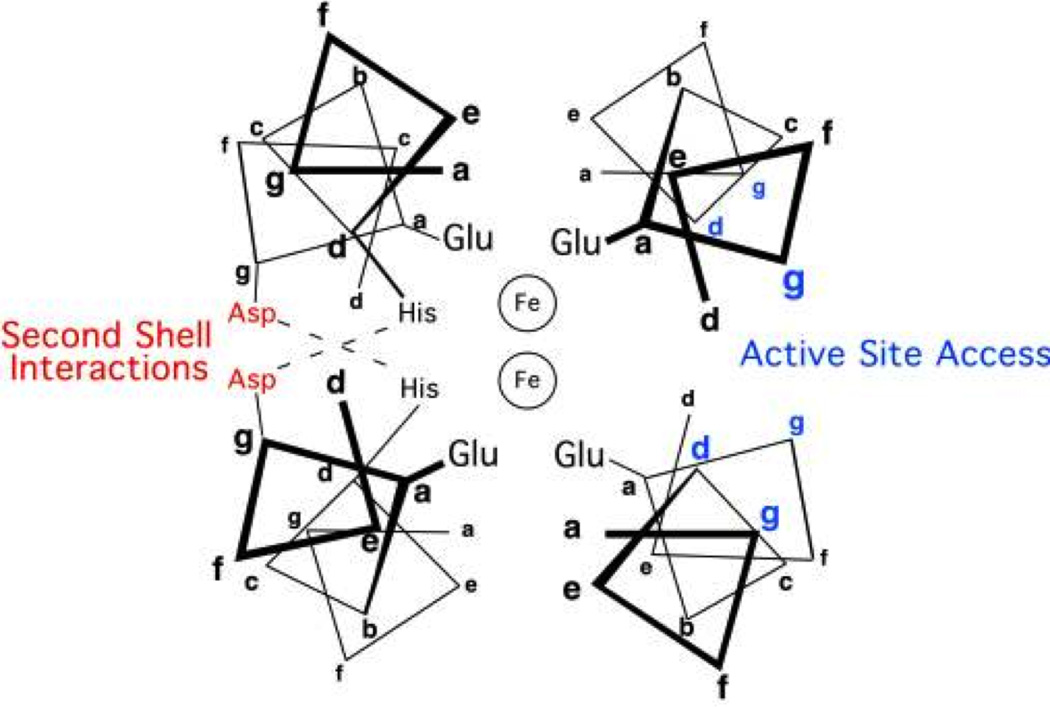
Helix-wheel representation of the antiparallel four-stranded coiled coil structure surrounding the active site of a diiron protein. Residue positions are labeled according to the heptad repeat generally applied to coiled coils. Figure was reproduced from ref. [11] with permission of the copyright holders.
DF1 was designed beginning with a D2-symmetric 4-helix bundle of unconnected 21 residue helices and is made up of a Glu4His2 active site, where two Glu residues bridge the metals, the other two Glu are non-bridging ligands, and the Nδ of each His is bound to a metal (sequence DYLRELLKLELQLIKQYREALEYVKLPVLAKILEDEEKHIEWLETILG) [11]. Additionally, second shell residues were added, which serve as geometric constraints and may be important to function. Asp35 forms a hydrogen bond with an active site His residue in a neighboring helix and Tyr7 (in a d position) forms a hydrogen bond with a non-bridging Glu residue from a neighboring helix. A combination of hydrophobic and polar residues were chosen for the interfaces, helix stabilizing polar residues were placed in the most solvent-accessible locations, and an interhelical loop was engineered in between each pair of helices. Structural analysis of di-ZnII-DF1 [11], di-MnII-DF1 [177], and the apo-version [178] of the protein revealed that the metal binding site and surrounding secondary interactions are very similar to the intended design and are largely preorganized for metal binding (Figure 19). Each iron is five-coordinate with a sixth vacant site lying on adjoining faces of the two metal ions, providing a potential binding site for exogenous ligands. Because the design of DF1 required the burial of six polar ionizable groups in the core of a four-helix bundle, the interior was packed with a large number of hydrophobic side chains to drive folding. These requirements for conformational stability are contrary to those for binding and enzymatic functions, as revealed by the difficulties in binding the metal co-factor (DF1 must be unfolded and refolded in the presence of metal to support binding) and the lack of any activity. In particular, analysis of the structure revealed that bulky Leu residues in the d positions opposite the His binding residues were likely blocking access of O2, which would limit its capability of supporting any activity. DF1 was redesigned to carve out a larger access site by mutating each of the Leu residues to either Ala or Gly (L13A-DF1 and L13G-DF1) [177–180]. These three designs were used to examine the thermodynamic costs of carving an active site access channel, with DF1 being the most stable and the Gly derivative being the least (a total loss of ~10 kcal/mol). To confirm the presence of this designed cavity, the crystal structures of di-MnII-L13A-DF1 and di-MnII-L13G-DF1 were solved [178]. The X-ray structure of di-MnII-L13A-DF1 revealed three dimers in the ASU, all nearly identical to those observed for the di-ZnII- and di-MnII-DF1 structures [180]. Further, an aqueous channel is formed by the Leu to Ala substitutions and is filled by ordered water molecules and evidence of a metal-bridging dimethyl sulfoxide (DMSO) molecule. The X-ray crystal structure of di-MnII-L13G-DF1 shows that with substitution by the smallest amino acid, Gly, the size of the aqueous channel increases even more [179]. There are now four independent dimers in the ASU, more water molecules present around the dimetal cofactor and no evidence of bound DMSO. More interesting is the presence of two different coordination environments around the di-MnII centers (Figure 20). Three of the crystallographically independent dimers in the ASU have bridging water molecules and an intermetal distance of 3.6 Å. However, the fourth has two terminal water molecules, one coordinated to each metal and trans to the liganding His residues. The intermetal distance in this dimer is 4.2 Å. Superimposition of the C-terminal helices (those containing the HXXE liganding site) reveal no difference in the four dimers but the N-terminal helices (those containing only a single liganding Glu residue) occupy different positions in the dimer containing the distinct metal coordination site. Particularly, two copies of the N-terminal helices shift in opposite directions; this sliding-helix induced shift is what leads to the increase in the metal-metal bond length. It is postulated that this may occur in native enzymes to accommodate coordination environment changes; however, it would likely be difficult to observe in a complex, large molecule. Structural analysis of DF1 and its mutants with bound di-ZnII and di-MnII demonstrate that the overall protein fold and general coordination environment around the dimetal site are retained with introduction of a metal site access channel. Although the DF1 constructs have proven useful for detailed structural characterization (as their stability facilitates crystal growth), they have only limited solubility and therefore cannot be fully characterized in solution. To improve the properties of DF1, some hydrophobic residues on the surface of L13A-DF1 were replaced with polar residues and a redesigned interhelical loop in DF2 and a longer interhelical loop was incorporated in DF2t [181–185].
Figure 19.
Ribbon diagrams of the structures of metal-bound and apo-DF1. (a) The X-ray crystal structure of di-ZnII-DF1 (PDB 1EC5) [11]. (b) The X-ray crystal structure of di- MnII-DF1 (PDB 1OVR) [177]. (c) The NMR structure of apo-DF1 (PDB 1NVO) [178]. The helix-loop-helix motif in front of the ligands is shown as transparent to give an improved view.
Figure 20.
2Fo-Fc electron density maps of the dinuclear metal-binding site of di-MnIIL13G- DF1 in the dimer (one of four in the asymmetric unit) with terminal waters bound to each metal (top) and in one of the dimers with a bridging water (bottom). Figure was reproduced from ref. [82] with permission of the copyright holders.
Next in searching for functionality, DFtet was designed by bridging rational design with a combinatorial approach to facilitate preparation of a variety of mutants [186]. The results obtained from the DF1 and DF2 models indicated three main aspects to be considered in order to obtain stable and functional models: interhelical turn design, residues responsible for primary and secondary ligands, and residues that define the active site access channel. In order to fully examine how systematic changes would affect these aspects, a large number of combinations of mutations would be required. DFtet was designed with four disconnected helices that could be separately prepared then combinatorially assembled. To this end, A2B2[186] and AA’B2[187] heterotetrameric complexes were assembled and renamed DFtet. Design of these sequences implemented the use of negative protein design by destabilizing both homooligomeric folds and undesired heterotetrameric topologies. DFtet-A and DFtet-B only fold and assemble with the desired stoichiometry when mixed in a 1:1 ratio. A substrate pocket was designed into the interior of this symmetrical DFtet that lead to the development of an O2-dependent phenol oxidase [4]. Specifically, G4-DFtet, which had Gly residues substituted for Leu15 and Ala19 in both A chains and as a result, the least steric restrictions of all the variants, showed rapid formation of the oxo-species with no detectable intermediates. G4-DFtet could perform the two-electron oxidation of 4-aminophenol (4AP) to benzoquinone monoamine with a 1000-fold rate enhancement over the background reaction and at least 100 turnovers (Table 5). O2 oxidizes the diferrous form of the protein and the diferric form then reacts with 4AP. The resulting diferrous form is oxidized again to complete the catalytic cycle. The rate was decreased 2.5- and 5-fold when either Gly residue at positions 19 or 15 was changed to Ala. The catalytic efficiency is therefore sensitive to changes the size of a methyl group, and demonstrates the correlation between catalytic activity and size of the active site cavity.
Table 5.
Comparisons of the kinetics of 4-aminophenol by designed diiron proteins.
| Catalyst | kcat (s−1) | KM (mM) | kcat/KM (M−1 s−1) |
|---|---|---|---|
| di-FeIII-DF3a | 0.045 ± 0.003 | 1.97 ± 0.27 | 23.0 |
| G4DFtet | 0.022 ± 0.002 | 0.83 ± 0.06 | 25.7 |
. Taken from reference [81], measured at pH 7.0 and 25° C. The protein can perform 50 turnovers over the course of one hour.
. Taken from reference [4], pH 7.0 and 25° C. The protein performed >100 turnovers. ~1,000-fold rate enhancement observed over the background reaction (from comparing initial rate in presence and absence of protein).
Despite this success with DFtet, its borderline stability, complex stoichiometry, and tendency to undergo ligand-exchange reactions limited full characterization. Unfortunately, DF1 is not stable enough to handle the analogous Leu15 and Ala19 mutations in DFtet (Leu9 and Leu13 to Gly) to accommodate the phenol substrate. The interhelical turn sequence was therefore modified to create more electrostatic interactions leading to DF3 [81]. The solution structure of di-ZnII-DF3 demonstrates that the structure remains nearly identical to the design, with the exception of a larger access channel (Figure 21). This site could also bind iron, undergo reversible oxidation-reduction cycles, and catalyze the oxidation of 4AP for at least 50 turnovers. The protein follows Michaelis-Menten kinetics in the presence of O2, with an efficiency of 23 M−1 s−1, KM = 1.97 ± 0.27 mM and kcat 0.045 s−1 (Table 5). The designed metalloprotein could also catalyze the oxidation of other related substrates, such as 3,5-di-tert-butylcatechol (3,5-DTBC) (with a five-fold higher kcat than for 4AP) and p-phenylene diamine (expected to bind more weakly, and had a 75-fold lower kcat/KM that 4AP). No activity was observed for o-phenylenediamine. These results are all consistent with the anticipated catalytic properties of di-FeIII-DF3. Analysis of the crystal structure of di-ZnII-DF3 revealed that the L9G and L13G mutations created an active site cleft which, at its narrowest, was the width of a phenyl ring. Substitutions at the 3 and 5 positions could be accommodated in wider regions of the cavity. Thermodynamic analysis of a series of DF1 mutants versus DF3 revealed that the required functional mutations were relatively destabilizing, although the redesigned turn sequence helped to compensate for this. These requirements for activity, which lead to dramatic destabilization of the protein structure, demonstrate the tradeoff between conformational stability and catalytic function, something that is particularly apparent in metalloproteins because of the additional requirement for balance between often opposing metal binding requirements and functional requirements.
Figure 21.
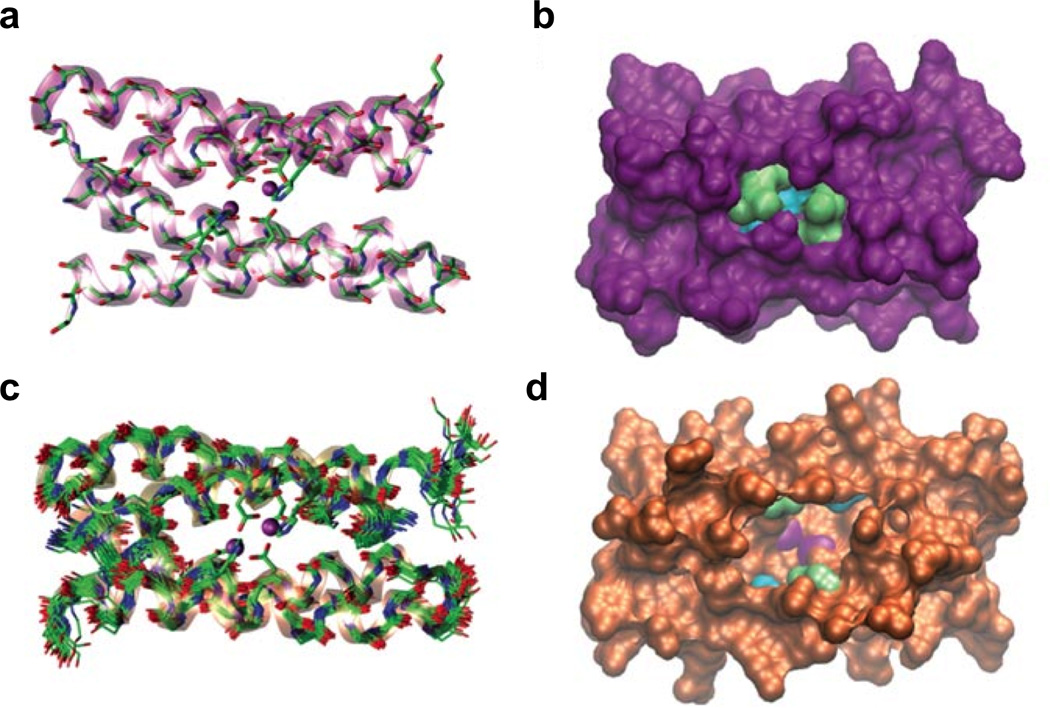
Comparison of DF1 and DF3 structures. (a) Crystal structure of di-ZnII-DF1 (PDB 1EC5). (b) Surface representation of the crystal structure of di-ZnII-DF1 to display the accessibility to the dimetal site. (c) NMR structure of di-ZnII-DF3 (PDB 2KIK). (d) Surface representation of the NMR structure of di-ZnII-DF3 to show the increased accessibility to the active site over DF1. The different residues in position 9 (lime) and 13 (cyan) are highlighted. Metal ions are colored magenta. Figure was reprinted by permission from Macmillan Publishers Ltd: Nature Chemical Biology [81], copyright 2009.
The success of these designs suggested it might be feasible not only to transfer an active site from a native enzyme onto a minimal de novo designed model, but also to alter its reactivity through a redesign approach. The target diiron enzyme for this reprogramming is AurF (a p-aminobenzoate N-oxygenase), which performs the N-oxygenation of anilines, and is the only structurally characterized N-oxygenase with a diiron center. In order to reproduce the structure of this enzyme, a third His residue needed to be incorporated into the active site. Since this would be an asymmetric substitution (single mutation), the single chain monomeric DFsc protein was used (designed by specifying 26 residues and allowing the remaining 88 to be modeled computationally) [188]. The NMR structure of di-ZnII-DFsc confirms that the fold of the protein is retained [189]. Analysis of DFsc in the context of ferroxidase activity revealed that while it demonstrated rapid formation of a diferric site in the presence of oxygen, it was not an oxygen intermediate as with the dimeric and tetrameric DF proteins but an off-pathway iron-tyrosinate complex [190,191]. Expanding the substrate access channel by preparing a quadruple Ala-Gly mutant (G4-DFsc; positions analogous to those described for DF3 and DFtet) lead to a minimization of formation of this complex and rapid oxidation to form a μ-oxo-bridged diferric cluster [80]. G4DFsc also catalyzes 4AP oxidation, like other members of the DF family with open active site access channels, and with a rate on the same order of magnitude as DFsc. Subsequently, His was substituted for Ile in position 100, structurally in the same position as in AurF. To alleviate the resulting steric clashes and balance out surrounding electrostatic interactions, three more substitutions were made adjacent to the His residue to form a polar network (Figure 22). Fortunately, 3His-G4DFsc is well-folded in the presence of divalent metal ions but, unlike G4DFsc, does not fold in their absence, as might be expected when incorporating a number of helix-destabilizing substitutions in addition to introduction of three polar substitutions within the hydrophobic, solvent-inaccessible core. Regardless, this new construct has 4AP activity reduced to near background levels, but can now catalyse the N-hydroxylation of arylamines such as p-anisidine (no N-hydroxylation reactivity is observed for G4DFsc) and other related substrates (Figure 23). This shift in reactivity is dramatic, especially considering that just four residues were mutated, and only one directly to the metal binding site. Although difficult to discern in complex native diiron proteins, this study on small de novo models suggests that it is the single His residue that directly contributes to the reactivity change. More broadly, this study is an elegant demonstration of how active site structure can be directly correlated with function, in the absence of the bulk of the surrounding protein matrix found in native metalloproteins. The loss in stability of the apo construct is unfortunate, however, the presence of divalent metals enables characterization and reactivity studies. This decrease in stability with the introduction of function is another demonstration of the tradeoff between function and stability. The fact that metal ions stabilize the fold illustrates this interplay and how de novo design can be used to better understand the relationships between metal binding, protein folding, and function.
Figure 22.
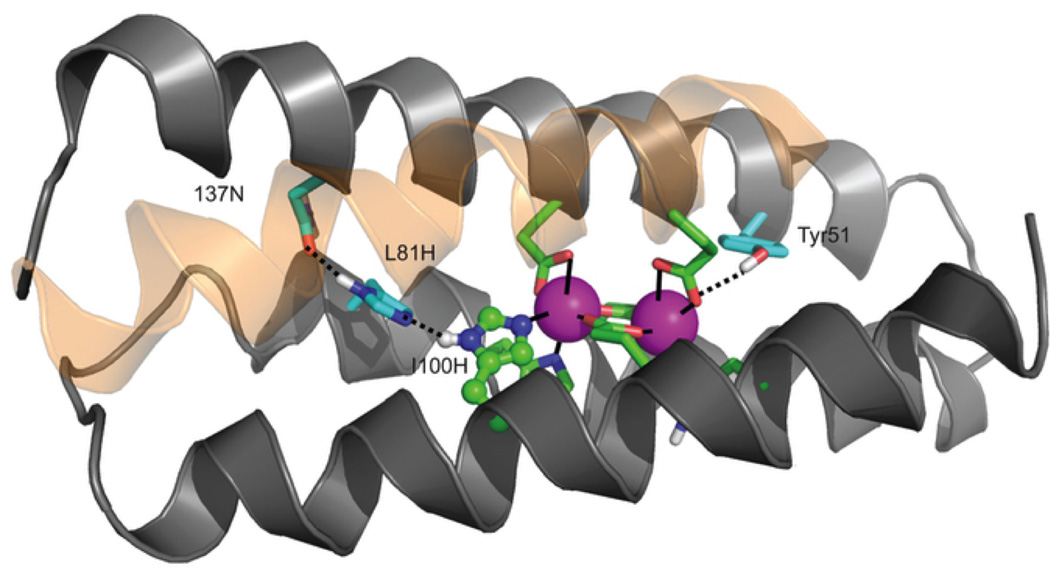
Ribbon diagram showing the structure of the 3His-G2DFsc variant (PDB 2LFD) with the added metal-binding His residue (H100) and supporting mutations (I37N, L81H). Although kinetic data is reported for the 3His-G4DFsc variant, the structure shown here contains only two Gly mutations because of increased stability during data collection (this variant also showed N-oxygenase activity, albeit slower than the G4-variant). Figure was reprinted by permission from Macmillan Publishers Ltd: Nature Chemistry [80], copyright 2012.
Figure 23.
Proposed reaction scheme for N-hydroxylation of p-anisidine. Figure was reprinted by permission from Macmillan Publishers Ltd: Nature Chemistry [80], copyright 2012.
Section 4.4. Dirhodium peptide catalysts for structure-selective side-chain modification
In the past few years, the Ball group has focused on the preparation of dirhodium-induced α-helices that form heterodimeric 2SCCs for a number of applications of interest to enzymatic function. Particularly attractive about their strategy is that they have incorporated organometallic fragments in order to prepare bioinspired catalysts not remotely similar to what is found in nature. This group showed that they could prepare peptide complexes with dirhodium that would be stabilized or induced to form α-helices upon binding to two carboxylate groups separated by four residues and in some cases, three [69,108]. Although other peptides have been examined (and sequences as short as 9 residues could be stabilized by dirhodium binding to a i, i + 4 carboxylate motif), much of the work has focused on the E3/K3 heterodimeric 2SCC originally reported by Hodges [192]. The E3/K3 coil has complementary charges at the positions flanking the helical interfaces, e and g, to direct heterodimerization. By incorporating a dirhodium complex into the K3 coil at an i, i + 7 Glu motif, coiled coil formation could be inhibited, demonstrating the importance of the spacing of the Glu residues. K3a,eRh2, in which Glu residues are present at i, i + 4 positions, can be prepared and forms a heterodimeric coiled coil with the E3 coil. By incorporating a reactive Trp residue at the complementary g position of E3 (E3gW) and combining this substrate with the dirhodium metallopeptide (catalyst) and a diazo reagent, a 103 rate enhancement for Trp modification over free Rh2(OAc)4 reagent was observed (Figure 24) [193,194]. Further controls demonstrate that this is a site-selective molecular recognition event. Specifically, modification only occurs when the target Trp residue is placed in a complementary position of the coiled coil. When Trp is placed in a random position of the complementary strand, reactivity shows no improvement over the control reaction with free Rh2(OAc)4. Further, attempts to modify E3eW or K3gW with K3a,eRH2 were not successful. This work has since been extended to include site-specific modification of a number of other amino acids (even in the presence of the more reactive Trp), some of which do not have any previous methods reported for their modification, demonstrating that molecular recognition through coiled coil formation can override the inherent chemical reactivity of the substrate [194,195]. Further, this work has been extended to demonstrate modification of larger natural targets in complex systems [196,197]. The Ball group has also shown that reactivity can be specifically inhibited through binding a His-containing substrate peptide to the metallopeptide catalyst [198]. Both of these mechanisms are important to the development of catalysts with enzyme-like characteristics including selectivity and sequence-specific inhibition. Overall, this work demonstrates the utility of designed peptide systems for performing organometallic chemistry in aqueous solutions. Related work on incorporating organometallic complexes into native proteins for this type of chemistry has also been reported [25–27], but this is the only example which exploits the known interactions in a de novo coiled coil system for site-specific organometallic chemistry.
Figure 24.
Representation of structure-selective peptide modification by a ruthenium-bound peptide catalyst. 1 is the diazo reagent. X is the amino acid to be modified (Trp is discussed in the text). Figure was reproduced from ref. [195] with permission of The Royal Society of Chemistry.
Conclusions
Recently, de novo metalloprotein design has proven successful in generating unique metalloenzyme models, with increasingly impressive catalytic efficiencies in biologically relevant environments. The role of the protein scaffold in tuning catalytic efficiencies is becoming more and more apparent as many of these designed proteins, even the most minimal, are “easily” more efficient than their synthetic model complex counterparts and avoid many of the drawbacks to small molecule model complex chemistry. Combining these advances with those obtained by taking a redesign approach, it is readily apparent how these strategies can complement each other ultimately to delineate a detailed relationship between specific structures and functions on a molecular level. Interestingly, the DeGrado group has shown that a very minimal approach with mutations of very few residues could be taken to confer an entirely new catalytic function. We have shown that simply exchanging the metal ion in the same peptide construct can convert a 3SCC from an efficient hydrolytic enzyme to a stable redox-active metalloenzyme. Further work of this nature should aid in understanding the roles of specific residues and microenvironment structures on function. It also may allow one to address how strictly necessary much of the complexity around many native proteins really is or if it is simply a result of evolution conservatively reusing well established scaffolds. Of course, functional designed metalloenzymes have only begun to come close to fruition and have not yet matched the efficiency of their native counterparts, but it is now more realistic than ever to assume these objectives can be realized. There are a number of future challenges to be met including the development of multiple metal sites with differing functions (e.g., electron transfer with a second catalytic site), substrate selective sites, well defined proton, water or substrate channels, controlled pKa and orientation of acid base catalysts and metal-binding selectivity.
Research highlights.
De novo metalloprotein design
Structural metal sites: Hg(II) binding to α-helical coiled coils
Hydrolytic catalysis by a de novo designed Zn(II) metalloprotein
Redox catalysis by a de novo designed Cu(I/II) metalloprotein
De novo designed binuclear metal sites
Acknowledgements
We thank the following Pecoraro group members, past and present, for their contributions to the work discussed in this review: Dr. Gregg R. Dieckmann, Dr. Debdip Ghosh, Dr. Brian T. Farrer, Dr. Manolis Matzapetakis, Dr. Olga Iranzo, Dr. Anna F. A. Peacock, Dr. Saumen Chakraborty, Dr. Matteo Tegoni, Fangting Yu. We thank our collaborators for their contributions and support of our work: Dr. Lars Hemmingsen, Dr. Jeanne A. Stuckey, Dr. James E. Penner-Hahn, Dr. William F. DeGrado. Finally, we acknowledge the National Institutes of Health (grant no. R01 ES0 12236), National Institutes of Health Chemistry-Biology Interface Training Program, and The University of Michigan Rackham Graduate School for financial support.
Abbreviations
- XSCC
x-stranded coiled coil where x = 2, 3, or 4
- CS
CoilSer
- DF
Due-Ferri
- Pal
pyridylalanine
- CD
circular dichroism
- Pen
Penicillamine
- PAC
perturbed angular correlation spectroscopy
- XAS
X-ray absorption spectroscopy
- GuHCl
guanidine hydrochloride
- StepAD
stepwise aggregation-deprotonation
- CO2
carbon dioxide
- MerR
mercury resistance regulator
- ASU
asymmetric unit
- CA
carbonic anhydrase
- pNPA
p-nitrophenyl acetate
- DECP
diethyl 7-hydroxy coumarinyl
- p-NPP
p-nitrophenyl phosphate
- NiR
nitrite reductase
- T2
Type 2
- XANES
X-ray absorption near-edge spectroscopy
- EXAFS
extended X-ray absorption fine structure
- EPR
electron paramagnetic resonance spectroscopy
- 4AP
4-aminophenol
- 3,5-DTBC
3,5-di-tert-butylcatechol
- AurF
p-aminobenzoate N-oxygenase.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Holm RH, Kennepohl P, Solomon EI. Chem. Rev. 1996;96:2239. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 2.Ragsdale SW. Chem. Rev. 2006;106:3317. doi: 10.1021/cr0503153. [DOI] [PubMed] [Google Scholar]
- 3.Dieckmann GR, McRorie DK, Lear JD, Sharp KA, DeGrado WF, Pecoraro VL. J. Mol. Biol. 1998;280:897. doi: 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan J, DeGrado WF. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11566. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeGrado WF, Raleigh DP, Handel T. Curr. Opin. Struct. Biol. 1991;1:984. [Google Scholar]
- 6.Nanda V, Koder RL. Nat. Chem. 2010;2:15. doi: 10.1038/nchem.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong J, Kharenko OA, Ogawa MY. Inorg. Chem. 2006;45:9974. doi: 10.1021/ic060222j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barker PD. Curr. Opin. Struct. Biol. 2003;13:490. doi: 10.1016/s0959-440x(03)00108-8. [DOI] [PubMed] [Google Scholar]
- 9.Lu Y, Yeung N, Sieracki N, Marshall NM. Nature. 2009;460:855. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yeung N, Lin Y-W, Gao Y-G, Zhao X, Russell BS, Lei L, Miner KD, Robinson H, Lu Y. Nature. 2009;462:1079. doi: 10.1038/nature08620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lombardi A, Summa CM, Geremia S, Randaccio L, Pavone V, DeGrado WF. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6298. doi: 10.1073/pnas.97.12.6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu Y. Inorg. Chem. 2006;45:9930. doi: 10.1021/ic052007t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Y, Berry SM, Pfister TD. Chem. Rev. 2001;101:3047. doi: 10.1021/cr0000574. [DOI] [PubMed] [Google Scholar]
- 14.Miner KD, Mukherjee A, Gao Y-G, Null EL, Petrik ID, Zhao X, Yeung N, Robinson H, Lu Y. Angew. Chem., Int. Ed. 2012;51:5589. doi: 10.1002/anie.201201981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, Yu Y, Hu C, Zhang W, Lu Y, Wang J. Angew. Chem., Int. Ed. 2012;51:4312. doi: 10.1002/anie.201108756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Der BS, Edwards DR, Kuhlman B. Biochemistry. 2012;51:3933. doi: 10.1021/bi201881p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khare SD, Kipnis Y, Greisen PJ, Takeuchi R, Ashani Y, Goldsmith M, Song Y, Gallaher JL, Silman I, Leader H, Sussman JL, Stoddard BL, Tawfik DS, Baker D. Nat. Chem. Biol. 2012;8:294. doi: 10.1038/nchembio.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong-Deyrup SW, Prasannan C, Dupureur CM, Franklin SJ. J. Biol. Inorg. Chem. 2012;17:387. doi: 10.1007/s00775-011-0861-0. [DOI] [PubMed] [Google Scholar]
- 19.Garner DK, Liang L, Barrios DA, Zhang J, Lu Y. ACS Catal. 2011;1:1083. doi: 10.1021/cs200258e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayer C, Gillingham DG, Ward TR, Hilvert D. Chem. Commun. (Cambridge, U. K.) 2011;47:12068. doi: 10.1039/c1cc15005g. [DOI] [PubMed] [Google Scholar]
- 21.Allard M, Dupont C, Muñoz Robles V, Doucet N, Lledós A, Maréchal J-D, Urvoas A, Mahy J-P, Ricoux R. Chem Bio Chem. 2012;13:240. doi: 10.1002/cbic.201100659. [DOI] [PubMed] [Google Scholar]
- 22.Bos J, Fusetti F, Driessen AJM, Roelfes G. Angew. Chem., Int. Ed. 2012;51:7472. doi: 10.1002/anie.201202070. [DOI] [PubMed] [Google Scholar]
- 23.Matsuo T, Imai C, Yoshida T, Saito T, Hayashi T, Hirota S. Chem. Commun. (Cambridge, U. K.) 2012;48:1662. doi: 10.1039/c2cc16898g. [DOI] [PubMed] [Google Scholar]
- 24.Harris KL, Lim S, Franklin SJ. Inorg. Chem. 2006;45:10002. doi: 10.1021/ic060877k. [DOI] [PubMed] [Google Scholar]
- 25.Ringenberg MR, Ward TR. Chem. Commun. (Cambridge, U. K.) 2011;47:8470. doi: 10.1039/c1cc11592h. [DOI] [PubMed] [Google Scholar]
- 26.Steinreiber J, Ward TR. Coord. Chem. Rev. 2008;252:751. [Google Scholar]
- 27.Ward TR. Acc. Chem. Res. 2011;44:47. doi: 10.1021/ar100099u. [DOI] [PubMed] [Google Scholar]
- 28.Hellinga HW. Fold Des. 1998;3:R1. doi: 10.1016/S1359-0278(98)00001-7. [DOI] [PubMed] [Google Scholar]
- 29.Benson DE, Wisz MS, Hellinga HW. Proc. Natl. Acad. Sci. U.S.A. 2000;97:6292. doi: 10.1073/pnas.97.12.6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. Annu. Rev. Biochem. 1999;68:779. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 31.DeGrado W, Wasserman Z. Science. 1989;243:622. doi: 10.1126/science.2464850. [DOI] [PubMed] [Google Scholar]
- 32.Bryson JW, Betz SF, Lu HS, Suich DJ, Zhou HX, O’Neil KT, DeGrado WF. Science. 1995;270:935. doi: 10.1126/science.270.5238.935. [DOI] [PubMed] [Google Scholar]
- 33.Park H-S, Nam S, Lee JK, Yoon CN, Mannervik B, Benkovic SJ, Kim H-S. Science. 2006;311:535. doi: 10.1126/science.1118953. [DOI] [PubMed] [Google Scholar]
- 34.Clark KM, Yu Y, Marshall NM, Sieracki Na, Nilges MJ, Blackburn NJ, van der Donk WA, Lu Y. J. Am. Chem. Soc. 2010;132:10093. doi: 10.1021/ja102632p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujieda N, Hasegawa A, Ishihama K-I, Itoh S. Chem.--Asian J. 2012;7:1203. doi: 10.1002/asia.201101014. [DOI] [PubMed] [Google Scholar]
- 36.Salgado EN, Radford RJ, Tezcan FA. Acc. Chem. Res. 2010;43:661. doi: 10.1021/ar900273t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radford RJ, Brodin JD, Salgado EN, Tezcan FA. Coord. Chem. Rev. 2011;255:790. [Google Scholar]
- 38.Brodin JD, Ambroggio XI, Tang C, Parent KN, Baker TS, Tezcan FA. Nat. Chem. 2012;4:375. doi: 10.1038/nchem.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Der BS, Machius M, Miley MJ, Mills JL, Szyperski T, Kuhlman B. J. Am. Chem. Soc. 2012;134:375. doi: 10.1021/ja208015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson JLR, Koder RL, Moser CC, Dutton PL. Biochem. Soc. Trans. 2008;36:1106. doi: 10.1042/BST0361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hodges RS. Biochem. Cell Biol. 1996;74:133. doi: 10.1139/o96-015. [DOI] [PubMed] [Google Scholar]
- 42.Apostolovic B, Danial M, Klok H-A. Chem. Soc. Rev. 2010;39:3541. doi: 10.1039/b914339b. [DOI] [PubMed] [Google Scholar]
- 43.Woolfson DN. Adv. Protein Chem. 2005;70:79. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- 44.Hodges RS, Saund AK, Chong PC, St-Pierre SA, Reid RE. J. Biol. Chem. 1981;256:1214. [PubMed] [Google Scholar]
- 45.Crick FHC. Acta Crystallogr. 1953;6:689. [Google Scholar]
- 46.DeGrado WF, Lear JD. J. Am. Chem. Soc. 1985;107:7684. [Google Scholar]
- 47.Kamtekar S, Schiffer J, Xiong H, Babik J, Hecht M. Science. 1993;262:1680. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 48.Lau SY, Taneja AK, Hodges RS. J. Biol. Chem. 1984;259:13253. [PubMed] [Google Scholar]
- 49.Handel TT, Williams S, DeGrado W. Science. 1993;261:879. doi: 10.1126/science.8346440. [DOI] [PubMed] [Google Scholar]
- 50.Lovejoy B, Choe S, Cascio D, McRorie D, DeGrado W, Eisenberg D. Science. 1993;259:1288. doi: 10.1126/science.8446897. [DOI] [PubMed] [Google Scholar]
- 51.Ogihara NL, Weiss MS, Degrado WF, Eisenberg D. Protein Sci. 1997;6:80. doi: 10.1002/pro.5560060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Betz S, Fairman R, O’Neil K, Lear J, Degrado W. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 1995;348:81. doi: 10.1098/rstb.1995.0048. [DOI] [PubMed] [Google Scholar]
- 53.Ghosh D, Pecoraro VL. Inorg. Chem. 2004;43:7902. doi: 10.1021/ic048939z. [DOI] [PubMed] [Google Scholar]
- 54.Kharenko OA, Ogawa MY. J. Inorg. Biochem. 2004;98:1971. doi: 10.1016/j.jinorgbio.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 55.Shiga D, Nakane D, Inomata T, Funahashi Y, Masuda H, Kikuchi A, Oda M, Noda M, Uchiyama S, Fukui K, Kanaori K, Tajima K, Takano Y, Nakamura H, Tanaka T. J. Am. Chem. Soc. 2010;132:18191. doi: 10.1021/ja106263y. [DOI] [PubMed] [Google Scholar]
- 56.Shiga D, Nakane D, Inomata T, Masuda H, Oda M, Noda M, Uchiyama S, Fukui K, Takano Y, Nakamura H, Mizuno T, Tanaka T. Biopolymers. 2009;91:907. doi: 10.1002/bip.21277. [DOI] [PubMed] [Google Scholar]
- 57.Shiga D, Funahashi Y, Masuda H, Kikuchi A, Noda M, Uchiyama S, Fukui K, Kanaori K, Tajima K, Takano Y, Nakamura H, Kamei M, Tanaka T. Biochemistry. 2012 doi: 10.1021/bi3007884. [DOI] [PubMed] [Google Scholar]
- 58.Xie F, Sutherland DEK, Stillman MJ, Ogawa MY. J. Inorg. Biochem. 2010;104:261. doi: 10.1016/j.jinorgbio.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 59.Peacock AFA, Iranzo O, Pecoraro VL. Dalton Trans. 2009;9226:2271. doi: 10.1039/b818306f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iranzo O, Chakraborty S, Hemmingsen L, Pecoraro VL. J. Am. Chem. Soc. 2011;133:239. doi: 10.1021/ja104433n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Matzapetakis M, Ghosh D, Weng T-C, Penner-Hahn JE, Pecoraro VL. J. Biol. Inorg. Chem. 2006;11:876. doi: 10.1007/s00775-006-0140-7. [DOI] [PubMed] [Google Scholar]
- 62.Neupane KP, Pecoraro VL. Angew. Chem., Int. Ed. 2010;49:8177. doi: 10.1002/anie.201004429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pecoraro VL, Peacock AFA, Iranzo O, Luczkowski M. ACS Symp. Ser. 2009;1012:183. [Google Scholar]
- 64.Ghosh D, Pecoraro VL. Curr. Opin. Chem. Biol. 2005;9:97. doi: 10.1016/j.cbpa.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 65.Iranzo O, Ghosh D, Pecoraro VL. Inorg. Chem. 2006;45:9959. doi: 10.1021/ic061183e. [DOI] [PubMed] [Google Scholar]
- 66.Iranzo O, Jakusch T, Lee K-H, Hemmingsen L, Pecoraro VL. Chem.--Eur. J. 2009;15:3761. doi: 10.1002/chem.200802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chakraborty S, Touw DS, Peacock AFA, Stuckey J, Pecoraro VL. J. Am. Chem. Soc. 2010;132:13240. doi: 10.1021/ja101812c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Touw DS, Nordman CE, Stuckey JA, Pecoraro VL. Proc. Natl. Acad. Sci. U.S.A. 2007;104:11969. doi: 10.1073/pnas.0701979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zaykov AN, MacKenzie KR, Ball ZT. Chem.--Eur. J. 2009;15:8961. doi: 10.1002/chem.200901266. [DOI] [PubMed] [Google Scholar]
- 70.Zastrow ML, Peacock AFA, Stuckey JA, Pecoraro VL. Nat. Chem. 2012;4:118. doi: 10.1038/nchem.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dieckmann GR, McRorie DK, Tierney DL, Utschig LM, Singer CP, O’Halloran TV, Penner-Hahn JE, DeGrado WF, Pecoraro VL. J. Am. Chem. Soc. 1997;119:6195. [Google Scholar]
- 72.Bryson JW, Desjarlais JR, Handel TM, DeGrado WF. Protein Sci. 1998;7:1404. doi: 10.1002/pro.5560070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walsh STR, Cheng H, Bryson JW, Roder H, DeGrado WF. Proc. Natl. Acad. Sci. U.S.A. 1999;96:5486. doi: 10.1073/pnas.96.10.5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Regan L, DeGrado WF. Science. 1988;241:976. doi: 10.1126/science.3043666. [DOI] [PubMed] [Google Scholar]
- 75.Handel T, DeGrado WF. J. Am. Chem. Soc. 1990;112:6710. [Google Scholar]
- 76.Hill RB, Raleigh DP, Lombardi A, DeGrado WF. Acc. Chem. Res. 2000;33:745. doi: 10.1021/ar970004h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schnepf R, Hörth P, Bill E, Wieghardt K, Hildebrandt P, Haehnel W. J. Am. Chem. Soc. 2001;123:2186. doi: 10.1021/ja001880k. [DOI] [PubMed] [Google Scholar]
- 78.Chakraborty S, Kravitz JY, Thulstrup PW, Hemmingsen L, DeGrado WF, Pecoraro VL. Angew. Chem., Int. Ed. 2011;50:2049. doi: 10.1002/anie.201006413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Regan L, Clarke ND. Biochemistry. 1990;29:10878. doi: 10.1021/bi00501a003. [DOI] [PubMed] [Google Scholar]
- 80.Reig AJ, Pires MM, Snyder RA, Wu Y, Jo H, Kulp DW, Butch SE, Calhoun JR, Szyperski T, Szyperski TG, Solomon EI, DeGrado WF. Nat. Chem. 2012;4:900. doi: 10.1038/nchem.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Faiella M, Andreozzi C, de Rosales RTM, Pavone V, Maglio O, Nastri F, DeGrado WF, Lombardi A. Nat. Chem. Biol. 2009;5:882. doi: 10.1038/nchembio.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maglio O, Nastri F, Martin de Rosales RT, Faiella M, Pavone V, DeGrado WF, Lombardi A, Chim CR. 2007;10:703. [Google Scholar]
- 83.Calhoun JR, Nastri F, Maglio O, Pavone V, Lombardi A, DeGrado WF. Biopolymers. 2005;80:264. doi: 10.1002/bip.20230. [DOI] [PubMed] [Google Scholar]
- 84.Cheng RP, Gellman SH, DeGrado WF. Chem. Rev. 2001;101:3219. doi: 10.1021/cr000045i. [DOI] [PubMed] [Google Scholar]
- 85.Lacroix E, Kortemme T. Curr. Opin. Struct. Biol. 1999;9:487. doi: 10.1016/s0959-440x(99)80069-4. [DOI] [PubMed] [Google Scholar]
- 86.Schneider JP, Kelly JW. J. Am. Chem. Soc. 1995;117:2533. [Google Scholar]
- 87.Platt G, Searle MS, Chung C-W. Chem. Commun. (Cambridge, U. K.) 2001:1162. [Google Scholar]
- 88.Venkatraman J, Naganagowda GA, Sudha R, Balaram P. Chem. Commun. (Cambridge, U. K.) 2001:2660. [Google Scholar]
- 89.Pessi A, Bianchi E, Crameri A, Venturini S, Tramontano A, Sollazzo M. Nature. 1993;362:367. doi: 10.1038/362367a0. [DOI] [PubMed] [Google Scholar]
- 90.Zhu C, Zhang C, Liang H, Lai L. Protein Cell. 2011;2:1006. doi: 10.1007/s13238-011-1121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Science. 2003;302:1364. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 92.Struthers MD, Cheng RP, Imperiali B. J. Am. Chem. Soc. 1996;118:3073. [Google Scholar]
- 93.Struthers MD, Cheng RP, Imperiali B. Science. 1996;271:342. doi: 10.1126/science.271.5247.342. [DOI] [PubMed] [Google Scholar]
- 94.Bonomo RP, Casella L, De Gioia L, Molinari H, Impellizzeri G, Jordan T, Pappalardo G, Purrello R, Rizzarelli E. Dalton Trans. 1997:2387. [Google Scholar]
- 95.Butterfield SM, Cooper WJ, Waters ML. J. Am. Chem. Soc. 2005;127:24. doi: 10.1021/ja045002o. [DOI] [PubMed] [Google Scholar]
- 96.Butterfield SM, Goodman CM, Rotello VM, Waters ML. Angew. Chem., Int. Ed. 2004;43:724. doi: 10.1002/anie.200352527. [DOI] [PubMed] [Google Scholar]
- 97.Hughes RM, Waters ML. Curr. Opin. Struct. Biol. 2006;16:514. doi: 10.1016/j.sbi.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 98.Cheng RP, Fisher SL, Imperiali B. J. Am. Chem. Soc. 1996;118:11349. [Google Scholar]
- 99.Daugherty RG, Wasowicz T, Gibney BR, DeRose VJ. Inorg. Chem. 2002;41:2623. doi: 10.1021/ic010555a. [DOI] [PubMed] [Google Scholar]
- 100.Kozísek M, Svatos A, Budesínský M, Muck A, Bauer MC, Kotrba P, Ruml T, Havlas Z, Linse S, Rulísek L. Chem.--Eur. J. 2008;14:7836. doi: 10.1002/chem.200800178. [DOI] [PubMed] [Google Scholar]
- 101.Nanda V, Rosenblatt MM, Osyczka A, Kono H, Getahun Z, Dutton PL, Saven JG, Degrado WF. J. Am. Chem. Soc. 2005;127:5804. doi: 10.1021/ja050553f. [DOI] [PubMed] [Google Scholar]
- 102.Turano P. Inorg. Chem. 2004;43:7945. doi: 10.1021/ic048962k. [DOI] [PubMed] [Google Scholar]
- 103.Maret W, Li Y. Chem. Rev. 2009;109:4682. doi: 10.1021/cr800556u. [DOI] [PubMed] [Google Scholar]
- 104.Finn BE, Drakenberg T. Adv. Inorg. Chem. 1999;46:441. [Google Scholar]
- 105.Doerr AJ, McLendon GL. Inorg. Chem. 2004;43:7916. doi: 10.1021/ic0490573. [DOI] [PubMed] [Google Scholar]
- 106.Ghadiri MR, Soares C, Choi C. J. Am. Chem. Soc. 1992;114:825. [Google Scholar]
- 107.Tsurkan MV, Ogawa MY. Inorg. Chem. 2007;46:6849. doi: 10.1021/ic700958h. [DOI] [PubMed] [Google Scholar]
- 108.Zaykov AN, V Popp B, Ball ZT. Chem.--Eur. J. 2010;16:6651. doi: 10.1002/chem.200903092. [DOI] [PubMed] [Google Scholar]
- 109.Suzuki K, Hiroaki H, Kohda D, Nakamura H, Tanaka T. J. Am. Chem. Soc. 1998;120:13008. [Google Scholar]
- 110.Li X, Suzuki K, Kanaori K, Tajima K, Kashiwada A, Hiroaki H, Kohda D, Tanaka T. Protein Sci. 2000;9:1327. doi: 10.1110/ps.9.7.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ghadiri MR, Choi C. J. Am. Chem. Soc. 1990;112:1630. [Google Scholar]
- 112.Ghadiri MR, Fernholz AK. J. Am. Chem. Soc. 1990;112:9633. [Google Scholar]
- 113.Ghadiri MR, Case MA. Angew. Chem., Int. Ed. 1993;32:1594. [Google Scholar]
- 114.Ghadiri MR, Soares C, Choi C. J. Am. Chem. Soc. 1992;114:4000. [Google Scholar]
- 115.Kharenko OA, Kennedy DC, Demeler B, Maroney MJ, Ogawa MY. J. Am. Chem. Soc. 2005;127:7678. doi: 10.1021/ja042757m. [DOI] [PubMed] [Google Scholar]
- 116.Kiyokawa T, Kanaori K, Tajima K, Koike M, Mizuno T, Oku J-I, Tanaka T. J. Pept. Res. 2004;63:347. doi: 10.1111/j.1399-3011.2004.00109.x. [DOI] [PubMed] [Google Scholar]
- 117.Murase S, Ishino S, Ishino Y, Tanaka T. J. Biol. Inorg. Chem. 2012;17:791. doi: 10.1007/s00775-012-0896-x. [DOI] [PubMed] [Google Scholar]
- 118.O’Neil K, DeGrado W. Science. 1990;250:646. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 119.Iranzo O, Cabello C, Pecoraro VL. Angew. Chem., Int. Ed. 2007;46:6688. doi: 10.1002/anie.200701729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee K-H, Cabello C, Hemmingsen L, Marsh ENG, Pecoraro VL. Angew. Chem., Int. Ed. 2006;45:2864. doi: 10.1002/anie.200504548. [DOI] [PubMed] [Google Scholar]
- 121.Peacock AFA, Hemmingsen L, Pecoraro VL. Proc. Natl. Acad. Sci. U.S.A. 2008;105:16566. doi: 10.1073/pnas.0806792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Peacock AFA, Stuckey JA, Pecoraro VL. Angew. Chem., Int. Ed. 2009;48:7371. doi: 10.1002/anie.200902166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Farrer BT, McClure CP, Penner-Hahn JE, Pecoraro VL. Inorg. Chem. 2000;39:5422. doi: 10.1021/ic0010149. [DOI] [PubMed] [Google Scholar]
- 124.Farrer BT, Harris NP, Balchus KE, Pecoraro VL. Biochemistry. 2001;40:14696. doi: 10.1021/bi015649a. [DOI] [PubMed] [Google Scholar]
- 125.Farrer BT, Pecoraro VL. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3760. doi: 10.1073/pnas.0336055100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Matzapetakis M, Farrer BT, Weng T-C, Hemmingsen L, Penner-Hahn JE, Pecoraro VL. J. Am. Chem. Soc. 2002;124:8042. doi: 10.1021/ja017520u. [DOI] [PubMed] [Google Scholar]
- 127.Matzapetakis M, Pecoraro VL. J. Am. Chem. Soc. 2005;127:18229. doi: 10.1021/ja055433m. [DOI] [PubMed] [Google Scholar]
- 128.Chakraborty S, Iranzo O, Zuiderweg ERP, Pecoraro VL. J. Am. Chem. Soc. 2012;134:6191. doi: 10.1021/ja210510g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vondrášek J. Journal of Inorganic Biochemistry. 1998;71:115. doi: 10.1016/s0162-0134(98)10042-9. [DOI] [PubMed] [Google Scholar]
- 130.Zheng H, Chruszcz M, Lasota P, Lebioda L, Minor W. J. Inorg. Biochem. 2008;102:1765. doi: 10.1016/j.jinorgbio.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wright JG, Tsang HT, Penner-Hahn JE, O’Halloran TV. J. Am. Chem. Soc. 1990;112:2434. [Google Scholar]
- 132.Faller P, Ctortecka B, Tröger W, Butz T, Vasák M. J. Biol. Inorg. Chem. 2000;5:393. doi: 10.1007/pl00010668. [DOI] [PubMed] [Google Scholar]
- 133.Łuczkowski M, Stachura M, Schirf V, Demeler B, Hemmingsen L, Pecoraro VL. Inorg. Chem. 2008;47:10875. doi: 10.1021/ic8009817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tanaka T, Mizuno T, Fukui S, Hiroaki H, Oku J-I, Kanaori K, Tajima K, Shirakawa M. J. Am. Chem. Soc. 2004;126:14023. doi: 10.1021/ja047945r. [DOI] [PubMed] [Google Scholar]
- 135.Christianson DW, Fierke CA. Acc. Chem. Res. 1996;29:331. [Google Scholar]
- 136.Gomis-Rüth FX, Kress LF, Bode W. EMBO J. 1993;12:4151. doi: 10.1002/j.1460-2075.1993.tb06099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kimura E, Shiota T, Koike T, Shiro M, Kodama M. J. Am. Chem. Soc. 1990;112:5805. [Google Scholar]
- 138.Koerner TB, Brown RS. Can. J. Chem. 2002;80:183. [Google Scholar]
- 139.Bazzicalupi C, Bencini A, Bianchi A, Fusi V, Giorgi C, Paoletti P, Valtancoli B, Zanchi D. Inorg. Chem. 1997;36:2784. doi: 10.1021/ic961521j. [DOI] [PubMed] [Google Scholar]
- 140.Verpoorte JA, Mehta S, Edsall JT. J. Biol. Chem. 1967;242:4221. [PubMed] [Google Scholar]
- 141.Innocenti A, Scozzafava A, Parkkila S, Puccetti L, De Simone G, Supuran CT. Bioorg. Med. Chem. Lett. 2008;18:2267. doi: 10.1016/j.bmcl.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 142.Dwyer MA, Looger LL, Hellinga HW. Science. 2004;304:1967. doi: 10.1126/science.1098432. [DOI] [PubMed] [Google Scholar]
- 143.Dwyer MA, Looger LL, Hellinga HW. Science. 2008;319:569. doi: 10.1126/science.319.5863.569b. [DOI] [PubMed] [Google Scholar]
- 144.Gomez-Tagle P, Vargas-Zúñiga I, Taran O, Yatsimirsky AK. J. Org. Chem. 2006;71:9713. doi: 10.1021/jo061780i. [DOI] [PubMed] [Google Scholar]
- 145.Woolley P. Nature. 1975;258:677. doi: 10.1038/258677a0. [DOI] [PubMed] [Google Scholar]
- 146.Huguet J, Brown RS. J. Am. Chem. Soc. 1980;102:7571. [Google Scholar]
- 147.Brown RS, Curtis NJ, Huguet J. J. Am. Chem. Soc. 1981;103:6953. [Google Scholar]
- 148.Brown RS, Salmon D, Curtis NJ, Kusuma S. J. Am. Chem. Soc. 1982;104:3188. [Google Scholar]
- 149.Slebocka-Tilk H, Cocho JL, Frackman Z, Brown RS. J. Am. Chem. Soc. 1984;106:2421. [Google Scholar]
- 150.Zhang X, van Eldik R, Koike T, Kimura E. Inorg. Chem. 1993;32:5749. [Google Scholar]
- 151.Zhang X, van Eldik R. Inorg. Chem. 1995;34:5606. [Google Scholar]
- 152.Nakata K, Shimomura N, Shiina N, Izumi M, Ichikawa K, Shiro M. J. Inorg. Biochem. 2002;89:255. doi: 10.1016/s0162-0134(01)00419-6. [DOI] [PubMed] [Google Scholar]
- 153.Liang Z, Xue Y, Behravan G, Jonsson BH, Lindskog S. Eur. J. Biochem. 1993;211:821. doi: 10.1111/j.1432-1033.1993.tb17614.x. [DOI] [PubMed] [Google Scholar]
- 154.Krebs JF, Ippolito JA, Christianson DW, Fierke CA. J. Biol. Chem. 1993;268:27458. [PubMed] [Google Scholar]
- 155.Averill BA. Chem. Rev. 1996;96:2951. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]
- 156.Suzuki S, Kataoka K, Yamaguchi K. Acc. Chem. Res. 2000;33:728. doi: 10.1021/ar9900257. [DOI] [PubMed] [Google Scholar]
- 157.Merkle AC, Lehnert N. Dalton Trans. 2012;41:3355. doi: 10.1039/c1dt11049g. [DOI] [PubMed] [Google Scholar]
- 158.Schnepf R, Haehnel W, Wieghardt K, Hildebrandt P. J. Am. Chem. Soc. 2004;126:14389. doi: 10.1021/ja0484294. [DOI] [PubMed] [Google Scholar]
- 159.Hong J, Kharenko OA, Fan J, Xie F, Petros AK, Gibney BR, Ogawa MY. Angew. Chem., Int. Ed. 2006;45:6137. doi: 10.1002/anie.200601517. [DOI] [PubMed] [Google Scholar]
- 160.Kiyokawa T, Kanaori K, Tajima K, Koike M, Mizuno T, Oku J-I, Tanaka T. J. Pept. Res. 2004;63:347. doi: 10.1111/j.1399-3011.2004.00109.x. [DOI] [PubMed] [Google Scholar]
- 161.Shiga D, Hamano Y, Kamei M, Funahashi Y, Masuda H, Sakaguchi M, Ogura T, Tanaka T. J. Biol. Inorg. Chem. 2012;17:1025. doi: 10.1007/s00775-012-0916-x. [DOI] [PubMed] [Google Scholar]
- 162.Tegoni M, Yu F, Bersellini M, Penner-Hahn JE, Pecoraro VL. Proc. Natl. Acad. Sci. U.S.A. 2012;109:21234. doi: 10.1073/pnas.1212893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Olesen K, Veselov A, Zhao Y, Wang Y, Danner B, Scholes CP, Shapleigh JP. Biochemistry. 1998;37:6086. doi: 10.1021/bi971603z. [DOI] [PubMed] [Google Scholar]
- 164.Jacobson F, Pistorius A, Farkas D, De Grip W, Hansson O, Sjölin L, Neutze R. J. Biol. Chem. 2007;282:6347. doi: 10.1074/jbc.M605746200. [DOI] [PubMed] [Google Scholar]
- 165.Suzuki S, Yamaguchi K, Kataoka K, Kobayashi K, Tagawa S, Kohzuma T, Shidara S, Iwasaki H. J. Biol. Inorg. Chem. 1997;2:265. [Google Scholar]
- 166.Wijma HJ, Jeuken LJC, Verbeet MP, Armstrong FA, Canters GW. J. Biol. Chem. 2006;281:16340. doi: 10.1074/jbc.M601610200. [DOI] [PubMed] [Google Scholar]
- 167.Wasser IM, de Vries S, Moënne-Loccoz P, Schröder I, Karlin KD. Chem. Rev. 2002;102:1201. doi: 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]
- 168.Monzani E, Anthony GJ, Koolhaas A, Spandre A, Leggieri E, Casella L, Gullotti M, Nardin G, Randaccio L, Fontani M, Zanello P, Reedijk J. J. Biol. Inorg. Chem. 2000;5:251. doi: 10.1007/s007750050369. [DOI] [PubMed] [Google Scholar]
- 169.Kujime M, Fujii H. Angew. Chem., Int. Ed. 2006;118:1107. doi: 10.1002/anie.200503555. [DOI] [PubMed] [Google Scholar]
- 170.Woollard-Shore JG, Holland JP, Jones MW, Dilworth JR. Dalton Trans. 2010;39:1576. doi: 10.1039/b913463h. [DOI] [PubMed] [Google Scholar]
- 171.Isoda N, Yokoyama H, Nojiri M, Suzuki S, Yamaguchi K. Bioelectrochemistry. 2010;77:82. doi: 10.1016/j.bioelechem.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 172.Komeda N, Nagao H, Kushi Y, Adachi G, Suzuki M, Uehara A, Tanaka K. Bull. Chem. Soc. Jpn. 1995;68:581. [Google Scholar]
- 173.Hiratsu T, Suzuki S, Yamaguchi K. Chem. Commun. (Cambridge, U. K.) 2005:4534. doi: 10.1039/b507932b. [DOI] [PubMed] [Google Scholar]
- 174.Yamaguchi K, Okada T, Suzuki S. Inorg. Chem. Commun. 2006;9:989. [Google Scholar]
- 175.Migita Y, Yokoyama H, Minami A, Mori T, Nojiri M, Suzuki S, Yamaguchi K. Electroanalysis. 2009;21:2441. [Google Scholar]
- 176.Nagao H, Komeda N, Mukaida M, Suzuki M, Tanaka K. Inorg. Chem. 1996;35:6809. doi: 10.1021/ic960303n. [DOI] [PubMed] [Google Scholar]
- 177.Geremia S, Di Costanzo L, Randaccio L, Engel DE, Lombardi A, Nastri F, DeGrado WF. J. Am. Chem. Soc. 2005;127:17266. doi: 10.1021/ja054199x. [DOI] [PubMed] [Google Scholar]
- 178.Maglio O, Nastri F, Pavone V, Lombardi A, DeGrado WF. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3772. doi: 10.1073/pnas.0730771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.DeGrado WF, Di Costanzo L, Geremia S, Lombardi A, Pavone V, Randaccio L. Angew. Chem., Int. Ed. 2003;42:417. doi: 10.1002/anie.200390127. [DOI] [PubMed] [Google Scholar]
- 180.Di Costanzo L, Wade H, Geremia S, Randaccio L, Pavone V, DeGrado WF, Lombardi a. J. Am. Chem. Soc. 2001;123:12749. doi: 10.1021/ja010506x. [DOI] [PubMed] [Google Scholar]
- 181.Pasternak A, Kaplan J, Lear JD, Degrado WF. Protein Sci. 2001;10:958. doi: 10.1110/ps.52101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Maglio O, Nastri F, Calhoun JR, Lahr S, Wade H, Pavone V, DeGrado WF, Lombardi A. J. Biol. Inorg. Chem. 2005;10:539. doi: 10.1007/s00775-005-0002-8. [DOI] [PubMed] [Google Scholar]
- 183.Lahr SJ, Engel DE, Stayrook SE, Maglio O, North B, Geremia S, Lombardi A, DeGrado WF. J. Mol. Biol. 2005;346:1441. doi: 10.1016/j.jmb.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 184.Wade H, Stayrook SE, Degrado WF. Angew. Chem., Int. Ed. 2006;45:4951. doi: 10.1002/anie.200600042. [DOI] [PubMed] [Google Scholar]
- 185.Wei P-P, Skulan AJ, Wade H, DeGrado WF, Solomon EI. J. Am. Chem. Soc. 2005;127:16098. doi: 10.1021/ja053661a. [DOI] [PubMed] [Google Scholar]
- 186.Summa CM, Rosenblatt MM, Hong J-K, Lear JD, DeGrado WF. J. Mol. Biol. 2002;321:923. doi: 10.1016/s0022-2836(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 187.Marsh ENG, DeGrado WF. Proc. Natl. Acad. Sci. U.S.A. 2002;99:5150. doi: 10.1073/pnas.052023199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. J. Mol. Biol. 2003;334:1101. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 189.Calhoun JR, Liu W, Spiegel K, Dal Peraro M, Klein ML, Valentine KG, Wand AJ, DeGrado WF. Structure. 2008;16:210. doi: 10.1016/j.str.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Calhoun JR, Bell CB, Smith TJ, Thamann TJ, DeGrado WF, Solomon EI. J. Am. Chem. Soc. 2008;130:9188. doi: 10.1021/ja801657y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bell CB, Calhoun JR, Bobyr E, Wei P-P, Hedman B, Hodgson KO, Degrado WF, Solomon EI. Biochemistry. 2009;48:59. doi: 10.1021/bi8016087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Litowski JR, Hodges RS. J. Biol. Chem. 2002;277:37272. doi: 10.1074/jbc.M204257200. [DOI] [PubMed] [Google Scholar]
- 193.Sambasivan R, Ball ZT. J. Am. Chem. Soc. 2010;132:9289. doi: 10.1021/ja103747h. [DOI] [PubMed] [Google Scholar]
- 194.Popp BV, Ball ZT. J. Am. Chem. Soc. 2010;132:6660. doi: 10.1021/ja101456c. [DOI] [PubMed] [Google Scholar]
- 195.Popp BV, Ball ZT. Chem. Sci. 2011;2:690. [Google Scholar]
- 196.Chen Z, Popp BV, Bovet CL, Ball ZT. ACS Chem. Biol. 2011;6:920. doi: 10.1021/cb2001523. [DOI] [PubMed] [Google Scholar]
- 197.Chen Z, Vohidov F, Coughlin JM, Stagg LJ, Arold ST, Ladbury JE, Ball ZT. J. Am. Chem. Soc. 2012;134:10138. doi: 10.1021/ja302284p. [DOI] [PubMed] [Google Scholar]
- 198.Popp BV, Chen Z, Ball ZT. Chem. Commun. (Cambridge, U. K.) 2012;48:7492. doi: 10.1039/c2cc33808d. [DOI] [PubMed] [Google Scholar]
- 199.Iranzo O, Thulstrup PW, Ryu S-B, Hemmingsen L, Pecoraro VL. Chem.--Eur. J. 2007;13:9178. doi: 10.1002/chem.200701208. [DOI] [PubMed] [Google Scholar]
- 200.Ghosh D, Lee K-H, Demeler B, Pecoraro VL. Biochemistry. 2005;44:10732. doi: 10.1021/bi0506674. [DOI] [PubMed] [Google Scholar]
- 201.Dieckmann G. Use of Metal-binding De Novo-designed Alpha-helical Peptides in the Study of Metalloprotein Structures. University of Michigan; 1995. [Google Scholar]
- 202.Jackman JE, Merz KM, Fierke Ca. Biochemistry. 1996;35:16421. doi: 10.1021/bi961786+. [DOI] [PubMed] [Google Scholar]
- 203.Wang X, Conway W, Burns R, McCann N, Maeder M. J Phys Chem A. 2010;114:1734. doi: 10.1021/jp909019u. [DOI] [PubMed] [Google Scholar]
- 204.Pocker Y, Bjorkquist DW. J. Am. Chem. Soc. 1977;99:6537. [Google Scholar]
- 205.Tocheva EI, Eltis LD, Murphy MEP. Biochemistry. 2008;47:4452. doi: 10.1021/bi7020537. [DOI] [PubMed] [Google Scholar]
- 206.Leferink NGH, Han C, Antonyuk SV, Heyes DJ, Rigby SEJ, Hough Ma, Eady RR, Scrutton NS, Hasnain SS. Biochemistry. 2011;50:4121. doi: 10.1021/bi200246f. [DOI] [PubMed] [Google Scholar]