

Abstract
Expression of long-lasting synaptic plasticity and long-term memory requires new protein synthesis, which can be repressed by phosphorylation of eukaryotic initiation factor 2α subunit (eIF2α). It was reported previously that eIF2α phosphorylation is elevated in the brains of Alzheimer’s disease (AD) patients and AD model mice. Therefore, we determined whether suppressing eIF2α kinases could alleviate synaptic plasticity and memory deficits in AD model mice. The genetic deletion of the eIF2α kinase PERK prevented enhanced eIF2α phosphorylation, as well as deficits in protein synthesis, synaptic plasticity, and spatial memory in APP/PS1 AD model mice. Similarly, deletion of another eIF2α kinase, GCN2, prevented impairments of synaptic plasticity and spatial memory defects displayed in the APP/PS1 mice. Our findings implicate aberrant eIF2α phosphorylation as a novel molecular mechanism underlying AD-related synaptic pathophysioloy and memory dysfunction and suggest that PERK and GCN2 are potential therapeutic targets for the treatment of individuals with AD.
Alzheimer’s disease (AD) is the most common cause of dementia and is without disease-modifying therapy. The incidence of AD has been escalating considerably with population aging and has the potential to evolve into a global public health crisis if it continues unchecked1–3. A multitude of studies in the past decade suggest that AD is a disorder of synaptic dysfunction due to the toxic effects of amyloid beta (Aβ), a short peptide derived from amyloid precursor protein (APP)4. On the other hand, it has been established that de novo protein synthesis is indispensable for long-lasting synaptic plasticity and the formation of long-term memory5–6. It is unknown whether dysregulated translation contributes to synaptic dysfunction and memory impairments associated with AD.
Eukaryotic initiation factor 2 (eIF2) plays a key role in the regulation of protein synthesis and alterations in the phosphorylation of the α subunit of eIF2 can cause memory impairments7–9. Four kinases, PKR, HRI, GCN2, and PERK, can phosphorylate eIF2α, which results in inhibition of general mRNA translation and enhanced translation of selective mRNAs such as the transcriptional regulator activating transcription factor 4 (ATF4), a repressor of long-term synaptic plasticity and memory8,10–11. Interestingly, elevated eIF2α phosphorylation has been correlated with AD pathogenesis12–14, but its link to synaptic failure and memory defects in AD is unknown. Therefore, in this study we asked whether reducing eIF2α phosphorylation by genetically removing either PERK or GCN2 could prevent AD-associated impairments in synaptic plasticity and memory. We found that suppressing the expression either PERK or GCN2 reduces elevated eIF2α phosphorylation, as well as synaptic plasticity deficits and memory impairments displayed by AD model mice. Our findings implicate elevated eIF2α phosphorylation as a contributor to AD-related synaptic pathophysiology and suggest that PERK and/or GCN2 could be novel therapeutic targets to ameliorate synaptic and memory dysfunction in individuals with AD.
RESULTS
Deletion of PERK prevents Aβ-induced impairment in LTP
We first examined the state of eIF2α phosphorylation in different brain regions from 10–12 month-old APP/PS1 AD model mice15. Western blots were performed to probe for eIF2α phosphorylation at serine51, which is targeted by all four eIF2α kinases11. We observed an increase in the levels of phosphorylated eIF2α in the hippocampus (150.60 ± 24.26 % of wild-type levels; Fig. 1a) and prefrontal cortex (data not shown), but not in the cerebellum (76.96 ± 10.71 % of wild-type levels, Fig. 1b), from APP/PS1 mice. We also examined the hippocampus in brain sections from postmortem human AD patients with Western blots and immunohistochemistry. Consistent with the above data from APP/PS1 mice and previous findings in other AD model mice5–7, we observed increased levels of phosphorylated eIF2α in the hippocampus from AD patients when compared to age-matched controls (273.30 ± 35.72 % of age-matched control levels; Fig. 1c, d). In agreement with the increased levels of phosphorylated eIF2α, we also observed an increase in ATF4, a repressor of long-lasting long-term potentiation (LTP) and long-term memory (LTM)8, in the hippocampus of APP/PS1 mice (184.42 ± 39.72 % of wild-type levels; Fig. 1e). Thus, eIF2α phosphorylation is abnormally elevated in AD.
Figure 1. Increased eIF2α phosphorylation in Alzheimer’s disease.

(a) eIF2α phosphorylation was increased in the hippocampus of APP/PS1 AD model mice. n=7 for both groups. p=0.041.(b) eIF2α phosphorylation was not altered in cerebellum of APP/PS1 AD model mice. n=11 for WT and n=10 for APP/PS1. p=0.080. (c) eIF2α phosphorylation was elevated in postmortem human AD brain samples. n=4 for AD and age-matched control groups. p=0.012. (d) DAB staining revealed increased eIF2α phosphorylation in the hippocampus of postmortem human AD brains, representative of three independent experiments. Scale bar, 150 μm. (e) ATF4 levels were increased in the hippocampus of APP/PS1 mice compared to WT littermates. n=6 for each group. p=0.044 Full-length blots/gels are presented in Supplementary Figure 7.
We subsequently investigated whether AD-associated impairments in synaptic plasticity and memory could be alleviated by reducing eIF2α phosphorylation. Among the kinases for eIF2α, PERK activity is usually associated with ER stress, which has been implicated in AD9. Therefore, we posited that PERK-induced eIF2α phosphorylation and the subsequent suppression of de novo protein synthesis may be a consequence of ER stress in AD. First, we bred mice harboring a floxed PERK gene16 with mice expressing a brain-specific Cre recombinase17 to generate mice in which PERK was conditionally removed in excitatory neurons in the forebrain and hippocampus late in development (Supplementary Fig. 1a)18. Next, using immunofluorescence combined with confocal microscopy, we observed punctate co-localization of PERK and phosphorylated eIF2α in dendrites of adult mouse hippocampal neurons in culture (Supplementary Fig. 1b). We proceeded to examine the Aβ-dependent regulation of eIF2α phosphorylation in wild-type (WT) mice. Consistent with recent findings18, in WT slices LTP-inducing high-frequency stimulation (HFS) caused dephosphorylation of eIF2α, which was prevented by Aβ (Fig. 2a). To acquire direct evidence for Aβ-mediated effects on protein synthesis, we utilized SUnSET, a non-radioactive method to monitor new protein synthesis19. In agreement with its ability to blunt eIF2α dephosphorylation, Aβ blocked HFS-induced increases in de novo protein synthesis (Fig. 2b). We then applied exogenous Aβ (1–42) (500 nM) to hippocampal slices and induced LTP in area CA1 by HFS. In the presence of Aβ, LTP was inhibited in WT slices, but in slices prepared from PERK cKO mice Aβ had no effect on LTP (Fig. 2c, d). Moreover, removal of PERK itself did not affect hippocampal synaptic plasticity, as HFS induced similar LTP in slices from either WT or PERK cKO mice (Fig. 2c, d). In contrast, co-application of Aβ and Sal003 (Sal), an inhibitor of eIF2α dephosphorylation8, resulted in LTP failure in PERK cKO mice (Fig. 2c, d). Taken together, these results indicate that Aβ-induced impairments in hippocampal synaptic plasticity are alleviated by deleting the eIF2α kinase PERK.
Figure 2. Aβ-induced impairment in LTP is alleviated by deleting the eIF2α kinase PERK.

(a) LTP-inducing stimulation decreased the phosphorylation of eIF2α (middle lane), which was reversed by Aβ(1–42) (right lane). Slices were harvested 30 minutes post-HFS and area CA1 was microdissected for Western blot analysis. n=8. *p<0.05. (b) De novo protein synthesis (assayed by SUnSET). LTP-inducing stimulation increased de novo protein synthesis, which was blunted by Aβ(1–42). Slices were harvested 30 minutes post-HFS and area CA1 was microdissected. n=4. *p<0.05. (c) Treatment of hippocampal slices from WT mice with 500 nM Aβ(1–42) resulted in impaired LTP (grey triangles, n=8) compared with LTP in vehicle-treated WT slices (open squares, n=7). In contrast, LTP was induced in slices from PERK cKO mice in the presence of Aβ(1–42) (half-filled triangles, n=6), which was blunted by application of 10 μM Sal003 (Sal, grey diamonds, n=7). In addition, HFS induced LTP in PERK cKO mice that was comparable to that in WT littermates (grey circles, n=7). (d) Cumulative data showing the mean fEPSP slope 80 min post-HFS from the LTP experiments in panel c. *p<0.05; **p<0.01. Full-length blots/gels are presented in Supplementary Figure 7.
Deletion of PERK corrects abnormalities in AD model mice
We then proceeded to generate a mutant mouse line that expressed both APPswe/PS1ΔE9 and homozygous Cre PERK−/−flox transgenes (APP/PS1/PERK cKO). The breeding strategy involved two stages: generation of female mice with the APPswe/PS1ΔE9 and heterozygous Cre PERK+/−flox transgenes followed by breeding with male heterozygous PERK+/−flox mice. All mice generated for these experiments were aged for 10–12 months, an age when APP/PS1 mice reliably display synaptic dysfunction and memory deficits20–21. After approximately 18 months of breeding we obtained a sufficient number of aged APP/PS1/PERK cKO mutant mice (APPswe/PS1ΔE9 and Cre PERK−/−flox), along with three other littermate groups for experiments (Fig. 3a): WT (Cre only), APP/PS1 (APPswe/PS1ΔE9), and PERK cKO (Cre PERK−/−flox).
Figure 3. Generation of AD model mice with reduced PERK-eIF2α signaling.

(a) Diagram depicting the creation of mice with AD-associated transgenes and reduced PERK/eIF2α signaling. (b) eIF2α phosphorylation was reduced in hippocampal area CA1 of APP/PS1/PERK cKO mice compared to the increased levels of eIF2α phosphosphorylation in APP/PS1 mice, which was correlated with the expression of PERK (c). n=10 for APP/PS1/PERK cKO, n=6 the other three groups. (d) Representative Western blot showing that de novo protein synthesis (assayed by SUnSET) was reduced in APP/PS1 mice compared to WT littermates. In addition, de novo protein synthesis in PERK cKO and APP/PERK cKO mice was not different from WT mice. (e) Cumulative data showing densitometric analysis of experiments in panel d. n=4. *p<0.05. (f) Elevated levels of ATF4 in APP/PS1 mice were reduced to WT levels in APP/PS1/PERK cKO mice. Western blots were performed on tissue from area CA1 of the hippocampus. n=10. All data for the densitometric analysis of the Western blots were presented as mean ± SEM. *p< 0.05, **p< 0.01, ***p<0.001. Full-length blots/gels are presented in Supplementary Figure 7.
Consistent with decreased expression of PERK, the increased phosphorylation of eIF2α in the APP/PS1 mice was reduced in hippocampus and prefrontal cortex of APP/PS1/PERK cKO mice (Fig. 3b, c and Supplementary Fig. 2a, b). In contrast, phosphorylation of eIF2α and the expression of PERK in the cerebellum were indistinguishable between the four groups of mice (Supplementary Fig. 2c, d). To examine the effects of PERK deletion on protein synthesis in aged AD model mice, we performed SUnSET experiments on hippocampal slices and observed that levels of newly synthesized proteins were reduced in APP/PS1 mice when compared to WT littermates (Fig. 3d, e). Consistent with the restoration of eIF2α phosphorylation (Fig. 3b), the reduction of protein synthesis was prevented in APP/PS1/PERK cKO mice (Fig. 3d, e). We also examined the expression of ATF4 and CHOP/GADD153, both of which have been shown to be downstream of eIF2α phosphorylation following cellular stress in non-neuronal systems22. We detected downregulation of ATF4 expression, but no alteration in CHOP levels in APP/PS1/PERK cKO mice when compared to APP/PS1 mice (Fig. 3f, Supplementary Fig. 2e), suggesting that expression of protein levels of ATF4 and CHOP might be differentially regulated in the mouse brains22–23. Thus, dysregulation of eIF2α phosphorylation and de novo protein synthesis in APP/PS1 mice are normalized by PERK deletion.
We next tested spatial learning and memory of the four genotypes of mice described above on three independent behavioral tasks: Morris water maze (MWM), object location (OL), and Y water maze (YWM). During acquisition of the hidden platform version of MWM, WT mice showed a day-to-day decrease in escape latency whereas APP/PS1 mice displayed significantly higher escape latencies (Fig. 4a). Furthermore, compared to WT mice, APP/PS1 mice spent significantly less time within target quadrant and crossed the platform location fewer times in probe tests when the platform was removed (Fig. 4b, c). Notably, the impaired learning and memory deficits exhibited by the APP/PS1 mice was not exhibited by the APP/PS1/PERK cKO mutant mice, as indicated by reduced escape latency, increased target quadrant occupation, and more platform crossings that were comparable to those displayed by WT mice (Fig. 4a–c). Of note and consistent with previous findings18, PERK cKO mice did not display any observable phenotype in the MWM test (Fig. 3a–c), suggesting that reduction of PERK does not alter hippocampus-dependent spatial memory in this task. To investigate the possibility that the learning and memory improvements displayed by the APP/PS1/PERK cKO mice were attributable to effects on vision, motivation, or swimming ability, we tested mice on the visible platform task and found no observable differences between the four groups of mice in their latency to find the visible platform (Fig. 3d). In agreement, the results from the OL and YWM tasks also indicate that spatial memory impairments associated with APP/PS1 mice were not displayed by APP/PS1/PERK cKO mice (Fig. 4e, f). All together, these behavioral studies indicate that spatial memory defects in APP/PS1 mice were prevented by suppressing/restoring PERK/eIF2α signaling.
Figure 4. Spatial memory deficits in APP/PS1 AD model mice are alleviated by suppressing PERK/eIF2α signaling.

(a) Escape latency in the Morris water maze plotted against the training days. WT n=14; APP/PS1, n=14; PERK cKO, n=16; APP/PS1/PERK cKO n=11. Repeated measures ANOVA followed by a post hoc Bonferroni multiple comparison test, p= 0.0028, F(3, 54)=8.440. APP/PS1 vs WT: p<0.01, t=4.815; APP/PS1 vs APP/PS1/PERK cKO: p<0.05, t=3.671. APP/PS1 vs PERK cKO: p>0.05, t=2.806. No difference was detected among WT, PERK cKO, and APP/PS1/PERK cKO groups. Repeated measures ANOVA. P=0.09, F(2,40)=3.302. (b) Percentage of time spent in the target quadrant during a 60 second probe trial of MWM test. One-way ANOVA, p = 0.0498, F=2.869. *p< 0.05. (c) Frequency of platform crossing during a 60 second probe trial of MWM test. One-way ANOVA, p =0.0109, F=4.171.*p< 0.05. (d) Visible platform test. Repeated measures ANOVA, p= 0.0943. F=5.653. (e) Object location task. Percentage of time interacting with the object at a new location (out of total time spent with objects) was calculated as discrimination ratio. WT, n=14; APP/PS1, n=10; PERK cKO, n=8; APP/PS1/PERK cKO, n=7. Independent t-test. *p< 0.05 (f) Y water maze task. Spatial memory was measured by percentage of correct arm choice. WT, n=18; APP/PS1, n=18; PERK cKO, n=15; APP/PS1/PERK cKO, Independent t-test. n=9. *p<0.05.
The onset of sporadic AD is age-dependent and it has been proposed that increased eIF2α phosphorylation in response to acute and/or early accumulation of Aβ could be protective14. Therefore, we determined whether reducing PERK/eIF2α signaling impacted the spatial memory of APP/PS1 mice at a younger age. We performed MWM tests on mice at 3–5 months of age and observed no significant differences in escape latency, target quadrant occupation, or platform crossings between the four groups of mice (Supplementary Fig. 3a–c). These findings suggest that suppressing PERK/eIF2α signaling does not significantly affect spatial learning and memory performance in young AD model mice.
We next determined whether deleting PERK could rescue synaptic plasticity deficits in aged APP/PS1 mice. LTP was dramatically reduced in APP/PS1 mice, and the removal of PERK resulted in normal LTP in the APP/PS1/PERK cKO mice (Fig. 5a, b). LTP was not altered in the PERK cKO mice (Fig. 5a, b). To determine whether restored LTP in the APP/PS1/PERK cKO mice was dependent on protein synthesis, we induced LTP in the presence of the general protein synthesis inhibitor anisomycin. Treatment of WT slices with anisomycin (40 μM) resulted in inhibition of LTP compared to vehicle-treated control slices (Fig. 5c). Importantly, LTP in the APP/PS1/PERK cKO mice also was inhibited by anisomycin (Fig. 5c), indicating that deletion of PERK resulted in restored LTP that was protein synthesis-dependent. We then investigated the effects of reducing PERK/eIF2α signaling on amyloidogenesis. Compared with APP/PS1 mice, which displayed considerable levels of Aβ deposits in the brain, Aβ levels were decreased in the hippocampus of APP/PS1/PERK cKO mice (Fig. 5d). The decrease in the brain Aβ load was not due to effects on APP expression, as levels of full-length APP were not changed (Fig. 5d). Moreover, beta C-terminal fragment (β-CTF), but not alpha C-terminal fragment (α-CTF) of APP was reduced in the hippocampus of APP/PS1/PERK cKO mice (Fig. 5e). There were no significant changes in the levels of the APP cleavage enzymes BACE1 or γ-secretase as reflected by unaltered PEN2, an essential component of γ-secretase complex (Supplementary Fig. 4a, b). Moreover, consistent with previous findings24, we observed a reduction of the Aβ degrading enzyme neprilysin in the hippocampus of the APP/PS1 mice, which was corrected in the APP/PS1/PERK cKO mice (Fig. 5f).
Figure 5. LTP impairments in APP/PS1 mice are rescued by decreasing PERK/eIF2α signaling.

(a) LTP was inhibited in APP/PS1 mice (n=6) compared to LTP in WT mice (n=9). LTP was sustained in APP/PS1/PERK cKO mice (n=4) and PERK cKO mice (n=8). (b) Cumulative data showing mean fEPSP slopes 80 min post-HFS from the LTP experiments in panel a. Data were presented as mean +SEM. (c) LTP in APP/PS1/PERK cKO mice (n=7) was inhibited by anisomycin (40 μM). n=8 for WT, n=5 for anisomycin/WT, and n=7 for anisomycin/APP/PS1. Anisomycin was applied into recording chamber 30 min before HFS and present throughout the experiments. (d) Brain Aβ levels were decreased in APP/PS1/PERK cKO mice compared to APP/PS1 mice. n=6 for each group. Levels of full-length APP were not changed. n=9 for APP/PS1/PERK cKO group and n=6 for each of other three groups. (e) β-CTF, but not α-CTF was reduced in the hippocampus of APP/PS1/PERK cKO mice, compared with APP/PS1 mice. n=6. (f) Neprilysin expression was reduced in APP/PS1 mice and was corrected in APP/PS1/PERK cKO mice. n=7 for each group. (g, h) Representative blots (g) and cumulative data of densitometric analysis (h) showing that levels of activity-regulated cytoskeleton-associated protein (Arc, n=5), protein kinase M zeta (PKMζ, n=11), and synaptophysin (n=5) were reduced in hippocampal area CA1 of APP/PS1 mice and was corrected in APP/PS1/PERK cKO mice. Levels of calcium/calmodulin-dependent protein kinase II (CaMKII, n=5) and AMPA receptor subunit GluA1 (n=4) were not significantly altered. *p<0.05; **p<0.01. Full-length blots/gels are presented in Supplementary Figure 7.
To further elucidate the molecular mechanisms associated with the effects of PERK deletion on APP/PS1 mice, we examined regulation of the levels of a series of proteins that have been implicated in synaptic plasticity and memory, including activity-regulated cytoskeleton-associated protein (Arc), calcium/calmodulin-dependent protein kinase II (CaMKII), AMPA receptor subunit GluA1, and protein kinase M zeta (PKMζ)25–27. We observed reduced levels of both Arc and PKMζ in hippocampal area CA1 in APP/PS1 mice, which was corrected by the deletion of PERK (Fig. 5g, h). However, levels of CaMKII and GluA1 were not significantly changed (Fig. 5g, h). Finally, we observed increased levels of PSD-95 and decreased levels of synaptophysin in the APP/PS1 mice, in agreement with previous studies28. The dysregulation of these synaptic proteins was restored in the APP/PS1/PERK cKO mice to levels indistinguishable from those in WT littermates (Fig. 5g, h and Supplementary Fig. 5e,f). We also examined the levels of other proteins that could be involved in the regulation of PERK/eIF2α signaling, such as type 1 protein phosphatase (PP1), which contributes to dephosphorylation of eIF2α29, transcription factor Nrf2, which has been shown in non-neuronal cells to be a substrate of PERK30, and the unfolded protein response (UPR) markers ATF6 and IRE1α31. However, none of the aforementioned proteins were significantly changed across the four mouse genotypes (Supplementary Fig. 5a–d). Thus, the reduction of PERK/eIF2α signaling in APP/PS1 mice decreased amyloidogenesis and restored physiological levels of neprilysin, as well as the plasticity- and memory-related proteins Arc and PKMζ.
Deletion of GCN2 corrects abnormalities in AD model mice
To further elucidate the role of eIF2α phosphorylation in AD-associated impairments of synaptic plasticity and memory, we performed LTP experiments on hippocampal slices from a mouse line in which GCN2, another eIF2α kinase, was globally and constitutively removed32. Similar to the experiments with PERK cKO mice, LTP in slices from GCN2 knockout (GCN2 KO) mice was still expressed in the presence of Ab (Fig. 6a, b). Of note, unlike PERK cKO mice, there was a trend of enhanced LTP in the GCN2 KO mice (Fig. 6a, b). In addition, consistent with the experiments with PERK cKO mice, co-application of Aβ and Sal003 (Sal) resulted in LTP failure in GCN2 KO mice (Fig. 6a, b). Taken together, these results suggest that inhibiting GCN2 prevents Ab-induced synaptic plasticity impairments by reducing eIF2α phosphorylation.
Figure 6. Removal of GCN2 reverses AD-associated LTP failure.

(a) Normal LTP was induced in slices from GCN2 KO mice in the presence of Aβ(1–42) (half-filled triangles, n=7), which was blunted by application of 10 μM Sal003 (Sal, grey diamonds, n=7). In contrast, slices of WT littermates treated with Aβ(1–42) (grey circles, n=5) exhibited impaired LTP. In addition, HFS induced hippocampal LTP in GCN2 KO mice that was enhanced (grey triangles, n=5) compared to WT littermates (open squares, n=5). (b) Cumulative data showing mean fEPSP slopes 80 min post-HFS from the LTP experiments in panel a. (c) HFS-induced hippocampal LTP was inhibited in APP/PS1 mice (light grey triangles, n=6) compared to LTP in WT mice (open squares, n=8). In contrast, hippocampal LTP was sustained in APP/PS1/GCN2 KO mice (half-filled triangles, n=5) and GCN2 KO mice (grey circles, n=6). (c) Representative fEPSP traces for data shown in panel b. (d) Cumulative data showing mean fEPSP slopes 80 min post-HFS from the LTP experiments in panel b. Data were presented as mean ± SEM. *p<0.05.
We proceeded to cross APP/PS1 mice with GCN2 KO mice to generate APP/PS1/GCN2 KO mice and aged them for 10–12 months before conducting the following experiments. First, we confirmed that the abnormally high levels of eIF2α phosphorylation and ATF4 expression in the hippocampus of APP/PS1 mice were restored in APP/PS1/GCN2 KO mice to the levels observed in WT mice (Supplementary Fig. 6a, b). Of interest, phosphorylation of eIF2α in GCN2 KO mice was not different from that in WT mice (Supplementary Fig. 6a, b). Consistent with the biochemical findings, we found that hippocampal LTP deficits in APP/PS1 mice were normalized in APP/PS1/GCN2 KO mice (Fig. 6c–e).
We then tested the behavioral effects of removing GCN2 in the APP/PS1 mice by performing the MWM task to access spatial learning and memory. Similar to the observations from the studies with the APP/PS1/PERK cKO mice, the impairments in spatial learning and memory displayed by the aged APP/PS1 mice were prevented in the APP/PS1/GCN2 KO mice as indicated by decreased escape latency, increased platform crossings as well as target quadrant occupancy that were close to those displayed by WT mice (Fig. 7a–c). In addition, the improvements in learning and memory displayed by the APP/PS1/GCN2 KO mice were not due to effects on vision, motivation, or swimming ability, as demonstrated by visible platform task which showed no difference between groups (Fig. 7d). Taken together, these findings indicate that genetic removal of the eIF2α kinase GCN2 prevents AD-associated LTP failure and spatial memory impairments.
Figure 7. Spatial memory deficits in APP/PS1 AD model mice are alleviated by deleting eIF2α kinase GCN2.
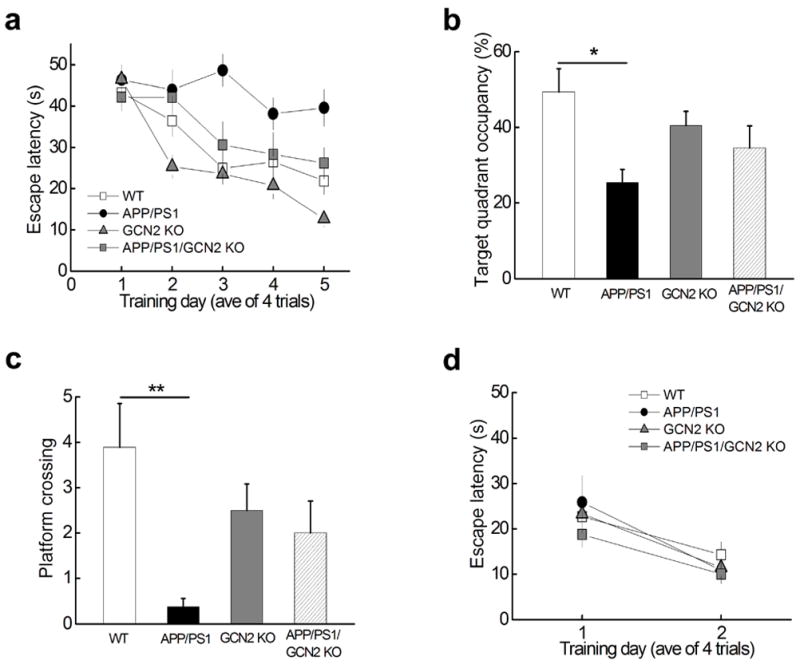
(a) Escape latency in the Morris water maze plotted against the training days. APP/PS1 mice (black circles, n=8) were slower to learn than WT mice (open squares, n=9), APP/PS1/GCN2 KO (grey squares, n=9) or GCN2 KO mice (grey triangles, n=12). Repeated measures ANOVA followed by a post hoc Bonferroni multiple comparison test, p= 0.0014, F(3, 37)=9.960. APP/PS1 vs WT: p<0.01, t=4.532; APP/PS1 vs APP/PS1/GCN2 KO: p<0.05, t=3.360; APP/PS1/GCN2 KO vs WT: p>0.05, t=1.172. (b) Percentage of time spent in the target quadrant during a 60 second probe trial of MWM test. One-way ANOVA, p = 0.0261, F=3.584. (c) Frequency of platform crossing during a 60 second probe trial of MWM test. One-way ANOVA, p = 0.0144, F=4.057. Number of platform crossing of APP/PS1/GCN2 KO mice is not significantly different from that of WT mice; APP/PS1 vs APP/PS1/GCN2 KO: p=0.0526. **p< 0.01. (d) In the visible platform test no difference was observed for escape latency among the four genotypes of mice. Repeated measures ANOVA, p=0.3735. F=1.500.
DISCUSSION
Identifying molecular mechanisms underlying synaptic failure and memory loss associated with AD could provide the basis for novel therapeutics for the treatment of this devastating neurodegenerative disease. Numerous studies over the past few decades have established a key role for de novo protein synthesis in long-lasting synaptic plasticity and long-term memory5–6,33. As a translational factor controlling general protein synthesis, eIF2α, through modulation of its phosphorylation state, is known to be important in maintaining long-lasting forms of synaptic plasticity, and long-term memory7–9. In the current study, we have shown that reducing eIF2α phosphorylation, via forebrain-specific conditional deletion PERK, one of its kinases, protects mice from AD-related deficits in synaptic plasticity and spatial memory, which was correlated with decreased amyloidogenesis and restoration of normal expression of plasticity-related proteins. Moreover, we have shown that deletion of GCN2, another eIF2α kinase, also prevents AD-related deficits in synaptic plasticity and spatial memory, which also is likely due to the resetting of eIF2α phosphorylation and normal translational homeostasis (Supplementary Fig. 8).
Phosphorylation of eIF2α by PERK: a double-edged sword
In normal physiological situations, phosphorylation of eIF2α usually is considered to be protective. In response to a wide range of cellular stressors, eIF2α is phosphorylated by one of its four kinases, which includes PERK, leading to inhibition of general mRNA translation, but also increased translation of selective mRNAs, including that encoding ATF4. The reduction of general protein synthesis allows cells to conserve energy resources and enhanced translation of selective mRNAs increases expression of stress-related proteins, thereby reconfiguring gene expression in order to manage the stress condition11,34–35. This type of mechanism might account for the late-onset of synaptic failure and memory deficits observed in sporadic AD (the majority of AD) and in most AD model mice. Therefore, we hypothesized that the deletion of PERK in APP/PS1 mice early in life might be detrimental to memory formation. However, we did not observe any change in the spatial memory of young APP/PS1/PERKcKO mice (Supplementary Fig. 3a–c), suggesting that another eIF2α kinase may be responsible for maintaining translational homeostasis. In contrast, in later stages of pathophysiologies associated with neurodegenerative diseases, abnormally high levels of stress-inducing agents such reactive oxygen species (ROS) are likely generated constantly, overwhelming the ability of the endogenous antioxidant mechanisms to remove them36–37, which then results in unusually high levels of eIF2α phosphorylation (Fig. 1a–d). In other words, the fine balance between new protein synthesis and the stress response, which is intact under normal physiological conditions, is likely to be disrupted permanently in later stages of AD, making the dysregulation of protein synthesis more pronounced and longer lasting than in the initial stages of the disease. This long-term disruption of translational homeostasis will inevitably hinder long-lasting synaptic plasticity and long-term memory because de novo protein synthesis is essential for both processes33,38. Notably, it was shown recently that mice treated with a compound that inhibits the downstream consequences of eIF2α phosphorylation display enhanced learning and memory39, consistent with the idea that eIF2α phosphorylation limits memory formation.
Phosphorylation of eIF2α in AD: PERK versus GCN2
The four kinases that phosphorylate eIF2α, PERK, GCN2, PKR, and HRI, typically are classified by their response to a specific type of cellular stress, each of which activates a single kinase. However, the idea that each eIF2α kinase responds to only one type of stressor is almost certainly oversimplified11,35. Studies have indicated that during conditions such as oxidative stress, multiple eIF2α kinases, especially PERK and GCN2, are recruited either simultaneously or sequentially to reset cellular homeostasis47–49. In agreement, we have shown here that deletion of either PERK or GCN2 is capable of correcting eIF2α hyperphosphorylation, presumably induced by several cellular stressors, as well as synaptic plasticity and memory deficits displayed by AD model mice. Interestingly our studies revealed that although PERK deletion by itself results in decreased basal phosphorylation of eIF2α (Fig. 3b), removal of GCN2 alone had no effect on basal eIF2α phosphorylation (Supplementary Fig. 6a). Moreover, genetic deletion of PKR also did not alter the basal phosphorylation of eIF2α (Supplementary Fig. 1d). Taken together, these findings suggest a dominant role for PERK in controlling the basal state of eIF2α phosphorylation in brain.
Although our current study focused on eIF2α kinases, it should be noted that dephosphorylation of eIF2α, by phosphatases such as protein phosphatase 129, might also play an important role in mediating the effects of AD-related deficits in protein synthesis-dependent synaptic plasticity and memory. Future studies will be necessary for a better understanding of how the disruption of translational homeostasis is involved in AD pathophysiology. In brief, given the general requirement of de novo protein synthesis for long-lasting synaptic plasticity and long-term memory consolidation, and in light of recent findings demonstrating that hyperphosphorylation of eIF2α is associated with synaptic deficits and neuronal loss in prion-disease model mice50, it is possible that the dysregulation of eIF2α phosphorylation represents a common molecular mechanism underlying diseases characterized by neurodegeneration and memory dysfunction. Thus, both PERK and GCN2 have potential as novel therapeutic targets not only for AD and prion-disease, but also for diseases such as frontotemporal dementia and Lewy body disease.
METHODS
Transgenic mice
All mice (C57BL/6) were housed in the Transgenic Mouse Facility of New York University, compliant with the NIH Guide for Care and Use of Laboratory Animals. The facility is kept on a 12 h light/dark cycle, with a regular feeding and cage-cleaning schedule. Mice of either sex were used. PERK forebrain conditional knockout mice (PERK cKO) were generated by crossing brain-specific Cre recombinase mouse line (T-29) kindly provided by Dr. S. Tonegawa51 with PERK (−/−) mice harboring floxed PERK gene16. The creation of APP/PS1/PERK cKO double mutant mice required two stages of breeding: generation of female mice with APPswe/PS1ΔE9 and heterozygous Cre PERK+/−flox transgenes followed by breeding with male heterozygous PERK+/−flox mice. The generation of APP/PS1/GCN2 KO double mutant mice were also involved in two steps of breeding: generation of male mice with APPswe/PS1ΔE9 and heterozygous GCN2+/− transgenes followed by breeding with female heterozygous GCN2+/− mice. All genotypes were determined by polymerase chain reaction (PCR).
Western blot and antibodies
Lysates were prepared as described52. Phosphatase inhibitor cocktail I & II (Sigma), and protease inhibitor cocktail (Sigma) were added to lysis buffer. Protein concentration was determined by the Bradford technique (Bio-Rad Laboratories), and equal amounts of protein from each sample were loaded on 4–12% Tris-Glycine SDS-PAGE (Invitrogen) gels. After transfer, membranes were blocked for at least 30 min at room temperature with blocking buffer [BB; 5% non fat dry milk in TBS containing 0.1% Tween 20 (TBS-T)], then probed overnight at 4°C using the following primary antibodies (dilution 1:1000) for: Aβ 6E10 (Covance, SIG-39300), ATF4 (generous gift from Dr. RC Wek, Indiana University School of Medicine), Arc (Santa Cruz, sc-17839), ATF6 (Abcam, ab37149), BACE1 (Abcam, ab2077), CaMKII (Millipore, 07–1496), CHOP/GADD153 (Abcam, ab11419), phospho-eIF2α (Ser51) (Invitrogen, 44728G), eIF2α (Cell Signaling, 2103), GluA1 (Cell Signaling, 8850), IRE1 (Cell Signaling, 3294), neprilysin/CD10 (Santa Cruz, sc-9149), p-Nrf2 and Nrf2 (Abcam, ab76026 and ab31163), PEN2 (Cell Signaling, 5451), PERK (Cell Signaling, 3192), PKMζ (Santa Cruz, sc-17781 ), PP1 (Cell Signaling, 2582), PSD95 (Millipore, ), Synaptophysin (Chemicon, MAB368), Actin (Sigma, A1978), GAPDH (Cell Signaling, 2118), and Tubulin (Sigma, T6074). The “n” refers to the number of slices, from at least 3 litters/groups of mice.
Densitometric analysis of the bands was performed using Scion Image software (Scion Corporation). Data were analyzed using one-tailed and (where specified) two-tailed t-tests or, where for multiple groups, one-way ANOVA followed by Bonferroni post-hoc tests, using Prism (Graphpad Software Inc.) and Origin (OriginLab Corp.). Summary data were presented as group means with standard error bars. Data collection and analysis were not performed blind for the Western blot experiments.
Drug treatment
Anisomycin stock solution (40 mM, Tocris) was prepared in DMSO and was diluted into ACSF to its final concentration 40 μM before use. Stock for anisomycin was kept for maximal three days. Puromycin stock (5 mg/ml, Sigma) was prepared in distilled water and diluted into ACSF to its final concentration 5μg/ml the day of experiment. Sal003 (Sal) stock (10 mM, Calbiochem) was prepared in DMSO and diluted into ACSF to its final concentration. Aβ(1–42) stock (100 μM, Bachem) was prepared in distilled water and stored at −20 °C for at least 24 hrs before use at a final concentration of 500 nM. This type of preparation protocol yields ample Aβ oligomers53. Incubation of hippocampal slices with drugs was performed in either recording chambers or maintenance chambers as needed.
Postmortem AD brain samples
Hippocampal sections of postmortem human brain samples were provided by the late Dr. Mark Smith from Case Western Reserve University. Samples were collected and prepared in accordance with the institution’s IRB approved protocols as described previously54. Briefly, hippocampal sections from cases of pathologically confirmed AD (n = 12; age 69–96 yr, mean 84 ± 8.2 yr) and age-matched controls (n = 7; age 66–86 yr, mean 71.5 ± 6.8 yr) were collected, fixed in methacarn (methanol: chloroform: acetic acid; 6:3:1), and embedded in paraffin. Subsequently, 6 μm-thick sections were cut and processed for immunohistochemistry. Briefly, sections were deparaffinized in xylene, rehydrated in a series of graded ethanol, and endogenous peroxidase activity was removed by incubating 30 min in 2% H2O2. Following a 10-min incubation in 10% normal goat serum, sections were incubated overnight at 4°C with phospho-eIF2α (Ser51) antibody (Invitrogen) at a dilution of 1:200 in 1% BSA containing 0.2% triton X-100. Following successive incubations in biotinylated goat anti-rabbit secondary antibody (Vector Laboratories) and peroxidase-anti-peroxidase complex, staining was developed using 3,3-diaminobenzidine as a cosubstrate and counterstained with 0.2% cresyl violet.
Immunoblotting
Soluble protein extracts from brain sections were provided by late Dr. Mark Smith from Case Western Reserve University. Samples were prepared as described previously54. Briefly, brain sections from AD (n = 4; age 72–89 yr) and control (n = 3; age 65–86 yr) were homogenized in 10 volume of lysis buffer equipped with protease and phosphatase inhibitors [50 mM Tris-HCl (pH 7.6), 0.02% sodium azide, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1% Nonidet P-40, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 1 mg/mL aprotinin, 2 mg/mL antipain, and 1 mM sodium orthovanadate]. Protein concentrations were determined by bicinchoninic acid assay (Pierce). 20 μg of soluble protein extracts were prepared from brain samples and standard Western blotting techniques were used as described in this section. The experimenter was blind for the data collection and analysis for these experiments.
Hippocampal slice preparation and electrophysiology
Acute 400 μm transverse hippocampal slices were prepared using a vibratome as described previously21. The slices were maintained at room temperature in a submersion chamber with artificial cerebrospinal fluid (ACSF) containing (in mM) 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 24 NaHCO3, and 15 glucose, bubbled with 95% O2/5% CO2. Slices were incubated for at least 2 hours before removal for experiments. For electrophysiology experiments, slices were transferred to recording chambers (preheated to 32 °C) where they were superfused with oxygenated ACSF. Monophasic, constant-current stimuli (100 μsec) were delivered with a bipolar silver electrode placed in the stratum radiatum of area CA3, and the field excitatory postsynaptic potentials (fEPSPs) were recorded in the stratum radiatum of area CA1. Baseline fEPSPs were monitored by delivering stimuli at 0.033 Hz. fEPSPs were acquired, and amplitudes and maximum initial slopes measured, using pClamp 10 (Axon Instruments, Foster City, CA). LTP was induced with a high-frequency stimulation (HFS) protocol consisting of two 1-second long 100 Hz trains, separated by 60 sec, delivered at 70–80 % of the intensity that evoked spiked fEPSPs. Data collection and analysis were not performed blind to the conditions of the experiments. The “n” refers to the number of slices, from at least 3 litters/group of mice.
Protein synthesis assay
Proteins were labeled using a protocol adapted from the SUnSET method19. Hippocampal slices were incubated with puromycin (5 μg/ml) 30 minutes before and throughout the electrophysiology experiments. Newly synthesized proteins were end-labeled with puromycin. At the end of physiology experiment, slices were harvested and frozen on dry ice. Area CA1 was microdissected and protein lysates were prepared for Western blotting. Puromycin-labeled proteins were identified using the mouse monoclonal antibody 12D10. Protein synthesis levels were determined by taking total lane density in the molecular weight range of 10 kDa to 250 kDa. Data collection and analysis were not performed blind for these experiments. The “n” refers to the number of slices, from at least 3 litters/groups of mice.
Mouse behavior
Morris water maze
Morris water maze test was performed as described52. The training paradigm for the hidden platform version of the Morris water maze consisted of 4 trials (60 sec maximum; interval 15 min) each day for 5 consecutive days. The probe trial was carried 2 hrs after the completion of training on day 5. The visible platform task consisted of 4 trials each day for 2 consecutive days with the escape platform marked by a visible cue and moved randomly between four locations. The trajectories were recorded with a video tracking systm (Ethovision XT).
Object location
Object location (OL) test was performed as described55. Two identical objects were placed in the far corners of the arena. Each mouse was allowed to explore the two objects for 10 minutes. After a delay of 60 minutes, the test phase began in which one of the objects was move to a new place and the mouse was allowed to explore the arena for 5 minutes. The mouse was placed into the arena facing the center of the opposite wall. Exploratory behavior was defined as the animal directing its nose toward the object closely.
Y water maze
Y water maze test was conducted as described previously18181818. At the day of training, the animal was trained to pick up one side of the maze, where a platform is hidden. The test phase began after a delay of 24 hours.
For all behavioral tasks, the experimenters were blind to the genotypes of the mice and no mouse was excluded. The “n” refers to the number of mice.
Immunofluorescence and confocal microscopy
Ice-cold 4% paraformaldehyde in PBS (pH 7.4) was used to fix primary cultured neurons (20 minutes) or hippocampal slices (overnight). Fixed slices were then washed with PBS before cutting into 40-μm thick sections on a vibratome. Free-floating slice sections or neurons were blocked with 10% normal goat serum, 1% BSA, and 0.1% Na azide in PBS for 2 hrs, and incubated with primary antibodies in 1% BSA. The following primary antibodies were used: phospho-eIF2α (Ser51) (Invitrogen, 44728G), PERK (Cell Signaling,3192), PSD95 (Millipore, 05–494). Alexa Fluor 568 or 488 secondary antibodies (Invitrogen) were used. Following extensive washing, sections were mounted onto pre-subbed slides with Vectashield mounting medium with or without DAPI (Vector laboratories). The sections were imaged using a Leica TCS SP5 confocal microscope (Leica) at 630X. All parameters (pinhole, contrast, gain, offset) were held constant for all sections from the same experiment. The “n” refers to the number of slices, from at least 3 litters/groups of mice.
Data analysis
Data are presented as mean + SEM. Summary data were presented as group means with standard error bars. For comparison between two groups, a two-tailed independent Student’s t-test was used. For comparisons between multiple groups, ANOVA was used followed by individual post hoc tests when applicable. Error probabilities of p < 0.05 were considered statistically significant. The data were collected and processed randomly. No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications 7,52. Data distribution was assumed to be normal but this was not formally tested.
Supplementary Material
Acknowledgments
We thank the late Dr. Mark A. Smith of Case Western Reserve University, for providing postmortem AD brain samples. We thank Dr. Ronald C. Wek of Indiana University School of Medicine for providing the ATF4 antibody. We thank Dr. Charles A. Hoeffer of New York University School of Medicine for help with the design of the mouse breeding. We thank Heather Bowling and Dr. Moses V. Chao of the New York University School of Medicine for providing primary cultured neurons. We thank Dr. Emanuela Santini for advice on the statistical analyses for the mouse behavioral tests. We thank Yiran Zhang for technical help. We thank Maggie Dorsey for keeping colonies of transgenic mice. We thank all Klann laboratory members for comments on the manuscript. This work was supported by National Institutes of Health grants NS034007 and NS047834, and Alzheimer’s Association Investigator grant to E.K. The authors declare no competing financial interests.
Footnotes
Author Contributions T.M. did the majority of the experimental work and data analysis. M.A.T performed Western blots and immunohistochemistry on postmortem human tissue. A.J.W. did OL scoring, performed Western blots for the GCN2 knock mice, and genotyped mice. C.B. and E.G. performed Western blots for the PKR knockout mice. P.P. provided puromycin antibody. P.P. and E.G. also were involved in the experimental design. D.R.C. provided breeders of PERK and GCN2 knockout mice. E.K. directed and supervised the project. T.M. and E.K. designed the experiments and wrote the paper. All authors contributed to the analysis of data, discussion of the results, and the final draft of the paper.
References
- 1.Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr71. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011;17:1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 4.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 5.Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26. doi: 10.1016/j.neuron.2008.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klann E, Dever TE. Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci. 2004;5:931–942. doi: 10.1038/nrn1557. [DOI] [PubMed] [Google Scholar]
- 7.Costa-Mattioli M, et al. Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature. 2005;436:1166–1173. doi: 10.1038/nature03897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costa-Mattioli M, et al. eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell. 2007;129:195–206. doi: 10.1016/j.cell.2007.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang Z, et al. eIF2alpha Phosphorylation-dependent translation in CA1 pyramidal cells impairs hippocampal memory consolidation without affecting general translation. J Neurosci. 2010;30:2582–2594. doi: 10.1523/JNEUROSCI.3971-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klann E, Antion MD, Banko JL, Hou L. Synaptic plasticity and translation initiation. Learn Mem. 2004;11:365–372. doi: 10.1101/lm.79004. [DOI] [PubMed] [Google Scholar]
- 11.Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 12.Chang R, Wong A, Ng H, Hugon J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport. 2002;13:2429–2432. doi: 10.1097/00001756-200212200-00011. [DOI] [PubMed] [Google Scholar]
- 13.Kim SM, et al. Activation of eukaryotic initiation factor-2 α-kinases in okadaic acid-treated neurons. Neuroscience. 2010;169:1831–1839. doi: 10.1016/j.neuroscience.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 14.O’Connor T, et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60:988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jankowsky JL, et al. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- 16.Zhang P, et al. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoeffer CA, et al. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron. 2008;60:832–845. doi: 10.1016/j.neuron.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trinh MA, et al. Brain-Specific Disruption of the eIF2α Kinase PERK Decreases ATF4 Expression and Impairs Behavioral Flexibility. Cell Rep. 2012;1:676–688. doi: 10.1016/j.celrep.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- 20.Cohen E, et al. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma T, et al. Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J Neurosci. 2011;31:5589–5595. doi: 10.1523/JNEUROSCI.6566-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palam LR, Baird TD, Wek RC. Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J Biol Chem. 2011;286:10939–10949. doi: 10.1074/jbc.M110.216093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. 2010;191:1113–1125. doi: 10.1083/jcb.201006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang S, et al. Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. J Neurochem. 2010;115:47–57. doi: 10.1111/j.1471-4159.2010.06899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sacktor TC. How does PKMζ maintain long-term memory? Nat Rev Neurosci. 2011;12:9–15. doi: 10.1038/nrn2949. [DOI] [PubMed] [Google Scholar]
- 26.Bramham CR, et al. The Arc of synaptic memory. Exp Brain Res. 2010;200:125–140. doi: 10.1007/s00221-009-1959-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 28.Leuba G, et al. Differential changes in synaptic proteins in the Alzheimer frontal cortex with marked increase in PSD-95 postsynaptic protein. J Alzheimers Dis. 2008;15:139–151. doi: 10.3233/jad-2008-15112. [DOI] [PubMed] [Google Scholar]
- 29.Tsaytler P, Bertolotti A. Exploiting the selectivity of protein phosphatase 1 for pharmacological intervention. FEBS J. 2013;280:766–770. doi: 10.1111/j.1742-4658.2012.08535.x. [DOI] [PubMed] [Google Scholar]
- 30.Cullinan SB, et al. Nrf2 Is a Direct PERK Substrate and Effector of PERK-Dependent Cell Survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 32.Zhang P, et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol. 2002;22:6681–6688. doi: 10.1128/MCB.22.19.6681-6688.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alberini C. The role of protein synthesis during the labile phases of memory: revisiting the skepticism. Neurobiol Learn Mem. 2008;89:234–246. doi: 10.1016/j.nlm.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paschen W, Proud CG, Mies G. Shut-down of translation, a global neuronal stress response: mechanisms and pathological relevance. Curr Pharm Des. 2007;13:1887–1902. doi: 10.2174/138161207780858401. [DOI] [PubMed] [Google Scholar]
- 35.Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal. 2007;9:2357–2371. doi: 10.1089/ars.2007.1764. [DOI] [PubMed] [Google Scholar]
- 36.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 37.Ma T, Klann E. Amyloid _: Linking Synaptic Plasticity Failure to Memory Disruption in Alzheimer’s Disease. J Neurochem. 2012;120 (Suppl 1):140–148. doi: 10.1111/j.1471-4159.2011.07506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klann E, Richter JD. Translational control of synaptic plasticity and learning and memory. In: Mathews MB, Sonenberg N, Hershey JWB, editors. Translational control in biology and medicine. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2007. pp. 485–506. [Google Scholar]
- 39.Sidrauski C, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife. 2013;2:e00498. doi: 10.7554/eLife.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glenner MDGG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120(3):885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 41.Masters CL, et al. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holtzman DM, Goate A, Kelly J, Sperling R. Mapping the road forward in Alzheimer’s disease. Sci Transl Med. 2011;3:114ps148. doi: 10.1126/scitranslmed.3003529. [DOI] [PubMed] [Google Scholar]
- 43.Miners J, Barua N, Kehoe P, Gill S, Love S. Aβ-degrading enzymes: potential for treatment of Alzheimer disease. J Neuropathol Exp Neurol. 2011;70:944–959. doi: 10.1097/NEN.0b013e3182345e46. [DOI] [PubMed] [Google Scholar]
- 44.Mawuenyega KG, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Apelt J, Ach K, Schliebs R. Aging-related down-regulation of neprilysin, a putative beta-amyloid-degrading enzyme, in transgenic Tg2576 Alzheimer-like mouse brain is accompanied by an astroglial upregulation in the vicinity of beta-amyloid plaques. Neurosci Lett. 2003;339:183–186. doi: 10.1016/s0304-3940(03)00030-2. [DOI] [PubMed] [Google Scholar]
- 46.Hersh LB, Rodgers DW. Neprilysin and amyloid beta peptide degradation. Curr Alzheimer Res. 2008;5:225–231. doi: 10.2174/156720508783954703. [DOI] [PubMed] [Google Scholar]
- 47.Zhan K, Narasimhan J, Wek RC. Differential activation of eIF2 kinases in response to cellular stresses in Schizosaccharomyces pombe. Genetics. 2004;168:1867–1875. doi: 10.1534/genetics.104.031443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamanaka RB, Bennett BS, Cullinan SB, Diehl JA. PERK and GCN2 contribute to eIF2alpha phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol Biol Cell. 2005;16:5493–5501. doi: 10.1091/mbc.E05-03-0268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang HY, et al. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol. 2004;24:1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moreno JA, et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature. 2012;485:507–511. doi: 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsien JZ, et al. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- 52.Banko JL, et al. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci. 2005;25(42):9581–9590. doi: 10.1523/JNEUROSCI.2423-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma T, et al. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS One. 2010;5:e12845. doi: 10.1371/journal.pone.0012845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bonda DJ, et al. Indoleamine 2,3-dioxygenase and 3-hydroxykynurenine modifications are found in the neuropathology of Alzheimer’s disease. Redox Rep. 2010;15:161–168. doi: 10.1179/174329210X12650506623645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barker GRI, Warburton EC. When Is the Hippocampus Involved in Recognition Memory? J Neurosci. 2011;31:10721–10731. doi: 10.1523/JNEUROSCI.6413-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.