

Previous studies show that the COPII coat is phosphorylated. The phosphorylated coat, however, cannot rebind to the ER to initiate a new round of vesicle budding. The present study shows that Sit4p/PP6, a Ser/Thr phosphatase, dephosphorylates the COPII coat. Consistent with a role in coat recycling, Sit4p/PP6 regulates ER-to-Golgi traffic.
Abstract
Traffic from the endoplasmic reticulum (ER) to the Golgi complex is initiated when the activated form of the GTPase Sar1p recruits the Sec23p-Sec24p complex to ER membranes. The Sec23p-Sec24p complex, which forms the inner shell of the COPII coat, sorts cargo into ER-derived vesicles. The coat inner shell recruits the Sec13p-Sec31p complex, leading to coat polymerization and vesicle budding. Recent studies revealed that the Sec23p subunit sequentially interacts with three different binding partners to direct a COPII vesicle to the Golgi. One of these binding partners is the serine/threonine kinase Hrr25p. Hrr25p phosphorylates the COPII coat, driving the membrane-bound pool into the cytosol. The phosphorylated coat cannot rebind to the ER to initiate a new round of vesicle budding unless it is dephosphorylated. Here we screen all known protein phosphatases in yeast to identify one whose loss of function alters the cellular distribution of COPII coat subunits. This screen identifies the PP2A-like phosphatase Sit4p as a regulator of COPII coat dephosphorylation. Hyperphosphorylated coat subunits accumulate in the sit4Δ mutant in vivo. In vitro, Sit4p dephosphorylates COPII coat subunits. Consistent with a role in coat recycling, Sit4p and its mammalian orthologue, PP6, regulate traffic from the ER to the Golgi complex.
INTRODUCTION
Proteins destined to traffic through the secretory pathway are sorted into transport vesicles before they tether and fuse to their acceptor membrane (Whyte and Munro, 2002). Tight regulation of these events is required for efficient cargo transport and the maintenance of organelle identity. Genetic and biochemical studies in the yeast Saccharomyces cerevisiae play a major role in identifying the highly conserved components of the secretory apparatus and elucidating the mechanistic details of the first step in the pathway, anterograde transport between endoplasmic reticulum (ER) and Golgi complex (Lord et al., 2013).
The generation of ER-derived transport vesicles begins when the activated form of the GTPase Sar1p recruits the inner shell of the COPII coat, the Sec23p-Sec24p complex. The coat inner shell sorts cargo into the vesicle before recruiting the outer shell of the coat, the Sec13p-Sec31p complex. These events lead to the hydrolysis of GTP on Sar1p and vesicle budding (Lee et al., 2005; Zanetti et al., 2011). After Sar1p is released from the vesicle, TRAPPI, a multimeric guanine nucleotide exchange factor (GEF), binds to the coat via interaction with Sec23p and recruits the Rab GTPase Ypt1p to the vesicle (Cai et al., 2007; Lord et al., 2011). Subsequently, activated Ypt1p binds to its effector Uso1p, a long coiled-coil tether that links the vesicle to the Golgi (Lord et al., 2011). Once the vesicle tethers to the Golgi, Hrr25p, a serine/threonine kinase, phosphorylates the inner shell of the COPII coat. Eventually, the coat is released and the vesicle fuses with the Golgi. Through sequential interactions with different binding partners (Sar1p-GTP, TRAPPI, and Hrr25p), the coat subunit Sec23p mediates the directionality of these events (Lord et al., 2011).
In vitro transport studies reveal that phosphorylation of the COPII coat is required for vesicle fusion, whereas its dephosphorylation is needed to initiate a new round of vesicle budding (Lord et al., 2011). In an effort to identify the phosphatase that dephosphorylates the COPII coat, we screened mutants in known protein phosphatases for defects in the intracellular distribution of COPII coat subunits. Here we report that Sit4p, a serine/threonine phosphatase, is a key regulator of COPII coat dephosphorylation. In the sit4Δ mutant, COPII coat subunits become hyperphosphorylated and their subcellular distribution is altered. In vitro, Sit4p dephosphorylates coat subunits. Consistent with a role in coat recycling, Sit4p and its mammalian orthologue, PP6, are required for ER-to-Golgi traffic.
RESULTS
Identification of a phosphatase that alters the intracellular distribution of COPII coat subunits
We previously showed, by differential fractionation, that phosphorylation of Sec23p drives it from membranes into the cytosol (Lord et al., 2011). In an attempt to identify a phosphatase that dephosphorylates the COPII coat, we screened known phosphatase mutants in the yeast deletion library for a change in the distribution of Sec23p, using differential fractionation as an assay.
The yeast phosphatome contains ∼32 members, which are largely divided into four families: serine/threonine phosphoprotein phosphatases (PPP), magnesium-dependent serine/threonine phosphatases (PPM), protein tyrosine phosphatases (PTP), and dual-specificity phosphatases (DSP), which can dephosphorylate phosphoserine/phosphothreonine, as well as phosphotyrosine residues (Breitkreutz et al., 2010). Only three (Glc7, Cdc14, and Ssu72) of these 32 phosphatases are essential for growth in yeast (Figure 1A). None of the temperature-sensitive (ts) mutants in the essential phosphatases, including Glc7p (Supplemental Figure S1A), which was previously implicated in ER-to-Golgi traffic (Bryant and James, 2003), showed a change in the distribution of Sec23p. Of the 29 nonessential phosphatases screened from the library, an increase in the cytosolic pool of Sec23p was observed only in the sit4Δ mutant. This phenotype was confirmed when SIT4 was deleted in our laboratory strain background (Figure 1B, left).
FIGURE 1:

Screening of the yeast phosphatome to identify a phosphatase that alters the intracellular distribution of COPII coat subunits. (A) Table of the protein phosphatase families in yeast that were screened to identify a phosphatase that regulates COPII coat dephosphorylation. (B) Left, total cell lysates (T) from wild type (SFNY1841; lanes 1–3) and the sit4Δ mutant (SFNY2045; lanes 4–6) centrifuged at 150,000 × g, separated into supernatant (S) and pellet (P) fractions, and analyzed by Western blot analysis on a 10% SDS–polyacrylamide gel. The SNARE Bos1p is shown as a fractionation control. Right, the same experiment for the pph21Δ mutant (SFNY2411). (C) Expression of Sit4p alters the migration of Lst1p and Sec31p. The sit4Δ mutant expressing SIT4 (pNRB530 GALpr-SIT4 URA3) under the GAL promoter was grown to early log phase in synthetic complete minus uracil medium with 2% raffinose. The overexpression of Sit4p, which was induced with 2% galactose, did not lead to a growth defect. Lysates were prepared at the indicated time points and analyzed by Western blot analysis.
Next we examined the distribution of the other known phosphorylated coat subunits, Sec24p and Sec31p, in the sit4Δ mutant (Salama et al., 1997; Lord et al., 2011). Sec24p, the cargo adaptor of the COPII coat, has two paralogues, Lst1p and Iss1p (Roberg et al., 1999; Shimoni et al., 2000). Available antibodies to Lst1p enabled us to also examine the distribution of this coat subunit. The cytosolic pools of Sec24p, Lst1p, and Sec31p all increased in sit4Δ cells (Figure 1B, left, and Supplemental Figure S1B) but not in pph21Δ cells, another phosphatase mutant (Figure 1B, right, and Supplemental Figure S1C). Like Sit4p, Pph21p is a member of the PPP phosphatase family (Figure 1A). Of interest, Lst1p and Sec31p, two highly phosphorylated coat subunits (Stark et al., 2010), appeared to be less stable and migrate more slowly in lysates prepared from the sit4Δ mutant (Figure 1B, compare lanes 1 and 4). When Sit4p was overexpressed, Lst1p (Figure 1C, left) and Sec31p (Figure 1C, right) migrated faster. This increase in mobility was most prominent as the level of Sit4p expression increased (Figure 1C, bottom). Together these findings show that Sit4p, a type 2A serine/threonine phosphatase, regulates the intracellular distribution of COPII coat subunits, as well as the mobility of Sec31p and Lst1p on SDS–polyacrylamide gels.
Sit4p dephosphorylates Lst1p and Sec31p in vivo and in vitro
To address directly whether the shift in mobility of Lst1p and Sec31p in the sit4Δ mutant is the consequence of hyperphosphorylation of these coat subunits, we treated sit4Δ lysates with calf intestinal alkaline phosphatase (CIP) and analyzed the mobility of these coat subunits on a low-percentage polyacrylamide gel. Both Lst1p and Sec31p migrated faster after CIP treatment (Figure 2A, left, compare lanes 4 and 5). This shift in mobility was not observed when EDTA, a known inhibitor of CIP (Whisnant and Gilman, 2002), was present during the incubation (Figure 2A, left, compare lanes 4–6), or when wild-type lysate was treated with CIP (Figure 2A, left, compare lanes 1–3). We also immunoprecipitated Lst1p and Sec31p from wild-type and sit4Δ-mutant lysates and treated them with CIP (Figure 2A, right, and Supplemental Figure S2A). Interestingly, although we could not easily detect phosphorylation of Lst1p in wild-type lysates, a small but reproducible shift was observed when we treated immunoprecipitated Lst1p with CIP (Figure 2A, right). This shift was more pronounced when Lst1p was immunoprecipitated from the sit4Δ mutant (Figure 2A, right, compare lanes 3 and 4). Together these findings support the proposal that Lst1p and Sec31p are hyperphosphorylated in the sit4Δ mutant.
FIGURE 2:

Sit4p dephosphorylates Lst1p and Sec31p in vitro. (A) Left, lysates prepared from wild type (SFNY 1841) and the sit4Δ mutant (SFNY 2045) were incubated at 37°C for 15 min (lanes 1, 4) with CIP (lanes 2, 5) or CIP and EDTA (lanes 3, 6), before they were analyzed on a 6% SDS–polyacrylamide gel by Western blot analysis. Right, same as left, except that the samples were immunoprecipitated before CIP treatment. The asterisk marks the hyperphosphorylated forms of Lst1p and Sec 31p. (B) Top, equimolar amounts (0.2 μM) of GST, GST-Lst1p, and GST-Sec13p were incubated with Sit4p (0.2 μM), washed, and analyzed by Western blot analysis using anti-His antibody. GST and GST-Sec13p served as negative controls. Bottom, equimolar amounts of GST, GST-Sec31p (aa 1–500, 501–878, 879–1114, 1115–1273), and GST-Sec13p were incubated with Sit4p (0.2 μM) and processed as described. (C) Glutathione–Sepharose beads containing GST-Lst1p and GST-Sec31p (aa 1–500) were incubated without (lane 1) or with His6–Hrr25p K38R (lane 2) or His6–Hrr25p (lanes 3–6) in kinase assay buffer at 30°C for 1 h. The beads were washed and resuspended in phosphatase assay buffer without (lanes 1–4) or with Sit4p-HA3 (lanes 5 and 6) for 1 h at 30°C. EDTA was added (lane 6) to inhibit phosphatase activity. The samples were then analyzed on a 6% SDS–polyacrylamide gel by Western blot analysis.
If Sit4p dephosphorylates Lst1p and Sec31p, it may directly bind to these coat subunits in vitro. As shown in Figure 2B, hexahistidine (His6)-Sit4p bound to glutathione S-transferase (GST) fusions of Lst1p and Sec31p (amino acids [aa] 1–500 and 1115–1273) but not to GST and GST-Sec31p (aa 501–878 and 879–1114). His6-Sit4p also failed to bind to the coat subunit Sec13p (Figure 2B), which is not phosphorylated (Salama et al., 1997). Thus Sit4p binds to coat subunits that are hyperphosphorylated in the sit4Δ mutant. To determine whether Sit4p dephosphorylates Sec31p and Lst1p in vitro, we first phosphorylated GST-Lst1p and GST-Sec31p (aa 1–500) with His6-Hrr25p. Slower-migrating bands were seen only when these fusion proteins were incubated with His6-Hrr25p but not kinase-dead Hrr25p K38R (Figure 2C, lanes 1–3). Both GST-Lst1p and GST-Sec31p (aa 1–500) are substrates for Hrr25p in vitro, as 32P was incorporated into these fusion proteins when they were incubated with the kinase and [γ-32P]ATP in kinase assay buffer (Supplemental Figure S2B). In addition, the more dramatic shift that we observed on SDS–polyacrylamide gels with Lst1p made it possible for us to use the hrr25-5 mutant to demonstrate that this coat subunit is also a substrate of Hrr25p in vivo. These data and the isolation of the hrr25-5 mutant are described in the Supplemental Material (Supplemental Figure S2C and Table S2). Because bacterially expressed Sit4p is not active in vitro (unpublished observations), we immunoprecipitated Sit4p-HA3 from a yeast lysate and used the immunopurified phosphatase to dephosphorylate GST-Lst1p and GST-Sec31p. Phosphorylated GST-Lst1p and GST-Sec31p both migrated faster after they were incubated with Sit4p-HA3 in the absence (Figure 2C, compare lanes 4 and 5) but not the presence of EDTA (compare lanes 4–6). These data indicate that Sit4p can dephosphorylate Lst1p and Sec31p that had been phosphorylated in vitro by Hrr25p. Sit4p is the catalytic subunit of a multimeric phosphatase known to form a complex with four regulatory Sit4p-associated proteins (SAPs): Sap4p, Sap155p, Sap185p, and Sap190p (Luke et al., 1996). Because yeast but not recombinant Sit4p is active in vitro, these data also support the hypothesis that the catalytic activity of Sit4p depends on its association with the Sap regulatory subunits (Luke et al., 1996). His6-Sit4p also bound to GST-Sec23p and GST-Sec24p in vitro (Supplemental Figure S2D), suggesting that these coat subunits are also substrates of this phosphatase. This proposal is consistent with the observation that the soluble pool of Sec23p and Sec24p increases in the sit4Δ mutant (Figure 1B, left).
The loss of Sit4p function disrupts COPII vesicle budding in vitro and ER-to-Golgi traffic in vivo
If Sit4p regulates the dephosphorylation of the COPII coat in vivo, the loss of Sit4p function should disrupt secretion. To begin to address this possibility, we compared the trafficking of proteins secreted into the medium in the sit4Δ mutant and wild type. Briefly, wild-type and mutant cells were pulse labeled with S35-ProMix for 6, 8, 10, and 15 min, and proteins secreted into the medium were analyzed on an SDS–polyacrylamide gel. Between 8 and 15 min, several bands were secreted into the medium in wild-type cells. Four of these bands (marked by asterisks) were absent in the sit4Δ mutant at 15 min (Figure 3A, left). Quantitation of one of these bands, p150, is shown in Figure 3A (right). Several other bands were either secreted normally (arrow) or more slowly (arrowheads) in the sit4Δ mutant (Figure 3A, left). Together these findings indicate that secretion of several different proteins is delayed in the sit4Δ mutant.
FIGURE 3:

ER-to-Golgi traffic is delayed in the sit4Δ mutant. (A) Left, wild type (lanes 1–4) and the sit4Δ mutant (lanes 5–8) were pulse labeled at the indicated time points, and proteins secreted into the supernatant were analyzed as described in Materials and Methods. Right, quantitation of the secretion of p150 from three independent experiments. Error bars represent SD. The p values for 8- and 10-min time points are 0.002 and 0.004, respectively. (B) Left, wild type (lanes 1–4) and the sit4Δ mutant (lanes 5–8) were pulse labeled and chased, and CPY processing was analyzed as described in Materials and Methods. The p1, p2, and m forms of CPY are marked. Right, quantification of the ratio of p1:p2 forms from three independent experiments. Error bars represent SEM. *p < 0.05, Student's t test. (C) Mutant sit4Δ fractions are defective in vesicle budding. Permeabilized yeast cells (PYCs) and S1 fractions prepared from wild type (SFNY2051) and the sit4Δ mutant (SFNY 2179) were assayed as before (Groesch et al., 1990). Vesicle budding (left) and membrane fusion (right) were quantitatively measured as described in Lian and Ferro-Novick (1993). **p < 0.01, ***p < 0.001, Student's t test.
To address whether the defect in secretion is a consequence of disrupting membrane traffic between the ER and Golgi complex, we monitored the trafficking of the vacuolar hydrolase carboxypeptidase Y (CPY). CPY traffics from the ER (p1 CPY) to the Golgi (p2 CPY) before it is proteolytically processed to the mature form (mCPY) in the vacuole (Stevens et al., 1982). When the processing of CPY was monitored in the sit4Δ mutant at 0, 5, 10, and 15 min and compared with wild type, a delay in the conversion of p1 to p2 CPY was observed (Figure 3B, left). Quantitation of this trafficking defect from three separate experiments revealed a significant delay in the conversion of p1 to p2 CPY at 5 min (Figure 3B, right). Whereas p1 CPY was not apparent at 10 and 15 min in the wild type, it lingered in the mutant. Together these findings indicate that membrane traffic between ER and Golgi complex is kinetically delayed in the absence of the phosphatase Sit4p.
It was reported that phosphorylated COPII coat proteins cannot bind to membranes (Dudognon et al., 2004). A prediction of this observation is that COPII vesicle budding should be disrupted in the sit4Δ mutant. To test this prediction, we measured the budding of COPII vesicles from the ER in vitro by monitoring the release of the cargo marker pro–α-factor from the ER (Ruohola et al. 1988; Lian and Ferro-Novick, 1993). Interestingly, sit4Δ-mutant fractions showed a decrease in vesicle budding when compared with wild type (Figure 3C, left). Fusion of COPII vesicles with the Golgi, which was quantitatively assessed with an antibody that only recognizes carbohydrate added to yeast glycoproteins in the Golgi, was also decreased in the sit4Δ mutant (Figure 3C, right). These findings are consistent with the proposal that dephosphorylation of the coat is required to initiate a new round of vesicle budding (Lord et al., 2011). The defect in membrane fusion may be the consequence of blocking the cycling of the coat on and off membranes. A similar result was obtained when phosphomimetic mutations in Sec23p were analyzed in vitro (Lord et al., 2011).
Components of the secretory apparatus that function in ER-to-Golgi traffic are generally essential for growth (Novick et al., 1980). Although loss of Sit4p leads to a severe growth defect, it is not essential for growth in our laboratory strain background. The growth defect associated with the loss of SIT4 varies in different strain backgrounds depending on which allele of the polymorphic gene SSD1 (Suppressor of SIT4 deletion) is expressed (Sutton et al., 1991). Some alleles of SSD1 allow for the growth of sit4Δ strains, whereas other alleles lead to their inviability. In our strain background, the loss of Sit4p leads to slow growth (Supplemental Figure S3A). When, however, we deleted both SSD1 and SIT4 (ssd1Δsit4Δ) in the same haploid strain, the cells died (Supplemental Figure S3B). This observation suggests that our laboratory strain contains an allele of SSD1 that supports growth and secretion in the sit4Δ mutant.
Sit4p interacts with Hrr25p in yeast and mammalian cells
Phosphatases are frequently found in complexes with kinases that function together in the same process (Breitkreutz et al., 2010). In differential fractionation studies, however, Hrr25p and Sit4p do not cofractionate with each other. Hrr25p largely fractionates with membranes (Figure 4A, left; Lord et al., 2011), whereas Sit4p is largely soluble (Figure 4A, left). These differential fractionation studies were performed in the absence of salt to maintain the association of Hrr25p with membranes. Therefore, to address whether Sit4p and Hrr25p interact with each other under physiological conditions, we immunoprecipitated Sit4p-HA3 from lysates prepared in the presence of 150 mM NaCl. As shown in Figure 4A (right), Hrr25p (lane 2) specifically coimmunoprecipitated with Sit4p-HA3. No Hrr25p was precipitated from lysates that lacked the hemagglutinin (HA) tag (lane 1), and the TRAPP subunit Trs33p was not observed in the precipitate. The interaction was also direct, as purified recombinant GST-Hrr25 bound to His6-Sit4p in vitro (Supplemental Figure S3C).
FIGURE 4:

Sit4p interacts with Hrr25p in yeast and mammalian cells. (A) Left, total cell lysates (T) prepared from wild type (SFNY1841; lanes 1–3) were centrifuged at 150,000 × g to generate supernatant (S) and pellet (P) fractions. The asterisk represents degradation products of Hrr25p. Bos1p is shown as a fractionation control. Right, lysates from untagged cells (SFNY1841; lanes 1, 3) or Sit4-HA–tagged cells (SFNY2070; lanes 2, 4) were incubated with anti-HA beads (IP) and immunoblotted for Hrr25p and Trs33p. The entire precipitate was immunoblotted for Hrr25p and Trs33p, whereas only 1% of the IP was immunoblotted for Sit4p. The input is 0.5% of the total lysate that was immunoprecipitated. Approximately 0.5–1.0% of the Hrr25p coprecipitated with Sit4p-HA. (B) Left, total (T) lysates prepared from HeLa (lanes 1–3) or COS-7 (lanes 4x6) cells were centrifuged at 150,000 × g to generate supernatant (S) and pellet (P) fractions and analyzed by Western blot analysis. Right, cell lysates of HeLa cells expressing myc-CK1δ were immunoprecipitated with either control rabbit IgG (lane 1) or anti-PP6 (lane 2) antibody. Whereas all of the precipitate was blotted for CK1δ and mTrs120, only 5% of the IP was analyzed for PP6. The input represents 0.5 and 1% of the lysate that was immunoprecipitated (lanes 3 and 4, respectively). Approximately 0.5–1.0% of the myc-CKIδ coprecipitated with PP6. (C) CK1δ colocalizes with early Golgi and pre-Golgi markers. HeLa cells were immunostained with anti-CK1δ (green), anti-GM130 (red), and anti-ERGIC-53 (red) antibodies. Scale bar, 20 μm.
Previous studies demonstrated that the role of CKIδ, the mammalian orthologue of Hrr25p, is conserved in mammalian cells (Yu and Roth, 2002; Lord et al., 2011). Next we wanted to determine whether the role of Sit4p is also conserved. As was shown for Sit4p, its orthologue, PP6, was largely cytosolic in both HeLa and COS-7 cells, whereas CK1δ was mostly membrane bound (Figure 4B, left, and Supplemental Figure S4A). Consistent with previous studies localizing CK1δ to the Golgi (Milne et al., 2001), CK1δ was found in the perinuclear region of cells and colocalized with the cis-Golgi marker GM130 (Figure 4C, top). Although the trafficking machinery that mediates ER-to-Golgi traffic is highly conserved from yeast to humans, the architecture of the pathway is different. In yeast, COPII vesicles fuse with the Golgi, whereas in mammalian cells, COPII vesicles fuse with a pre-Golgi compartment that matures into the Golgi (Lord et al., 2013). Consistent with the proposal that CKIδ is required for COPII vesicle fusion (Lord et al., 2011), we found it localized to a greater degree with ERGIC-53, which marks the pre-Golgi compartment (Figure 4C, bottom). To determine whether CK1δ and PP6 interact with each other, we prepared lysates in the presence of 150 mM NaCl and precipitated the endogenous copy of PP6 with anti-PP6 antibody from HeLa cell lysates transiently expressing myc-CK1δ. When the precipitates were analyzed by Western blot analysis, PP6 antibody, but not immunoglobulin G (IgG), precipitated CK1δ (Figure 4B, right). The TRAPPII subunit mTrs120 was not present in the precipitate (Figure 4B, right), indicating that CK1δ specifically coprecipitates with PP6.
PP6, the orthologue of Sit4p, is required for ER-to-Golgi traffic in mammalian cells
To address whether PP6 is required for the subcellular distribution of COPII subunits, we used small interfering RNA (siRNA) to deplete cells of PP6 and then examined the distribution of Sec31A. Differential fractionation was performed on COS-7 cells (Figure 5A, middle) transfected with mock or siRNA targeted against PP6 mRNA (Figure 5A, left, and Supplemental Figure S4B), and lysates were separated into supernatant and pellet fractions. As shown in Figure 5A (middle; quantitated on the right), Sec31A distributed between the supernatant and membrane fractions in mock-treated cells (lanes 1–3), whereas in depleted cells it was largely present in the supernatant (lanes 4–6), suggesting that PP6 regulates the recycling of Sec31A on membranes. The loss of PP6 caused a more dispersed punctate localization pattern for the COPI coat (Figure 5B; compare Mock with siPP6-08, described in Materials and Methods; Zeng et al., 2010). This defect was specifically due to the loss of PP6, as it was rescued by the expression of a knockdown-resistant construct of PP6 (Figure 5B; compare siPP6-08 with Rescue). Similar results were obtained with a second siRNA duplex, siPP6-07 (Supplemental Figure S4C).
FIGURE 5:

PP6 regulates the intracellular distribution of Sec31. (A) Left, COS-7 cells transfected with mock or PP6 siRNA (siPP6-08) were harvested 72 h posttransfection and immunoblotted with anti-PP6 antibody (top). Actin (bottom) was used as a loading control. Quantitation of PP6 depletion in three separate experiments is shown in Supplemental Figure S4B. Error bars represent SEM. N = 3. Middle, total (T) lysates from either mock (lanes 1–3) or PP6-depleted (lanes 4–6) cells were centrifuged at 150,000 × g to generate supernatant (S) and pellet (P) fractions. Calnexin was used as a fractionation control. Right, quantitation of the supernatant:pellet ratio of Sec31A from three separate experiments. Error bars represent SEM. N = 3. *p < 0.05, Student's t test. (B) COPI fragments in PP6-depleted cells. COS-7 cells were transfected with mock, PP6 siRNA-08, or PP6 siRNA-08 and pCMV-Myc-hPPP6C-m3-08 (Materials and Methods). The cells were immunostained with anti-COPI antibody (left) and the number of fragmented structures quantified (right). Error bars represent SEM. N > 100 cells in three independent experiments. ***p < 0.001, Student's t test. Scale bar, 20 μm.
To determine whether PP6 regulates membrane traffic, we monitored the trafficking of a temperature-sensitive form of vesicular stomatitis virus G (VSV-G) protein (tsO45 VSV-G–green fluorescent protein [GFP]) to the cell surface in mock and PP6 COS-7–depleted cells (target sequence, siPP6-08; Supplemental Figure S4D and Figure 6A). To accumulate misfolded tsO45 VSV-G–GFP in the ER, we shifted cells to 40°C and added cycloheximide before shifting the cells to 32°C, a temperature that allows tsO45 VSV-G–GFP to fold into a conformation that is compatible with export from the ER. Trafficking of VSV-G to the cell surface was then monitored by immunofluorescence for 30, 60, and 90 min, using an antibody specific for the ectodomain of VSV-G. As shown in Figure 6A, delivery of VSV-G to the cell surface was delayed in PP6-depleted but not mock-treated cells. Figure 6B shows representative images of surface VSV-G and total VSV-G at 60 min.
FIGURE 6:
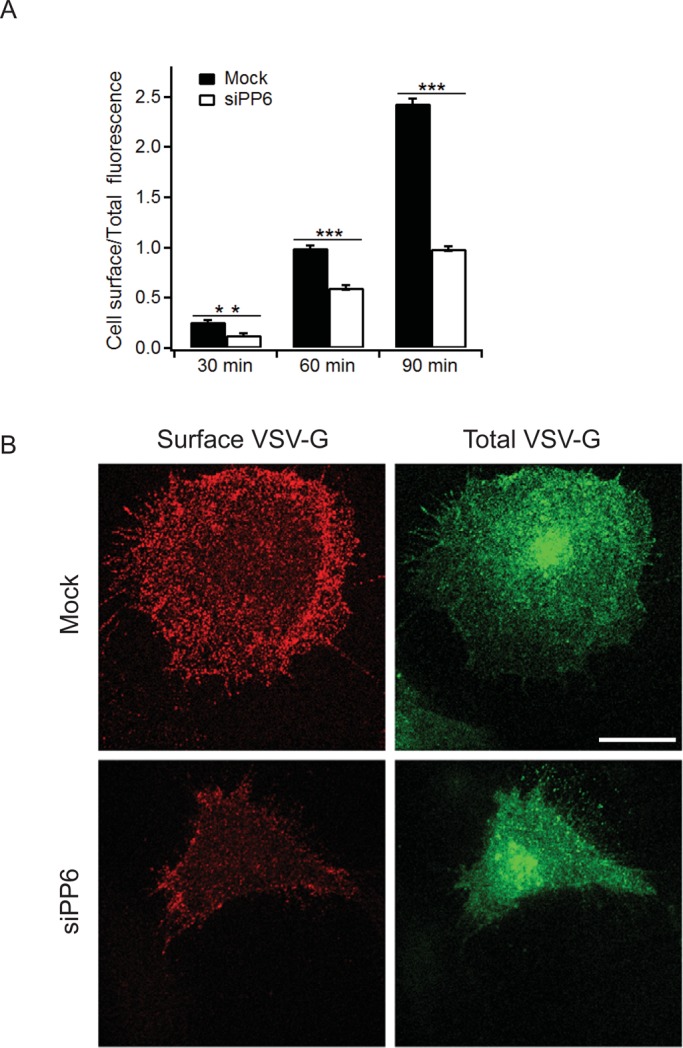
PP6 is required for the delivery of VSV-G to the cell surface. (A) COS-7 cells transfected with mock or PP6 siRNA (siPP6-08) were transfected with tsO45VSV-G-GFP and incubated at 40°C for 20 h. Cells were then treated with cycloheximide (100 μg/ml final concentration) for 30 min and shifted to 32°C for 30, 60, and 90 min. To detect cell surface VSV-G–GFP, cells were fixed without permeabilization and immunostained with anti–VSV-G ectodomain antibody. The 60-min time point was set to 1. Error bars represent SEM. N > 90 cells from three separate experiments. The data shown were quantitated as described in Materials and Methods. **p < 0.01, ***p < 0.001, Student's t test. (B) Cells at the 60-min time point were immunostained with anti–VSV-G ectodomain antibody to analyze cell surface VSV-G (red). Total VSV-G was analyzed with VSV-G–GFP (green). Scale bar, 20 μm.
To address whether VSV-G traffic was delayed between the ER and Golgi complex in depleted cells, we examined the intracellular pool of VSV-G at 0, 5, and 10 min after shifting the cells to 32°C. At 0 min, VSV-G resided in the ER in mock and depleted cells. By 5 min, some VSV-G reached the Golgi in the mock-treated cells, but delivery was delayed in the depleted cells. By 10 min, most of the VSV-G had reached the Golgi in mock-treated but not depleted cells (Figure 7A, left). Quantitation of the increasing ratio of juxtanuclear to peripheral VSV-G (Figure 7A, right) revealed a significant delay in the depleted cells at both 5 and 10 min. Similar results were obtained with a second siRNAi duplex, siPP6-07 (unpublished observations). During these experiments we noted that GM130 did not fragment in the PP6-depleted cells. Other integral membrane Golgi markers discussed later also did not show any fragmentation. Thus depletion of PP6 may indirectly affect COPI dynamics by disrupting ER-to-Golgi traffic without significantly affecting Golgi morphology.
FIGURE 7:

PP6 is required for ER-to-Golgi traffic in mammalian cells. (A) Left, COS-7 cells transfected with mock or PP6 siRNA (siPP6-08) were transfected with tsO45VSV-G-GFP and incubated at 40°C for 20 h. Cells were then treated with cycloheximide (100 μg/ml final concentration) for 30 min and shifted to 32°C for 0, 5, and 10 min, permeabilized with 0.1% Triton X-100, and immunostained with anti-GM130 antibody (red). Right, data shown on the left quantitated as described in Thayanidhi et al. (2010). Error bars represent the SEM. N = 3. More than 20 cells were examined for each time point in three separate experiments in which ∼89% of the PP6 was depleted. ***p < 0.001, Student's t test. Scale bar, 20 μm. (B) COS-7 cells were transfected with PP6 siRNA or mock transfected with Lipofectamine reagent alone and grown for 3 d. Cells were then either fixed directly (untreated) or incubated 1 h with BFA, followed by fixation (0-min washout) or recovery in medium without BFA for 75 min before fixation (75-min washout). Quantitation of pronounced giantin Golgi intensity (Materials and Methods) from >125 randomly selected cells for each plotted value. Y-axis value = (Golgi intensity at 75 min washout − Golgi intensity at 0-min washout)/(Golgi intensity of mock cells − Golgi intensity at 0 min) × 100%. Error bars represent SEM. Results are from a single representative experiment. The experiment was performed three times with similar outcomes. **p < 0.01, Student's t test. Knockdown of PP6 was 90% as assessed by immunoblotting of duplicate coverslips.
To determine whether PP6 is required for the transport of endogenous cargoes between the ER and Golgi complex, we examined the recovery of two integral Golgi membrane proteins after washout of brefeldin A (BFA), an assay that recapitulates all steps in ER-to-Golgi transport and Golgi biogenesis. Two Golgi markers were simultaneously monitored: GOS-28, a Golgi-restricted soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) that is present in all cisternae, as well as in intra-Golgi vesicles (Orci et al., 2000); and giantin, a tail-anchored, cis-Golgi membrane protein that interacts with the Golgi matrix but redistributes to the ER in BFA (Miles et al., 2001). Both Golgi markers displayed compact juxtanuclear localization in untreated COS-7 cells and dispersed into punctate and fine cytoplasmic reticular structures subsequent to a 1-h incubation with BFA. These behaviors were indistinguishable in mock and PP6-depleted cells (Supplemental Figure S5, untreated and 0-min washout). On washout of BFA, the pronounced juxtanuclear accumulation returned, with giantin reaccumulating more quickly and completely than GOS-28. Of importance, for both markers, mock-transfected cells reaccumulated juxtanuclear labeling more rapidly than their siPP6-transfected counterparts (Supplemental Figure S5, 75-min washout). This difference was quantifiable for giantin by measuring an objective parameter, the pronounced concentration of Golgi fluorescence (Figure 7B). Because GOS-28 labeling produced lower-contrast juxtanuclear accumulations, objective quantification was more difficult. The BFA results demonstrate that the requirement for PP6 in ER-to-Golgi transport extends to the constitutive export of endogenous cargoes. Together these findings imply that the role of Sit4 in ER-to-Golgi traffic is evolutionarily conserved.
DISCUSSION
The serine/threonine kinase Hrr25p was first implicated in membrane traffic when a mutation in hrr25 was identified as a suppressor of the growth and secretion defect of the temperature-sensitive sec12-4 mutant, which blocks the budding of COPII vesicles from the ER (Murakami et al., 1999). This led to the proposal that Hrr25p is a negative regulator of vesicle budding. More recently, we showed that Sec23p and Sec24p are substrates of Hrr25p and phosphorylation of the coat drives the membrane-bound pool into the cytosol (Lord et al., 2011). When Hrr25p activity is disrupted in vitro with the ATP competitive inhibitor IC261, vesicle budding is stimulated, suggesting that an increase in the unphosphorylated pool of the coat enhances budding. Together these findings imply that dephosphorylation of the COPII coat by a phosphatase is needed to recycle the coat for a new round of membrane traffic. To achieve a complete understanding of how phosphorylation and dephosphorylation regulate the membrane association of the COPII coat complex, it is important to identify the phosphatase that dephosphorylates the coat.
To identify this phosphatase, we screened ts and deletion mutants of the known phosphatases in yeast for changes in the distribution of the COPII coat subunit Sec23p. This screen identified one phosphatase, Sit4p, a member of the serine-threonine PPP family. These phosphatase family members contain one catalytic subunit and several regulatory subunits. Sit4p is the catalytic subunit of a multisubunit phosphatase complex that contains four Sit4p-associated proteins, called Sap4p, Sap155p, Sap185p, and Sap190p, which function positively with Sit4p (Luke et al., 1996). We found that the catalytic subunit of the complex, Sit4p, binds directly to COPII coat subunits. Phosphatase activity, however, may depend on its association with the Saps, as we were unable to express active Sit4p in bacteria. To show that Sit4p dephosphorylates the COPII coat, we focused on two of the more heavily phosphorylated COPII components, Lst1p and Sec31p (PhosphoGRID; Stark et al., 2000). We found that hyperphosphorylated forms of Lst1p and Sec31p accumulate in the sit4Δ mutant in vivo and demonstrated that Sit4p dephosphorylates Hrr25p-phosphorylated forms of Lst1p and Sec31p in vitro. Sit4p also binds directly to Sec23p and Sec24p, and loss of Sit4p increases the cytosolic pool of these coat subunits, suggesting that Sit4p may dephosphorylate both Sec23p and Sec24p.
COPII coat subunits are present in the cytosol and on ER-derived vesicles. It was postulated that the vesicular pool is selectively phosphorylated by Hrr25p, an orthologue of CKIδ (Lord et al., 2011). How this selectivity is achieved is unknown. The cytosolic phosphatase Sit4p may play a role in ensuring that coat subunits, prematurely phosphorylated in the cytosol, are rapidly dephosphorylated. Consistent with previously published high-throughput studies (Ho et al., 2000), which suggested a physical interaction between Sit4p and Hrr25p, we showed that Hrr25p and Sit4p directly bind to each other in vitro. Previous studies suggested that Hrr25p mediates phosphorylation of the COPII coat after it tethers to the Golgi, an event that is required to uncoat the vesicle (Lord et al., 2011). Although Hrr25p primarily resides on membranes, a small fraction is also present in the cytosol (Lord et al., 2011; Figure 4A, left) and may access the COPII coat before it can bind to membranes or before COPII-coated vesicles tether to the Golgi. Cytosolic phosphorylation of coat subunits could be counterbalanced by dephosphorylation via Sit4p. Another possibility is that Sit4p may bind to and dephosphorylate Hrr25p, a known phosphoprotein (Li et al., 2007; Holt et al., 2009). Although both hypotheses are possible, additional experiments will be needed to determine the physiological relevance of the Hrr25p/Sit4p interaction.
Consistent with the finding that Sit4p is the phosphatase that recycles the COPII coat, we found that the loss of Sit4p disrupts traffic from the ER to the Golgi complex in vivo and in vitro. In addition, the role of Sit4p in COPII coat recycling appears to be evolutionarily conserved. Depletion of the mammalian orthologue of Sit4p, PP6, by siRNA altered the subcellular distribution of Sec31 and delayed traffic between the ER and Golgi complex. PP6 and CKIδ appear to have the same intracellular distribution as their orthologues, and, like their orthologues, they interact with each other (Figure 4; Lord et al., 2011). Sit4p/PP6 previously was shown to function in the transition from G1 to S phase of the cell cycle (Sutton et al., 1991) and during mitotic spindle formation (Zeng et al., 2010). Therefore our findings uncover a new role for this phosphatase in COPII coat dephosphorylation and the regulation of ER-to-Golgi traffic.
MATERIALS AND METHODS
Strains and growth conditions
Yeast cells and plasmids used in this study are listed in Table 1 and Supplemental Table S1, respectively. Genomic disruptions and integrations were constructed using the method of Longtine et al. (1998) and confirmed by colony PCR and/or Western blot analysis. Cells were grown in yeast extract peptone, synthetic complete, or minimal medium supplemented with the appropriate amino acids. All media contained 2% glucose as the carbon source unless indicated otherwise.
TABLE 1:
Yeast strains used in this study.
| Strain number | Genotype | Source |
|---|---|---|
| SFNY1308 | MATa GAL+ ade2-1 his3-11 ura3-1 can1-100 ssd1-d2 trp1-1 leu2-3 glc7::LEU2 glc7-10::TRP1 | Michael Stark, School of Life Sciences, University of Dundee, Dundee, UK |
| SFNY1309 | MATa GAL+ ade2-1 his3-11 ura3-1 can1-100 ssd1-d2 trp1-1 leu2-3 glc7::LEU2 GLC7::TRP1 | Michael Stark |
| SFNY1841 | MATa GAL+ ura3-52 leu2-3, 112 his3Δ200 | Laboratory strain |
| SFNY1842 | MATα GAL+ ura3-52 leu2-3, 112 his3Δ200 | Laboratory strain |
| SFNY2028 | MATa ura3-52 leu2-3, 112 his3Δ200 | Michael Hampsey, Department of Biochemistry, Robert Wood Johnson Medical School, Piscataway, NJ |
| SFNY2029 | MATa ura3-52 leu2-3, 112 his3Δ200 ssu72-2 | Michael Hampsey |
| SFNY2030 | MATa GAL+ ade2-1 his3-11 ura3-1 can1-100 trp1-1 leu2-3 bar1::hisG | David Morgan, Department of Physiology, University of California, San Francisco, San Francisco, CA |
| SFNY2031 | MATa GAL+ ade2-1 his3-11 ura3-1 can1-100 trp1-1 leu2-3 bar1::hisG cdc14-1 | David Morgan |
| SFNY2045 | MATa GAL+ ura3-52 leu2-3, 112 his3Δ200 sit4Δ::His3MX6 | This study |
| SFNY2046 | MATα GAL+ ura3-52 leu2-3, 112 his3Δ200 sit4Δ::His3MX6 | This study |
| SFNY2051 | MATα ura3Δ0 leu2Δ0 met15Δ0 his3Δ1 hrr25Δ::KanMX6 pRS315-HRR25(CEN, LEU2+) | This study |
| SFNY2070 | MATa GAL+ ura3-52 leu2-3, 112 his3Δ200 SIT4-3xHA::His3MX6 | This study |
| SFNY2179 | MATα ura3Δ0 leu2Δ0 met15Δ0 his3Δ1 hrr25Δ::KanMX6 pRS315-HRR25(CEN, LEU2+) sit4Δ::His3MX6 | This study |
| SFNY2219 | MATa GAL+ ura3-52 leu2-3, 112 his3Δ200 sit4Δ::His3MX6 ssd1Δ::KanMX6 pRS316-SIT4(CEN, URA3+) | This study |
| SFNY2226 | MATα GAL+ ura3-52 leu2-3, 112 his3Δ200 sit4Δ::His3MX6 GAL1pr-SIT4::URA3 | This study |
| SFNY2411 | MATa GAL+ ura3-52 leu2-3, 112 his3Δ200 pph21Δ::KanMX6 | This study |
Antibodies
Monoclonal antibodies directed against GM130 and Sec31A were purchased from BD Biosciences (San Jose, CA). Polyclonal anti-PP6 antibody was purchased from Bethyl Laboratories (Montgomery, TX). Polyclonal anti-GST and monoclonal anti-HA and anti-His antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), Covance (Berkeley, CA), and Thermo Scientific (Waltham, MA), respectively. Mouse monoclonal anti–GOS-28 antibody was purchased from Stressgen (San Diego, CA). Rabbit polyclonal anti-giantin antibody was purchased from Covance. Monoclonal antibodies directed against COPI (CM1A10) and VSV-G ectodomain were obtained from G. Warren (Max F. Perutz Laboratories, Vienna, Austria). Anti–ERGIC-53 and anti-Lst1p antibodies were obtained from H.-P. Hauri (University of Basel, Basel, Switzerland) and Randy Schekman (University of California, Berkeley, CA), respectively. Polyclonal antibody directed against CKIδ was prepared to full-length CKIδ fused to GST and affinity purified on an Affi-Gel column (Bio-Rad, Hercules, CA) preloaded with GST-CKIδ. The flowthrough was discarded and the bound IgG was eluted with 0.2M glycine (pH 2.8) followed by rapid neutralization with 1 M potassium phosphate (pH 7.5). The antibody was then buffer exchanged with phosphate-buffered saline (PBS), concentrated, and stored at −80°C in 30% glycerol. Fluorescently labeled secondary antibodies were purchased from Life Technologies (Carlsbad, CA).
In vitro kinase assay
Purified GST-fusion proteins (2 μg) immobilized on glutathione–Sepharose beads were incubated with 250 ng of His6-Hrr25p (aa 1–394) or catalytically inactive His6-Hrr25p K38R (aa 1–394) for 1 h at 30°C in kinase assay buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.4, 2 mM EDTA, 10 mM MgCl2, 1 mM dithiothreitol [DTT], 5 mM cold ATP, 2.5 μCi of [32P]ATP, 100 μM sodium orthovanadate, 10 mM sodium fluoride, 10 mM sodium pyrophosphate, and protease inhibitors). The beads were washed twice with 1× PBS and eluted in 25 μl of sample buffer by heating to 100°C for 5 min. The samples were analyzed by autoradiography.
Differential centrifugation
A total of 100 OD600 units of yeast cells was pelleted, resuspended in 2 ml of buffer (1.4 M sorbitol, 100 mM sodium phosphate, pH 7.5, 0.35% 2-mercaptoethanol, and 0.5 mg/ml Zymolyase) and incubated for 30 min at 37°C. The spheroplasts were then centrifuged over a 4-ml sorbitol cushion (1.7 M sorbitol, 100 mM HEPES, pH 7.2) for 5 min at 2100 × g at 4°C. The supernatant was removed, and the pellet was resuspended in 1 ml of cold lysis buffer (100 mM HEPES, pH 7.2, 1 mM EDTA, 0.2 mM DTT, protease and phosphatase inhibitors) and lysed using a Dounce homogenizer. The lysate was centrifuged for 2 min at 500 × g at 4°C, and the supernatant was transferred to a new tube. An aliquot (100 μl) was mixed with 50 μl of 3× sample buffer (total fraction [T]) and heated at 100°C for 5 min, and 500 μl of the remaining portion was centrifuged for 90 min at 150,000 × g at 4°C. The lipid layer was removed, and 100 μl of the supernatant (S) was mixed with 50 μl of 3× sample buffer and heated at 100°C for 5 min. The pellet (P) was resuspended in 500 μl of lysis buffer, and 100 μl was mixed with 50 μl of 3× sample buffer and heated at 100°C for 5 min.
For mammalian cells, a fully confluent 15-cm dish of COS-7/HeLa cells was washed twice with cold PBS. The cells were harvested, resuspended in 300 μl of cold lysis buffer, and lysed by passing through a 25-gauge needle 10 times. The cell debris was pelleted at 500 × g, and 100-μl fractions (T, S, and P) were saved as described above.
Immunoprecipitation from yeast and mammalian lysates
Spheroplasts prepared from 200 OD600 of yeast cells were lysed in 1 ml of IP buffer (25 mM HEPES, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM DTT, 1 mM EDTA, protease inhibitors). Cell lysates were centrifuged at 16, 000 × g for 15 min, and the cleared supernatants (4 mg of total protein) were incubated with 20 μl of a 50% slurry of anti-HA beads (Sigma-Aldrich, St. Louis, MO) at 4°C for 3 h.
For mammalian cells, HeLa cells (10-cm confluent plate) transiently expressing myc-CKIδ were lysed in 1 ml of the IP buffer. The lysates were clarified by centrifugation at 16,000 × g for 15 min. An equal volume of the supernatant was incubated with 2 μg of either control IgG or anti-PP6 antibody for 3 h. The immunocomplexes were then bound to 40 μl of protein A beads (50% slurry in IP buffer) for 1 h at 4°C. The beads were washed 3× with IP buffer and eluted in sample buffer by heating to 100°C for 5 min. Immunoprecipitates were then subjected to Western blot analysis.
In vitro phosphatase assays
For Sit4p phosphatase assays, Sit4p-HA3 was immunoprecipitated from a yeast lysate as described and incubated with phosphorylated recombinant proteins. To phosphorylate GST-Lst1p and GST-Sec31p (aa 1–500) in vitro, immobilized proteins were phosphorylated with recombinant His6-Hrr25 1–394 as described (except that [32P]ATP was omitted), and the beads were washed 3× with 700 μl of phosphatase buffer (100 mM Tris, pH 8.0, 2 mM DTT, 1 mM MnCl2). The phosphorylated proteins (250 ng) were then aliquoted into tubes containing 20 μl of phosphatase buffer, empty beads, or beads coated with Sit4-HA3, and the samples were incubated at 30°C for 1 h.
For CIP assays, spheroplasts prepared from 100 OD600 of yeast cells were lysed in lysis buffer (50 mM Tris, pH 7.9, 100 mM NaCl, 10 mM MgCl2, 1 mM DTT, protease inhibitors), and the cleared supernatant (500 μg of total protein) was incubated at 37°C for 10–15 min without or with CIP (0.5 U/μl final concentration) or with CIP and 50 mM EDTA. After the incubation, sample buffer was added and samples were heated to 100°C for 5 min. Samples were then subjected to Western blot analysis.
CPY pulse chase assay
Cells were grown overnight at 25°C in minimal medium to early log phase. A total of 16 OD600 units was pelleted, resuspended in 3.6 ml of fresh minimal medium, incubated at 37°C for 20 min, and pulse labeled with 400 Ci of [35S]methionine for 4 min at 37°C. An aliquot (700 μl) was removed as the 0-min time point, centrifuged, and washed twice with ice-cold 10 mM sodium fluoride/sodium azide. Chase mix (150 μl of 250 mM methionine) was added to the remaining sample. At the desired time points, 700 μl of the cell suspension was removed and processed as described above. The cells were converted to spheroplasts, pelleted at 4000 × g for 3 min, resuspended in 100 μl of 1% SDS, and heated for 5 min at 95°C. The samples were diluted with 900 μl of 1× PBS containing 1% Triton X-100 and centrifuged at 16,000 × g for 15 min, and the supernatant was incubated with 3 μl of anti-CPY antibody for 1 h at 4°C. A 50% slurry of protein A–Sepharose (60 μl) was added, and the samples were incubated for an additional hour. The beads were washed, and protein was solubilized in 1× sample buffer and electrophoresed on an 8% SDS–polyacrylamide gel.
General secretion assay
General secretion was measured as described before (Grote et al., 2000). Briefly, cells were grown overnight to early log phase in synthetic complete medium without methionine. The next day, 1.5 OD600 units of cells were resuspended in 400 μl of methionine-free medium supplemented with 0.06 mg/ml bovine serum albumin and incubated at 37°C for 20 min. A total of 150 μCi of 35S-ProMix was added to each sample, and aliquots of cells were removed and spun for 5 s at the indicated time points. The medium (300 μl) was transferred to an ice-cold tube containing 30 μl of stop mix (500 mM NaN3, 500 mM NaF), followed by another spin at 14,000 × g for 1 min. The supernatant was then transferred to a fresh tube containing 20 μl of 100% trichloroacetic acid (TCA) with 1 mg/ml sodium deoxycholate and incubated on ice for 1 h. The TCA precipitated proteins were pelleted (14,000 × g for 5 min) and washed twice with ice-cold acetone. The acetone-washed pellets were air dried, resuspended in sample buffer, heated to 100°C for 5 min, and electrophoresed on an 8% SDS–polyacrylamide gel.
In vitro binding assays with recombinant proteins
Bacterially expressed and purified His6-Sit4p (0.2 μM) was incubated with equimolar amounts of GST-fusion proteins or GST (0.2 μM) in binding buffer (25 mM HEPES, pH 7.2, 150 mM NaCl, 0.5% Triton X-100, 1 mM DTT, 1 mM EDTA, 1 mM MnCl2, and protease inhibitors) for 3–4 h at 4°C. The beads were washed 3× with binding buffer and eluted in 25 μl of sample buffer by heating to 100°C for 5 min.
RNA interference
HeLa and COS-7 cells were grown in DMEM supplemented with 10% fetal bovine serum at 37°C in a 5% CO2 incubator. siRNAs targeting human PP6 (siPP6-07, CGCUAGACCUGGACAAGUA; siPP6-08, GUUUGGAGACCUUCACUUA) were obtained from Dharmacon (Denver, CO). Transfections (cDNA and siRNA) were performed using Lipofectamine 2000 (Life Technologies) as per manufacturer's instructions.
For the rescue experiments, COS-7 cells transfected with PP6 siRNA-08 were cotransfected with pCMV-Myc-hPPP6C-m3-08, an expression vector that expresses myc-PP6 containing three silent mutations (267 T to C, 268 T to C, and 282 T to C) in the target sequence of si-PP6-08. Cells transfected with PP6 siRNA-07 were cotransfected with pCMV-Myc-hPPP6C-m3-07, an expression vector containing three silent mutations (18 G to A, 21 C to T, and 24 G to A) in the target sequence of si-PP6-07.
Immunofluorescence microscopy
Cells were grown and processed for immunofluorescence as described previously (Yamasaki et al., 2009). The secondary antibody used to localize CK1δ was anti-rabbit IgG conjugated to Alexa 488. Anti-mouse IgG conjugated to Alexa 594 was used to localize ERGIC-53, GM130, COPI, and Sec31A. Images were taken with an LSM 510 confocal microscope using a 100× objective and captured with a digital AxioCam MRm camera (Carl Zeiss Microimaging, Thornwood, NY). For Figure 7B and Supplemental Figure S5, anti-rabbit IgG conjugated to fluorescein isothiocyanate was used to label giantin, and anti-mouse IgG conjugated to Cy3 was used to label GOS-28. Wide-field fluorescence images were captured using a 60× objective on a Nikon E800 microscope (Nikon, Melville, NY) with excitation and emission filter wheels (Chroma Technology, Bellows Falls, VT), a Hamamatsu Orca 2 camera (Hamamatsu, Hamamatsu, Japan), Nikon Z-drive, and OpenLab 5.0 software (Improvision; PerkinElmer, Waltham, MA).
VSV-G trafficking assay
Approximately 24 h after transfecting COS-7 cells with mock/PP6 siRNA at 37°C, we transfected cells with pEGFPdKA206K-N1-VSVG tsO45. The next day, the cells were shifted to 40°C, and after 20 h they were treated with 100 μg/ml cycloheximide. After 30 min, the cells were shifted to 32°C for various times and processed as described previously (Seemann et al., 2000; Yamasaki et al., 2009). For the data in Figure 6, the cells were fixed in 3.7% paraformaldehyde for 15 min and stained with anti–VSV-G ectodomain antibody (primary antibody) and anti-mouse IgG conjugated to Alexa 594 (secondary antibody). Total VSV-G–GFP fluorescence was quantified as before (Seemann et al., 2000). All images were captured at the same setting and exposure. To quantitate the data, the cell area (A) was defined manually and the mean of the fluorescence intensity (I) in that area was measured using ImageJ (National Institutes of Health, Bethesda, MD). The integrated optical density (IOD) was determined by the formula IOD = A × I. Expression levels were normalized by determining the ratio of cell surface to total VSV-G IOD. The data were quantified from three independent experiments, and >30 cells were examined for each time point in every experiment. The zero time point was subtracted from each time point shown. For the data in Figure 7A, the cells were permeabilized in 0.1% Triton X-100 and quantified as described in Thayanidhi et al. (2010) from three independent experiments. More than 20 cells were examined for each time point in every experiment.
BFA recovery assay
The BFA recovery experiments were performed as follows. Three days after transfection with PP6 siRNA or mock transfection with Lipofectamine reagent alone, the medium was replaced with medium containing 2.5 μg/ml BFA. After 1-h incubation at 37°C in BFA, coverslips were dipped several times in PBS and placed into wells of fresh, prewarmed medium containing 10 μg/ml cycloheximide. Coverslips were then fixed at various times and processed for double-label immunofluorescence using anti–GOS-28 and anti-giantin antibodies. Imaging of cells was conducted using a fixed exposure time for each color channel. To quantify giantin Golgi intensity, we used Openlab software. Regions of interest (ROIs) were drawn tightly around each Golgi region and the maximum intensity recorded. A second ROI was drawn abutting outside and following the edge of the first and its mean intensity recorded. Our measure of pronounced “Golgi intensity” (Figure 7B) is defined as the maximum intensity of the first ROI divided by the mean intensity of the second ROI, calculated individually for each cell in every assayed field. For cells without a clear juxtanuclear labeling (e.g., at 0-min washout), the ROI was drawn around the brightest section of the perinuclear area. Image capture, drawing of ROIs, and calculation of intensities were performed doubly blind.
Supplementary Material
Acknowledgments
We thank Elizabeth Miller, Seema Mattoo, and Vincent Tagliabracci for advice and M. Stark, D. Morgan, and M. Hampsey for strains. We also thank Y. Jiang, G. Warren, H.-P. Hauri, and R. Schekman for antibodies and Wenyun Zhou for technical support. Salary support for D.B., J.Z, S.M., S.C., and S.F.-N. was provided by the Howard Hughes Medical Institute. S.F.-N. is an Investigator of the Howard Hughes Medical Institute. K.D.C. is supported by the Ludwig Institute for Cancer Research and a grant from the National Institutes of Health (RO1 GM104141). Work at the University of Montana was supported by National Institutes of Health Grant GM106323 (to J.C.H.) and by the Center for Structural and Functional Neuroscience.
Abbreviations used:
- COPII
coat protein complex II
- GTP
guanosine triphosphate
- IP
immunoprecipitation
- PP2A
protein phosphatase 2A
- PP6
protein phosphatase 6
- TRAPP
transport protein particle
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-02-0114) on July 17, 2013.
*Present address: Department of Cell and Developmental Biology, Vanderbilt University School of Medicine, Nashville, TN 37232.
REFERENCES
- Breitkreutz A, et al. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043–1046. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant NJ, James DE. The Sec1p/Munc18 (SM) protein, Vsp45p, cycles on and off membranes during vesicle transport. J Cell Biol. 2003;161:691–696. doi: 10.1083/jcb.200212078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Yu S, Menon S, Cai Y, Lazarova D, Fu C, Reinisch K, Hay JC, Ferro-Novick S. TRAPPI tethers COPII vesicles by binding the coat subunit Sec23. Nature. 2007;445:941–944. doi: 10.1038/nature05527. [DOI] [PubMed] [Google Scholar]
- Dudognon P, Maeder-Garavaglia C, Carpentier J-L, Paccaud J-P. Regulation of a COPII component by cytosolic O-glycosylation during mitosis. FEBS Lett. 2004;561:44–50. doi: 10.1016/S0014-5793(04)00109-7. [DOI] [PubMed] [Google Scholar]
- Groesch ME, Ruohola H, Bacon R, Rossi G, Ferro-Novick S. Isolation of a functional vesicular intermediate that mediates ER to Golgi transport in yeast. J Cell Biol. 1990;111:45–53. doi: 10.1083/jcb.111.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote E, Carr CM, Novick PJ. Ordering the final events in yeast exocytosis. J Cell Biol. 2000;151:439–451. doi: 10.1083/jcb.151.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, Morgan DO. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 2009;325:1682–1686. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MCS, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122:605–617. doi: 10.1016/j.cell.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Li X, Gerber SA, Rudner AD, Beausoleil SA, Haas W, Villén J, Elias JE, Gygi SP. Large-scale phosphorylation analysis of α-factor-arrested Saccharomyces cerevisiae. J Proteome Res. 2007;6:1190–1197. doi: 10.1021/pr060559j. [DOI] [PubMed] [Google Scholar]
- Lian JP, Ferro-Novick S. Bos1p, an integral membrane protein of the endoplasmic reticulum to Golgi transport vesicles, is required for their fusion competence. Cell. 1993;73:735–745. doi: 10.1016/0092-8674(93)90253-m. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lord C, Bhandari D, Menon S, Ghassemian M, Nycz D, Hay J, Ghosh P, Ferro-Novick S. Sequential interactions with Sec23 control the direction of vesicle traffic. Nature. 2011;473:181–186. doi: 10.1038/nature09969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Ferro-Novick S, Miller EA. The highly conserved COPII coat complex sorts cargo from the endoplasmic reticulum and targets it to the Golgi. Cold Spring Harb Perspect Biol. 2013;5:a013367. doi: 10.1101/cshperspect.a013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke MM, Della Seta F, Di Como CJ, Sugimoto H, Kobayashi R, Arndt KT. The SAP, a new family of proteins, associate and function positively with the Sit4 phosphatase. Mol Cell Biol. 1996;16:2744–2755. doi: 10.1128/mcb.16.6.2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles S, McManus H, Forsten KE, Storrie B. Evidence that the entire Golgi apparatus cycles in interphase HeLa cells: sensitivity of Golgi matrix proteins to an ER exit block. J Cell Biol. 2001;155:543–55. doi: 10.1083/jcb.200103104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne DM, Looby P, Meek DW. Catalytic activity of protein kinase CK1δ (casein kinase 1δ) Is essential for its normal subcellular localization. Exp Cell Res. 2001;263:43–54. doi: 10.1006/excr.2000.5100. [DOI] [PubMed] [Google Scholar]
- Murakami A, Kimura K, Nakano A. The inactive form of a yeast casein kinase I suppresses the secretory defect of the sec12 mutant. J Biol Chem. 1999;274:3804–3810. doi: 10.1074/jbc.274.6.3804. [DOI] [PubMed] [Google Scholar]
- Novick P, Field C, Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell. 1980;21:205–215. doi: 10.1016/0092-8674(80)90128-2. [DOI] [PubMed] [Google Scholar]
- Orci L, Ravazzola M, Volchuk A, Engel T, Gmachl M, Amherdt M, Perrelet A, Sollner TH, Rothman JE. Anterograde flow of cargo across the Golgi stack potentially mediated via bidirectional “percolating” COPI vesicles. Proc Natl Acad Sci USA. 2000;97:10400–10405. doi: 10.1073/pnas.190292497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberg KJ, Crotwell M, Espenshade P, Gimeno R, Kaiser CA. LST1 is a SEC24 homologue used for selective export of the plasma membrane ATPase from the endoplasmic reticulum. J Cell Biol. 1999;145:659–672. doi: 10.1083/jcb.145.4.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruohola H, Kabcenell AK, Ferro-Novick S. Reconstitution of protein transport from the endoplasmic reticulum to the Golgi complex in yeast: the acceptor Golgi compartment is defective in the sec23 mutant. J Cell Biol. 1988;107:1465–1476. doi: 10.1083/jcb.107.4.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama NR, Chuang JS, Schekman RW. Sec31 encodes an essential component of the COPII coat required for transport vesicle budding from the endoplasmic reticulum. Mol Biol Cell. 1997;8:205–217. doi: 10.1091/mbc.8.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann J, Jokitalo EJ, Warren G. The role of the tethering proteins p115 and GM130 in transport through the Golgi apparatus in vivo. Mol Biol Cell. 2000;11:635–645. doi: 10.1091/mbc.11.2.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoni Y, Kurihara T, Ravazzola M, Amherdt M, Orci L, Schekman R. Lst1p and Sec24p cooperate in sorting of the plasma membrane ATPase into COPII vesicles in Saccharomyces cerevisiae. J Cell Biol. 2000;151:973–984. doi: 10.1083/jcb.151.5.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Su T-C, Breitkreutz A, Lourenco P, Dahabieh M, Breikreutz B-J, Tyers M, Sadowski I. PhosphoGRID: a database of experimentally verified in vivo phosphorylation sites from budding yeast Saccharomyces cerevisiae. Database (Oxford) 2010. 2010:bap026. doi: 10.1093/database/bap026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens T, Esmon B, Schekman R. Early stages in the yeast secretory pathway are required for transport of carboxypeptidase Y to the vacuole. Cell. 1982;30:439–448. doi: 10.1016/0092-8674(82)90241-0. [DOI] [PubMed] [Google Scholar]
- Sutton A, Immanuel D, Arndt KT. The SIT4 protein phosphatase functions in late G1 for progression into S phase. Mol Cell Biol. 1991;11:2133–2148. doi: 10.1128/mcb.11.4.2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayanidhi N, Helm JR, Nycz DC, Bentley M, Liang Y, Hay JC. α-Synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol Cell Biol. 2010;21:1850–1863. doi: 10.1091/mbc.E09-09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whisnant AR, Gilman SD. Studies of reversible inhibition, irreversible inhibition, and activation of alkaline phosphatase by capillary electrophoresis. Anal Biochem. 2002;307:226–234. doi: 10.1016/s0003-2697(02)00062-3. [DOI] [PubMed] [Google Scholar]
- Whyte JR, Munro S. Vesicle tethering complexes in membrane traffic. J Cell Sci. 2002;115:2627–2637. doi: 10.1242/jcs.115.13.2627. [DOI] [PubMed] [Google Scholar]
- Yamasaki A, Menon S, Yu S, Barrowman J, Meerloo T, Oorschot V, Klumperman J, Satoh A, Ferro-Novick S. mTrs130 is a component of a mammalian TRAPPII complex, a Rab1 GEF that binds to COPI-coated vesicles. Mol Biol Cell. 2009;20:4205–4215. doi: 10.1091/mbc.E09-05-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Roth MG. Casein kinase I regulates membrane binding by ARF GAP1. Mol Biol Cell. 2002;13:2559–2570. doi: 10.1091/mbc.E02-04-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R. COPII and the regulation of protein sorting in mammals. Nat Cell Biol. 2011;14:20–28. doi: 10.1038/ncb2390. [DOI] [PubMed] [Google Scholar]
- Zeng K, Bastos RN, Barr FA, Gruneberg U. Protein phosphatase 6 regulates mitotic spindle formation by controlling the T-loop phosphorylation state of Aurora A bound to its activator TPX2. J Cell Biol. 2010;191:1315–1332. doi: 10.1083/jcb.201008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.