

The N-glycans promote solubility in the ER even for mutant glycoproteins, such as mutant α1-antitrypsin. This study shows that enzymatic monoglucosylation activity of the enzyme UGGT1 and lectin chaperone abundance are required for N-glycans to provide maximum solubility to the misfolded substrate.
Abstract
Protein folding in the endoplasmic reticulum (ER) is error prone, and ER quality control (ERQC) processes ensure that only correctly folded proteins are exported from the ER. Glycoproteins can be retained in the ER by ERQC, and this retention contributes to multiple human diseases, termed ER storage diseases. UDP-glucose:glycoprotein glucosyltransferase (UGGT1) acts as a central component of glycoprotein ERQC, monoglucosylating deglucosylated N-glycans of incompletely folded glycoproteins and promoting subsequent reassociation with the lectin-like chaperones calreticulin and calnexin. The extent to which UGGT1 influences glycoprotein folding, however, has only been investigated for a few selected substrates. Using mouse embryonic fibroblasts lacking UGGT1 or those with UGGT1 complementation, we investigated the effect of monoglucosylation on the soluble/insoluble distribution of two misfolded α1-antitrypsin (AAT) variants responsible for AAT deficiency disease: null Hong Kong (NHK) and Z allele. Whereas substrate solubility increases directly with the number of N-linked glycosylation sites, our results indicate that additional solubility is conferred by UGGT1 enzymatic activity. Monoglucosylation-dependent solubility decreases both BiP association with NHK and unfolded protein response activation, and the solubility increase is blocked in cells deficient for calreticulin. These results suggest that UGGT1-dependent monoglucosylation of N-linked glycoproteins promotes substrate solubility in the ER.
INTRODUCTION
Although the amino acid sequence of a protein has all the information required for it to fold into its native functional conformation (Anfinsen, 1973), folding pathways and efficiency are dramatically influenced by the environment (Braakman and Bulleid, 2011). Newly made secretory and transmembrane proteins in the endoplasmic reticulum (ER) require the assistance of chaperones and enzymes to fold into their native conformation (Braakman and Bulleid, 2011). Recognition, retention, refolding, and degradation of misfolded protein substrates in the ER are referred to collectively as ER quality control (Ellgaard et al., 1999; Trombetta and Parodi, 2003; Hebert and Molinari, 2007; Anelli and Sitia, 2008; Braakman and Bulleid, 2011). Protein folding failure in the ER can lead to insolubility by either formation of protein aggregates or ordered polymerization. Once rendered insoluble in the ER, entrapped proteins are degraded (through autophagy) or can persist as undegraded insoluble complexes possibly contributing to cell toxicity, or they may be resolubilized (Ishida and Nagata, 2009; Hidvegi et al., 2010; Rampelt et al., 2012). The mechanisms that promote protein solubility in the ER are poorly understood.
The folding, trafficking, and degradation of glycoproteins is coupled with the modification of N-linked oligosaccharides (Helenius and Aebi, 2004; Hebert and Molinari, 2012). On translocation into the ER, a preassembled oligosaccharide core (Glc3Man9GlcNAc2) is transferred to asparagine residues within the acceptor sequence Asn-Xxx-Ser/Thr. Glucosidases I and II sequentially trim this core to a monoglucosylated glycan that can mediate binding to lectin-like ER chaperones calreticulin (CRT) and calnexin (CNX). Interaction with CRT and CNX serves to retain improperly folded substrates in the ER as a key step in glycoprotein quality control, as well as to promote folding through interactions with ERp57 (Zapun et al., 1998). On release from CRT/CNX, glucosidase II can remove the remaining glucose to produce a product that, if folded properly, traffics to the Golgi or, if folded improperly, is directed to ER-associated degradation (ERAD) or is subject to reglucosylation for interaction with CRT/CNX. Uridine diphosphate (UDP)-glucose:glycoprotein glucosyltransferase (UGGT1) is proposed to be a central gatekeeper for ER quality control for glycoproteins, as it recognizes the partially folded status as a substrate to reglucosylate the deglucosylated N-glycan to direct another round of interaction with CRT/CNX (Maattanen et al., 2010; Hebert and Molinari, 2012).
There are two UGGT genes in mouse, Uggt1 and Uggt2 (Arnold et al., 2000). The product of Uggt1 has reglucosylation activity in vitro, and its deletion destroys reglucosylation activity in cells and is embryonic lethal at day E13 in mice (Molinari et al., 2005). In contrast, the product of Uggt2 has not been demonstrated to have reglucosylation activity, and its function is unknown. Of interest, exchange of the UGGT1 80% amino-terminal substrate recognition domain into UGGT2 partially restores reglucosylation activity in vitro, demonstrating that the carboxy-terminal 20% of UGGT2 can function as a glucosyltransferase in the proper context (Arnold and Kaufman, 2003).
Whereas the purported mechanism for UGGT1-mediated quality control—reiterative monoglucosylation of deglucosylated N-glycans on incompletely folded glycoproteins, and subsequent rounds of association of these glycoproteins with CRT/CNX—has been extensively described (Sousa et al., 1992; Sousa and Parodi, 1995; Parodi, 2000; Hebert et al., 1995; D'Alessio et al., 2010), the extent to which UGGT1 influences the susceptibility of ER substrate proteins to form insoluble protein aggregates or polymers is unclear. In this study, we use two different α1-antitrypsin (AAT) variants prone to misfolding to investigate the effect of UGGT1 on the solubility of these glycoprotein substrates. One variant is the well-studied ERAD substrate known as null Hong Kong variant (α1-antitrypsin–null Hong Kong variant [NHK]); the other is the more common mutant Z allele (α1-antitrypsin Z allele variant [ATZ]). Both alleles cause AAT deficiency (the most common genetic liver disease in children), which is a representative of the class of “conformational diseases” caused by retention of misfolded glycoproteins in the ER (Kim and Arvan, 1998; Rutishauser and Spiess, 2002; Schroder and Kaufman, 2005; Perlmutter, 2006). In this article, using Uggt1−/− mouse embryonic fibroblasts (MEFs) expressing NHK or ATZ and complemented with plasmid-encoded UGGT1, we demonstrate that UGGT1 enzymatic activity, in conjunction with lectin-like chaperones, helps to limit aggregation and polymer formation of misfolded glycoprotein substrates, decreasing their BiP association and ER stress.
RESULTS
UGGT1 increases the solubility of AAT mutants NHK and ATZ
Uggt1−/− MEFs have undetectable levels of UGGT1 protein and enzymatic monoglucosylation activity (Molinari et al., 2005). UGGT1 in the ER is associated with misfolded NHK and ATZ variants of AAT (Choudhury et al., 1997; Schmidt and Perlmutter, 2005). ATZ is predisposed to form insoluble ordered polymers (Gooptu et al., 2009), whereas ER-entrapped NHK can be recovered in a detergent-soluble pool in UGGT1-expressing cells (Liu et al., 1997).
To directly investigate the effect of UGGT1 on NHK, ATZ, or wild-type AAT (wt-AAT) solubility, we metabolically labeled Uggt1−/− MEFs (or those complemented with plasmid-encoded UGGT1) expressing these substrates with [35S]methionine/cysteine to approach steady state. By quantitative immunoprecipitation (IP; Supplemental Figure S1A), we determined that over the course of the metabolic labeling period (24 h), a fraction of mutant NHK molecules was secreted; a second portion was recovered in the intracellular detergent-soluble supernatant fraction; and a third was recovered as aggregates that failed to be solubilized from Uggt1−/− MEFs lysed in nonionic detergents (Figure 1A). This “detergent-insoluble fraction” was indeed solubilized in SDS-containing detergent extracts.
FIGURE 1:

UGGT1 increases the solubility of α1-antitrypsin mutants NHK and ATZ. (A–C) The 24-h metabolic labeling of Uggt1−/− MEFs transfected with (A) NHK, (B) ATZ, or (C) wt-AAT ± UGGT1 cotransfection. Uggt1−/− MEFs were transfected with expression vectors encoding the misfolded α1-antitrypsin variants NHK or ATZ or wild-type AAT (wt-AAT) and cotransfected with either empty vector or UGGT1 expression vector. Cells were radiolabeled from 24 to 48 h posttransfection in complete medium plus 10 μCi/ml [35S]methionine/cysteine. Extracellular, soluble, and insoluble fractions were produced (Materials and Methods), and TCA-precipitation normalized volumes of each fraction were subjected to double–anti-AAT quantitative IP and PNGaseF digestion and analyzed by reducing SDS–PAGE and autoradiography. Substrate solubility was calculated by dividing total soluble (extracellular + intracellular soluble) substrate amount by insoluble substrate amount. Middle, Student's t test used for statistical testing; right, paired Student's t test used for statistical testing (*p ≤ 0.05). Error bars represent SEM. For ATZ, all UGGT1+ bands were darkened with imaging software to make synthesis (data not shown) ± UGGT1 appear equal.
Whereas endoglycosidase H (EndoH)-resistant NHK molecules were found in the secreted fraction, both the soluble and detergent-insoluble intracellular fractions were EndoH sensitive, indicating that neither the soluble nor the insoluble intracellular pool had arrived in a Golgi compartment from which they could be further modified by Golgi N-glycan processing enzymes (Figure 2D). We confirmed by several independent approaches that a fraction of mutant NHK molecules forms detergent-insoluble protein aggregates. First, pulse-chase experiments demonstrated that newly synthesized NHK was not initially recovered in the detergent-insoluble fraction but became so as a function of chase, indicating time-dependent specificity (Figure 2C). Second, we compared solubility of NHK using three standard detergent lysis methods that have been used for analysis of AAT and its variants (Schmidt and Perlmutter, 2005; Kroeger et al., 2009; Galli et al., 2011): 1) 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate (CHAPS; 2% wt/vol); 2) Nonidet P-40 (1% vol/vol); and 3) Triton X-100 (0.5% wt/vol) and sodium deoxycholate (0.5% wt/vol). In each case, a detergent-insoluble fraction of NHK was recovered (Supplemental Figure S1B). Finally, intracellular wt-AAT was always undetectable in the insoluble fraction (Figures 1C and 2A), indicating that recovery in the detergent-insoluble pool was not caused by failure to disrupt cellular membranes from Uggt1−/− MEFs. These findings, together with other results (Greenblatt et al., 2011), provide strong evidence that NHK is predisposed to aberrant protein aggregation.
FIGURE 2:

NHK and ATZ, but not wt-AAT, become insoluble over time. (A) Transient transfection of Uggt1−/− MEFs with wt-AAT ± UGGT1 and analysis by pulse chase at the indicated time points (Materials and Methods). No wt-AAT was detected in the insoluble fraction at any time point. (B) Same as A for ATZ. Here ATZ progressively accumulates in the insoluble fraction over time. (C) Same as A for NHK. Here NHK progressively accumulates in the insoluble fraction over time. Soluble NHK was run on a nonreducing gel because under reducing conditions, the reduced IP antibody physically pushes all NHK bands down to the same molecular weight. For A–C, four times the TCA-normalized amount of insoluble fraction was used for IP compared with soluble fraction (actual relative amount of insoluble ATZ and NHK is 25% of what is seen in the gel image). (D) EndoH digest of extracellular, soluble, and insoluble NHK after steady-state labeling. The majority of extracellular NHK is EndoH resistant, evidence of having Golgi-modified N-glycans typically found on secreted glycoproteins. NHK in the soluble and insoluble fractions is completely EndoH sensitive, indicating that these forms are not modified by Golgi-localized N-glycan–modifying enzymes.
Complementation of Uggt1−/− MEFs with UGGT1 greatly expanded the steady-state intracellular pool of NHK molecules (Figure 1A). Whereas only half as many NHK molecules were secreted, there was a great expansion of the pool of soluble intracellular NHK molecules (Figure 1A, middle, red bar) and a decrease in the pool of detergent-insoluble NHK (Figure 1A, middle, blue bar). In addition, the total soluble (extracellular plus soluble)/insoluble NHK ratio increased with UGGT1 cotransfection (Figure 1A, right). Thus the net effect of coexpressing UGGT1 was to increase the solubility of NHK.
The same experimental analysis was used to investigate ATZ trafficking in the ER. Whereas NHK cannot undergo loop-sheet polymerization, recovery of detergent-insoluble ATZ is believed to reflect the formation of ordered polymers (Gooptu et al., 2009). Here as well, UGGT1 expression increased the total soluble/insoluble ratio of ATZ almost twofold (with little effect on the secreted fraction; Figure 1B). In addition, similar to NHK, in pulse-chase experiments, ATZ was not detected in the detergent-insoluble fraction initially but accumulated in the insoluble fraction over time (Figure 2B). Furthermore, to directly detect ATZ polymers in transfected cells, we used a recently developed monoclonal antibody (2C1) that specifically recognizes ATZ polymers (Miranda et al., 2010). Immunofluorescence costaining with 2C1 and polyclonal anti-AAT antibody and subsequent quantitation of signal intensity in the entire cell volume through Z-stack analysis revealed a decrease in the ratio of 2C1 to AAT signal in cells cotransfected with UGGT1 (Supplemental Figure S2, A and B).
Thus, for mutant glycoproteins that are prone to misfolding in two distinctly different ways (formation of ordered polymers [ATZ] or formation of disordered aggregates [NHK]), UGGT1 increased substrate solubility. By contrast, UGGT1 had no significant effect on the behavior of wt-AAT, consistent with the observation that wt-AAT molecules were soluble in Uggt1−/− MEFs even before complementation with UGGT1 (Figure 1C).
UGGT1 promotes NHK solubility via monoglucosylation activity
To investigate whether increased NHK solubility was due to UGGT1 chaperone function or enzymatic reglucosylation activity (Parodi, 2000), we eliminated the enzymatic contribution of UGGT1 either by glucosidase inhibition (with castanospermine [CAS], which prevents formation of unglucosylated N-glycans that serve as substrates for UGGT1-mediated monoglucosylation) or complementation with UGGT1 variants lacking monoglucosylation activity. When CAS was included throughout the labeling period, UGGT1 did not increase solubility of NHK (Figure 3A). Furthermore, a catalytically inactive UGGT1 (lacking six amino acids within the catalytic site, called ΔUGGT1; Arnold et al., 2000) was similarly unable to augment the solubility of NHK (Figure 3A). A UGGT1/UGGT2 chimera (in which the C-terminal 20% of UGGT2 replaced the C-terminal 20% of UGGT1), however, bearing approximately half of the enzymatic activity of native UGGT1 (Arnold and Kaufman, 2003) increased NHK solubility, and this effect was similarly eliminated by CAS treatment (Figure 3A). Of importance, these various UGGT1 constructs were expressed at comparable levels (Supplemental Figure S4). In parallel to these NHK results, we found that enhanced solubility of ATZ also correlated with the presence of enzymatic UGGT1 monoglucosylation activity (Supplemental Figure S3).
FIGURE 3:

UGGT1 promotes solubility of NHK through monoglucosylation activity. (A) Cotransfection with NHK and UGGT1 engineered variants and analysis of NHK. After 24 h metabolic labeling in the presence or absence of glucosidase inhibitor castanospermine, total soluble/insoluble ratios of NHK were determined. An increased total soluble/insoluble ratio was observed only when UGGT1 reglucosylation activity was expected, and this effect was blocked by glucosidase inhibition. Error bars represent SEM. (B) Estimate of the proportion of soluble NHK associated with CNX in the various experimental conditions from A. Soluble material was subjected to anti-CNX/anti-AAT antibody tandem IP and anti-AAT antibody double IP. Appreciable NHK was only detected after anti-CNX/anti-AAT tandem IP when UGGT1 reglucosylation activity and monoglucosylated substrate was expected. (C) JBM and UGGT1 activity. (D) JBM digestion of N-glycans on NHK immunopurified from 24-h-labeled soluble and insoluble fractions. Bands corresponding to monoglucosylated or unglucosylated N-glycans are as indicated. Soluble and insoluble fractions were run on the same gel, but insoluble bands were darkened with imaging software to appear similarly dense to soluble bands.
Because lectin-like chaperones CRT and CNX recognize monoglucosylated N-glycans, as an indirect measure of monoglucosylation we examined NHK association with CNX at the end of the 24-h metabolic labeling of Uggt1−/− MEFs ± UGGT1 complementation. As in previous experiments, 2% CHAPS was included in the cell lysis buffer to maintain CNX–substrate interactions (Solda et al., 2007). Only when Uggt1−/− MEFs were complemented with enzymatically active glucosyltransferase, however, was the CNX–NHK association detected (Figure 3B).
To directly establish that increased NHK solubility in UGGT1-expressing cells is caused by enzymatic monoglucosylation activity, we used jack-bean α-mannosidase (JBM; an exomannosidase that cleaves eight mannose residues from unglucosylated ER-type N-glycans but only five mannose residues from monoglucosylated ER-type N-glycans; Figure 3C) to create diagnostic digests to assess the glycosylation status of steady-state soluble and detergent-insoluble pools. On JBM digestion, intracellular soluble NHK from UGGT1-expressing MEFs produced a predominant higher–molecular mass digestion product distinct from the primary form recovered in Uggt1−/− MEFs (Figure 3D, left). By contrast, in spite of UGGT1 expression, intracellular detergent-insoluble NHK, upon JBM digestion, produced a predominant lower–molecular mass digestion product that comigrated with that from UGGT1-deficient MEFs (Figure 3D, right). Strikingly, upon UGGT1 complementation, the majority of detergent-insoluble NHK lacks monoglucosylated N-glycans, whereas the majority of soluble NHK from the same cells bear monoglucosylated N-glycans. The findings strongly suggest that increased solubility of the substrate is a consequence of its N-glycan monoglucosylation.
UGGT1 maintains solubility of NHK over time
As noted, all newly synthesized NHK, ATZ, and wt-AAT molecules begin in the soluble fraction; thus the accumulation of NHK and ATZ molecules takes time. Although steady-state labeling provides useful conditions for determining the sizes of soluble and insoluble intracellular pools, it does not provide insight into the entry or exit of molecules in these fractions. Therefore, after steady-state labeling, we followed pools of NHK for up to 6 h in the absence of further metabolic labeling. As previously observed (Figure 1A), intracellular NHK solubility was enhanced in UGGT1-expressing cells (Figure 4, A and B). At 3 and 6 h after the steady-state labeling period, the pool of soluble NHK decreased in Uggt1−/− MEFs both without and with UGGT1 complementation. Without UGGT1 complementation, however, some of the molecules formerly in the soluble pool entered and expanded the detergent-insoluble pool (Figure 4, A and B), whereas in the presence of UGGT1, expansion of the detergent-insoluble pool was prevented. Indeed, despite a decrease in soluble NHK as a function of chase time, the relative increase in NHK solubility as a consequence of UGGT1 activity was maintained at all times (Figure 4C). Thus the results suggest that UGGT1 activity limits misfolded glycoprotein entry into detergent-insoluble complexes.
FIGURE 4:
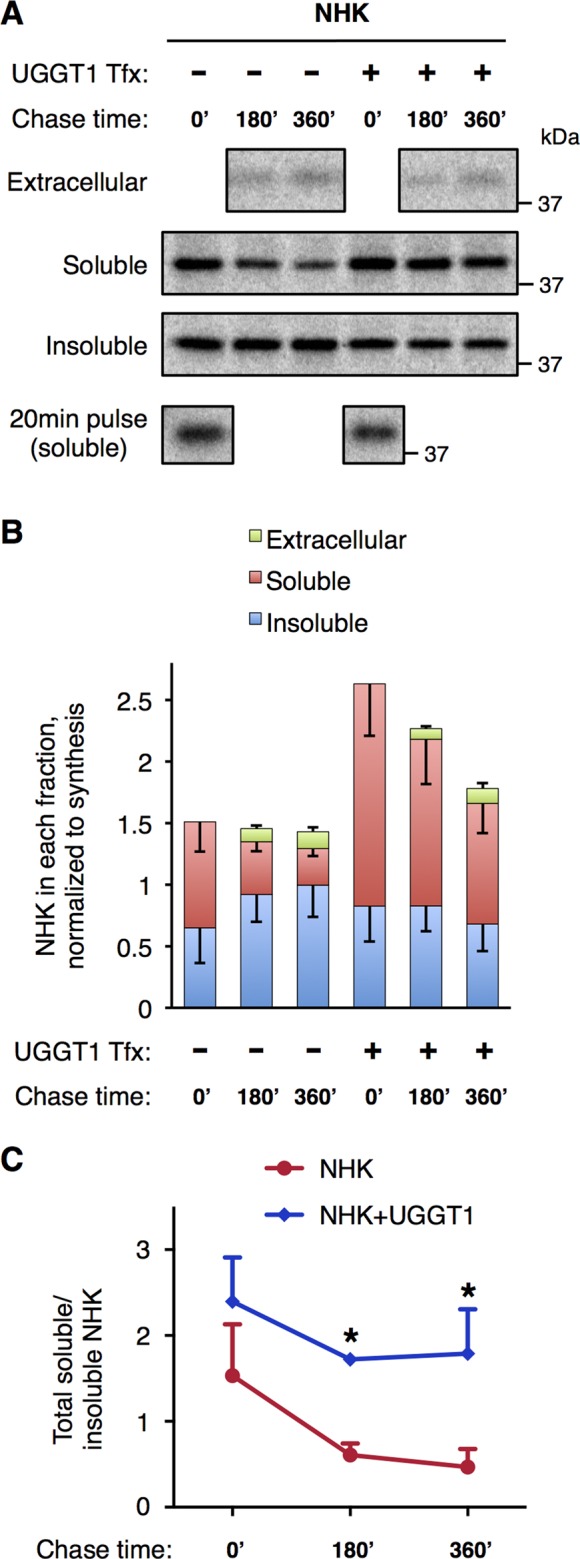
UGGT1 maintains increased total soluble/insoluble ratio for NHK during 6-h chase after 24-h labeling. (A) The 24-h labeling and chase analysis of NHK. After 24-h labeling, cells were chased with unlabeled media for 3 or 6 h, and then extracellular, soluble, and insoluble fractions were subjected to anti-AAT double IP. The amount of newly synthesized NHK recovered from 20-min pulse labeling was used to quantify NHK synthesis for normalization. (B) Total amount of NHK remaining in each fraction after chase, normalized to synthesis. Error bars represent SEM. (C) Plot of total soluble/insoluble NHK at 0-, 3-, and 6-h chase points. Student's t test was used for statistical testing (*p ≤ 0.05). Error bars represent SEM.
ERAD inhibition increases UGGT1-dependent NHK solubility
To investigate whether NHK is degraded by ERAD during the course of our experiments, we treated NHK-transfected cells with kifunensine (KIF; a mannosidase inhibitor) for the entire 24-h metabolic labeling period. KIF potently inhibits glycoprotein ERAD (Tokunaga et al., 2000), including the degradation of NHK (Brodsky, 2007; Hosokawa et al., 2007). By measuring the amount of steady-state NHK recovered in all fractions and normalizing this total to its synthesis in the same cells, we could deduce the amount of NHK degraded under each experimental condition (Figure 5, A and B). When KIF was present, the amount of NHK recovered in the insoluble fraction increased, both with and without UGGT1 cotransfection (Figure 5, A and B). In the presence of UGGT1, however, KIF treatment also promoted an increase in total soluble NHK (Figure 5, A–C). Similarly, a pulse-chase technique showed that when UGGT1 was present, KIF treatment stabilized soluble NHK in cells (Figure 5D). Of interest, this KIF-induced increase in soluble NHK was not seen in Uggt1−/− MEFs cotransfected with empty vector (Figure 5B). Given that ERAD inhibition expands NHK solubility caused by UGGT1 activity, the effect of UGGT1 to promote the solubility of NHK in the absence of ERAD inhibition is likely to be larger than the magnitude shown in Figures 1–3.
FIGURE 5:
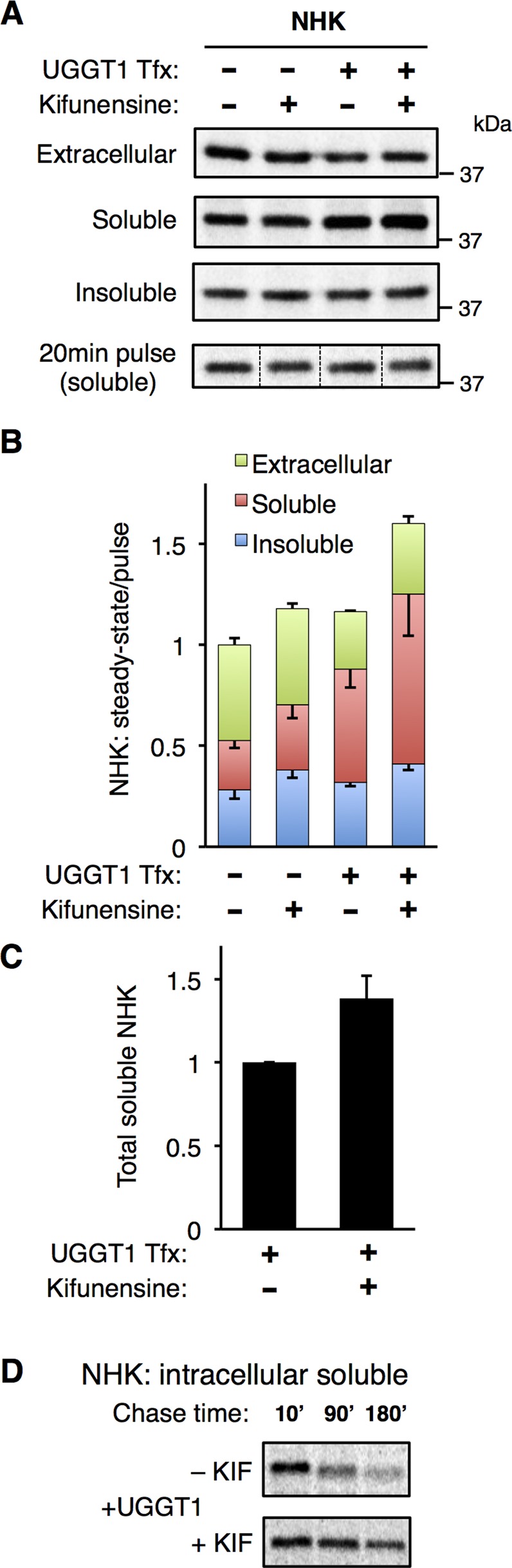
ERAD inhibition increases UGGT1-dependent NHK solubility. (A) Effect of kifunensine on intracellular NHK. After cotransfection with NHK and UGGT1, 24-h metabolic labeling, and treatment with the mannosidase inhibitor kifunensine, amounts of NHK in extracellular, soluble, and insoluble fractions were determined. Amount of newly synthesized NHK recovered from 20-min pulse labeling was used to quantify NHK synthesis for normalization. (B) Quantification of NHK in extracellular, soluble, and insoluble fractions, normalized to synthesis. Error bars represent SEM. (C) Total soluble NHK from B, with UGGT1 cotransfection, with and without kifunensine treatment. Error bars represent SEM. (D) Pulse-chase experiment demonstrating effect of kifunensine to limit degradation of soluble NHK when UGGT1 is present.
Relative contribution of N-glycan addition versus that of monoglucosylation to the solubility of misfolded mutant AAT
NHK, ATZ, and wt-AAT all have three N-linked glycosylation sites at positions N46, N83, and N247. To investigate the contribution of each of these N-glycans to NHK solubility, we eliminated N-glycosylation at these sites by mutagenizing each Asn acceptor to Gln and prepared all possible combinations of one-site or two-site N-glycosylation mutants. We then measured the total soluble/insoluble ratio for each of the seven constructs with or without UGGT1 complementation (Figure 6, A and B). The majority of unglycosylated NHK (NHK-N46Q_N83Q_N247Q or NHKQQQ) was insoluble, and UGGT1 cotransfection had no effect on the solubility of this construct (Figure 6B, bars 1 and 2). For the NHK constructs with only one N-glycosylation site (NHK-N83Q_N247Q, NHK-N46Q_N247Q, and NHK-N46Q_N83Q), in the absence of UGGT1, solubility did not increase, but solubility was enhanced by expression of UGGT1 (Figure 6B, bars 3–8). The largest UGGT1-mediated solubility increase occurred with the NHK-N46Q_N247Q mutant, perhaps indicating particular importance of the N83 N-glycan (Figure 6B, bars 5 and 6). For the NHK constructs with two N-glycosylation sites (NHK-N247Q, NHK-N83Q, NHK-N46Q), solubility increased both with and without UGGT1, but the increase was greater when UGGT1 was present (Figure 6B, bars 9–14), and each of the double-glycan mutants behaved comparably. Similar to Figure 1A, the fully glycosylated protein was the most soluble NHK construct, but UGGT1 expression conferred additional solubility (Figure 6B, bars 15 and 16). The results indicate that each N-linked glycan contributes to the solubility of NHK, but maximal solubility is achieved only in UGGT1-expressing cells.
FIGURE 6:

UGGT1-mediated solubility of NHK requires N-glycan on NHK. (A) Analysis of NHK N-glycosylation mutants in transfected Uggt1−/− MEFs. Uggt1−/− MEFs were transfected with expression vectors encoding NHK N-glycosylation–site mutants with all combinations of the three N-glycosylation sites mutated (Asp(N) to Gln(Q); 1, NHK-N46,83,247QQQ; 2, NHK-N83,247QQ; 3, NHK-N46,247QQ; 4, NHK-N46,83QQ; 5, NHK-N247Q; 6, NHK-N83Q; 7, NHK-N46Q), cotransfected with either empty vector or UGGT1 expression vector, and subjected to steady-state labeling analysis. (B) Quantification of three replicates of experiment in A. A significant portion of the increased solubility of NHK with increasing number of N-glycans added is due to UGGT1 reglucosylation activity. Student's t test was used for statistical testing (*p ≤ 0.05). Asterisks directly above bars indicate a significant difference in total soluble/insoluble ratio between that bar and nonglycosylated NHK (NHKQQQ), either with or without UGGT1 cotransfection. Error bars represent SEM.
UGGT1 cotransfection reduces NHK association with BiP and BiP-luciferase response
The binding of unfolded and misfolded proteins in the ER to BiP activates the unfolded protein response (UPR; Dorner et al., 1987, 1989; Malhotra and Kaufman, 2007). To examine BiP association with NHK, we analyzed dithiobis succinimidyl propionate (DSP)–cross-linked cell lysates by immunoprecipitation with anti-AAT and subjected the immunoprecipitates to reducing SDS–PAGE and immunoblotting with anti-BiP and anti-AAT. Consistent with the metabolic labeling results of Figure 1A, there was increased NHK in the soluble fraction of Uggt1−/− MEFs complemented with UGGT1 (Figure 7A, bottom). Despite the fact that there was more NHK present in the soluble pool, however, the actual amount of BiP associated with NHK in that pool decreased (Figure 7A; quantified in B). To investigate UPR activation in cells expressing NHK, ATZ, or wt-AAT, we coexpressed BiP-luciferase and normalized the signal from the ER stress response reporter directly to the abundance of the misfolded protein substrate. Of interest, neither wt-AAT nor mutant ATZ expression (data not shown) increased BiP-luciferase activity, regardless of the presence or absence of UGGT1 expression (Figure 7C, bars 1 and 2), consistent with previous reports (Graham et al., 1990; Hidvegi et al., 2005, 2007). By contrast, NHK expression induced an appreciable BiP-luciferase response that was attenuated in cells complemented with UGGT1 (Figure 7C, bars 3 and 4). Of note, UGGT1 complementation did not diminish (and actually augmented) UPR activation to tunicamycin treatment, presumably because of the increased protein load in the ER and the fact that reglucosylation activity is irrelevant in cells in which N-linked glycosylation has been blocked by tunicamycin (Figure 7C, bars 5 and 6). Taken together, the results of Figure 7 indicate that BiP association with a misfolded glycoprotein substrate decreases as a consequence of substrate monoglucosylation, which reduces UPR activation in these cells.
FIGURE 7:
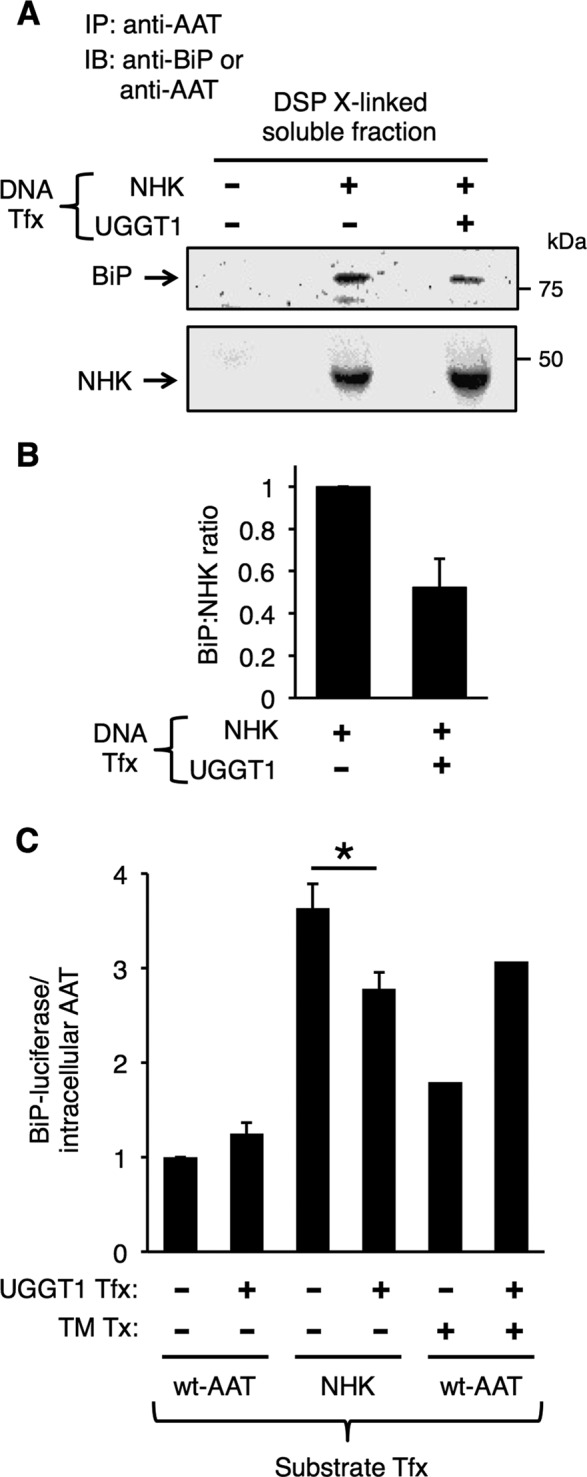
UGGT1 cotransfection reduces NHK association with BiP and reduces expression from the BiP promoter. (A) Amount of BiP associated with NHK determined by immunoblotting of soluble material from DSP-cross-linked Uggt1−/− MEFs, with or without UGGT1 cotransfection, with anti-BiP or anti-AAT antibodies. (B) Quantification of three replicates of experiment in A. UGGT1 cotransfection reduces the amount of BiP associated with NHK per molecule. Error bars represent SEM. (C) BiP-luciferase response to transient transfection of NHK or wt-AAT normalized to total intracellular (soluble + insoluble) amount of AAT (we determined that expression of a cotransfected Renilla luciferase is an inappropriate tool for BiP-luciferase normalization). Tunicamycin treatment (6 h, 5 μg/ml) was included in cells transfected with AAT as a control for UPR activation. Student's t test was used for statistical testing (*p ≤ 0.05). Error bars represent SEM.
The ER lectin calreticulin is linked to increased solubility of monoglucosylated glycoprotein substrate
To explore whether UGGT1-mediated solubility of NHK is due to increased interaction with lectin-like chaperones, we performed CRT small interfering RNA (siRNA)–mediated knockdown in Uggt1−/− MEFs (CRT, the most abundant soluble ER lectin, is likely to interact with a high fraction of soluble glycoprotein substrates). Because calreticulin–substrate association is weak, we used DSP cross-linking to stabilize NHK–CRT interactions that might otherwise be lost when washing the immunoprecipitated beads (Patil et al., 2000). In both DSP cross-linked and non–cross-linked cell lysates, interaction between NHK and CRT was readily demonstrated (Figure 8A). CRT siRNA knockdown (KD) was > 50% efficient in Uggt1−/− MEFs at 48 h post-siRNA treatment, and this did not up-regulate other ER chaperones, as well as CNX (Figure 8B). On NHK transfection in these cells, however, CRT KD largely blocked the increase of NHK solubility caused by UGGT1 complementation (Figure 8, C–E). In addition, CRT KD increased the proportion of insoluble NHK and blocked the effect of UGGT1 to decrease the proportion of insoluble NHK (Figure 8D). The data indicate that abundance of CRT is a key factor in promoting solubility of monoglucosylated N-linked glycoprotein substrates in the ER.
FIGURE 8:

Calreticulin siRNA knockdown reduces UGGT1-mediated increased solubility of NHK. (A) Tandem anti-AAT/anti-CRT IP of soluble material from Uggt1−/− MEFs cotransfected with NHK and UGGT1 and treated with or without DSP cross-linking. The results demonstrate association between NHK and CRT. (B) Effect of CRT knockdown on chaperone expression. CRT siRNA transfection greatly reduces the amount of CRT protein without any compensatory increase in CNX, BiP, or GRP94 protein. (C) Steady-state labeling analysis of Uggt1−/− MEFs transfected with CRT or NegCtrl siRNAs and cotransfected with NHK and either UGGT1 or empty vector (Materials and Methods). (D) Quantification of C, each fraction represented as proportion of total. (E) The effect of UGGT1 on the total soluble/insoluble ratio of NHK upon CRT knockdown. Student's t test was used for statistical testing (*p ≤ 0.05). Error bars represent SEM.
DISCUSSION
Protein solubility, or, more important, the formation of insoluble protein aggregates or polymers, is believed to play a fundamental role in the pathology of a wide array of human diseases (Balch et al., 2008). Mutations in secreted proteins can disrupt their normal ER folding program, resulting in aggregation and cell stress/death (Wu and Kaufman, 2006; Wang and Kaufman, 2012; Menon et al., 2007). Regulation of aggregate/polymer formation and the role of chaperone interaction in this process are incompletely understood. In this article we describe how UGGT1 enzymatic activity correlates with increased solubility of two model glycoproteins susceptible to aggregation and polymerization.
A large body of work has delineated the key role of UGGT1 in reglucosylation of nonnative glycoprotein folding intermediates and subsequent reassociation with the lectin-like chaperones CRT and CNX (Sousa et al., 1992; Sousa and Parodi, 1995; Parodi, 2000; Hammond et al., 1994; Hebert et al., 1995; Cannon and Helenius, 1999; D'Alessio et al., 2010). Here we show that the ATZ allele of AAT, which is prone to misfolding, formed more insoluble polymers in UGGT1-deficient cells (Figure 1B). The NHK variant of AAT also accumulated in the insoluble fraction, and the proportion detected in the insoluble pool after 24 h of labeling decreased in the presence of UGGT1 activity (Figure 1A). Of interest, UGGT1 maintained the solubility of both nonnative ER forms of these AAT variants, but the consequences of this increased solubility were different for NHK and ATZ. For NHK, soluble nonnative forms were retained in the ER, and the secretion rate was lower upon UGGT1 complementation; thus UGGT1 increases quality control for secretion of nonnative NHK. For ATZ, UGGT1 retained soluble nonnative forms in the ER and decreased the rate of entry into the insoluble fraction without altering secretion. In parallel with these biochemical studies, the amount of ATZ recognized by a polymer-specific antibody also decreased upon UGGT1 complementation (Supplemental Figure S2, A and B). The reason for solubility affecting trafficking of NHK and ATZ in different ways was not elucidated by this study but likely involves different maturation pathways for the two proteins, possibly dependent on retentive ER chaperone interactions and/or interactions with ER exit complexes.
Of importance, our results demonstrate from multiple independent approaches that UGGT1 requires its reglucosylation activity to promote NHK solubility. First, castanospermine treatment, which blocks glucose removal from N-glycans and prevents the formation of unglucosylated N-glycan substrates for UGGT1, decreased UGGT1-mediated solubility. Second, overexpression of a catalytically inactive UGGT1 enzyme did not increase NHK solubility. In contrast, NHK solubility was increased by expression of a chimeric UGGT1/UGGT2 protein that contains the 20% carboxy-terminal catalytic domain of UGGT2 (Arnold and Kaufman, 2003). Of interest, although UGGT2 does not have reglucosylation activity in vitro, the chimeric protein harboring the catalytic domain of UGGT2 can function to promote substrate solubility (Figure 3A). Finally, UGGT1 had no effect on the solubility of NHK with all three N-glycosylation sites disrupted, which prevents formation of the UGGT1 substrate (Figure 6, A and B).
To characterize the eventual fate of NHK molecules in the soluble and insoluble fractions, we performed experiments with 6-h chase after 24-h labeling. Of interest, insoluble NHK was quite stable over a 6-h chase and actually increased in amount with time in the absence of UGGT1, with soluble molecules entering the insoluble fraction (Figure 4, A and B). Theoretically, the amount of NHK in the insoluble fraction after 24 h is determined by three processes: 1) entry from the soluble fraction, 2) degradation of insoluble forms, and 3) solubilization of insoluble forms to enter the soluble fraction. Of interest, monoglucosylated NHK was not detected in the insoluble fraction (Figure 3D), indicating that UGGT1 cannot reglucosylate molecules that entered the insoluble fraction, and therefore UGGT1 may not promote actual resolubilization. We cannot rule out the possibility, however, that insoluble molecules that are reglucosylated may immediately reenter the soluble fraction and thus are not detected in the insoluble fraction based on jack-bean α-mannosidase digestion. Another possibility for the absence of monoglucosylated NHK in the insoluble fraction, based on a previous finding that NHK is found in ER subcompartments called inclusion bodies (Zuber et al., 2007), is that NHK may be physically sequestered away from UGGT1. Finally, perhaps the insoluble NHK forms a tight aggregate such that the recognition site for UGGT1 is not exposed.
Because ERAD is believed to require soluble substrates for degradation (Nakatsukasa and Brodsky, 2008), the finding that UGGT1 promotes glycoprotein solubility could have implications for the degradation of glycoproteins in the ER. Indeed, upon ERAD inhibition with kifunensine, total soluble NHK increased, and this effect was most prominent when UGGT1 was present (Figure 5, A–C). Insoluble NHK also seemed to increase upon ERAD inhibition, presumably due to the greater amount of soluble NHK available for entry into the insoluble fraction (although we cannot rule out that kifunensine may directly inhibit degradation of insoluble NHK).
Another finding from this study involves the contribution of the three N-glycans of NHK to its solubility. First, we found that although solubility of NHK did increase directly by adding hydrophilic N-glycans to the polypeptide, a major fraction of this solubilization was UGGT1 dependent (Figure 6, A and B). This indicates that, at least for NHK, UGGT1-mediated reglucosylation contributes to substrate solubility, in addition to solubility afforded by adding hydrophilic N-glycans to the hydrophobic polypeptide. CRT knockdown greatly decreased NHK solubilization in response to UGGT1 enzymatic activity (Figure 8, C–E). From this, we infer that this UGGT1-dependent solubilization effect is due to reassociation with CRT/CNX and is not a direct contribution from the glucose added to the N-glycan A-chain.
The CNX cycle hypothesis (Hammond et al., 1994) proposes that UGGT1 reglucosylates nonnative glycoprotein folding intermediates and promotes their reassociation with CRT/CNX for further folding attempts. We directly tested how BiP interaction is involved in UGGT1-enhanced NHK solubility. We showed that, whereas the amount of soluble NHK increased with UGGT1 overexpression, the amount of BiP bound to soluble NHK clearly decreased (Figure 7, A and B). This appears to be an example of BiP acting as a second-level ER quality controller for glycoproteins when interaction with their primary set of chaperones is disturbed (Molinari et al., 2002, 2005).
The role of UGGT1 in glycoprotein trafficking is intimately linked with the lectin-like chaperones CNX and CRT (D'Alessio et al., 2010). Previous studies implied that NHK does not interact with calreticulin (Oda et al., 2003; Cameron et al., 2009); we found, however, that NHK extensively associates with CRT in Uggt1−/− MEFs cotransfected with NHK and UGGT1 (Figure 8A). siRNA-mediated CRT knockdown strikingly reduced the ability of UGGT1 to increase NHK solubility (Figure 8, C–E). The fact that >99% of intracellular CRT resides in the soluble fraction (data not shown) presumably explains its ability to promote the solubility of monoglucosylated NHK.
The mechanisms governing protein solubility and quality control in the ER are incompletely understood, but this study highlights how the ER enzyme UGGT1 can improve glycoprotein quality control by promoting solubility of glycoprotein substrates that are prone to misfolding (Figure 9). Our results contribute to the CNX cycle theory by demonstrating that UGGT1-dependent lectin interaction can increase glycoprotein substrate solubility in the ER. Further studies on the effect of glycan modifications and chaperone interactions should provide additional clues to the mechanism(s) maintaining solubility of glycoproteins during ER folding.
FIGURE 9:

UGGT1 promotes solubility of glycoproteins by promoting interaction with CNX and CRT. Model depicting the role of UGGT1 in promoting solubility of glycoproteins by promoting interaction with CNX and CRT. Monoglucosylated NHK is found only in the soluble fraction and not in the insoluble fraction. NHK is associated with CRT and CNX in the presence of UGGT1, and this interaction may be a mechanism for maintaining solubility of NHK. Therefore, at the hypothesized decision point at which unglucosylated NHK can exit the ER, be degraded by ERAD, form insoluble aggregates, or be monoglucosylated by UGGT1, we hypothesize that UGGT1 promotes a decision by NHK to remain soluble.
MATERIALS AND METHODS
Reagents and antibodies
Castanospermine was from Tocris Bioscience (Ellisville, MO). Kifunensine was from Cayman Chemical (Ann Arbor, MI). Protein A–Sepharose, CHAPS, N-ethylmaleimide (NEM), phenylmethanesulfonyl fluoride (PMSF), leupeptin, antipain, pepstatin A, DSP, and JBM were from Sigma-Aldrich (St. Louis, MO). Chymostatin was from VWR (Radnor, PA). EndoH and PNGaseF were from New England BioLabs (Ipswich, MA). Rabbit anti-AAT antibody was from DAKO (Carpinteria, CA). Mouse monoclonal antibody (mAb) anti-KDEL and rabbit anti-CNX (used for immunoblotting) were from Enzo Life Sciences (Plymouth Meeting, PA). Rabbit anti-CRT was from Thermo Scientific (Waltham, MA). Rabbit mAb anti–glyceraldehyde-3-phosphate dehydrogenase was from Cell Signaling (Beverly, MA). Rabbit anti-BiP was from P. Arvan. Rabbit anti-CNX (used for IP) antibody was a kind gift from A. Helenius (Institute of Biochemistry, ETH Zürich, Zurich, Switzerland). Rabbit polyclonal anti-UGGT1 antibody was a kind gift from A. Parodi (Laboratory of Glycobiology, Fundación Instituto Leloir, Buenos Aires, Argentina). Mouse monoclonal anti-antitrypsin polymer antibody (2C1) was a kind gift from E. Miranda (Dipartimento di Biologia e Biotecnologie ‘‘Charles Darwin’’ and Istituto Pasteur-Fondazione Cenci Bolognetti, Universita di Roma ‘‘La Sapienza,’’ Rome, Italy) and D. Lomas (Department of Medicine, University of Cambridge, Cambridge Institute for Medical Research, Cambridge, UK).
cDNAs and constructs
NHK and ATZ mutant constructs were made by QuikChange mutagenesis (Stratagene, Santa Clara, CA) from wt-AAT in pcDNA3.1 using standard methods. wt-AAT, NHK, and ATZ constructs were modified by S. H. Back (School of Biological Sciences, University of Ulsan, Ulsan, Republic of Korea) to express AAT and enhanced green fluorescent protein as two separate proteins from a single mRNA by cap-dependent and internal ribosome entry site–dependent translation. UGGT1 and variants were previously published (Arnold and Kaufman, 2003). NHK N-glycosylation–site mutants were constructed using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Stratagene).
Cell culture
Uggt1−/− MEFs (Molinari et al., 2005) were maintained in DMEM (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum and Glutamax (Life Technologies). Cells were cultured at 37°C in a 5% CO2 incubator.
Steady-state metabolic labeling/pulse-labeling analysis
Uggt1−/− MEFs were transiently transfected using Lipofectamine 2000 transfection reagent (Life Technologies) with AAT constructs and either UGGT1 (1:3 M ratio AAT:UGGT1) or an equal microgram amount of empty vector (pEDΔC). The next day, cells were trypsinized and plated in separate six-well plates for pulse labeling or steady-state analysis. Complete medium supplemented with 0.1 mCi/ml [35S]methionine and [35S]cysteine (Tran35S-label; MP Biomedicals, Solon, OH) was added to steady-state wells 24 h posttransfection, and media and cells were collected 48 h posttransfection (steady-state labeling). Cells in the remaining wells were starved for 20 min in media lacking Met/Cys, pulsed for 20 min with starvation medium supplemented with 0.1 mCi/ml [35S]Met/Sys, and collected in the same manner as described next for steady-state labeling.
For each well, medium was collected on ice and centrifuged at 10,000 × g for 10 min at 4°C, and this cleared supernatant was called the extracellular fraction. Cells were lysed in 550 μl 2%CHAPS/HBS (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 200 nM NaCl), pH 6.8, plus 200 mM PMSF, 20 mM NEM, and protease inhibitors (10 μg/ml final concentration chymostatin, leupeptin, antipain, and pepstatin A) for 20 min on ice. Cells were scraped from the wells with a rubber policeman, and lysate was transferred to a centrifuge tube. Then 100 μl of lysis buffer was used to wash the well (to collect as much cellular material as possible), and this wash was added to the same centrifuge tube. Lysates were centrifuged at 10,000 × g for 10 min at 4°C, and the supernatant was transferred to a new tube and called the soluble fraction. Pellets from this centrifugation step were washed with 150 μl of lysis buffer and centrifuged at 10,000 × g for 10 min at 4°C, and then the wash was removed. Pellets were then solubilized by addition of 100 μl of 1% SDS, sonicated, and boiled at 95°C for 5 min. One milliliter of 1% Triton X-100 was added to the pellet/SDS mixture, followed by centrifugation at 10,000 × g for 10 min at room temperature. Unless indicated otherwise, equal proportions of each fraction were used for IP.
Pulse-chase analysis
Uggt1−/− MEFs were transfected as for steady-state labeling, but the starvation, pulse, and chase were performed 24 h after transfection. Preparation of soluble and insoluble fractions and double anti-AAT IP were similar to steady-state labeling analysis, but samples were not subjected to PNGaseF digestion.
Immunoprecipitation/gel electrophoresis
Trichloroacetic acid (TCA)-normalized volumes of extracellular, soluble, and insoluble fractions were incubated at 4°C overnight with anti-AAT antibody plus protein A–Sepharose. Beads were washed three times with ice-cold 0.5% CHAPS/HBS, pH 7.4, and then the wash was removed completely by careful aspiration with a small-bore pipette tip. To reduce background, antigen–antibody complexes were disrupted using the previously described 1% SDS/1% Triton X-100 resuspension method, and 1 ml of this supernatant was then incubated overnight at 4°C with anti-AAT antibody plus protein A–Sepharose, then washed and dried by aspiration (double IP). In most cases, beads were then subjected to PNGaseF digestion for 1 h, and then 3 μl of 5× sample buffer plus dithiothreitol was added, and this bead suspension was boiled at 95°C for 5 min. Bead supernatants were analyzed by SDS–PAGE on 10% Tris-HCl Criterion gels or Mini-PROTEAN gels (Bio-Rad, Hercules, CA) at 200 V for 40 min. Gels were then Coomassie stained, destained, and dried in a gel-vacuum dryer for 2 h at 80°C. Finally, dried gels were exposed to a phosphor screen overnight and imaged on a Typhoon imager (GE Healthcare, Piscataway, NJ), and bands were quantified with ImageQuant (GE Healthcare).
DSP cross-linking
At 48 h posttransfection, NHK transfected cells were cross-linked in 2 mM DSP in 4% DMSO/96% PBS for 30 min at room temperature. Soluble and insoluble fractions were prepared, and TCA-normalized volumes were subjected to anti-AAT double IP and analyzed by reducing SDS–PAGE (breaks DSP cross-link).
BiP-luciferase assay
ER stress response was measured 48 h posttransfection using BiP-luciferase expression plasmid (Tirasophon et al., 1998) and a Dual-Luciferase Reporter Assay System (Promega, Madison, WI), and a parallel set of wells was steady-state labeled to determine total intracellular AAT in each transfection group.
Immunofluorescence
Uggt1−/− MEFs were transfected with ATZ ± UGGT1 cotransfection, plated on chamber slides, and fixed with 4% paraformaldehyde solution 48 h posttransfection. Cells were permeabilized and blocked in blocking buffer (1% bovine serum albumin, 0.1% Triton X-100, 0.01% NaN3 in PBS) for 1 h and incubated overnight with 2C1 culture media supernatant (1:10) and anti-AAT antibody (1:1000) in blocking buffer. Secondary antibody (Alexa 350, 555; 1:1000; Invitrogen, Carlsbad, CA) incubation was at room temperature for 1 h. Slides were imaged on an Olympus FV500 confocal microscope with FluoView software (Olympus, Tokyo, Japan). Z-stack analysis was performed using MetaMorph software (Molecular Devices, Sunnyvale, CA).
siRNA knockdown experiment
Uggt1−/− MEFs were transfected with Lipofectamine RNAiMAX transfection reagent (Life Technologies) at 0 h with either an equal mixture of two nonoverlapping CRT siRNAs (Silencer Select; Life Technologies) or nontargeting negative control no. 1 siRNA at a 10 nM final concentration in transfection media. Each transfection group was then transfected with Lipofectamine 2000 at 48 h with DNA expression plasmids plus the same siRNAs as the 0-h transfection. Cells were trypsinized and split into two groups. One group was steady-state labeled from 72 to 96 h, and the other group was collected for immunoblot analysis at 96 h.
Statistical analysis
Student's t test was used throughout with Prism software (GraphPad Software, La Jolla, CA).
Supplementary Material
Acknowledgments
We gratefully thank S. Lentz at the Morphology and Image Analysis Core, Michigan Diabetes Research and Training Center, University of Michigan, Ann Arbor, MI. This work was supported in whole or in part by National Institutes of Health Grants DK042394, DK088227, HL052173, HL057346 (R.J.K.), and DK40344 (P.A.). M.M. is supported by Signora Alessandra and by grants from the Foundation for Research on Neurodegenerative Diseases, the Swiss National Science Foundation, the Association Française contre les Myopathies, the Novartis Stiftung für Medizinisch-Biologische Forschung, and the Gabriele Foundation.
Abbreviations used:
- AAT
α1-antitrypsin
- ATZ
α1-antitrypsin Z allele variant
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate hydrate
- CNX
calnexin
- CRT
calreticulin
- DSP
dithiobis succinimidyl propionate
- EndoH
endoglycosidase H
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- JBM
jack-bean α-mannosidase
- NHK
α1-antitrypsin–null Hong Kong variant
- UGGT1
UDP-glucose:glycoprotein glucosyltransferase
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-02-0101) on July 17, 2013.
REFERENCES
- Anelli T, Sitia R. Protein quality control in the early secretory pathway. EMBO J. 2008;27:315–327. doi: 10.1038/sj.emboj.7601974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- Arnold SM, Fessler LI, Fessler JH, Kaufman RJ. Two homologues encoding human UDP-glucose:glycoprotein glucosyltransferase differ in mRNA expression and enzymatic activity. Biochemistry. 2000;39:2149–2163. doi: 10.1021/bi9916473. [DOI] [PubMed] [Google Scholar]
- Arnold SM, Kaufman RJ. The noncatalytic portion of human UDP-glucose: glycoprotein glucosyltransferase I confers UDP-glucose binding and transferase function to the catalytic domain. J Biol Chem. 2003;278:43320–43328. doi: 10.1074/jbc.M305800200. [DOI] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- Brodsky JL. The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation) Biochem J. 2007;404:353–363. doi: 10.1042/BJ20061890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron PH, Chevet E, Pluquet O, Thomas DY, Bergeron JJ. Calnexin phosphorylation attenuates the release of partially misfolded alpha1-antitrypsin to the secretory pathway. J Biol Chem. 2009;284:34570–34579. doi: 10.1074/jbc.M109.053165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon KS, Helenius A. Trimming and readdition of glucose to N-linked oligosaccharides determines calnexin association of a substrate glycoprotein in living cells. J Biol Chem. 1999;274:7537–7544. doi: 10.1074/jbc.274.11.7537. [DOI] [PubMed] [Google Scholar]
- Choudhury P, Liu Y, Bick RJ, Sifers RN. Intracellular association between UDP-glucose:glycoprotein glucosyltransferase and an incompletely folded variant of alpha1-antitrypsin. J Biol Chem. 1997;272:13446–13451. doi: 10.1074/jbc.272.20.13446. [DOI] [PubMed] [Google Scholar]
- D'Alessio C, Caramelo JJ, Parodi AJ. UDP-GlC:glycoprotein glucosyltransferase-glucosidase II, the ying-yang of the ER quality control. Semin Cell Dev Biol. 2010;21:491–499. doi: 10.1016/j.semcdb.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner AJ, Bole DG, Kaufman RJ. The relationship of N-linked glycosylation and heavy chain-binding protein association with the secretion of glycoproteins. J Cell Biol. 1987;105:2665–2674. doi: 10.1083/jcb.105.6.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner AJ, Wasley LC, Kaufman RJ. Increased synthesis of secreted proteins induces expression of glucose-regulated proteins in butyrate-treated Chinese hamster ovary cells. J Biol Chem. 1989;264:20602–20607. [PubMed] [Google Scholar]
- Ellgaard L, Molinari M, Helenius A. Setting the standards: quality control in the secretory pathway. Science. 1999;286:1882–1888. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- Galli C, Bernasconi R, Solda T, Calanca V, Molinari M. Malectin participates in a backup glycoprotein quality control pathway in the mammalian ER. PLoS One. 2011;6:e16304. doi: 10.1371/journal.pone.0016304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooptu B, Ekeowa UI, Lomas DA. Mechanisms of emphysema in alpha1-antitrypsin deficiency: molecular and cellular insights. Eur Respir J. 2009;34:475–488. doi: 10.1183/09031936.00096508. [DOI] [PubMed] [Google Scholar]
- Graham KS, Le A, Sifers RN. Accumulation of the insoluble PiZ variant of human alpha 1-antitrypsin within the hepatic endoplasmic reticulum does not elevate the steady-state level of grp78/BiP. J Biol Chem. 1990;265:20463–20468. [PubMed] [Google Scholar]
- Greenblatt EJ, Olzmann JA, Kopito RR. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant alpha-1 antitrypsin from the endoplasmic reticulum. Nat Struct Mol Biol. 2011;18:1147–1152. doi: 10.1038/nsmb.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond C, Braakman I, Helenius A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc Natl Acad Sci USA. 1994;91:913–917. doi: 10.1073/pnas.91.3.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert DN, Foellmer B, Helenius A. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell. 1995;81:425–433. doi: 10.1016/0092-8674(95)90395-x. [DOI] [PubMed] [Google Scholar]
- Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2007;87:1377–1408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- Hebert DN, Molinari M. Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem Sci. 2012;37:404–410. doi: 10.1016/j.tibs.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- Hidvegi T, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- Hidvegi T, Mirnics K, Hale P, Ewing M, Beckett C, Perlmutter DH. Regulator of G Signaling 16 is a marker for the distinct endoplasmic reticulum stress state associated with aggregated mutant alpha1-antitrypsin Z in the classical form of alpha1-antitrypsin deficiency. J Biol Chem. 2007;282:27769–27780. doi: 10.1074/jbc.M704330200. [DOI] [PubMed] [Google Scholar]
- Hidvegi T, Schmidt BZ, Hale P, Perlmutter DH. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J Biol Chem. 2005;280:39002–39015. doi: 10.1074/jbc.M508652200. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, You Z, Tremblay LO, Nagata K, Herscovics A. Stimulation of ERAD of misfolded null Hong Kong alpha1-antitrypsin by Golgi alpha1,2-mannosidases. Biochem Biophys Res Commun. 2007;362:626–632. doi: 10.1016/j.bbrc.2007.08.057. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Nagata K. Autophagy eliminates a specific species of misfolded procollagen and plays a protective role in cell survival against ER stress. Autophagy. 2009;5:1217–1219. doi: 10.4161/auto.5.8.10168. [DOI] [PubMed] [Google Scholar]
- Kim PS, Arvan P. Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones. Endocr Rev. 1998;19:173–202. doi: 10.1210/edrv.19.2.0327. [DOI] [PubMed] [Google Scholar]
- Kroeger H, Miranda E, MacLeod I, Perez J, Crowther DC, Marciniak SJ, Lomas DA. Endoplasmic reticulum-associated degradation (ERAD) and autophagy cooperate to degrade polymerogenic mutant serpins. J Biol Chem. 2009;284:22793–22802. doi: 10.1074/jbc.M109.027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Choudhury P, Cabral CM, Sifers RN. Intracellular disposal of incompletely folded human alpha1-antitrypsin involves release from calnexin and post-translational trimming of asparagine-linked oligosaccharides. J Biol Chem. 1997;272:7946–7951. doi: 10.1074/jbc.272.12.7946. [DOI] [PubMed] [Google Scholar]
- Maattanen P, Gehring K, Bergeron JJ, Thomas DY. Protein quality control in the ER: the recognition of misfolded proteins. Semin Cell Dev Biol. 2010;21:500–511. doi: 10.1016/j.semcdb.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Lee J, Abplanalp WA, Yoo SE, Agui T, Furudate S, Kim PS, Arvan P. Oxidoreductase interactions include a role for ERp72 engagement with mutant thyroglobulin from the rdw/rdw rat dwarf. J Biol Chem. 2007;282:6183–6191. doi: 10.1074/jbc.M608863200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda E, et al. A novel monoclonal antibody to characterize pathogenic polymers in liver disease associated with alpha1-antitrypsin deficiency. Hepatology. 2010;52:1078–1088. doi: 10.1002/hep.23760. [DOI] [PubMed] [Google Scholar]
- Molinari M, Galli C, Piccaluga V, Pieren M, Paganetti P. Sequential assistance of molecular chaperones and transient formation of covalent complexes during protein degradation from the ER. J Cell Biol. 2002;158:247–257. doi: 10.1083/jcb.200204122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari M, Galli C, Vanoni O, Arnold SM, Kaufman RJ. Persistent glycoprotein misfolding activates the glucosidase II/UGT1-driven calnexin cycle to delay aggregation and loss of folding competence. Mol Cell. 2005;20:503–512. doi: 10.1016/j.molcel.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Nakatsukasa K, Brodsky JL. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic. 2008;9:861–870. doi: 10.1111/j.1600-0854.2008.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda Y, Hosokawa N, Wada I, Nagata K. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science. 2003;299:1394–1397. doi: 10.1126/science.1079181. [DOI] [PubMed] [Google Scholar]
- Parodi AJ. Protein glucosylation and its role in protein folding. Annu Rev Biochem. 2000;69:69–93. doi: 10.1146/annurev.biochem.69.1.69. [DOI] [PubMed] [Google Scholar]
- Patil AR, Thomas CJ, Surolia A. Kinetics and the mechanism of interaction of the endoplasmic reticulum chaperone, calreticulin, with monoglucosylated (Glc1Man9GlcNAc2) substrate. J Biol Chem. 2000;275:24348–24356. doi: 10.1074/jbc.M003102200. [DOI] [PubMed] [Google Scholar]
- Perlmutter DH. Pathogenesis of chronic liver injury and hepatocellular carcinoma in alpha-1-antitrypsin deficiency. Pediatr Res. 2006;60:233–238. doi: 10.1203/01.pdr.0000228350.61496.90. [DOI] [PubMed] [Google Scholar]
- Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI, Bukau B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012;31:4221–4235. doi: 10.1038/emboj.2012.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutishauser J, Spiess M. Endoplasmic reticulum storage diseases. Swiss Med Wkly. 2002;132:211–222. doi: 10.4414/smw.2002.09861. [DOI] [PubMed] [Google Scholar]
- Schmidt BZ, Perlmutter DH. Grp78, Grp94, and Grp170 interact with alpha1-antitrypsin mutants that are retained in the endoplasmic reticulum. Am J Physiol Gastrointest Liver Physiol. 2005;289:G444–G455. doi: 10.1152/ajpgi.00237.2004. [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Solda T, Galli C, Kaufman RJ, Molinari M. Substrate-specific requirements for UGT1-dependent release from calnexin. Mol Cell. 2007;27:238–249. doi: 10.1016/j.molcel.2007.05.032. [DOI] [PubMed] [Google Scholar]
- Sousa M, Parodi AJ. The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J. 1995;14:4196–4203. doi: 10.1002/j.1460-2075.1995.tb00093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa MC, Ferrero-Garcia MA, Parodi AJ. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. Biochemistry. 1992;31:97–105. doi: 10.1021/bi00116a015. [DOI] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–1824. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga F, Brostrom C, Koide T, Arvan P. Endoplasmic reticulum (ER)-associated degradation of misfolded N-linked glycoproteins is suppressed upon inhibition of ER mannosidase I. J Biol Chem. 2000;275:40757–40764. doi: 10.1074/jbc.M001073200. [DOI] [PubMed] [Google Scholar]
- Trombetta ES, Parodi AJ. Quality control and protein folding in the secretory pathway. Annu Rev Cell Dev Biol. 2003;19:649–676. doi: 10.1146/annurev.cellbio.19.110701.153949. [DOI] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Kaufman RJ. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 2006;13:374–384. doi: 10.1038/sj.cdd.4401840. [DOI] [PubMed] [Google Scholar]
- Zapun A, Darby NJ, Tessier DC, Michalak M, Bergeron JJ, Thomas DY. Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. J Biol Chem. 1998;273:6009–6012. doi: 10.1074/jbc.273.11.6009. [DOI] [PubMed] [Google Scholar]
- Zuber C, Cormier JH, Guhl B, Santimaria R, Hebert DN, Roth J. EDEM1 reveals a quality control vesicular transport pathway out of the endoplasmic reticulum not involving the COPII exit sites. Proc Natl Acad Sci USA. 2007;104:4407–4412. doi: 10.1073/pnas.0700154104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.