

This study describes the novel role of Rap1 as a molecular switch for down-regulation of the Rho-dependent pathway of agonist-induced endothelial hyperpermeability. The Rho-Rap-Rac autoregulation loop may represent a fundamental mechanism of homeostasis and be critical for reestablishment of cell monolayer integrity in pathological conditions.
Abstract
Activation of the Rho GTPase pathway determines endothelial cell (EC) hyperpermeability after injurious stimuli. To date, feedback mechanisms of Rho down-regulation critical for barrier restoration remain poorly understood. We tested a hypothesis that Rho down-regulation and barrier recovery of agonist-stimulated ECs is mediated by the Ras family GTPase Rap1. Thrombin-induced EC permeability driven by rapid activation of the Rho GTPase pathway was followed by Src kinase–dependent phosphorylation of the Rap1-specific guanine nucleotide exchange factor (GEF) C3G, activation of Rap1, and initiation of EC barrier recovery. Knockdown experiments showed that Rap1 activation was essential for down-regulation of Rho signaling and actin stress fiber dissolution. Rap1 activation also enhanced interaction between adherens junction (AJ) proteins VE-cadherin and p120-catenin and stimulated AJ reannealing mediated by the Rap1 effector afadin. This mechanism also included Rap1-dependent membrane translocation of the Rac1-specific GEF Tiam1 and activation of Rac1-dependent peripheral cytoskeletal dynamics, leading to resealing of intercellular gaps. These data demonstrate that activation of the Rap1-afadin axis is a physiological mechanism driving restoration of barrier integrity in agonist-stimulated EC monolayers via negative-feedback regulation of Rho signaling, stimulation of actin peripheral dynamics, and reestablishment of cell–cell adhesive complexes.
INTRODUCTION
The integrity of specialized cell monolayers lining internal organs and vasculature (epithelial, mesothelial, and endothelial cells [ECs]) is a critical feature for normal functions of the organism. Increased vascular endothelial permeability is a cardinal feature of many pathological conditions, including brain and lung edema, atherosclerosis, cancer, and other syndromes. However, molecular mechanisms stimulating restoration of cell monolayer integrity, including endothelial barriers, remain largely unexplored.
Rho family small GTPases represent a group of monomeric 20- to 30-kDa GTP-binding proteins (Bishop and Hall, 2000). Small GTPases act as molecular switches, cycling between an active GTP-bound state and an inactive GDP-bound state; this cycle is regulated by guanine nucleotide exchange factors (GEFs) facilitating the exchange of GDP for GTP. GTPase-activating proteins (GAPs) increase the intrinsic rate of GTP hydrolysis by Rho GTPases and by guanine nucleotide dissociation inhibitors (RhoGDI), which associate with inactivated Rho and Rac (Boguski and McCormick, 1993; Bishop and Hall, 2000; Zheng, 2001). The Rho family members Rho, Rac, and Cdc42 exhibit distinct effects on the actin cytoskeleton, cell adhesions, and cell motility (Machesky and Hall, 1997; Kiosses et al., 1999; Maekawa et al., 1999; Sells et al., 1999; Timpson et al., 2001). Activated Rho, via its effector, Rho-associated kinase (Rho-kinase), induces assembly of stress fibers and focal adhesions (Kaibuchi et al., 1999; Geiger and Bershadsky, 2001; Katsumi et al., 2004). Rho-kinase may directly catalyze myosin light chain (MLC) phosphorylation or may act indirectly via inactivation of MLC phosphatase (MYPT) by phosphorylation at Thr-695, Ser-894, and Thr-850 (van Nieuw Amerongen et al., 2000; Fukata et al., 2001). Together these mechanisms cause actomyosin stress fiber formation, MLC phosphorylation, and cell contraction leading to increased EC permeability.
The serine protease thrombin is produced on the surface of injured endothelium from prothrombin circulating in blood. Thrombin not only induces blood coagulation but also directly increases pulmonary vascular permeability and plays a major role in the pathogenesis of brain and pulmonary edema (Bogatcheva et al., 2002; Mehta and Malik, 2006) by activating the RhoA cascade of endothelial permeability (van Nieuw Amerongen et al., 2000; Burridge and Wennerberg, 2004). Although permeability increase caused by agonists such as thrombin is reversible, the mechanisms to switch from hyperpermeability to barrier recovery in stimulated cell monolayers remain unclear.
Rap1 is a member of the Ras family of small GTPases. Rap1 may be activated by various signals and functions in diverse processes, ranging from cell differentiation to enhancement of cell adhesions and control of lung endothelial barrier in vivo (Bos et al., 2001; Fukuhara et al., 2005; Kooistra et al., 2005; Birukova et al., 2009). Rap1 activity is stimulated by the Rap1 GEFs Epac, C3G, and PDZ-GEF, which become activated by receptor-mediated mechanisms or changes in cell–cell and cell–substrate adhesive complexes (Bos, 2005; Cullere et al., 2005; Kooistra et al., 2007). Inhibition of Rap1 by Rap-specific GAP protein (RapGAP) abolishes Rap1-dependent attachment to the matrix and intercellular adhesion in the epithelial monolayer (Rangarajan et al., 2003; Fukuyama et al., 2006). Afadin, also known as AF6, is a Rap1 effector and scaffold protein containing an actin-binding domain and Ras-binding domains (Hoshino et al., 2005; Sato et al., 2006; Kooistra et al., 2007). Afadin mediates Rap1-dependent assembly of cell–cell junctions (Tawa et al., 2010) and endothelial barrier enhancement induced by barrier protective agents (Birukova et al., 2012a).
Via activation of the Rac-specific GEFs Tiam1 and Vav2, Rap1 may also engage the Rac1 pathway of EC barrier enforcement upon stimulation with barrier-enhancing agonists (Fukuhara et al., 2005; Birukova et al., 2007; Tawa et al., 2010). This link suggests a potential role for Rap1 as an integrator of barrier-enhancing signaling.
Although the contrasting effects of Rho, Rap1, and Rac1 on cell monolayer barrier properties are now well recognized, their roles in agonist-challenged cell monolayers and reversal of barrier dysfunction remain unclear. This study tested Rap1 as a molecular switch from barrier-disruptive to barrier recovery response in thrombin-stimulated endothelial monolayers. We characterized Rap1-dependent mechanisms involved in both the down-regulation of barrier-disruptive Rho signaling and in activation of molecular mechanisms of EC barrier restoration.
RESULTS
Thrombin-induced activation of Rho and EC barrier compromise is followed by activation of Rap1
Thrombin induces rapid and potent increases in endothelial permeability driven by activation of Rho GTPase–dependent signaling, increased phosphorylation of Rho kinase target, an increase of myosin light chain phosphatase (MYPT), and accumulation of di-phosphorylated MLCs, the molecular events that trigger stress fiber formation, actomyosin contractility, and EC monolayer barrier compromise (reviewed in Burridge and Wennerberg, 2004; Beckers et al., 2010; Ivanov et al., 2010). The initial barrier-disruptive EC response to thrombin is followed by a recovery phase initiated after 15–30 min of agonist challenge and characterized by gradual dissolution of central actin stress fibers, resealing of intercellular gaps, and formation of lamellipodia-like processes (Figure 1A). Interestingly, thrombin-induced rapid activation of Rho contributing to EC barrier disruption is followed by activation of the Ras family GTPase, Rap1 (Figure 1B).
FIGURE 1:
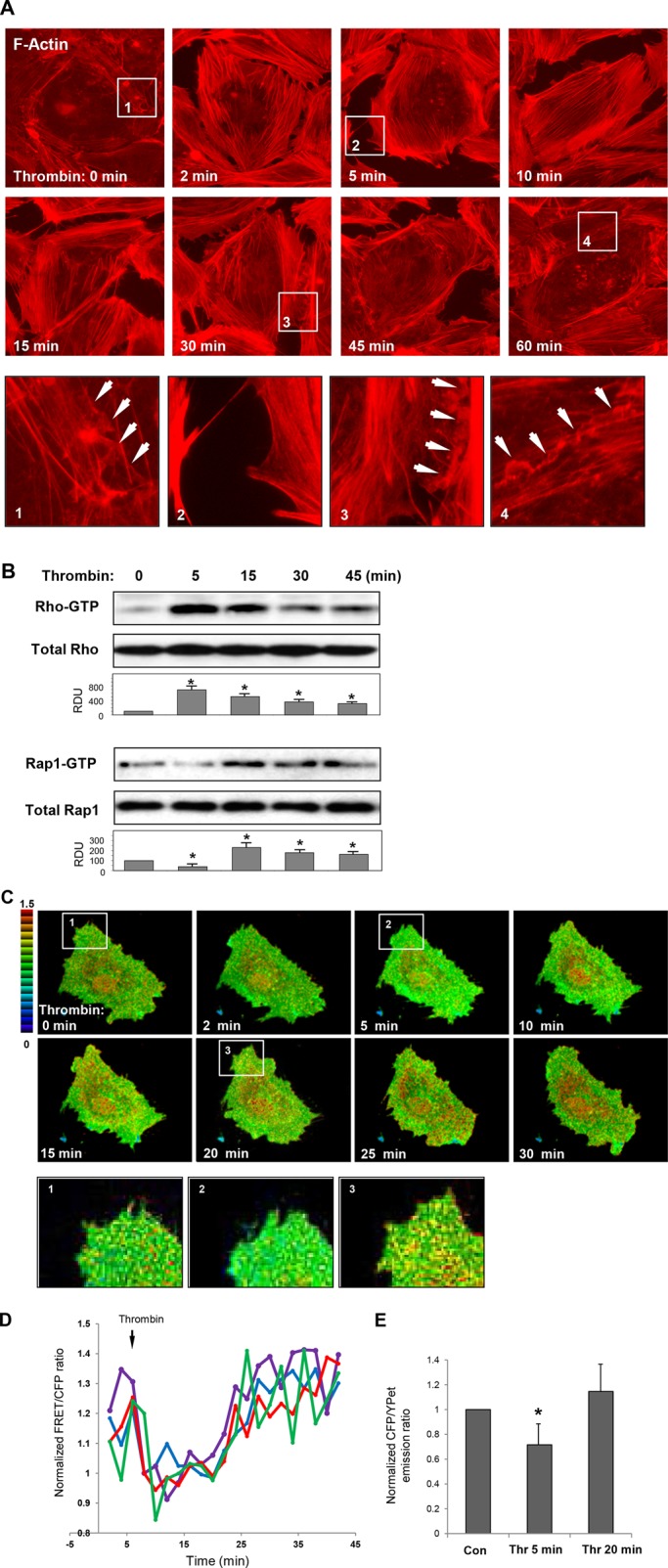
Effect of thrombin on F-actin remodeling and activation of Rho and Rap1 GTPases in human pulmonary ECs. (A) ECs grown on glass coverslips were stimulated with thrombin (0.5 U/ml) for different time periods; this was followed by immunofluorescence staining with Texas Red phalloidin to detect actin filaments. Insets depicting higher-magnification images of F-actin structures at the cell periphery are shown below. Arrows indicate cell–cell interface areas. (B) Time course of thrombin-induced activation of Rho and Rap1 evaluated by pull-down assays with agarose-conjugated rhotekin and Ral-GDS, respectively. Bottom panels show total Rho and Rap1 content in EC lysates used as normalization control. Bar graphs depict results of membrane densitometry analysis; data are expressed as mean ± SD; *, p < 0.05 vs. control. (C) FRET analysis of thrombin-induced Rap1 activation in live cells. HPAECs were transfected with CFP/YPet-Rap-Raichu biosensor for 24 h. Images represent ratio of activated Rap1 to the total Rap1 content. Areas of Rap1 activation appear in yellow and red. (D) Quantitative analysis of thrombin-induced Rap1 activation in single cells. Graphs represent time-dependent changes in normalized CFP/YPet emission ratio in four different cells. (E) Bar graphs depict average Rap1 activity before and after 5 min and 20 min of thrombin stimulation. Data are expressed as mean ± SD of five independent measurements; *, p < 0.05 vs. control.
We analyzed the spatial Rap1 activation in thrombin-stimulated confluent pulmonary EC monolayers, using Rap1 biosensor (Sakurai et al., 2006). Thrombin rapidly decreased total Rap1 activity in the first minutes, the time points corresponding to Rho activation and gap formation (Figure 1B); this was followed by up-regulation of Rap1 activity after 10–30 min. Rap1 activation was observed as a dot-like pattern throughout the cell and specifically in peripheral compartments involved in reestablishment of EC monolayer integrity (Figure 1C). Insets depict higher-magnification images of EC peripheral areas at different time points after thrombin treatment. Tracking of time-dependent Rap1 activity in single cells and analysis of average Rap activity at different time points are shown in Figure 1, D and E.
Thrombin-induced Rap1 activation and EC barrier recovery is associated with activation of Src and Src-dependent phosphorylation of the Rap1 GEF C3G
Thrombin-induced activation of the tyrosine kinase Src is also involved in control of EC permeability (Tiruppathi et al., 2001; Vouret-Craviari et al., 2002; Liu et al., 2010). Because activity of the Rap1-specific GEF C3G, is controlled by tyrosine phosphorylation (Fukuyama et al., 2005), we followed Src activation and C3G phosphorylation patterns in thrombin-stimulated pulmonary ECs. Thrombin-induced Src phosphorylation at Tyr-416, the site reflecting Src activation, was detected 5 min after stimulation and reached peak levels by 10–20 min (Figure 2A). The role of Src in the EC barrier recovery was tested in experiments with administration of the Src inhibitor PP2 5 min after thrombin addition. At this point, ECs develop maximal Rho activation and permeability response. Inhibition of Src by PP2 posttreatment after thrombin challenge abolished C3G phosphorylation (Figure 2B), which led to inhibition of Rap1 activation (Figure 2C). Inhibition of Src, C3G phosphorylation, and Rap1 activation by PP2 posttreatment after thrombin dramatically attenuated EC barrier recovery as evaluated by measurements of transendothelial electrical resistance (Figure 2D).
FIGURE 2:
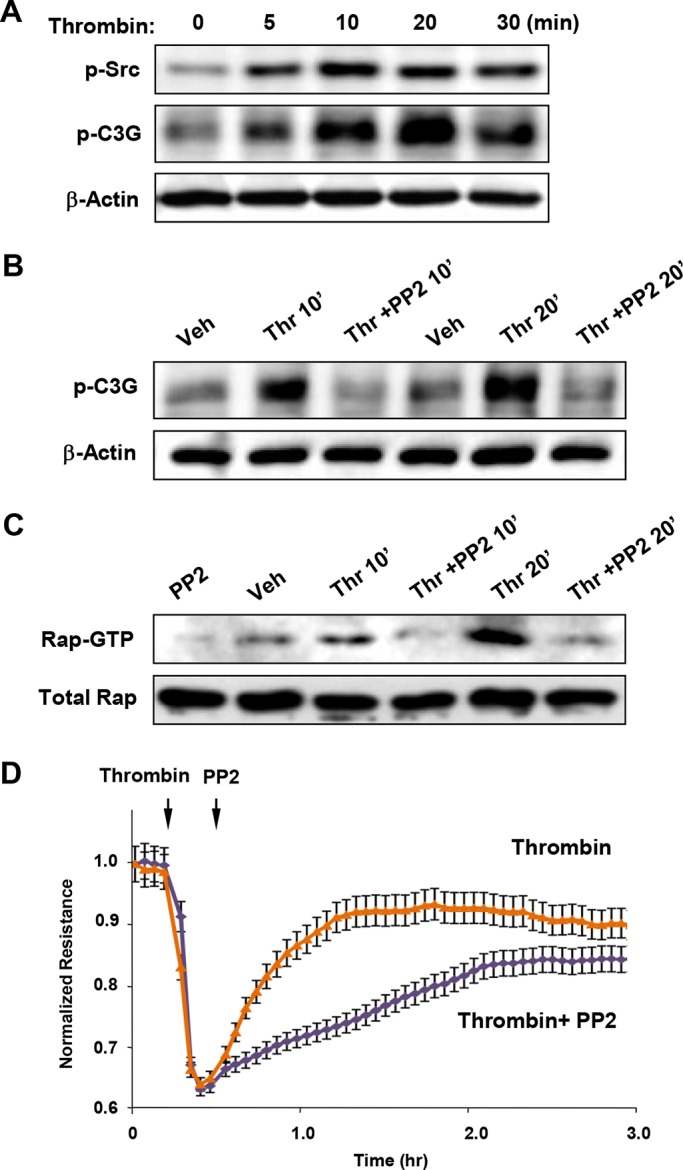
Role of Src and C3G phosphorylation in Rap1 activation and EC barrier restoration after thrombin. (A) Time-dependent Src activation was monitored by immunoblotting with p-Y416–specific antibody reflecting the Src-activated state. (B) ECs were stimulated with thrombin (0.5 U/ml, 5 min); this was followed by addition of vehicle or the Src kinase inhibitor PP2 (5 μM). C3G tyrosine phosphorylation was detected by Western blot with phosphospecific antibody. Reprobing with β-actin antibody was used as normalization control. (C) ECs were stimulated with thrombin (0.5 U/ml, 5 min); this was followed by addition of vehicle or the Src kinase inhibitor PP2 (5 μM). Rap1 activation was evaluated using Rap1-GTP pull-down assay and normalized to the total Rap1 content in cell lysates. (D) HPAECs plated on microelectrodes were treated with thrombin (5 min); this was followed by addition of PP2 (5 μM). Measurements of TER were performed over 3 h. Arrows indicate times of thrombin and PP2 addition.
Rap1 depletion compromises endothelial monolayer recovery after thrombin
Small interfering RNA (siRNA)-induced Rap1 depletion in EC monolayers significantly suppressed the restoration of transendothelial electrical resistance (TER) to control levels (Figure 3A), suggesting the direct role of Rap1 in EC barrier recovery after agonist stimulation. Functional permeability effects were linked to morphological changes. Immunofluorescence analysis showed slight but statistically significant decrease in VE-cadherin accumulation at cell junctions in Rap1-depleted EC monolayers under basal conditions (Figure 3, B and C). Thrombin induced robust stress fiber formation and disruption of continuous peripheral VE-cadherin in control cells, and Rap1 depleted ECs (Figure 3B). These changes were reversible in control ECs and returned to original patterns 1 h after thrombin challenge, thus reflecting completion of EC monolayer recovery (Figure 3B, top panels). In contrast, dissolution of stress fibers and restoration of adherens junctions (AJ) in Rap1-depleted EC monolayers was impaired (Figure 3B, bottom panels). Inhibition of EC monolayer recovery by Rap1 depletion was documented by dramatic attenuation of intercellular gap resealing and reappearance of VE-cadherin at cell junction areas (Figure 3C).
FIGURE 3:
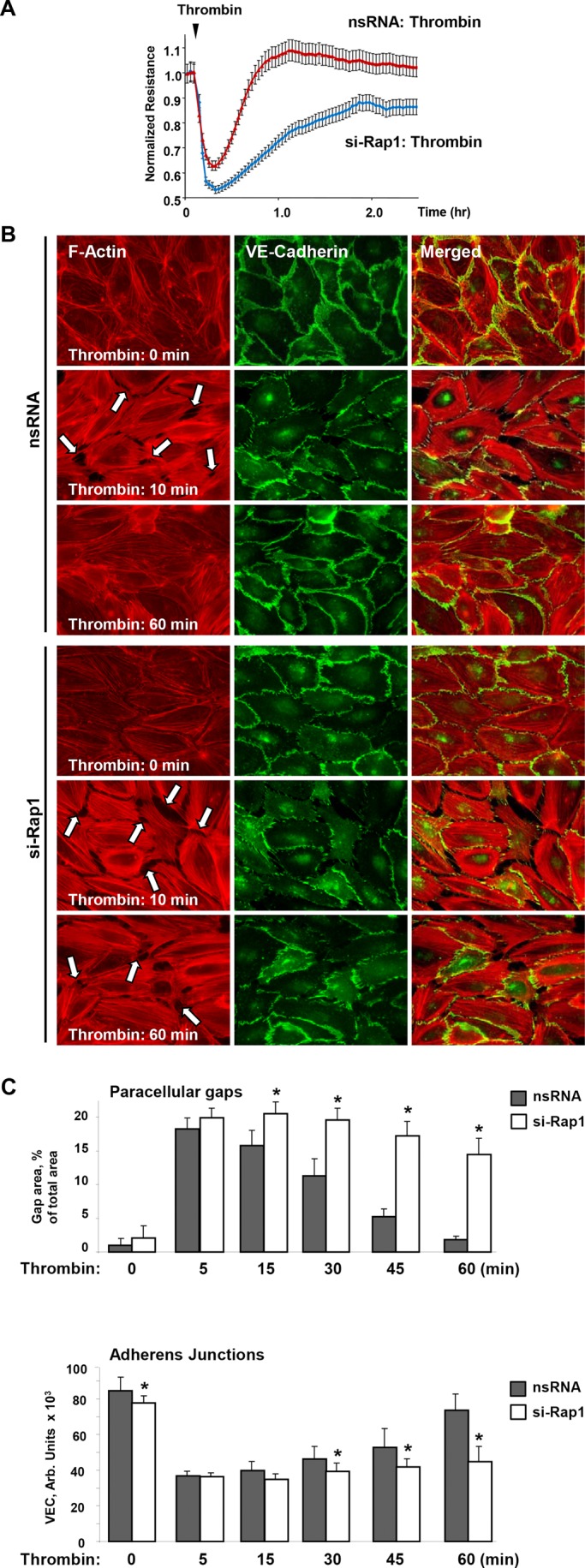
Effects of Rap1 knockdown on functional and structural barrier restoration of pulmonary EC monolayer after thrombin. (A) ECs plated on microelectrodes were transfected with Rap1-specific siRNA or nonspecific RNA 48 h prior to TER measurements. Control and Rap1-specific siRNA-treated ECs were stimulated with thrombin at the time indicated by the arrow, and TER changes were monitored over time. (B) ECs plated on glass coverslips were transfected with Rap1-specific siRNA or nonspecific RNA prior to stimulation with thrombin. Immunofluorescence staining of F-actin (left) and VE-cadherin (middle) was performed using Texas Red phalloidin and VE-cadherin specific antibody, respectively. Right, merged images of F-actin and VE-cadherin staining. Arrows indicate areas of thrombin-induced intercellular gap formation. (C) Quantitative analysis of gap formation and cell junction VE-cadherin localization in control and Rap1-depleted ECs at different times after thrombin treatment. Data are expressed as mean ± SD; *, p < 0.05.
Rap1 controls Rho signaling during endothelial recovery after thrombin
We analyzed effects of Rap1 knockdown on thrombin-induced activation of Rho and phosphorylation of MYPT1 and MLC as the downstream effectors of the Rho pathway (Burridge and Wennerberg, 2004). Decline of Rho activity at 30 min and return to baseline levels by 45 min after thrombin stimulation observed in ECs treated with nonspecific RNA was significantly delayed in ECs with Rap1 knockdown (Figure 4A). Sustained Rho activation resulted in prolonged Rho-kinase–dependent phosphorylation of MYPT and MLC in pulmonary ECs with depleted Rap1 (Figure 4B). An alternative approach to Rap1 depletion, the expression of dominant-negative Rap1 mutant (Rap1A-DN), yielded similar results. Cells expressing Rap1A-DN demonstrated prolonged MLC phosphorylation after thrombin treatment (Figure 4C).
FIGURE 4:
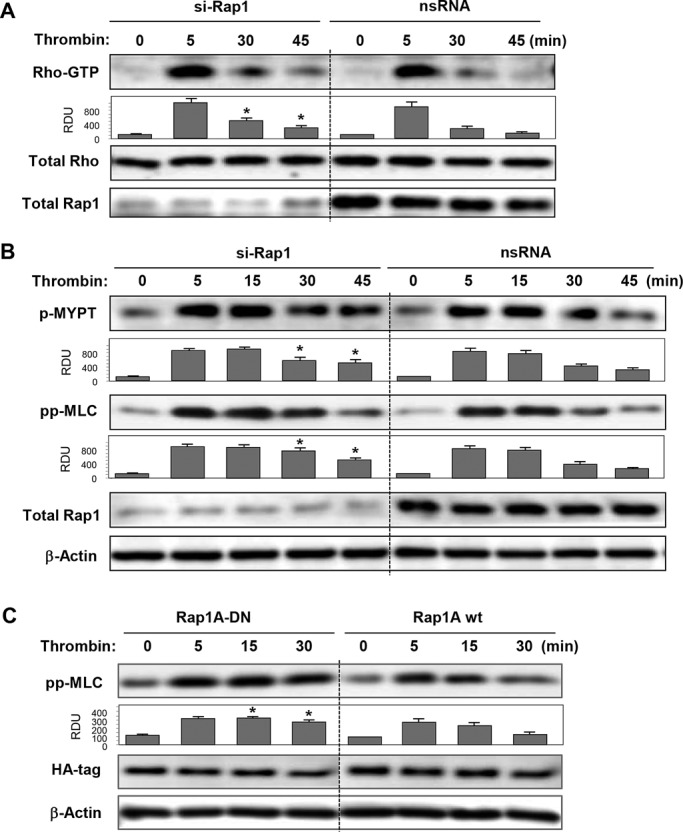
Effects of Rap1 knockdown on thrombin-induced Rho signaling. (A) Cells treated with nonspecific or Rap1-specific siRNA were stimulated with thrombin for the indicated time periods. Rho activation was evaluated by RhoGTP pull-down assay (top) and normalized to total Rho content in cell lysates (middle). Rap1 depletion was verified by Western blot (bottom). (B) Cells treated with nonspecific or Rap1-specific siRNA were stimulated with thrombin, and activation of Rho signaling was evaluated by Western blot with phospho-MYPT and diphospho-MLC antibody. (C) Cells transfected with HA-tagged wild-type (Rap1A wt) and dominant negative (Rap1A-DN) Rap1A mutant were treated with thrombin. Activation of Rho pathway was evaluated by immunoblotting with diphospho-MLC antibody. Reprobing with β-actin antibody was used as the normalization control. Rap1 depletion was verified by Western blot. Bar graphs depict results of membrane densitometry analysis; data are expressed as mean ± SD; *, p < 0.05 vs. control.
Alternative inactivation of Rap1 by expression of negative regulator of Rap1 (Rap1-GAP) showed that, similar to Rap1 depletion, expression of RapGAP prevented complete restoration of EC barrier after thrombin (Figure 5A). Expression of Rap1-GAP also prevented the disappearance of actin stress fibers in ECs at time points beyond 45 min post–thrombin challenge (Figure 5B).
FIGURE 5:
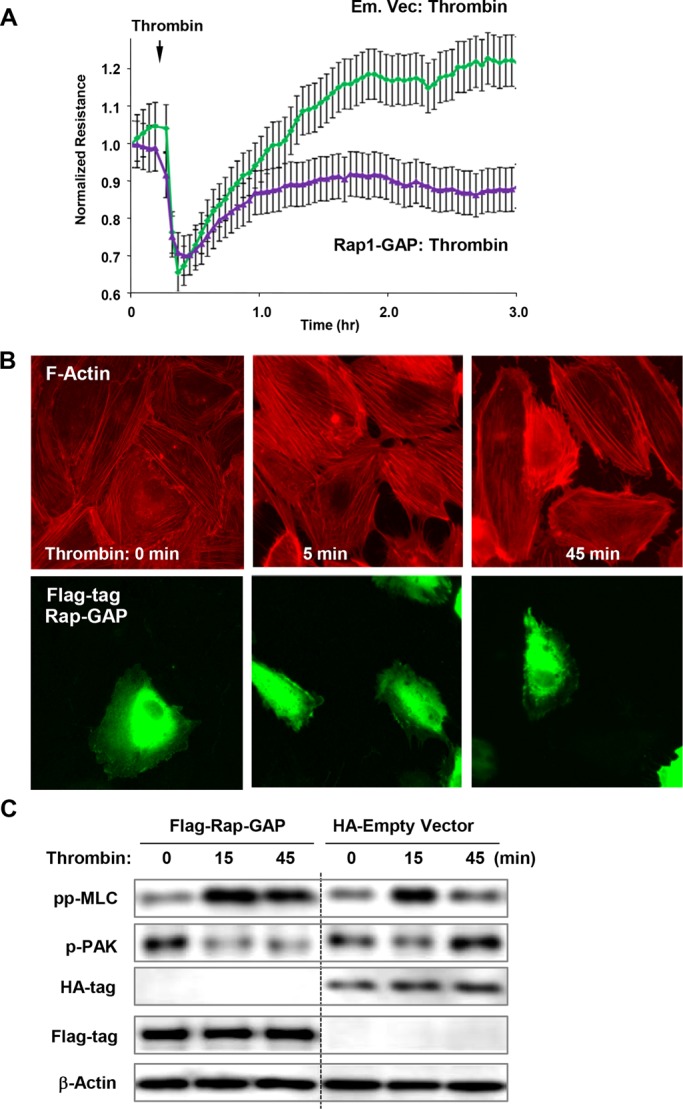
Effects of Rap1-GAP and inactive Rap1 on EC barrier recovery, Rac activation, and Rho down-regulation after thrombin. (A) Measurements of TER were performed in thrombin-stimulated cells transfected with Rap1-GAP or control vector. (B) Cells were transfected with Flag-tagged Rap1-GAP and treated with thrombin. Actin stress fiber formation was evaluated by Texas Red phalloidin staining (top). Staining with anti-Flag antibody indicates Rap1-GAP–overexpressing cells (bottom). (C) Cells were transfected with Flag-tagged Rap1-GAP or empty vector expressing HA-tag and treated with thrombin. Activation of Rac signaling was evaluated by immunoblotting with phospho-PAK1 antibody. Reprobing with β-actin antibody was used as the normalization control.
Because Rho and Rac signaling play reciprocal roles in EC barrier regulation, we examined the effects of Rap1 inhibition on parameters of Rac signaling. Rac-mediated autophosphorylation of PAK1 at Thr-423 has been previously observed during EC barrier recovery (Birukova et al., 2012b). Thrombin stimulation of control ECs decreased PAK1 phosphorylation at an early time point (15 min), but significantly increased PAK1 phosphorylation at 45 min, the time point corresponding to the monolayer recovery (Figure 5C, right lanes). In contrast, overexpression of Rap1-GAP abolished PAK1 phosphorylation increase after 45 min of thrombin, suggesting depressed Rac up-regulation during recovery after thrombin in Rap1-GAP–expressing cells (Figure 5C, left lanes).
Rap1-induced Rac signaling contributes to cytoskeletal dynamics and EC barrier recovery after thrombin challenge
Several cytoskeletal Rac effectors, such as the Arp2/3 complex, p21Arc, PAK1, and cortactin, control cortical actin structure (Borisy and Svitkina, 2000; Weed and Parsons, 2001). Rap1 involvement in the cytoskeletal dynamics associated with EC monolayer recovery was evaluated in live microscopy studies. Pairs of neighbor cells expressing green fluorescent protein (GFP)-cortactin were used for analysis. Disruption of the cell–cell connection and formation of an intercellular gap were seen after 5 min of thrombin challenge, while formation of cortactin-positive lamellipodia-like structures was observed as early as 15 min and became more prominent at later time points (25–30 min). Expansion of peripheral cortactin-positive cell areas resulted in resealing of intercellular gaps by 30 min of thrombin stimulation (Figure 6A, top panels and high-magnification insets). Rap1 knockdown did not block thrombin-induced intercellular gap formation and cell retraction, but abolished peripheral cytoskeletal dynamics and the reestablishment of cell contacts (Figure 6A, bottom panels).
FIGURE 6:
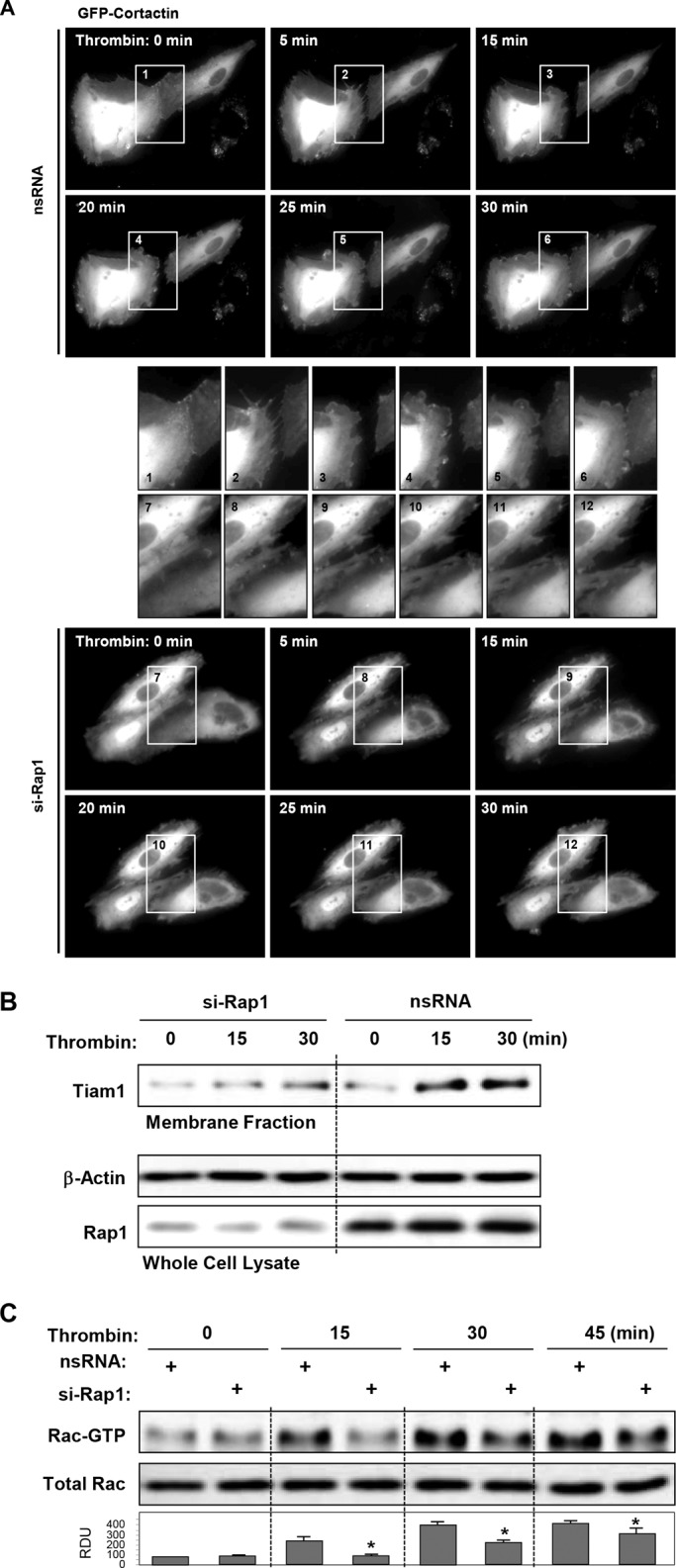
Role of Rac pathway in Rap1-dependent EC recovery after thrombin. (A) Live-cell imaging of HPAECs transfected with Rap1 siRNA or nonspecific RNA and expressing GFP-cortactin. Snapshots depict extension of peripheral cortactin-positive cell areas leading to resealing of intercellular gaps by 30 min of thrombin stimulation (top panels). Rap1 knockdown abolished this effect (bottom panels). Insets depict higher-magnification cell areas marked by rectangles. (B) Cells transfected with nonspecific RNA or Rap1-specific siRNA were treated with thrombin for 15 min or 30 min, and membrane translocation of the Rac-specific GEF Tiam1 was analyzed by Western blot analysis of EC membrane fractions. (C) Analysis of delayed Rac activation in thrombin-stimulated cells transfected with nonspecific RNA or Rap1-specific siRNA. Reprobing with β-actin antibody was used as the normalization control. Bar graphs depict results of membrane densitometry analysis; data are expressed as mean ± SD; *, p < 0.05 vs. nsRNA.
Precise mechanisms of Rac activation during EC barrier recovery remain to be delineated. Activity of the Rac-specific GEF Tiam1 may be regulated by receptor-mediated mechanisms, AJ complexes, and interaction with Rap1 (Noren et al., 2001; Arthur et al., 2004; Bos, 2006; Gonzalez et al., 2006). We tested whether Rap activation during cell recovery after thrombin could be associated with activation of Tiam1 evaluated by membrane translocation. Thrombin-induced Tiam1 membrane translocation after 15–30 min of stimulation was abolished by Rap1 knockdown (Figure 6B). Rap1 knockdown also abolished activation of Rac GTPase during recovery phase (Figure 6C), observed after 15, 30, and 45 min of thrombin stimulation.
Rap1 stimulates Rac-independent AJ reannealing and VE-cadherin–p120-catenin complex reassembly via Rap1–afadin mechanism
We used pulmonary ECs, transiently transfected with GFP-β-catenin, for live microscopy analysis of AJ restoration after thrombin challenge. Disappearance of GFP-β-catenin from cell–cell junction areas was observed after 5–15 min of thrombin challenge and reflects agonist-induced AJ disruption. Reappearance of a continuous GFP-β-catenin signal at the cell–cell boundary was observed after 20–30 min of thrombin challenge (Figure 7A, top panels). Rap1 depletion prevented formation of GFP-β-catenin–positive cell–cell junctions during the time of experiment (Figure 7A, bottom panels).
FIGURE 7:
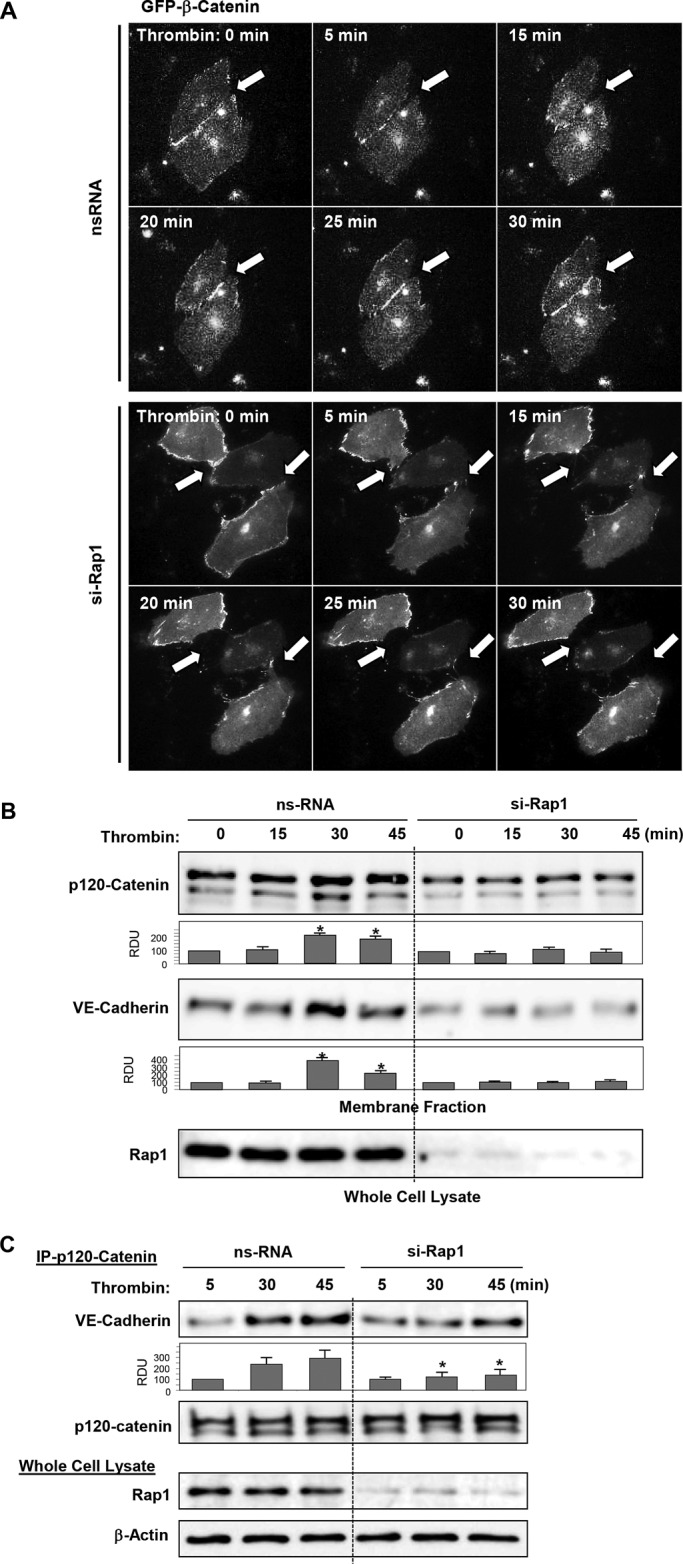
Role of Rap1 in reannealing of AJs and increased p120-catenin–VE-cadherin interactions during recovery after thrombin. (A) Live-cell imaging of HPAECs transfected with Rap1 siRNA or nonspecific RNA and expressing GFP-β-catenin. Snapshots depict reestablishment of β-catenin–positive AJ previously disrupted by thrombin challenge. (top panels, shown by arrow). Rap1 knockdown abolished this effect (bottom panels, shown by arrow). (B) Increased colocalization of p120-catenin and VE-cadherin in cell membrane fraction after 30 min of thrombin stimulation was abolished by Rap1 knockdown. Rap1 depletion was verified by Western blot. (C) Cells were transfected with nonspecific RNA or Rap1-specific siRNA; this was followed by thrombin stimulation. Coimmunoprecipitation assays using antibody to p120-catenin were performed, and VE-cadherin and p120-catenin content in the immunoprecipitates was detected using specific antibodies. Equal protein loading was confirmed by membrane reprobing with antibodies to p120-catenin. Bottom panels depict siRNA-based depletion of endogenous Rap1. Reprobing with β-actin antibody was used as the normalization control. Bar graphs depict results of membrane densitometry analysis; data are expressed as mean ± SD; *, p < 0.05 vs. control.
Accumulation of AJ protein in plasma membrane fractions reflects increased assembly of AJ complexes. In agreement with results described above, increasing levels of the AJ proteins p120-catenin and VE-cadherin were observed in cell membrane fractions of cells incubated with thrombin during 30–45 min. p120-catenin and VE-cadherin membrane translocation at later times after thrombin treatment was abolished by knockdown of Rap1 (Figure 7B). Coimmunoprecipitation studies showed increased interaction between p120-catenin and VE-cadherin after 30 min and 45 min of thrombin challenge that was abolished by Rap1 depletion (Figure 7C).
A recent study shows that the AJ-associated Rap1 effector afadin promotes AJ complex assembly and mediates agonist-induced EC barrier enhancement (Birukova et al., 2012a). The potential role of afadin in EC barrier recovery after thrombin was further tested. Thrombin caused rapid disappearance of afadin from cell–cell junction areas within 5 min of stimulation, and reappearance after 30–60 min (Figure 8A). Afadin depletion prevented the reassembly of junctions and restoration of actin cytoskeletal pattern after 60 min of thrombin challenge (Figure 8B) and significantly delayed resealing of paracellular gaps in thrombin-challenged EC monolayers (Figure 8C).
FIGURE 8:
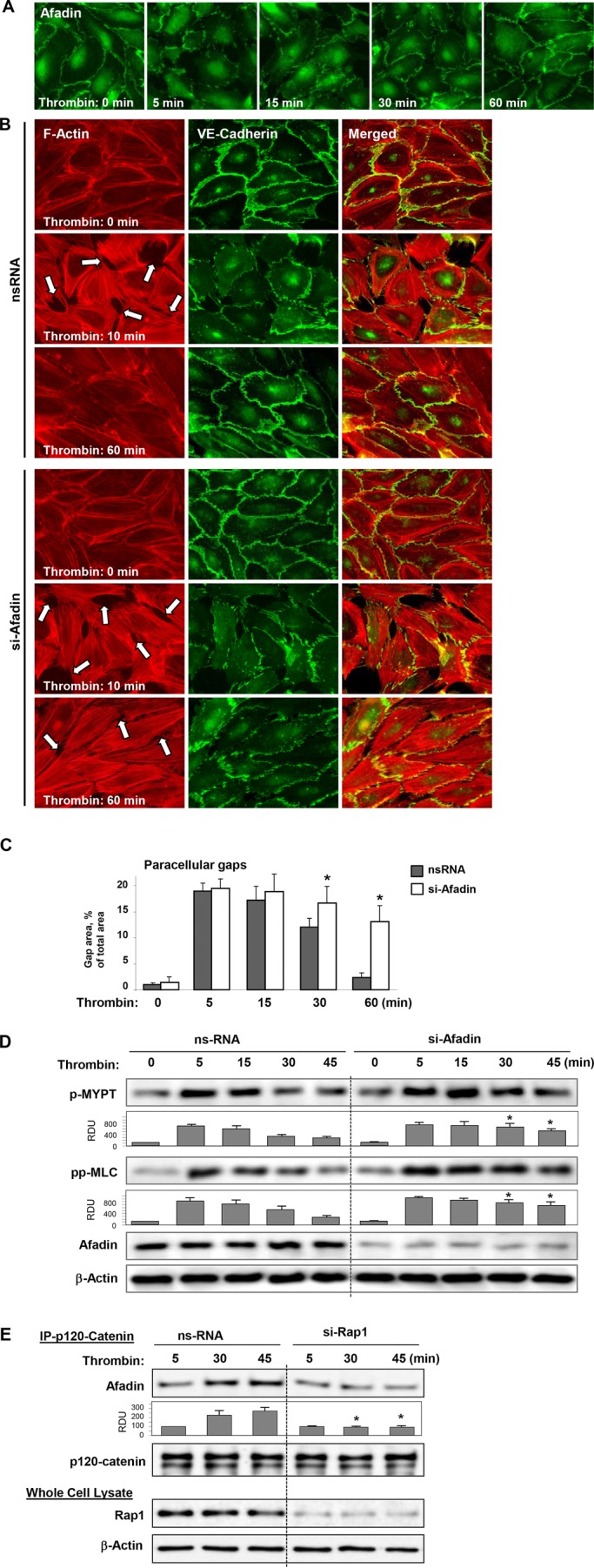
Role of afadin in EC barrier restoration and reversal of Rho signaling after thrombin. (A) HPAEC monolayers were stimulated with thrombin (0.5 U/ml) for the indicated periods of time. Peripheral localization of afadin was evaluated by immunofluorescence staining with afadin antibody. (B) HPAEC monolayers transfected with control RNA or afadin-specific siRNA were stimulated with thrombin (0.5 U/ml) for the indicated periods of time. Stress fiber formation and integrity of AJs was monitored by immunofluorescence staining with Texas Red phalloidin (left) and VE-cadherin (middle) antibody, respectively. Right, merged images of F-actin and VE-cadherin staining. (C) Quantitative analysis of gap formation in control and afadin-depleted ECs at different times after thrombin treatment. Data are expressed as mean ± SD; *, p < 0.05. (D) Activation of Rho signaling was evaluated by Western blot with phospho-MYPT and diphospho-MLC antibody. Reprobing with β-actin antibody was used as the normalization control. Afadin depletion was verified by Western blot. (E) Cells were transfected with nonspecific RNA or Rap1-specific siRNA; this was followed by thrombin stimulation. Coimmunoprecipitation assays using antibody to p120-catenin were performed, and afadin and p120-catenin content in the immunoprecipitates was detected using specific antibodies. Equal protein loading was confirmed by membrane reprobing with antibodies to p120-catenin. Bottom panels depict siRNA-based depletion of endogenous Rap1. Reprobing with β-actin antibody was used as the normalization control. Bar graphs depict results of membrane densitometry analysis; data are expressed as mean ± SD; *, p < 0.05 vs. control.
Afadin depletion also prevented down-regulation of Rho-dependent MYPT and MLC phosphorylation observed at time points beyond 30 min post–thrombin treatment (Figure 8C).
Rap1 dependence of afadin-mediated restoration of the EC cytoskeleton, AJ structure, and monolayer barrier properties, and down-regulation of Rho signaling was further investigated. Rap1 knockdown prevented increased association of afadin with p120-catenin (Figure 8D), which is important for enhancement of cell junctions and barrier function. Rap1 knockdown also abolished accumulation of afadin in the plasma membrane fraction after 30–45 min of thrombin treatment, the time frame corresponding to EC barrier restoration (Figure 9A). Expression of an afadin mutant lacking the Rap1-binding domain (afadin-ΔRBD) significantly attenuated EC barrier recovery monitored by TER measurements of thrombin-stimulated EC monolayers (Figure 9B). Expression of afadin-ΔRBD also delayed the dissolution of thrombin-induced stress fibers in ECs normally observed after 45 min of agonist treatment (Figure 9C). Because the Rap1-binding domain is required for afadin activation by Rap1, these data suggest a role of Rap1 in regulation of afadin functions during EC barrier recovery.
FIGURE 9:
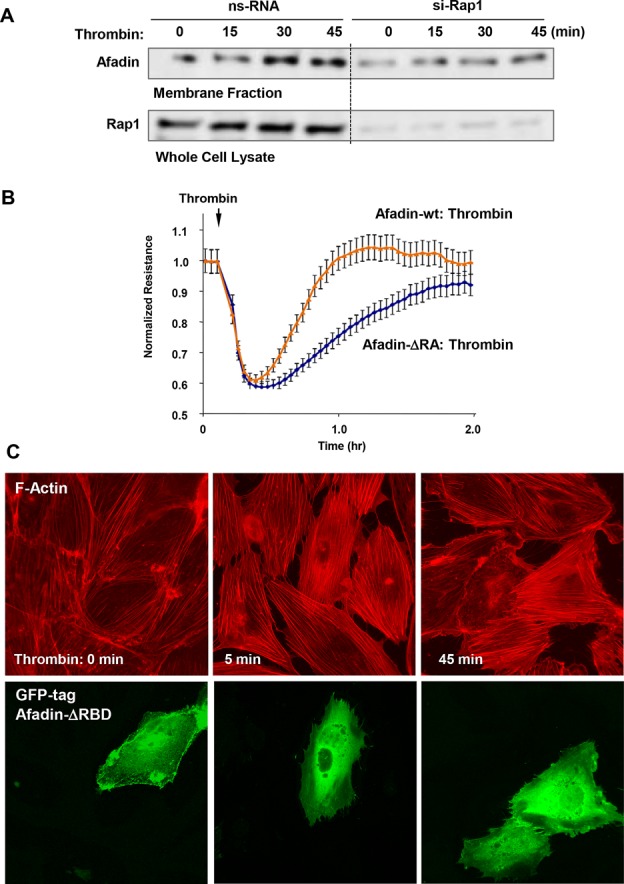
Afadin activation by Rap1 promotes EC barrier restoration and stress fiber disassembly after thrombin. (A) Cell membrane accumulation of afadin during HPAEC recovery after thrombin was abolished by Rap1 knockdown. Rap1 depletion was verified by Western blot. (B) ECs plated on microelectrodes were transfected with wild-type afadin or afadin-ΔRBD mutant 24 h prior to TER measurements. Stimulation with thrombin was performed at the time indicated by the arrow, and TER changes were monitored over 2 h. (C) Cells were transfected with GFP-tagged afadin-ΔRBD mutant and treated with thrombin. Actin stress fiber formation was evaluated by Texas Red phalloidin staining (top). Staining with anti-Flag antibody indicates afadin-ΔRBD–overexpressing cells (bottom).
DISCUSSION
Recovery of the vascular endothelial barrier after chemical and mechanical insults associated with various pathologies, including thromboembolism, ischemia/reperfusion, sepsis, acute respiratory distress syndrome, and trauma or ventilator-induced lung injury, is critical for survival and restoration of normal organ function. However, the molecular mechanisms underlying EC barrier restoration remain obscure. This study demonstrates a key role for Rap1 in down-regulation of Rho-mediated barrier-disruptive signaling and reestablishment of barrier integrity of agonist challenged endothelial monolayers.
Thrombin-induced cytoskeletal remodeling, intercellular gap formation, and EC barrier disruption are associated with rapid and transient activation of Rho GTPase. This study shows that Rho activation is followed by stimulation of Rap1. The direct role of Rap1 in recovery of EC barrier after thrombin challenge was shown by experiments with Rap1 knockdown or ectopic expression of an inhibitor of Rap1 activity, Rap1-GAP, which abolished rebound of basal transendothelial resistance reflecting EC barrier recovery. Spatial analysis of Rap1 activation using a fluorescence resonance energy transfer (FRET)-based Rap1 biosensor demonstrated increased Rap1 activity in two major locations: peripheral EC lamellipodia-like structures and intracellular vesicles. The time course of integral Rap1 activation in single cells corresponded to Rap1 activation measurements by GTP-Rap1 pull-down assays.
Previous reports described two alternative mechanisms of Rap1 activation. In one mechanism, Rap1 activation occurs upon AJ disassembly and is triggered by E-cadherin internalization and trafficking along the endocytic pathway (Balzac et al., 2005). In an alternative mechanism, Rap1 activation requires establishment of nectin or E-cadherin containing AJs (Fukuyama et al., 2005; Sakurai et al., 2006). Other data show Rap1 activation upon AJ disruption and disengagement of E-cadherin from AJs (Asuri et al., 2008). Visualization of activated Rap1 in primary ECs performed in our study suggests that activated Rap1 is likely localized in both submembrane and intracellular vesicle compartments. These data suggest the existence of both mechanisms in recovering ECs and resolve the apparent conflict between previous observations.
Our study shows that Rap1 activation in pulmonary ECs is dependent on Src-mediated C3G activation. Src-dependent C3G activation also controls cell adhesion and spreading of epithelial monolayers (Ohba et al., 2001). Our results show that subsequent treatment of thrombin-challenged ECs with Src inhibitor PP2 abolished C3G phosphorylation leading to compromised EC barrier recovery after thrombin.
The details of the Rap1–Rho cross-talk mechanism remain to be elucidated. In agreement with effects on barrier recovery discussed above, molecular inhibition of Rap1 markedly prolonged activation of the Rho pathway in thrombin-stimulated EC monolayers. Analysis of signaling cascades affected by Rap1 inhibition suggests two mechanisms involved in down-regulation of Rho. First, Rap1 knockdown prevented relocalization of afadin to cell junctions after thrombin challenge. Afadin plays a key role in establishment of nectin- and cadherin-containing AJs in endothelial and epithelial cell types (Takai and Nakanishi, 2003). We recently reported that Rap1-activated afadin promotes interactions between AJs and tight junctions that contribute to barrier enhancement of EC monolayers in vitro and in the animal model of lung vascular leakage caused by excessive mechanical ventilation (Birukova et al., 2012a). Importantly, inhibition of Rap1 prevented association of afadin with its AJ binding partner, p120-catenin, and their translocation to the cell membrane during recovery after thrombin. Studies in lung ECs (Birukova et al., 2011) and in epithelial cells (Wildenberg et al., 2006) show that membrane-associated p120-catenin interacts with a negative regulator of RhoA, p190RhoGAP, thus leading to down-regulation of the RhoA pathway and barrier recovery. Therefore, Rap1/afadin–dependent membrane accumulation of p120-catenin–p190RhoGAP complex may represent a negative-feedback mechanism of RhoA regulation during EC monolayer recovery. This mechanism is further supported by the data showing more pronounced and delayed activation of the Rho pathway reflected by increased MYPT and MLC phosphorylation in ECs with depleted afadin (Figure 8C).
The second potential mechanism of barrier restoration and RhoA down-regulation by Rap1 may be triggered by Rap1-dependent activation of the Rac1-specific GEF Tiam1 (Arthur et al., 2004) and stimulation of the Rac1 pathway. Depletion of Rap1 in our study prevented membrane association of Tiam1 during recovery after thrombin; this was linked to attenuation of Rac activation and Rac-mediated activation of the Rac effectors PAK1 and cortactin. Rap1 knockdown prevented Rac-dependent activation of peripheral cytoskeletal remodeling and cortactin accumulation at the cell periphery, the events essential for EC spreading and resealing the paracellular gaps.
In turn, Rac may down-regulate Rho activity in several ways, among them direct Rac interaction with RhoGDI (Wong et al., 2006), PAK1-dependent inhibition of the Rho-specific GEF p115RhoGEF (Rosenfeldt et al., 2006), and Rac stimulation of the Rho-specific GTPase-activating protein p190RhoGAP (Herbrand and Ahmadian, 2006). These mechanisms of Rac-Rho cross-talk may be critical for vascular barrier restoration, as well as for protective effects induced by established barrier-enhancing agonists, such as prostacyclin, sphingosine 1-phosphate, hepatocyte growth factor, oxidized phospholipids, and angiopoietin.
In summary, we propose a model of Rap1-Rac–dependent down-regulation of Rho signaling and restoration of EC barrier after thrombin challenge (Figure 10). While activating the RhoA pathway of barrier dysfunction, thrombin also stimulates other signaling mechanisms, including activation of Src kinase. Activated Src phosphorylates the Rap1-specific GEF C3G and activates Rap1. Rap1 engages afadin to promote reassembly of cell–cell junctions (AJs and tight junctions), recruits the p120-catenin–p190RhoGAP complex to the submembrane domain, and down-regulates the Rho pathway of EC permeability. In addition, Rap1 activates Tiam1 and Rac1, which trigger cortical cytoskeletal dynamics and cell spreading to reseal the paracellular gaps. Rac1 may also down-regulate Rho activity via activation of p190RhoGAP or inhibition of p115RhoGEF.
FIGURE 10:
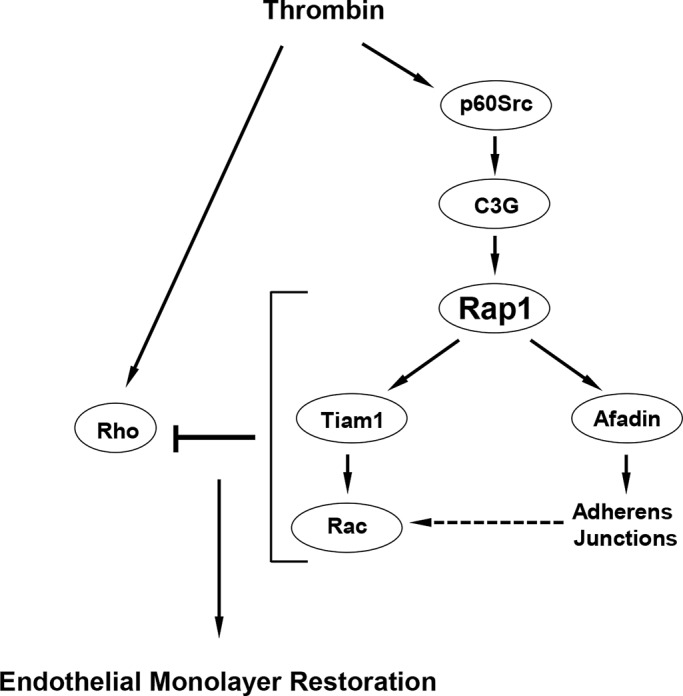
Role of Rap1 in EC barrier restoration after thrombin. In parallel with rapid activation of the Rho pathway leading to increased EC permeability, thrombin causes activation of Src kinase, which stimulates the Rap1-specific GEF C3G and, via Rap1-Tiam1, turns on the Rac1 signaling. Activation of Rap1-Rac1 signaling axis down-regulates the Rho pathway of barrier disruption and promotes reassembly of AJ complexes and endothelial monolayer barrier restoration.
These results demonstrate a key role of Rap1 as a master regulator of endothelial barrier restoration acting on key cellular processes such as down-regulation of barrier-disruptive Rho pathway and stimulation of cytoskeletal and cell junction recovery. Such an autoregulatory loop may represent a fundamental mechanism of homeostasis and may be critical in pathological conditions associated with compromised endothelial and epithelial barrier function.
MATERIALS AND METHODS
Reagents and cell culture
Unless specified, biochemical reagents were obtained from Sigma-Aldrich (St. Louis, MO). Reagents for immunofluorescence were purchased from Molecular Probes (Eugene, OR). Antibodies to Rho, Tiam1, hemagglutinin (HA), Flag, VE-cadherin, and p-C3G Tyr514 were from Santa Cruz Biotechnology (Santa Cruz, CA); p-cortactin Tyr421, p-Src Tyr416, p-MYPT Thr-696, pp-MLC Thr-18/Ser-19, and p-PAK Thr-423 antibodies were from Cell Signaling (Beverly, MA); Rac1, Rap1, and p120-catenin were from BD Transduction Laboratories (San Diego, CA). Human pulmonary artery ECs (HPAECs) were obtained from Lonza (Allendale, NJ), maintained in a complete culture medium according to the manufacturer's recommendations and used for experiments at passages 5–7.
DNA and siRNA transfections
Predesigned Rap1A- and afadin-specific human Stealth Select siRNA sets of standard purity were ordered from Invitrogen (Carlsbad, CA). Transfection of ECs with siRNA was performed as previously described (Singleton et al., 2009). Nonspecific, nontargeting siRNA was used as a control treatment. After 48 h of transfection, cells were used for experiments or harvested for Western blot verification of specific protein depletion. Test experiments using control fluorescently labeled RNA oligonucleotides showed ∼90% efficiency of siRNA transfection of ECs (unpublished data). Plasmids encoding HA-tagged Rap1A wild-type and Flag-tagged Rap1-GAP were provided by L. Quilliam (Indiana University, Indianapois, IN). Plasmid encoding HA-tagged dominant-negative Rap1A-S17N was obtained from Missouri S&T cDNA Resource Center (Rolla, MO). Plasmids encoding afadin-wild-type and afadin-ΔRBD (Ras/Rap1-binding domain) bearing GFP tags were provided by Y. Takai (Kobe University, Kobe, Japan). Plasmid encoding GFP-LifeAct was provided by L. Phillipson (University of Chicago, Chicago, IL). Plasmids encoding GFP-tagged wild-type cortactin and β-catenin were obtained from Addgene (Cambridge, MA). ECs were used for transient transfections according to a protocol described previously (Birukova et al., 2011). Control transfections were performed with empty vectors. For permeability measurements, to ensure more effective introduction of cDNA into the cell, nucleofection of HPAECs was performed using a kit from Amaxa Biosystems (Lonza). An optimized protocol of nucleofection was provided by the manufacturer.
Measurement of EC permeability
Measurements of TER across confluent HPAEC monolayers were performed using the electrical cell–substrate impedance sensing system (ECIS; Applied Biophysics, Troy, NY), as previously described (Birukova et al., 2011). In the current studies, we did not observe significant effects of nonspecific RNA or specific siRNA or DNA constructs on cell viability and monolayer integrity. Initial testing of nontransfected, siRNA- and DNA-transfected EC monolayers did not reveal significant differences in basal TER levels.
GTPase activation assay
Activation of Rac, Rap, or Rho GTPase in pulmonary EC culture was analyzed using GTPase in vitro pull-down assay kit available from Millipore (Billerica, MA), as previously described (Birukova et al., 2007).
FRET
FRET analysis of Rap1 activation was performed using CFP/YPet-Rap-Raichu biosensor, as described elsewhere (Sakurai et al., 2006). Cells were seeded on a glass-bottom dish coated with gelatin. Twenty-four hours after transfection, medium was changed to 2% fetal bovine serum endothelial basic medium (EBM) for 2 h. For detection of FRET, the cells were maintained on the microscope stage at 37°C. To minimize the photobleaching effect, the time interval for each imaging acquisition was set to be 30 s, and images were captured for 15 min using Olympus (Olympus America, Center Valley, PA) Model IX71 Microscope System equipped with a 63× oil-immersion objective and a CCD camera. MetaMorph software was used to control the filter wheel and data analysis. The ratiometric images of ECFP/YPet were computed and generated by the MetaMorph software to represent the spatiotemporal FRET signals.
Live-cell imaging
Cells were plated on MatTek dishes (MatTek, Ashland, MA) and transfected with GFP-cortactin or GFP-β-catenin. Images were acquired with 100×/1.45 numerical aperture oil objective in a 3I Marianas Yokogawa-type spinning-disk (Olympus America) confocal system equipped with a CO2 chamber and a heated stage. Time-lapse images were taken at 2-s intervals for 40–60 s.
Immunofluorescence
Endothelial monolayers plated on glass coverslips were subjected to immunofluorescence staining with the appropriate antibody. Texas Red phalloidin (Molecular Probes) was used to visualize F-actin. After being immunostained, slides were analyzed using a Nikon video imaging system (Nikon Instech, Tokyo, Japan). Images were processed with ImageJ (National Institutes of Health, Bethesda, MD) software and Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA) software.
Differential protein fractionation and immunoblotting
Confluent HPAECs were stimulated with thrombin; cytosolic and membrane fractions were isolated using a subcellular protein fractionation kit (Thermo Fisher Scientific, Rockford, IL) according to the manufacturer's protocol. For analysis of the protein phosphorylation profile, cells were stimulated, then lysed, and protein extracts were separated by SDS–PAGE, transferred to polyvinylidene fluoride membrane, and probed with specific antibodies. Equal protein loading was verified by reprobing membranes with antibody to β-actin or the specific protein of interest. The relative intensities of immunoreactive protein bands (relative density units) were analyzed and quantified by scanning densitometry using Image Quant software (Molecular Dynamics, Sunnyvale, CA).
Coimmunoprecipitation
After agonist stimulation, cells were washed in cold phosphate-buffered saline and lysed on ice with cold TBS-NP40 lysis buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 1% NP40) supplemented with protease and phosphatase inhibitor cocktails (Roche, Indianapolis, IN). Clarified lysates were then incubated with antibodies to p120-catenin (BD Transduction Laboratories) overnight at 4°C and washed three to four times with TBS-NP40 lysis buffer, and the complexes were analyzed by Western blotting using the appropriate antibodies.
Statistical analysis
Results are expressed as mean ± SD of four to six independent experiments. Experimental samples were compared with controls by unpaired Student's t test. For multiple-group comparisons, a one-way analysis of variance and post hoc multiple-comparison tests were used. p < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by Public Health Service grants HL87823 and HL76259 to K.G.B. and HL89257 and HL107920 to A.A.B. from the National Heart, Lung, and Blood Institute. The authors thank Shigetomo Fukuhara and Naoki Mochizuki, National Cerebral and Cardiovascular Center, Osaka, Japan, for providing the CFP/YPet-Rap-Raichu biosensor.
Abbreviations used:
- afadin-ΔRBD
afadin mutant lacking the Rap1-binding domain
- AJ
adherens junction
- EC
endothelial cell
- FRET
fluorescence resonance energy transfer
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- GFP
green fluorescent protein
- HA
hemagglutinin
- HPAEC
human pulmonary artery endothelial cell
- MLC
myosin light chain
- MYPT
myosin light chain phosphatase
- Rap1A-DN
dominant-negative Rap1 mutant
- Rap1-GAP
negative regulator of Rap1
- RapGAP
Rap-specific GAP protein
- siRNA
small interfering RNA
- TER
transendothelial electrical resistance
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-02-0098) on July 17, 2013.
REFERENCES
- Arthur WT, Quilliam LA, Cooper JA. Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. J Cell Biol. 2004;167:111–122. doi: 10.1083/jcb.200404068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuri S, Yan J, Paranavitana NC, Quilliam LA. E-cadherin dis-engagement activates the Rap1 GTPase. J Cell Biochem. 2008;105:1027–1037. doi: 10.1002/jcb.21902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzac F, Avolio M, Degani S, Kaverina I, Torti M, Silengo L, Small JV, Retta SF. E-cadherin endocytosis regulates the activity of Rap1: a traffic light GTPase at the crossroads between cadherin and integrin function. J Cell Sci. 2005;118:4765–4783. doi: 10.1242/jcs.02584. [DOI] [PubMed] [Google Scholar]
- Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost. 2010;103:40–55. doi: 10.1160/TH09-06-0403. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Fu P, Wu T, Dubrovskyi O, Sarich N, Poroyko V, Birukov KG. Afadin controls p120-catenin-ZO-1 interactions leading to endothelial barrier enhancement by oxidized phospholipids. J Cell Physiol. 2012a;227:1883–1890. doi: 10.1002/jcp.22916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Fu P, Xing J, Birukov KG. Rap1 mediates protective effects of iloprost against ventilator induced lung injury. J Appl Physiol. 2009;107:1900–1910. doi: 10.1152/japplphysiol.00462.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Tian Y, Dubrovskyi O, Zebda N, Sarich N, Tian X, Wang Y, Birukov KG. VE-cadherin trans-interactions modulate Rac activation and enhancement of lung endothelial barrier by iloprost. J Cell Physiol. 2012b;227:3405–3416. doi: 10.1002/jcp.24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Zagranichnaya T, Alekseeva E, Fu P, Chen W, Jacobson JR, Birukov KG. Prostaglandins PGE2 and PGI2 promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res. 2007;313:2504–2520. doi: 10.1016/j.yexcr.2007.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Zebda N, Cokic I, Fu P, Wu T, Dubrovskyi O, Birukov KG. p190RhoGAP mediates protective effects of oxidized phospholipids in the models of ventilator-induced lung injury. Exp Cell Res. 2011;317:859–872. doi: 10.1016/j.yexcr.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348:241–255. [PMC free article] [PubMed] [Google Scholar]
- Bogatcheva NV, Garcia JG, Verin AD. Molecular mechanisms of thrombin-induced endothelial cell permeability. Biochemistry (Mosc) 2002;67:75–84. doi: 10.1023/a:1013904231324. [DOI] [PubMed] [Google Scholar]
- Boguski MS, McCormick F. Proteins regulating Ras and its relatives. Nature. 1993;366:643–654. doi: 10.1038/366643a0. [DOI] [PubMed] [Google Scholar]
- Borisy GG, Svitkina TM. Actin machinery: pushing the envelope. Curr Opin Cell Biol. 2000;12:104–112. doi: 10.1016/s0955-0674(99)00063-0. [DOI] [PubMed] [Google Scholar]
- Bos JL. Linking Rap to cell adhesion. Curr Opin Cell Biol. 2005;17:123–128. doi: 10.1016/j.ceb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31:680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Bos JL, de Rooij J, Reedquist KA. Rap1 signalling: adhering to new models. Nat Rev Mol Cell Biol. 2001;2:369–377. doi: 10.1038/35073073. [DOI] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- Cullere X, Shaw SK, Andersson L, Hirahashi J, Luscinskas FW, Mayadas TN. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood. 2005;105:1950–1955. doi: 10.1182/blood-2004-05-1987. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Amano M, Kaibuchi K. Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol Sci. 2001;22:32–39. doi: 10.1016/s0165-6147(00)01596-0. [DOI] [PubMed] [Google Scholar]
- Fukuhara S, Sakurai A, Sano H, Yamagishi A, Somekawa S, Takakura N, Saito Y, Kangawa K, Mochizuki N. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol Cell Biol. 2005;25:136–146. doi: 10.1128/MCB.25.1.136-146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuyama T, Ogita H, Kawakatsu T, Fukuhara T, Yamada T, Sato T, Shimizu K, Nakamura T, Matsuda M, Takai Y. Involvement of the c-Src-Crk-C3G-Rap1 signaling in the nectin-induced activation of Cdc42 and formation of adherens junctions. J Biol Chem. 2005;280:815–825. doi: 10.1074/jbc.M411099200. [DOI] [PubMed] [Google Scholar]
- Fukuyama T, Ogita H, Kawakatsu T, Inagaki M, Takai Y. Activation of Rac by cadherin through the c-Src-Rap1-phosphatidylinositol 3-kinase-Vav2 pathway. Oncogene. 2006;25:8–19. doi: 10.1038/sj.onc.1209010. [DOI] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A. Assembly and mechanosensory function of focal contacts. Curr Opin Cell Biol. 2001;13:584–592. doi: 10.1016/s0955-0674(00)00255-6. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Kou R, Michel T. Rac1 modulates sphingosine 1-phosphate-mediated activation of phosphoinositide 3-kinase/Akt signaling pathways in vascular endothelial cells. J Biol Chem. 2006;281:3210–3216. doi: 10.1074/jbc.M510434200. [DOI] [PubMed] [Google Scholar]
- Herbrand U, Ahmadian MR. p190-RhoGAP as an integral component of the Tiam1/Rac1-induced downregulation of Rho. Biol Chem. 2006;387:311–317. doi: 10.1515/BC.2006.041. [DOI] [PubMed] [Google Scholar]
- Hoshino T, Sakisaka T, Baba T, Yamada T, Kimura T, Takai Y. Regulation of E-cadherin endocytosis by nectin through afadin, Rap1, and p120ctn. J Biol Chem. 2005;280:24095–24103. doi: 10.1074/jbc.M414447200. [DOI] [PubMed] [Google Scholar]
- Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. 2010;177:512–524. doi: 10.2353/ajpath.2010.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem. 1999;68:459–486. doi: 10.1146/annurev.biochem.68.1.459. [DOI] [PubMed] [Google Scholar]
- Katsumi A, Orr AW, Tzima E, Schwartz MA. Integrins in mechanotransduction. J Biol Chem. 2004;279:12001–12004. doi: 10.1074/jbc.R300038200. [DOI] [PubMed] [Google Scholar]
- Kiosses WB, Daniels RH, Otey C, Bokoch GM, Schwartz MA. A role for p21-activated kinase in endothelial cell migration. J Cell Biol. 1999;147:831–844. doi: 10.1083/jcb.147.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra MR, Corada M, Dejana E, Bos JL. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005;579:4966–4972. doi: 10.1016/j.febslet.2005.07.080. [DOI] [PubMed] [Google Scholar]
- Kooistra MR, Dube N, Bos JL. Rap1: a key regulator in cell-cell junction formation. J Cell Sci. 2007;120:17–22. doi: 10.1242/jcs.03306. [DOI] [PubMed] [Google Scholar]
- Liu DZ, Ander BP, Xu H, Shen Y, Kaur P, Deng W, Sharp FR. Blood-brain barrier breakdown and repair by Src after thrombin-induced injury. Ann Neurol. 2010;67:526–533. doi: 10.1002/ana.21924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky LM, Hall A. Role of actin polymerization and adhesion to extracellular matrix in Rac- and Rho-induced cytoskeletal reorganization. J Cell Biol. 1997;138:913–926. doi: 10.1083/jcb.138.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science. 1999;285:895–898. doi: 10.1126/science.285.5429.895. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- Noren NK, Niessen CM, Gumbiner BM, Burridge K. Cadherin engagement regulates Rho family GTPases. J Biol Chem. 2001;276:33305–33308. doi: 10.1074/jbc.C100306200. [DOI] [PubMed] [Google Scholar]
- Ohba Y, et al. Requirement for C3G-dependent Rap1 activation for cell adhesion and embryogenesis. EMBO J. 2001;20:3333–3341. doi: 10.1093/emboj/20.13.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan S, Enserink JM, Kuiperij HB, de Rooij J, Price LS, Schwede F, Bos JL. Cyclic AMP induces integrin-mediated cell adhesion through Epac and Rap1 upon stimulation of the β2-adrenergic receptor. J Cell Biol. 2003;160:487–493. doi: 10.1083/jcb.200209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeldt H, Castellone MD, Randazzo PA, Gutkind JS. Rac inhibits thrombin-induced Rho activation: evidence of a Pak-dependent GTPase crosstalk. J Mol Signal. 2006;1:8. doi: 10.1186/1750-2187-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai A, Fukuhara S, Yamagishi A, Sako K, Kamioka Y, Masuda M, Nakaoka Y, Mochizuki N. MAGI-1 is required for Rap1 activation upon cell-cell contact and for enhancement of vascular endothelial cadherin-mediated cell adhesion. Mol Biol Cell. 2006;17:966–976. doi: 10.1091/mbc.E05-07-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Fujita N, Yamada A, Ooshio T, Okamoto R, Irie K, Takai Y. Regulation of the assembly and adhesion activity of E-cadherin by nectin and afadin for the formation of adherens junctions in Madin-Darby canine kidney cells. J Biol Chem. 2006;281:5288–5299. doi: 10.1074/jbc.M510070200. [DOI] [PubMed] [Google Scholar]
- Sells MA, Boyd JT, Chernoff J. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J Cell Biol. 1999;145:837–849. doi: 10.1083/jcb.145.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton PA, Chatchavalvanich S, Fu P, Xing J, Birukova AA, Fortune JA, Klibanov AM, Garcia JG, Birukov KG. Akt-mediated transactivation of the S1P1 receptor in caveolin-enriched microdomains regulates endothelial barrier enhancement by oxidized phospholipids. Circ Res. 2009;104:978–986. doi: 10.1161/CIRCRESAHA.108.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai Y, Nakanishi H. Nectin and afadin: novel organizers of intercellular junctions. J Cell Sci. 2003;116:17–27. doi: 10.1242/jcs.00167. [DOI] [PubMed] [Google Scholar]
- Tawa H, et al. Role of afadin in vascular endothelial growth factor- and sphingosine 1-phosphate-induced angiogenesis. Circ Res. 2010;106:1731–1742. doi: 10.1161/CIRCRESAHA.110.216747. [DOI] [PubMed] [Google Scholar]
- Timpson P, Jones GE, Frame MC, Brunton VG. Coordination of cell polarization and migration by the Rho family GTPases requires Src tyrosine kinase activity. Curr Biol. 2001;11:1836–1846. doi: 10.1016/s0960-9822(01)00583-8. [DOI] [PubMed] [Google Scholar]
- Tiruppathi C, Naqvi T, Sandoval R, Mehta D, Malik AB. Synergistic effects of tumor necrosis factor-α and thrombin in increasing endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2001;281:L958–L968. doi: 10.1152/ajplung.2001.281.4.L958. [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res. 2000;87:335–340. doi: 10.1161/01.res.87.4.335. [DOI] [PubMed] [Google Scholar]
- Vouret-Craviari V, Bourcier C, Boulter E, van Obberghen-Schilling E. Distinct signals via Rho GTPases and Src drive shape changes by thrombin and sphingosine-1-phosphate in endothelial cells. J Cell Sci. 2002;115:2475–2484. doi: 10.1242/jcs.115.12.2475. [DOI] [PubMed] [Google Scholar]
- Weed SA, Parsons JT. Cortactin: coupling membrane dynamics to cortical actin assembly. Oncogene. 2001;20:6418–6434. doi: 10.1038/sj.onc.1204783. [DOI] [PubMed] [Google Scholar]
- Wildenberg GA, Dohn MR, Carnahan RH, Davis MA, Lobdell NA, Settleman J, Reynolds AB. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127:1027–1039. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- Wong KW, Mohammadi S, Isberg RR. Disruption of RhoGDI and RhoA regulation by a Rac1 specificity switch mutant. J Biol Chem. 2006;281:40379–40388. doi: 10.1074/jbc.M605387200. [DOI] [PubMed] [Google Scholar]
- Zheng Y. Dbl family guanine nucleotide exchange factors. Trends Biochem Sci. 2001;26:724–732. doi: 10.1016/s0968-0004(01)01973-9. [DOI] [PubMed] [Google Scholar]