

Abstract
Cryptosporidium parvum is an enteric protozoan parasite that has emerged as a major cause of diarrhea, malnutrition and gastroenteritis as well as posing a potential bioterrorism threat. C. parvum synthesizes guanine nucleotides from host adenosine in a streamlined pathway that relies on inosine 5′-monophosphate dehydrogenase (IMPDH). We have previously identified several parasite-selective C. parvum IMPDH (CpIMPDH) inhibitors by high-throughput screening. In this paper, we report the structure-activity relationship (SAR) for a series of benzoxazole derivatives with many compounds demonstrating CpIMPDH IC50 values in the nanomolar range and > 500-fold selectivity over human IMPDH (hIMPDH). Unlike previously reported CpIMPDH inhibitors, these compounds are competitive inhibitors versus NAD+. The SAR study reveals that pyridine and other small heteroaromatic substituents are required at the 2-position of the benzoxazole for potent inhibitory activity. In addition, several other SAR conclusions are highlighted with regard to the benzoxazole and the amide portion of the inhibitor, including preferred stereochemistry. An x-ray crystal structure of a representative E•IMP•inhibitor complex is also presented. Overall, the secondary amine derivative 15a (Q67) demonstrated excellent CpIMPDH inhibitory activity (IC50 = 0.5 ± 0.1 nM) and moderate stability (t1/2 = 44 min) in mouse liver microsomes. Compound 73, the racemic version of 15a, also displayed superb antiparasitic activity in a Toxoplasma gondii strain that relies on CpIMPDH (EC50 = 20 ± 20 nM), and selectivity versus a wild-type T. gondii strain (200-fold). No toxicity was observed (LD50 > 50 μM) against a panel of four mammalian cells lines.
Introduction
Cryptosporidiosis is an intestinal diarrheal disease most commonly caused by Cryptosporidium parvum and hominis. These protozoan parasites are widely distributed in both the developing and developed worlds1 and a major cause of severe diarrhea and malnutrition in children2. Infection is self-limiting in immune-competent adults, but chronic in immunocompromised individuals and children. Infections are transmitted by the fecal to oral route through drinking and recreational waters3. The infectious oocysts are resistant to standard water treatment methods like bleach and filtration. C. parvum oocysts are readily obtained from infected calves, and could potentially be used as a bio-weapon 4. Hence, C. parvum is categorized as a class B bio-warfare agent. Furthermore, currently available drugs are not effective for treating cryptosporidiosis and vaccine therapy is lacking5, so new drugs are needed.
Cryptosporidium are intracellular parasites. Genomic analysis revealed that these parasites cannot synthesize purine nucleotides de novo, but instead salvage adenosine from the host6. Adenosine is converted into guanine nucleotides in a streamlined pathway that relies on inosine 5′-monophosphate dehydrogenase (IMPDH) catalyzing the conversion of inosine 5′-monophosphate (IMP) to xanthosine 5′-monophosphate (XMP) 7. The parasite enzyme (CpIMPDH) is structurally distinct from mammalian IMPDHs. Cryptosporidium appears to have obtained its IMPDH gene from bacteria via lateral gene transfer6b, 6c, 8. Therefore, CpIMPDH has emerged as an attractive molecular target for the development of effective therapeutics for the treatment of diseases associated with this recalcitrant organism.
As previously reported, a high throughput screening campaign identified structurally diverse selective CpIMPDH inhibitors9. Several of these compound series have been optimized to produce low nanomolar CpIMPDH inhibitors such as A110, C64, C97, P96 and D48 (Figure 1)10. Herein, we report the structure-activity relationship for the benzoxazole-based inhibitor 1, a co-crystal structure of a representative derivative of this compound series with CpIMPDH and anti-parasitic activity for a subset of compounds in a Toxoplasma gondii surrogate model.
Figure 1.

Optimized CpIMPDH inhibitors A110, C64, C97, P96 and D48 and CpIMPDH inhibitor 1 identified by HTS.
Results and Discussion
Chemistry
Various derivatives of 1 were synthesized using the methods depicted in Scheme 1. 5-Nitro-2-arylbenzoxazoles 3 were prepared from 2-amino-4-nitrophenols 2 and aromatic aldehydes in the presence of activated carbon (DarcoKB)11. Reductive hydrogenation of 3 in the presence of 10% Pd-C provided 5-amine-2-arylbenzoxazoles 4. Substituted phenols and anilines were converted to the corresponding ethers and amines 6 upon treatment with ethyl 2-bromopropionates in the presence of K2CO3. Enantiomerically pure phenyl ethers were synthesized by using Mitsunobu reaction conditions with (+)-methyl D-lactate or (-)-methyl L-lactate10a. Ester hydrolysis was carried out in THF with 3M sodium hydroxide for racemic esters and with 3N HCl in THF for enantiomerically pure esters to yield acids 7. Subsequently, 7 was coupled with 5-amine-2-arylbenzoxazoles 4 with the aid of EDCI•HCl in anhydrous DMF yielding amides 8. N-alkylation of the amide was carried using sodium hydride and iodomethane to give 9 in moderate yield.
Scheme 1.
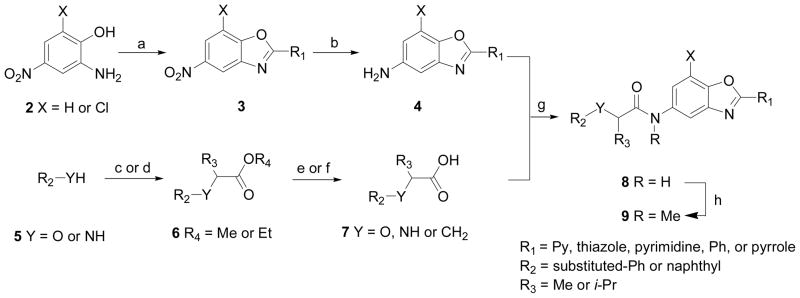
General procedure for the synthesis of benzoxazole derivatives.
Reagents and conditions: (a) R1-CHO, O2, DarcoKB, xylene, 120 °C, 6 h, 70–80% (b) H2 (1 atm), 10% Pd-C, EtOAc, 6 h, 80–86% (c) R3CHBrCOOR4, K2CO3, DMF, rt, 5 h, 85–92% when Y = O (or 70 °C, 3 h, 36% when Y = NH) (d) (R)- or (S)- R3CH(OH)COOR4, 0 °C, PPh3, 10 min, DEAD, rt, 12 h, 75–84% (e) 3 M NaOH, THF:H2O (2:1), 80 °C, 3 h, 85–95% (f) 3N HCl, THF, 70 °C, 6 h, 60–70% (g) EDCI•HCl, DMF, 0 °C - rt, 12 h, 63–79% (h) MeI, NaH, THF, 0 °C - rt, 1.5 h, 48%.
Enantiomerically pure benzoxazole amines were prepared using the method outlined in Scheme 2. L-alanine (10) was allowed to react with 2,3-di-chloroiodobenzene in the presence of CuI and Cs2CO3 to give 11 12. The acid was coupled with a benzoxazole amine derivative with the aid of EDCI•HCl to give 15a. Copper (II) acetate mediated coupling of L-alanine methyl ester with 1-naphthyl boronic acid (12) yielded 1313. Hydrolysis under mild basic conditions generated acid 14, which was subsequently coupled with a 5-amine-2-(4-pyridyl)benoxazole to yield 15b.
Scheme 2.

General procedure for the synthesis of enantiomerically pure secondary amines of 5-benzoxazoles.
Reagents and conditions: (a) 2,3-di-ClPhI, Cs2CO3, CuI, DMF, 90 °C, 48 h, 57% (b) L-alanine methyl ester hydrochloride, Cu(OAc)2, DCM, TEA, oxygen, 4Å molecular sieves, rt, 48 h, 34% (c) 1 M NaOH, MeOH, rt, 12 h, 67% (d) 4, EDCI•HCl, DMF, 0 °C - rt, 12 h, 60–70%.
An analogue of 1 lacking the ether oxygen was prepared following the synthetic procedure outlined in Scheme 3. 2,3-Dichlorophenylacetic acid (16) was treated with thionyl chloride to give the corresponding acid chloride 17. (R)-4-Benzyl-2-oxazolidinone (18) was deprotonated with n-butyllithium at −78 °C, and then the generated anion was quenched with 17 to give 1914. Diastereoselective methylation of 19 was carried out by treating this substrate with 1M solution of sodium bis(trimethylsilyl)amide at −78 °C, followed by the addition of iodomethane to give 20 with excellent diastereoselectivity14. Hydrolysis of 20 with lithium peroxide at lower temperature gave acid 21, which was subsequently converted to amide 22.
Scheme 3.

Synthesis of a 2-phenylpropionamide benzoxazole derivative.
Reagents and conditions: (a) SOCl2, 90 °C, 2 h (b) 18, n-BuLi (2.5 M), THF, −78 °C to 0 °C, 1 h, 80% (c) (i) NaN(TMS)2 in THF, −78 °C, 1 h (ii) MeI, THF, −78 °C, 2 h (iii) rt, 5 h, 77% (d) Li2O2, THF, H2O, 0 °C, 1 h, 87% (e) 4 (R1 = 4-Py, X = H), EDCI•HCl, DMF, 0 °C - rt, 12 h, 70%.
Replacement of the amide functional group of 1 with a bioisosteric 1,2,3-triazole was also investigated. This strategy had previously been successful with one structural class of CpIMPDH inhibitors10a, but not with another.10d The 1,2,3-triazole derivative of 1 was prepared according to the method depicted in Scheme 5. Propargyl ether 28 was synthesized using a Mitsunobu procedure. Then 2-(thiazol-5-yl)benzo[d]oxazol-5-amine (4, R1 = 5-thiazole, X = H) was converted to the corresponding diazonium chloride followed by quenching with sodium azide to yield 29. Finally, a CuI mediated reaction of alkyne 28 with azide 29 gave 30 15.
Scheme 5.
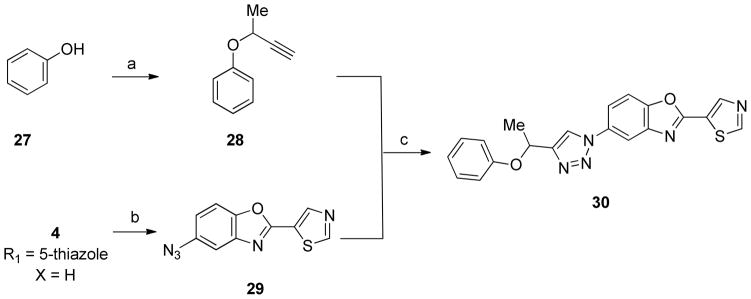
Synthesis of a 1,2,3-triazole derivative.
Reagents and conditions: (a) CH3CH(OH)C CH, PPh3, 0 °C, 10 min then DEAD, THF, 70 °C, 20 h, 69% (b) NaNO2, HCl, H2O, NaN3, -5 °C – 0 °C, 1.5 h, 89% (c) CH3CN, DIPEA, CuI, rt, 50 min, 84%.
An analogue in which the benzoxazole ring was replaced with a 1,3-diamide was generated using the method outlined in Scheme 6. 3-Nitroaniline 31 was coupled with isonicotinic acid to yield amide 32. Tin mediated reduction of 32 gave 33, which was coupled with (S)-2-(3,4-di-chlorophenoxy) propionic acid to generate 34.
Scheme 6.
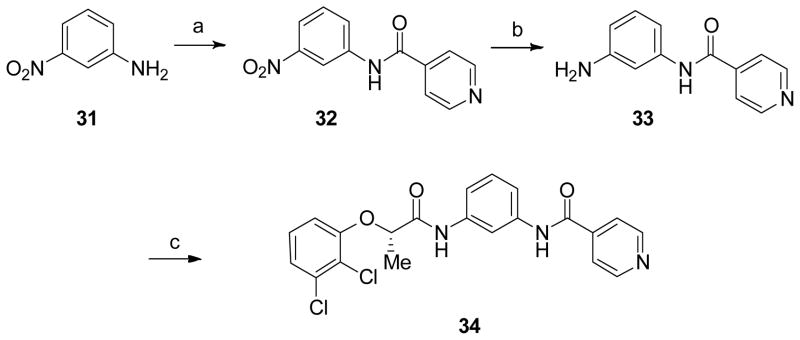
Synthesis of an 1,3-diamide derivative.
Reagents and conditions: (a) 4-PyCO2H, EDCI•HCl, DCM, 0 °C - rt, 12 h, 68% (b) SnCl2•H2O, EtOH, 70 °C, 4 h, 62% (c) (S)- 2,3-di-Cl-PhOCH(CH3)COOH, EDCI•HCl, DCM, 0 °C - rt, 12 h, 63%.
Regioisomers of 1 were prepared using the procedures outlined in Scheme 8. 2-Amino-5-nitrophenol (37) and aromatic aldehydes were condensed and oxidized in the presence of activated carbon (DarcoKB) to give 3811. Reductive hydrogenation of 38 in presence of 10% Pd-C provided 6-amino-2-aryl benzoxazoles 39. Subsequently, amines 39 were coupled with (S)-2-(2,3-di-chlorophenoxy) propionic acid with the aid of EDCI•HCl to yield 40.
Scheme 8.

General procedure for the synthesis of 6-substituted benzoxazole derivatives.
Reagents and conditions: (a) RCHO, O2, DarcoKB, xylene, 120 °C, 6 h, 75% (b) H2 (1 atm), 10% Pd-C, EtOAc, rt, 6 h, 85% (c) (S)-2,3-di-Cl-PhOCH(CH3)COOH, EDCI•HCl, DMF, 0 °C - rt, 12 h, 76%.
Evaluation of CpIMPDH inhibition
CpIMPDH and hIMPDH2 were expressed and purified as previously described (note that C. hominis IMPDH is identical to CpIMPDH)16. Enzymatic activity was assayed by monitoring the production of NADH9. IC50 values reported herein were determined from the average of three independent experiments, unless otherwise noted. IC50 values were also measured in the presence of 0.05% fatty acid free bovine serum albumin (BSA) in order to assess nonspecific protein binding17. Our previous experience indicates that IC50 values in the presence of BSA are a better predictor of antiparasitic activity18. None of the compounds significantly inhibited hIMPDH2 (IC50 >5μM).
Initially the aryl ether substituent was evaluated (Table 1). Removal of the 4-chloro group (41) resulted in a two-fold increase in inhibitory activity, whereas removal of the 2-chloro group (42) gave a two-fold loss in activity. Removal of both chlorine atoms resulted in activity comparable to the parent compound 1. The electron donating substitution 4-OMe (44) demonstrated comparable activity to 41. Translocation of the chlorine atom in 41 from the 2-position to the 3-position (45) was well tolerated. Combining these changes by utilizing a 2,3-di-chloro substituted phenyl ether (46) gave a five-fold increase in activity compared to 41 and 45. However, the 2,6-di-chloro phenyl ether derivative 47 was devoid of activity. Transforming the 2,3-dichlorophenyl into a 1-naphthyl resulted in a potent inhibitor (48) with an IC50 value of 9 nM, which only increased slightly in the presence of BSA. These results suggest that the sterics of this group are an important determinant for binding. However, addition of a chlorine atom to the naphthyl group in the 4-position (49) was detrimental.
Table 1.
SAR of phenyl ring of 1 for CpIMPDH inhibition.
| |||
|---|---|---|---|
| ID | R1 | IC50 (nM)
|
|
| (−) BSA | (+) BSA | ||
| 1 | 2,4-di-ClPh | 44 ± 8 | 120 ± 10 |
| 41 | 2-ClPh | 19 ± 2 | 50 ± 10 |
| 42 | 4-ClPh | 105 ± 17 | 118 ± 3 |
| 43 | Ph | 40 ± 5 | 50 ± 20 |
| 44 | 4-OMePh | 28 ± 2 | 34 ± 5 |
| 45 | 3-ClPh | 20 ± 7 | 32 ± 8 |
| 46 | 2,3-di-ClPh | 3 ± 1 | 11 ± 1 |
| 47 | 2,6-di-ClPh | >5000* | n.d |
| 48 | 1-naphthyl | 9 ± 3 | 14 ± 6 |
| 49 | 1-(4-Cl-naphthyl) | 27 ± 1 | 53 ± 9 |
One determination, n.d. = not determined.
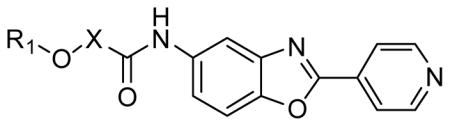
Next, the importance of amide methylene group was examined (Table 2). Removal of the α-methyl (50) or addition of another α-methyl (51) at this position resulted in complete loss of activity. Replacing the methyl with a larger isopropyl (52) resulted in approximately a three-fold loss of activity compared to 48. The enantiomers of 48 were also investigated and found to demonstrate a stereochemical preference. The (S)-isomer (54, IC50 = 6.1 nM, IC50 = 8 nM in the presence of BSA) was significantly more potent than the (R)-isomer (53, IC50 = 400 nM). Again the (S)-isomers of several other derivatives containing electron withdrawing groups at the 2,3-positions of the phenyl ether (55, 56 and 57) demonstrated excellent CpIMPDH inhibitory activities (IC50 < 10 nM). However, the (S)-isomer of the 2,3-di-methoxy derivative 58 was less active (IC50 = 50 nM).
Table 2.
SAR of amide α-position of 1 for CpIMPDH inhibition.
| ||||
|---|---|---|---|---|
| ID | X | R1 | IC50 (nM)
|
|
| (−) BSA | (+) BSA | |||
| 50 | CH2 | 2,4-di-ClPh | >5000* | n.d |
| 51 | CMe2 | 4-ClPh | >5000* | n.d |
| 52 | CH-i-Pr | 1-naphthyl | 24 ± 2 | 57 ± 5 |
| 53 | (R)-CHMe | 1-naphthyl | 400 ± 70 | 380 ± 60 |
| 54 | (S)-CHMe | 1-naphthyl | 6.1 ± 0.5 | 8 ± 3 |
| 55 | (S)-CHMe | 2,3-di-ClPh | 1.2 ± 0.2 | 3.5 ± 0.7 |
| 56 | (S)-CHMe | 2-Cl,3-CF3Ph | 9 ± 1 | 52 ± 5 |
| 57 | (S)-CHMe | 2-Cl,3-NO2Ph | 2.3 ± 0.9 | 6 ± 4 |
| 58 | (S)-CHMe | 2,3-di-OMePh | 50 ± 10 | 60 ± 20 |
One determination, n.d. = not determined.
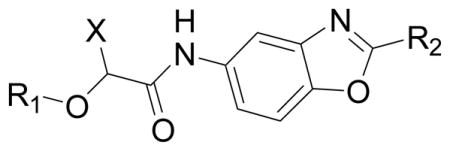
The importance of the pyridine substituent was subsequently examined (Table 3). Replacing the pyridine with phenyl (59) resulted in complete loss of activity (IC50 > 5 YM). Translocation of the nitrogen to the 2-or 3-positions (60 and 61) resulted in a 3- to 4-fold loss in potency. However, evaluation of derivatives 62 and 63 indicated that a 5-thiazole was an excellent replacement of the 4-pyridyl, while the 2-thiazole (65) and 2-pyrrole (66) derivatives resulted in loss of activity compared to 55 and 54, respectively. Finally, the 2-pyrimidine derivative 67 proved to be inactive.
Table 3.
SAR of the pyridine ring of 1 for CpIMPDH inhibition.
| |||||
|---|---|---|---|---|---|
| ID | X | R1 | R2 | IC50 (nM)
|
|
| (−) BSA | (+) BSA | ||||
| 59 | Me | 2,4-di-ClPh | Ph | >5000* | n.d |
| 60 | Me | 2,4-di-ClPh | 3-Py | 150 ± 20 | 600 ± 200 |
| 61 | Me | 2,4-di-ClPh | 2-Py | 210 ± 20 | 800 ± 200 |
| 62 | (S)-Me | 2,3-di-ClPh |
|
1.4 ± 0.3 | 2.3 ± 0.4 |
| 63 | (S)-Me | 1-naphthyl |
|
2.7 ± 0.7 | 4.0 ± 0.9 |
| 64 | Me | 1-(4-Cl-naphthyl) |
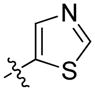
|
28 ± 6 | 70 ± 10 |
| 65 | (S)-Me | 2,3-di-ClPh |
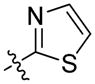
|
17 ± 6 | 30 ± 10 |
| 66 | (S)-Me | 1-naphthyl |
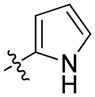
|
26 ± 5 | 50 ± 5 |
| 67 | Me | 1-naphthyl |
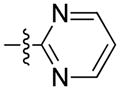
|
>5000* | n.d |
One determination, n.d. = not determined.
Following initial assessments of the phenyl ether, the amide α-methylene and the pyridine moieties in inhibitor 1, a variety of other changes were explored for the amide and central heterocycle (Table 4). For example, bioisosteric replacement of the amide with a 1,2,3-triazole (30), N-methylation of the amide nitrogen (9) or inversion of the amide (36) all resulted in loss of inhibitory activity. However, regioisomers produced by transposing the amide from the 5-position of the benzoxazole to the 6-position (40a and 40b), surprisingly, resulted in compounds with excellent CpIMPDH inhibitory activity (IC50 < 10 nM), although the relative effect of BSA appeared to be slightly greater compared to 55 and 62, respectively. Addition of a chlorine atom to the 7-position of the benzoxazole of 55 resulted in 68 and significantly eroded CpIMPDH inhibitory activity. Finally, replacing the benzoxazole with a benzimidazole (26) or opening of the oxazole ring (34) lead to loss of inhibitory activity.
Table 4.
Other miscellaneous changes to explore the SAR of 1 for CpIMPDH inhibition.
| ID | Structure | IC50 (nM)
|
|
|---|---|---|---|
| (−) BSA | (+) BSA | ||
| 30 |
|
>5000* | n.d. |
| 9 |
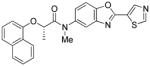
|
>5000* | n.d. |
| 36 |
|
>5000* | n.d. |
| 40a |
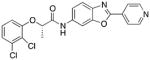
|
0.6 ± 0.5 | 4 ± 2 |
| 40b |
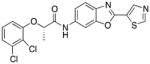
|
7 ± 1 | 60 ± 20 |
| 68 |
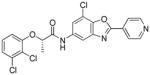
|
240 ± 90 | 600 ± 300 |
| 26 |
|
>5000* | n.d. |
| 34 |
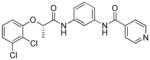
|
>5000* | n.d. |
One determination, n.d. = not determined.
The next perturbation examined was to replace the ether C-O bond with a C-C bond (Table 5). In the first iteration, a methylene substituted for the oxygen atom (69), which resulted in loss of inhibitory activity. However, deletion of the oxygen atom, generating a phenyl acetamide derivative, resulted in moderately potent inhibition (70, IC50 = 120 nM). Interestingly, the stereochemical preference of the phenyl acetamide derivatives was the opposite of the ether derivatives. For example, the (S)-isomer 71 was inactive, while the (R)-isomer 72 displayed an IC50 value of 22 nM. Another example was investigated to confirm this observation. The (R)-2,3-di-chlorophenyl derivative 22 was found to be a moderately potent CpIMPDH inhibitor, although it was less active than the (S)-ether 55.
Table 5.
SAR of benzyl amide derivatives of 1 for CpIMPDH inhibition.
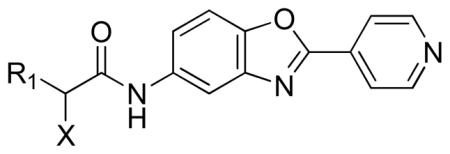
| ||||
|---|---|---|---|---|
| ID | X | R1 | IC50 (nM)
|
|
| (-) BSA | (+) BSA | |||
| 69 | Me | CH2Ph | > 5000 | n.d. |
| 70 | Me | Ph | 120 ± 20 | 130 ± 30 |
| 71 | (S)-Me | Ph | >5000* | n.d. |
| 72 | (R)-Me | Ph | 22.3 ± 5.9 | 28.0 ± 3.5 |
| 22 | (R)-Me | 2,3-di-ClPh | 60 ± 3 | |
Given the interesting results obtained with replacing the C-O bond of 1 with a C-C bond, the ether was exchanged with a secondary amine (Table 6). The 2,3-dichlorophenyl amine derivative 73 demonstrated slightly improved activity compared to the corresponding 2,3-dichlorophenyl ether 46. In addition, the (S)-isomer 15a proved to be a very potent CpIMPDH inhibitor (IC50 = 0.5 nM), although the (S)-naphthyl amine derivative 15b was less active (IC50 = 14 nM).
Table 6.
SAR of secondary amine derivatives of 1 for CpIMPDH inhibition.
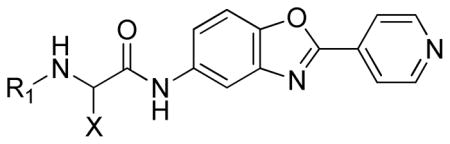
| ||||
|---|---|---|---|---|
| ID | X | R1 | IC50 (nM)
|
|
| (−) BSA | (+) BSA | |||
| 73 | Me | 2,3-di-ClPh | 2 ± 1 | 1.8 ± 0.5 |
| 15a | (S)- Me | 2,3-di-ClPh | 0.5 ± 0.1 | 1.1 ± 0.1 |
| 15b | (S)- Me | 1-naphthyl | 14 ± 6 | 15 ± 3 |
Evaluation of kinetic mechanism for CpIMPDH inhibition
The high throughput screen was designed to target the cofactor site since this site is the most diverged, and therefore most likely to yield inhibitors selective for the parasite enzyme.9 CpIMPDH, like other IMPDHs characterized to date, has a kinetic mechanism wherein substrates bind randomly and hydride transfer occurs forming a covalent E-XMP* intermediate and NADH.7 Products dissociate in an ordered fashion, with NADH release occurring before the hydrolysis and release of E-XMP*. In principle, IMPDH inhibitors that bind in the cofactor site can be competitive, uncompetitive or noncompetitive, depending on their relative affinities for the E, E•IMP and E-XMP* complexes. In practice, most such inhibitors are noncompetitive, suggesting comparable affinities for E•IMP and E-XMP*. Uncompetitive inhibition is also commonly observed, indicating a strong preference for E-XMP*. The inhibition mechanisms of four representative inhibitors (1, 63, 68 and 72) were evaluated. Surprisingly, the inhibition data with respect to NAD+ for all four compounds were best fit by competitive mechanism (Figure 3 and Table 7). However, the fit to a noncompetitve/mixed inhibition was not significantly inferior. This ambiguity is a consequence of NAD+ substrate inhibition, which prevents the use of saturating NAD+ concentrations.7 This observation suggests that these compounds have a strong preference for E•IMP. All four compounds are noncompetitive inhibitors with respect to IMP.
Figure 3.
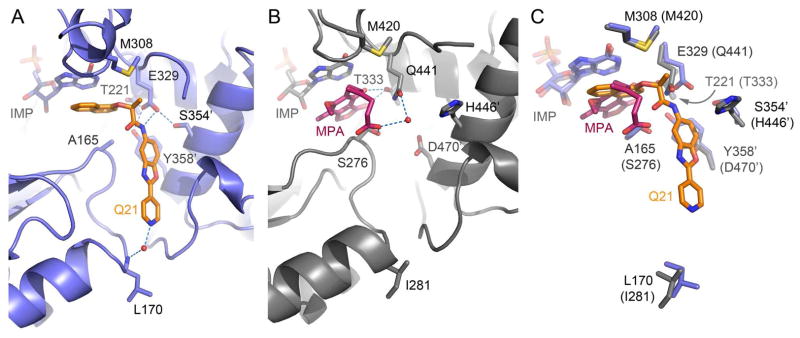
The inhibitor binding site in IMPDH. (A) Structure of CpIMPDH in complex with IMP and 54 (violet-blue; PDB ID code 4IXH). (B) Structure of Chinese hamster IMPDH (gray; PDB ID code)19 in complex with IMP and MPA. (C) Overlay of C. parvum and Chinese hamster active sites. IMP, 54, MPA and residues interacting with the inhibitors are shown as sticks. Numbers in parentheses in panel C indicate Chinese hamster labeling. Water molecules are shown as red spheres and hydrogen bonding interactions are shown as blue dashed lines. An apostrophe indicates a residue from the adjacent monomer.
Table 7.
Mechanism of inhibition of CpIMPDH by selected compounds.
| Cmpd | Substrate | Mechanism | Kis (nM) |
|---|---|---|---|
| 1 | IMP | NC | 58 ± 5 |
| NAD | C | 33 ± 8 | |
| 63a | IMP | NC | 1.6 ± 0.7 |
| NAD | C | 0.9 ± 0.3 | |
| 68 | IMP | NC | 210 ± 10 |
| NAD | C | 150 ± 30 | |
| 72 | IMP | NC | 100 ± 20 |
| NAD | C | 40 ± 10 |
Q26 was analyzed using tight binding treatment;
NC: noncompetitive; C: competitive.
Mouse liver microsomal stability
A selected set of the CpIMPDH inhibitors was evaluated for metabolic stability in mouse liver microsomes (Table 8). Compounds were incubated with microsomes at 37 °C in the presence and absence of NADPH. The percentage of compound remaining at various time points was determined and then the data were fit to a first-order decay model to determine half-life. Three ether derivatives (55, 62 and 40a) demonstrated poor stability in both the presence and absence of NADPH (t½ ≤ 12 min), whereas 54 was moderately more stable (t½ = 30 min). In the case of two phenyl acetamide derivatives, the unsubstituted inhibitor 72 proved more stable (t½ = 43 min) compared to the 2,3-di-chlorophenyl inhibitor 22 (t½ = 25 min) in the presence of NADPH. Both compounds were quite stable in the absence of NADPH. For the amine derivatives, the 2,3-di-chlorophenyl inhibitor 15a also displayed moderate stability in the presence of NADPH (t½ = 44 min), whereas the naphthyl derivative 15b was less stable in the presence or absence of NADPH (t½ = 18 min and 27 min, respectively).
Table 8.
NADPH-dependent mouse liver microsomal stability.
| |||||||
|---|---|---|---|---|---|---|---|
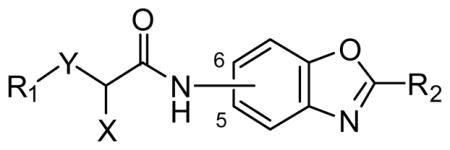
| ID | Connection | R1 | R2 | X | Y | t1/2 (min) | |
| + NADPH | − NADPH | ||||||
| 54 | 5 | 1-naphthyl | 4-Py | (S)-Me | O | 30 | -- |
| 55 | 5 | 2,3-di-ClPh | 4-Py | (S)-Me | O | 9.0 | 12 |
| 62 | 5 | 2,3-di-ClPh | 5-thiazolyl | (S)-Me | O | 7.0 | 11 |
| 72 | 5 | Ph | 4-Py | (R)-Me | --- | 43 | 130a |
| 22 | 5 | 2,3-di-ClPh | 4-Py | (R)-Me | --- | 25 | 130 |
| 15a | 5 | 2,3-di-ClPh | 4-Py | (S)-Me | NH | 44 | 110 |
| 15b | 5 | 1-naphthyl | 4-Py | (S)-Me | NH | 18 | 27 |
| 40a | 6 | 2,3-di-ClPh | 4-Py | (S)-Me | NH | 10 | 9 |
Estimated from a single time point at 45 min
The structure of CpIMPDH in complex with 54 and IMP
The structure of CpIMPDH in complex with IMP and 54 was solved at 2.10 Å resolution using molecular replacement with the structure of apo CpIMPDH (PDB ID code 3FFS) as the search model (Table S1). The structure (Figure 3A) revealed that the inhibitor binds in the active site and interacts with residues from two adjacent subunits, similarly as previously observed in the structure of CpIMPDH with C64 (PDB ID code 3KHJ)10b. This binding mode is significantly different compared to hIMPDH inhibitors and it likely accounts for the selectivity of the benzoxazole compounds. Inhibitors of mammalian IMPDH such as mycophenolic acid (MPA) bind in the nicotinamide subsite and interact directly with the purine ring of the substrate, IMP. In addition, the MPA interaction extends into the adenine subside. However, the interactions for MPA are limited to the residues within the same monomer (Figure 3B). In contrast, the naphthalene of 54 stacks against the purine ring of IMP, in a manner similar to the 2-thiazole of C64, with the remainder of 54 extending across the subunit interface into the pocket in the adjacent monomer. Unlike C64, the 2-(4-pyridyl)benzoxazole of 54 fills much of this cavity and the pyridine nitrogen of 54 participates in a water-mediated hydrogen bond with the L170 main chain amide nitrogen (Figures 3A and 4). This hydrogen bond can account for the requirement of the 4-pyridyl or 5-thiazolyl substituents. These additional interactions are likely to explain the increased affinity of 54 relative to C64 (IC50 = 28 nM and 6 nM for C64 and 54, respectively). The structure also reveals that the (S)-methyl group of 54 forms a hydrophobic interaction with M308. Inverting this center to the (R)-isomer would likely introduce a steric clash with E329, which forms a hydrogen bond with the amide NH of 54 and along with S354′ (where ′ denotes a residue from the adjacent subunit) and T221 forms a hydrogen bonding network with Y358′ (Figures 3A and 4). These observations provide a rationale for the observed stereochemical preference with regard to the substituent on the amide α-position in the ether and amine derivatives. In addition, the benzo-portion of the benzoxazole ring of 54 appears to interact with the side-chain of Y358′.
Figure 4.

Overlay of CpIMPDH structures with 54 (violet-blue; PDB ID code 4IXH) and C64 (PDB ID code 3KHJ).10b IMP, 54, C64 and the interaction residues are shown as sticks. A water molecule is shown as a red sphere and hydrogen bonding interactions are shown as blue dashed lines. An apostrophe denotes a residue from the adjacent monomer.
Evaluation of antiparasitic activity
Although the generation of potent CpIMPDH inhibitors has been accomplished with several structurally distinct compound classes, achieving antiparasitic activity in C. parvum remains a challenge. This organism cannot be continuously cultured in vitro, so such assays are poor mimics of in vivo infection in addition to having a poor dynamic range. However, the related intracellular parasite T. gondii has proven to be a well behaved organism that can be engineered to express fluorescent markers, facilitating its use in screening.20 Previously, we genetically engineered a T. gondii strain that relies on CpIMPDH (Toxo/CpIMPDH) to synthesize guanine nucleotides.18 In contrast, the wild type T. gondii strain RH (Toxo/WT) contains a eukaryotic IMPDH that is resistant to CpIMPDH inhibitors, thus providing target validation as well as a measure of host cell cytotoxicity18.
A set of twenty-two CpIMPDH inhibitors were evaluated for activity in both Toxo/CpIMPDH and Toxo/WT assays (Table 9). Four compounds (46, 48, 40a and 73) demonstrated EC50 values ≤ 250 nM in the Toxo/CpIMPDH assay and selectivity > 30-fold versus Toxo/WT. Thus, the 2,3-dichlorophenyl or 1-naphthyl ethers or amines at either the 5- or -6-positions of the 2-(4-pyridyl)- or 2-(thiazolyl)benzoxazoles translated into encouraging antiparasitic activity. Furthermore, two of these compounds (40a and 73) demonstrated EC50 values ≤ 30 nM and selectivity > 150-fold, indicating that the 2,3-dichlorophenyl ether or amine at either the 5- or 6-positions of the 2-(4-pyridyl)benzoxazole might be preferable. Based on the in vitro and cellular properties, these compounds are candidates for evaluation in an animal model of cryptosporidiosis.
Table 9.
Antiparasitic activity of select compounds. Assays as described in Methods18. Unless otherwise stated all values are the average of three independent determinations.
| Compound | EC50 (μM) | Selectivitya | |
|---|---|---|---|
| Toxo/WT | Toxo/CpIMPDH | ||
| 1 | 1.3 ± 0.1 b | 0.65 ± 0.03 b | 2 |
| 26 | 9 ± 3 | 9 ± 3 | 1 |
| 40a | 3 ± 1 | 0.02 ± 0.02 | 150 |
| 41 | 3.0 ± 1.0 | 0.9 ± 0.4 | 3 |
| 43 | 7 ± 4 | 4 ± 2 | 2 |
| 44 | >25 | 0.5 ± 0.2 | >50 |
| 45 | 1.1 ± 0.4 | 0.5 ± 0.1 | 2 |
| 46 | 8 ± 2 | 0.22 ± 0.04 | 40 |
| 48 | 2.4 ± 0.3 | 0.20 ± 0.09 b | 34 |
| 54 | 2.2 ± 0.6 | 0.4 ± 0.3 | 5 |
| 55 | 5 ± 2 | 0.3 ± 0.1 | 16 |
| 56 | 1.7 ± 0.6 | 0.8 ± 0.4 | 2 |
| 57 | 3 ± 1 | 0.19 ± 0.03 | 14 |
| 58 | 21 ± 6 | 0.7 ± 0.1 | 30 |
| 62 | 3.3 ± 0.5 | 0.2 ± 0.01 | 15 |
| 63 | 3.2 ± 0.8 | 0.30 ± 0.3 | 11 |
| 64 | 5 ± 4 | 2 ± 2 | 3 |
| 65 | 2.2 ± 0.8 | 0.3 ± 0.1 | 7 |
| 66 | 1.7 ± 0.7 | 1.0 ± 0.1 | 1.7 |
| 70 | 5 ± 3 | 2.5 ± 0.2 | 1.9 |
| 72 | 2.1 ± 0.5 | 0.4 ± 0.1 | 5 |
| 73 | 4 ± 2 | 0.02 ± 0.02 | 200 |
Selectivity is the ratio of EC50 Toxo/CpIMPDH to EC50 Toxo/WT.
Two determinations.
Evaluation of mammalian cytotoxicity activity
A subset of compounds (15a, 40a, 41, 44, 46 and 57) were also evaluated for cytotoxicity against four mammalian cell lines (HeLa, HEK293, COS and CHO). Viability was determined by monitoring metabolic activity with alamarBlue® assay. None of the compounds displayed significant toxicity (LD50 > 50 μM) against all four cell lines, except 41 which exhibited an LD50 ~ 12.5 μM in HEK293 cells.
Conclusions
A SAR study of CpIMPDH inhibitor 1 revealed that 2,3-di-substituted phenyl ethers improved potency, while a 4-pyridyl or 5-thiazolyl group was necessary at the 2-position of the benzoxazole. In addition, the (S)-isomers were significantly more potent compared to the corresponding (R)-isomers, but the connectivity of the amide could be transposed from the 5-position of the benzoxazole to the 6-position. The ether oxygen atom could be deleted generating α-arylamides where the chiral preference switched with the (R)-isomer being more potent. Furthermore, the ether oxygen atom could be replaced with an NH without jeopardizing potent CpIMPDH inhibitory activity and again the (S)-isomer was preferred. The secondary amine derivative 15a (Q67) or its racemic version 73 demonstrated excellent CpIMPDH inhibitory activity in the presence or absence of BSA and moderate stability in the presence of mouse liver microsomes, as well as superb activity and selectivity in a T. gondii surrogate cell assay of C. parvum infection and no significant cytotoxicity against a panel of four mammalian cells. Finally, a co-crystal structure of CpIMPDH with IMP and 54 (Q21) allowed for a more complete understanding of the structure-activity relationships observed with the benzoxazole inhibitors and also demonstrated that this compound series adopts a similar binding mode as a structurally distinct and previously reported CpIMPDH inhibitor C64.10b This information should assist in the continuing development of CpIMPDH inhibitors that will allow for further understanding of this apicomplexan parasite and its host interactions, advancing the treatment of cryptosporidiosis.
Experimental Section
Chemistry Material and Methods
Unless otherwise noted, all reagents and solvents were purchased from commercial sources and used without further purification. Compounds 1, 50, 51, 60 and 61 were purchased from Chembridge Corporation (San Diego, CA 92121, USA). All reactions were performed under a nitrogen atmosphere in dried glassware unless otherwise noted. All NMR spectra were obtained using a 400 MHz spectrometer and conducted in CDCl3, unless otherwise indicated. For 1H NMR, all chemical shifts are reported in δ units ppm and are referenced to tetramethylsilane (TMS). All chemical shift values are also reported with multiplicity, coupling constants and proton count. Likewise, for 13C NMR, all chemical shifts are reported in δ units ppm and are referenced to the central line of the triplet at 77.23 ppm for those conducted in CDCl3. Coupling constants (J values) are reported in hertz. Column chromatography was carried out on SILICYCLE SiliaFlash silica gel F60 (40–63 μm, mesh 230–400). High-resolution mass spectra (HRMS) were obtained using a Q-tof UE521 mass spectrometer (University of Illinois, SCS, and Mass Spectrometry Lab).
HPLC conditions
All final compounds have a chemical purity >98% as determined by analysis using a Agilent 1100 HPLC instrument equipped with a quaternary pump and a Zorbax® SB-C8 column (30 × 4.6 mm, 3.5 mm). UV absorption was monitored at 254 nm. The injection volume was 5 μL. HPLC gradient was 5 % acetonitrile and 95 % water (both solvents contain 0.1% trifluoroacetic acid) with a total run time of 2.5 min and a flow rate of 3.0 mL/min.
Enantiomeric purity was determined using HPLC analysis on a Agilent 1100 Series instrument equipped with a quaternary pump using a Chiralpak AS-H Column (250 × 4.6 mm) for all chiral compounds unless noted. The UV absorption was monitored at 254 nm and the injection volume was 10 μL. A Chiralpak OD-H column was used for 22, 40a, 40b and 58, and an OJ-H column was used for 15b, 73 and 74. For these latter two columns the UV absorption was monitored at 220 nm and an injection volume 20 μL was used. HPLC gradient for all compounds was 70 % n-hexane and 30 % i-propanol and a flow rate of 1.0 mL/min.
General procedure for the preparation of 5-nitro-2-arylbenzo[d]oxazoles (3). Exemplified for 5-nitro-2-(pyridine-4-yl)benzo[d]oxazole (3, R1 = 4-Py, X = H)
To a solution of 2-amino-4-nitrophenol (500 mg, 3.24 mmol) in anhydrous xylene (10 mL) in a 100 mL three-neck flask under an oxygen atmosphere was added 4-pyridine carboxaldehyde (347 mg, 3.23 mmol) and Darco KB (600 mg). The solution was stirred at 120 °C for 4 h. The reaction mixture was allowed to cool to room temperature and then filtered with the aid of Celite. The filtrate was concentrated and the residue was purified by silica gel column chromatography using a mixture of ethyl acetate/n-hexane (50:50) to give 5-nitro-2-(pyridine-4-yl)benzo[d]oxazole (600 mg, 77%) as a yellow solid. 1H NMR (CDCl3, 400 MHz) δ 7.76 (d, J = 9.2 Hz, 1H), 8.11 (d, J = 6 Hz, 2H), 8.40 (d, J1 = 12 Hz, J2 = 2.4 Hz, 1H), 8.73 (d, J = 2 Hz, 1H), 8.89 (d, J = 6 Hz, 2H).
General procedure for the preparation of 5-amino-2-aryl-benzo[d]oxazoles (4). Exemplified for 5-amino-2-(pyridine-4-yl)benzo[d]oxazole (4, R1 = 4-Py, X = H)
To a solution of 5-nitro-2-(pyridine-4-yl)benzo[d]oxazole (600 mg, 2.48 mmol) in ethyl acetate/MeOH (1:1, 10 mL) was added a catalytic amount of 10% Pd-C. The reaction mixture was placed under a hydrogen atmosphere and stirred for 6 h at room temperature. The mixture was then filtered, concentrated and the residue purified by flash column chromatography on silica gel using 100% ethyl acetate as eluent to afford 5-amino-2-(pyridine-4-yl)benzo[d]oxazole (450 mg, 86%) as a yellow solid. 1H NMR (CDCl3, 400 MHz) δ 6.78 (dd, J1 = 12 Hz, J2 = 2.4 Hz, 1H), 7.07 (d, J = 2 Hz, 1H), 7.40 (d, J = 4.8 Hz, 1H), 8.05 (dd, J1 = 8 Hz, J2 = 1.6 Hz, 2H), 8.79 (dd, J1 = 4 Hz, J2 = 1.6 Hz, 2H), 3.78 (s, 2H).
General procedure for the preparation of (±)-7. Exemplified for 2-(2,3-dichlorophenoxy)propionic acid (7, R2 = 2,3-di-ClPh, R3 = Me, Y = O)
To a solution of 2,3-dichlorophenol (200 mg, 1.22mmol) in anhydrous DMF (15 mL) was added K2CO3 (505 mg, 3.66 mmol) and ethyl 2-bromopropionate (287.1 mg, 1.58 mmol). The reaction mixture was stirred at room temperature for 5 h under a nitrogen atmosphere, diluted with water (50 mL) and extracted with ethyl acetate (3 X 50 mL). Note, when Y = NH this reaction was conducted at 70 °C for 3 h. The organic extracts were combined, washed with brine, dried over anhydrous MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography using a mixture of ethyl acetate and n-hexane (10:90) to give ethyl 2-(2,3-dichlorophenoxy)propionate (290 mg, 1.2 mmol, 90%) as a colorless liquid. The ester (290 mg, 1.60 mmol) was dissolved in THF: H2O (2:1), and then NaOH (132 mg, 3.3 mmol) was added. The reaction mixture was heated at 80 °C for 3 h. After the reaction mixture was allowed to cool to room temperature, 1N HCl was added until a pH of 7 was reached and then the mixture was extracted with DCM. The organic layers were dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by silica gel column chromatography using a mixture of ethyl acetate/n-hexane (60:40) to afford 2-(2,3-dichlorophenoxy)propionic acid (270 mg, 95%) as a white solid.
General procedure for the preparation of (R)- and (S)-7. Exemplified for (S)-2-(2,3-dichlorophenoxy)propionic acid (7, R2 = 2,3-di-ClPh, R3 = (S)-Me, Y = O)
2,3-Dichlorophenol (150 mg, 0.92 mmol) was dissolved in anhydrous DCM (6 mL) under a nitrogen atmosphere and then methyl (R)-(+)-2-(4-hydroxyphenoxy)propionate (143.7 mg, 1.38 mmol) was added. After the reaction mixture was cooled to 0 °C, PPh3 (289.4 mg, 1.10 mmol) was added portion-wise and the reaction mixture was stirred for 10 minutes. Then DEAD (240 mg, 1.37 mmol) was slowly added. The reaction mixture was stirred for 12 h at room temperature and then 10 mL of water was added and the mixture extracted with DCM. The organic layers were washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by column chromatography on silica gel using ethyl acetate/n-hexane (10:90) to yield methyl (S)-2-(2,3-dichlorophenoxy)propionate (180 mg, 0.76 mmol, 83%) as a colorless liquid. The ester (180 mg, 0.72 mmol) was added to a mixture of 6 mL THF in 3N HCl (2:8), and then heated at 70 °C for 6 h. The reaction mixture was allowed to cool to room temperature then extracted with DCM. Organic layers were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by silica gel column chromatography using ethyl acetate/n-hexane (60:40) to afford (S)-2-(2,3-dichlorophenoxy)propionic acid (110 mg, 62%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 1.51(d, J = 7.2 Hz, 3H), 4.27 (q, J = 7.2 Hz, 1H), 7.16 – 7.24 (m, 2H), 7.37 (dd, J1 = 3.6 Hz, J2 = 1.6 Hz, 1H).
General procedure for the preparation of benzoxazoles (8). Exemplified for (S)-2-(2,3-dichlorophenoxy)-N-(2-(pyridin-4-yl)benzo[d]oxazol-5-yl)propionamide (55)
To a solution of 5-amino-2-(pyridine-4-yl)benzo[d]oxazole (266.7 mg, 1.27 mmol) and (S)-2-(2,3-dichlorophenoxy)propionic acid (300 mg, 1.27 mmol) were dissolved in anhydrous DMF (5 mL) under a nitrogen atmosphere. The reaction mixture was cooled to 0 °C and then EDCI•HCl (489.6 mg, 2.55 mmol) was added. The reaction mixture was stirred for 12 h at room temperature. Volatiles were removed under reduced pressure and the mixture was dissolved in 20 mL DCM. The organic layer was washed sequentially with a saturated aqueous solution of sodium bicarbonate and brine. The organic layers were dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by silica gel column chromatography using ethyl acetate/n-hexane (50:50) to give 55 (390 mg, 72%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 1.76 (d, J = 6.8 Hz, 3H), 4.90 (q, J = 6.8 Hz, 1H), 6.93 (dd, J1 = 7.2 Hz, J2 = 2.4 Hz, 1H), 7.17 – 7.23 (m, 2H), 7.56 – 7.61 (m, 2H), 8.06 (dd, J1 = 4 Hz, J2 = 1.6 Hz, 2H), 8.17 (s, 1H), 8.81 – 8.83 (m, 3H); 13C NMR (CDCl3, 100 MHz) δ 18.6, 76.9, 111.2, 112.1, 113.2, 119.2, 121.2, 122.9, 124.3, 128.1, 134.3, 134.5, 134.7, 142.4, 148.0, 150.9, 153.7, 161.8, 169.2; ESIHRMS for C21H16N3O3Cl2 (M+H)+ calcd. 428.0569, found 428.0568, Chiral purity (% ee > 98, tR=12.84 min).
The following compounds were prepared in a similar manner:
2-(2-chlorophenoxy)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (41)
71%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.77 (d, J = 6.4 Hz, 3H), 4.90 (q, J = 6.8 Hz, 1H), 7.02 (t, J = 7.6 Hz, 2H), 7.25 – 7.29 (m, 1H), 7.45 (d, J = 8 Hz, 1H), 7.57 (d, J = 8.4 Hz, 1H), 7.64 (dd, J1 = 8 Hz, J2 = 2 Hz, 1H), 8.06 (d, J = 5.2 Hz, 2H), 8.17 (s, 1H), 8.82 (d, J = 4.4 Hz, 2H), 8.95 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 18.7, 76.8, 111.1, 111.9, 115.6, 119.2, 121.1, 123.5, 123.8, 128.4, 130.8, 134.3, 134.9, 142.3, 147.9, 150.9, 152.4, 161.7, 169.5; ESI-HRMS for C21H17N3O3Cl (M+H)+ calcd. 394.0958 found 394.0961.
2-(4-chlorophenoxy)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (42)
72%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.69 (d, J = 6.8 Hz, 3H), 4.79 (q, J = 6.8 Hz, 1H), 6.94 (d, J = 8.8 Hz, 2H), 7.30 (d, J = 8.4 Hz, 2H), 7.51 – 7.58 (m, 2H), 8.08 (dd, J1 = 10 Hz, J2 = 2 Hz, 3H), 8.29 (s, 1H), 8.82 (d = 4.8 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 18.8, 76.0, 111.2, 112.5, 117.3, 119.5, 121.2, 127.8, 130.1, 134.3, 134.5, 142.4, 148.1, 150.9, 155.3, 161.8, 170.1; ESI-HRMS for C21H17N3O3Cl (M+H)+ calcd. 394.0958 found 394.0961.
2-phenoxy-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (43)
63%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.69 (d, J = 6.4 Hz, 3H), 4.84 (q, J = 6.4 Hz, 1H), 7.07 – 6.99 (m, 3H), 7.34 (t, J = 8 Hz, 2H), 7.54 (t, J = 8.4 Hz, 2H), 8.08 (d, J = 13.6 Hz, 3H), 8.42 (s, 1H), 8.82 (s, 2H); 13C NMR (CDCl3, 100 MHz) δ 18.9, 75.6, 111.1, 112.4, 115.9, 119.6, 120.5, 121.2, 121.2, 121.3, 122.7, 128.1, 130.1, 134.3, 134.6, 142.4, 148.0, 150.9, 156.8, 161.7, 170.6; ESI-HRMS for C21H18N3O3 (M+H)+ calcd. 360.1348, found 360.1350.
2-(4-methoxyphenoxy)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (44)
73%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.66 (d, J = 6.8 Hz, 3H), 3.78 (s, 3H), 4.72 (q, J = 6.8 Hz, 1H), 6.86 (d, J = 9.2 Hz, 2H), 6.94 (d, J = 9.2 Hz, 2H), 7.56 (s, 2H), 8.07 (d, J = 6 Hz, 2H), 8.11 (s, 1H), 8.44 (s, 1H), 8.81 (d, J = 6 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 18.8, 55.8, 76.6, 94.6, 111.1, 112.3, 115.1, 117.4, 119.5, 121.2, 134.3, 134.7, 142.4, 148.0, 150.9, 155.2, 161.7, 170.8; ESI-HRMS for C22H20N3O4 (M+H)+ calcd. 390.1454 found 390.1457.
2-(3-chlorophenoxy)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (45)
73%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.68 (d, J = 6.8 Hz, 3H), 4.82 (q, J = 6.8 Hz, 1H), 6.86 – 6.89 (m, 1H), 7.02 (d, J = 7.6 Hz, 2H), 7.23 – 7.27 (m, 1H), 7.51 – 7.57 (m, 2H), 8.05 (d, J = 5.2 Hz, 2H), 8.09 (s, 1H), 8.37 (s, 1H), 8.81 (s, 2H); 13C NMR (CDCl3, 100 MHz) δ 18.7, 75.8, 111.1, 112.5, 113.8, 116.8, 119.6, 121.2, 123.0, 130.9, 134.2, 134.5, 135.5, 142.4, 148.1, 150.9, 157.4, 161.7, 170.0; ESIHRMS for C21H17N3O3Cl (M+H)+ calcd. 394.0958, found 394.0959.
2-(2,3-dichlorophenoxy)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (46)
71%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.77 (d, J = 6.8 Hz, 3H), 4.91 (q, J = 6.8 Hz, 1H), 6.93 (dd, J1 = 6.6 Hz, J2 = 2.8 Hz, 1H), 7.20 – 7.23 (m, 2H), 7.60 (d, J = 1.2 Hz, 2H), 8.08 (d, J = 4.4 Hz, 2H), 8.18 (s, 1H), 8.83 (s, 3H); 13C NMR (CDCl3, 100 MHz) δ 18.3, 76.6, 111.0, 111.8, 113.0, 119.0, 120.9, 122.7, 124.1, 127.8, 134.1, 134.3, 134.5, 142.2, 147.8, 150.7, 153.5, 161.5, 168.9; ESI-HRMS for C21H16N3O3Cl2 (M+H)+ calcd. 428.0569, found 428.0569.
2-(2,6-dichlorophenoxy)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (47)
67%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.61 (d, J = 6.8 Hz, 3H), 5.11 (q, J = 6.8 Hz, 1H), 7.09 (t, J = 7.6 Hz, 1H), 7.37 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.8 Hz, 1H), 7.18 (dd, J1 = 8.8 Hz, J2 = 1.6 Hz, 1H), 8.08 (d, J = 5.2 Hz, 2H), 8.20 (s, 1H), 8.82 (d, J = 5.2 Hz, 2H), 8.99 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 18.2, 79.5, 111.1, 112.1, 119.4, 121.2, 126.1, 129.5, 129.6, 134.3, 135.0, 142.4, 148.0, 148.7, 150.9, 169.5, 195.9; ESI-HRMS for C21H16N3O3Cl2 (M+H)+ calcd. 428.0569, found 428.0567.
2-(naphthalene-1-yloxy)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (48)
73%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.84 (d, J = 6.4 Hz, 3H), 5.04 (q, J = 6.8 Hz, 1H), 6.92 (d, J = 7.6 Hz, 1H), 7.39 (t, J = 8 Hz, 1H), 7.61 – 7.48 (m, 5H), 7.89 – 7.86 (m,1H), 8.05 – 8.08 (m, 3H), 8.33 – 8.36 (m, 1H), 8.41 (s, 1H), 8.80 (s, 2H); 13C NMR (CDCl3, 100 MHz) δ 19.1, 76.2, 107.3, 111.1, 112.4, 119.6, 121.21, 121.25, 121.5, 122.4, 125.8, 126.0, 126.1, 127.0, 128.1, 134.2, 134.6, 134.9, 142.4, 148.0, 150.9, 152.6, 170.6; ESI-HRMS for C25H20N3O3 (M+H)+ calcd. 410.1505, found 410.1508.
2-(4-chloronaphthalen-1-yloxy)-N-(2-(pyridin-4-yl)benzo[d]oxazol-5-yl)propionamide (49)
68%; white solid. 1H NMR (CDCl3, 400 MHz) δ 1.83 (d, J = 6.8 Hz, 3H), 5.0 (q, J = 6.8 Hz, 1H), 6.84 (d, J = 8.4 Hz, 1H), 7.46 – 7.50 (m, 2H), 7.54 (d, J = 8.8 Hz, 1H), 7.63 – 7.71 (m, 2H), 8.04 – 8.07 (m, 3H), 8.27 (d, J = 8 Hz, 1H), 8.36 (d, J = 8 Hz, 2H), 8.81 (d, J = 4 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 19.0, 76.5, 107.3, 111.1, 112.5, 119.6, 121.1, 122.0, 124.9, 125.5, 125.9, 126.9, 128.1, 131.8, 134.2, 134.4, 134.4, 134.7, 142.4, 148.1, 150.9, 151.7, 161.8, 170.3; ESI-HRMS for C25H19N3O3Cl (M+H)+ calcd. 444.1115, found 444.1119.
3-methyl-2-(naphthalene-1-yloxy)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)butanamide (52)
67%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.18 (d, J = 6.8 Hz, 3H), 1.25 (d, J = 6.8 Hz, 3H), 2.51 – 2.59 (m, 1H), 4.69 (d, J = 4 Hz, 1H), 6.84 (d, J = 7.6 Hz, 1H), 7.31 (t, J = 8 Hz, 1H), 7.37 – 7.47 (m, 3H), 7.51 – 7.54 (m, 2H), 7.80 (t, J = 6.4 Hz, 1H), 7.95 – 7.97 (m, 3H), 8.34 (d, J = 5.6 Hz, 2H), 8.72 (d, J = 5.2 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 17.5, 19.6, 32.5, 84.5, 106.7, 111.0, 112.7, 119.9, 121.1, 121.5, 122.1, 125.7, 126.0, 127.0, 128.1, 134.2, 134.4, 134.8, 142.2, 148.0, 150.7, 153.5, 161.6, 169.8; ESI-HRMS for C27H24N3O3 (M+H)+ calcd. 438.1818, found 438.1822.
(R)-2-(naphthalene-1-yloxy))-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (53)
76%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.83 (d, J = 6.8 Hz, 3H), 5.03 (q, J = 6.8 Hz, 1H), 6.91 (d, J = 7.6 Hz, 1H), 7.38 (t, J = 8 Hz, 1H), 7.50 – 7.58 (m, 5H), 7.85 – 7.87 (m, 1H), 8.03 – 8.07 (m, 3H), 8.34 (t, J = 2.8 Hz, 1H), 8.47 (s, 1H), 8.80 (bs, 2H); 13C NMR (CDCl3, 100 MHz) δ 19.1, 67.3, 76.2, 107.3, 111.1, 112.4, 119.6, 121.2, 121.5, 122.4, 125.8, 126.0, 126.1, 127.0, 128.1, 134.3, 134.6, 134.9, 142.3, 148.0, 150.9, 152.6, 161.7, 170.6; ESI-HRMS for C25H20N3O3 (M+H)+ calcd. 410.1505, found 410.1508, Chiral purity (% ee 98.1, tR=22.87).
(S)-2-(naphthalene-1-yloxy))-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (54)
71%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.78 (d, J = 6.8 Hz, 3H), 4.96 (q, J = 6.8 Hz, 1H), 6.85 (d, J = 7.6 Hz, 1H), 7.30 (t, J = 8 Hz, 1H), 7.50 – 7.42 (m, 5H), 7.77 – 7.79 (m, 1H), 7.92 (dd, J1 = 4.4 Hz, J2 = 1.6 Hz, 2H), 8.02 (s, 1H), 8.26 – 8.23 (m, 1H), 8.60 (s, 1H), 8.70 (d, J = 4.4 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 19.1, 76.2, 107.2, 111.0, 112.5, 119.7, 121.1, 121.5, 122.3, 125.0, 125.8, 126.0, 126.9, 128.0, 134.1, 134.7, 134.8, 142.2, 147.9, 150.8, 152.7, 161.6, 170.7; ESI-HRMS for C25H20N3O3 (M+H)+ calcd. 410.1505, found 410.1507, Chiral purity (% ee 98.9, tR=12.08).
(S)-2-(2-chloro-3-(trifluoromethyl)phenoxy)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (56)
70%; cream colored solid; 1H NMR (CDCl3, 400 MHz) δ 1.78 (d, J = 6.8 Hz, 3H), 4.94 (q, J = 6.4 Hz, 1H), 7.21 (d, J = 7.6 Hz, 1H), 7.37 – 7.44 (m, 2H), 7.59 (s, 2H), 8.07 (d, J = 5.2 Hz 2H), 8.19 (s, 1H), 8.82 (d, J = 5.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 18.5, 76.9, 111.2, 112.1, 118.4, 119.2, 121.1, 122.3, 124.0, 130.4, 134.2, 134.6, 142.4, 148.0, 150.9, 153.5, 161.8, 168.9; ESI-HRMS for C22H16N3O3ClF3 (M+H)+ calcd. 462.0832, found 462.0834, Chiral purity (% ee 99.4, tR=11.83).
(S)-2-(2-chloro-3-nitrophenoxy)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (57)
77%; cream colored solid; 1H NMR (CDCl3, 400 MHz) δ 1.80 (d, J = 6.8 Hz, 3H), 4.97 (q, J = 6.8 Hz, 1H), 7.24 (d, J = 8.4 Hz, 1H), 7.44 (t, J = 8.4 Hz, 1H), 7.53 – 7.62 (m, 3H), 8.07 – 8.09 (m, 2H), 8.20 (s, 1H), 8.68 (s, 1H), 8.82 – 8.84 9m, 2H); 13C NMR (CDCl3, 100 MHz) δ 18.5, 76.9, 111.3, 112.2, 117.2, 118.0, 118.7, 119.3, 121.2, 128.4, 134.2, 134.4, 142.4, 148.1, 149.9, 150.9, 153.7, 161.9, 168.6; ESI-HRMS for C21H16N4O5Cl (M+H)+ calcd. 439.0809, found 439.0805, Chiral purity (% ee >99, tR=35.93).
(S)-2-(2,3-dimethoxyphenoxy)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (58)
79%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.78 (d, J = 6.8 Hz, 3H), 3.89 (s, 3H), 4.01 (s, 3H), 4.78 (q, J = 6.8 Hz, 1H), 6.67 – 6.71 (m, 2H), 7.04 (t, J = 5.6 Hz, 1H), 7.55 (d, J = 8.8 Hz, 1H), 7.68 (dd, J1 = 9.2 Hz, J2 = 2.4 Hz, 1H), 8.07 (dd, J1 = 4.4 Hz, J2 = 1.6 Hz, 2H), 8.20 (d, J = 2 Hz, 1H), 8.82 (dd, J1 = 4.4 Hz, J2 = 1.2 Hz, 2H), 9.58 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 20.5, 57.0, 62.5, 80.3, 108.3, 111.4, 111.8, 112.7, 120.1, 122.0, 125.7, 135.3, 136.4, 140.4, 143.1, 148.6, 151.7, 152.8, 154.9, 162.4, 171.4; ESI-HRMS for C23H22N3O5 (M+H)+ calcd. 420.1559, found 420.1557, Chiral purity (% ee >99, tR=17.41).
2-(2,4-dichlorophenoxy)-N-(2-(phenylbenzo[d]oxazol-5-yl)propionamide (59)
73%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.74 (d, J = 6.8 Hz, 3H), 4.84 (q, J = 6.8 Hz, 1H), 6.94 (d, J = 8.8 Hz, 1H), 7.23 (dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 1H), 7.44 (d, J = 2.4 Hz, 1H), 7.54 – 7.51 (m,4H), 7.56 (d, J = 2 Hz, 1H), 7.59 (d, J = 2 Hz, 1H), 8.05 (d, J = 1.6 Hz, 1H), 8.24 (dd, J = 8 Hz, 2H), 8.75 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 18.6, 76.9, 110.8, 111.6, 116.4, 118.0, 124.7, 127.1, 127.8, 128.0, 128.4, 129.1, 130.6, 131.9, 134.2, 142.8, 148.0, 151.3, 164.2, 169.1; ESI-HRMS for C22H17N2O3Cl2 (M+H)+ calcd. 427.0616 found 427.0619.
(S)-2-(2,3-dichlorophenoxy)-N-(2-(thiazol-5-yl)benzo[d]oxazol-5-yl)propionamide (62)
76%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.76 (d, J = 6.8 Hz, 3H), 4.89 (q, J = 6.8 Hz, 1H), 6.92 (dd, J1 = 7.2 Hz, J2 = 2.4 Hz, 1H), 7.17 – 7.22 (m, 2H), 7.55 (q, J = 8.8 Hz, 2H), 8.09 (s, 1H), 8.64 (s, 1H), 8.81 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 18.6, 76.9, 110.9, 111.6, 113.2, 118.6, 120.5, 122.9, 124.3, 125.7, 128.1, 134.7, 142.4, 145.7, 147.6, 153.7, 156.4, 158.0, 169.1; ESI-HRMS for C19H14N3O3SCl2 (M+H)+ calcd. 434.0133, found 434.0136, Chiral purity (% ee 99.1, tR=15.5).
(S)-2-(naphthalene-1-yloxy)-N-(2-(thiazol-5-yl)benzo[d]oxazol-5-yl)propionamide (63)
78%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.81 (d, J = 6.8 Hz, 3H), 5.01 (q, J = 6.4 Hz, 1H), 6.89 (d, J = 7.6 Hz, 1H), 7.35 (t, J = 7.6 Hz, 1H), 7.45 – 7.54 (m, 5H), 7.82 (t, J = 4.8 Hz, 1H), 7.96 (s, 1H), 8.29 (d, J = 5.2 Hz, 1H), 8.55 (d, J = 21.6 Hz, 2H), 8.91 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 19.1, 76.2, 107.3, 110.7, 112.0, 119.0, 121.5, 122.3, 125.6, 125.8, 126.0, 126.1, 127.0, 128.0, 134.6, 134.8, 142.2, 145.6, 147.6, 152.6, 156.4, 157.8, 170.6; ESI-HRMS for C23H18N3O3S (M+H)+ calcd. 416.1069, found 416.1075, Chiral purity (% ee 98.7, tR=12.53).
2-(4-chloronaphthalen-1-yloxy)-N-(2-(thiazol-5-yl)benzo[d]oxazol-5-yl)propionamide (64)
64%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.82 (d, J = 6.8 Hz, 3H), 4.99 (q, J = 6.8 Hz, 1H), 6.83 (d, J = 8.4 Hz, 1H), 7.44 – 7.50 (m, 3H), 7.62 – 7.71 (m, 2H), 7.98 (d, J = 2 Hz, 1H), 8.25 – 8.37 (m, 3H), 8.62 (s, 1H), 8.96 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 19.1, 76.6, 107.4, 110.9, 112.1, 119.0, 122.0, 125.0, 125.6, 126.0, 126.9, 127.0, 128.2, 131.8, 134.5, 142.4, 145.8, 147.8, 151.7, 156.5, 161.8, 166.4, 170.3; ESI-HRMS for C23H17N3O3SCl (M+H)+ calcd. 450.0679, found 450.0680.
(S)-2-(2,3-dichlorophenoxy)-N-(2-(thiazol-2-yl)benzo[d]oxazol-5-yl)propionamide (65)
69%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.72 (d, J = 6.8 Hz, 3H), 4.85 (q, J = 6.8 Hz, 1H), 6.89 (dd, J1 = 8 Hz, J2 = 2 Hz, 1H), 7.12 – 7.18 (m, 2H), 7.55 (bs, 2H), 7.59 (d, J = 2.8 Hz, 1H), 8.03 (d, J = 3.2 Hz, 1H), 8.14 (d, J = 1.2 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 18.6, 77.0, 111.5, 112.0, 113.2, 119.5, 122.9, 123.5, 124.3, 128.0, 134.5, 134.9, 142.0, 145.3, 147.8, 153.7, 154.7, 158.0, 169.2; ESI-HRMS for C19H14N3O3SCl2 (M+H)+ calcd. 434.0133, found 434.0132, Chiral purity (% ee >99, tR=12.88).
N-(2-(1H-pyrrol-2-yl)benzo[d]oxazol-5-yl)2-(naphthalene-1-yloxy)propionamide (66)
70%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.82 (d, J = 6.8 Hz, 3H), 5.02 (q, J = 6.4 Hz, 1H), 6.36 (dd, J1 = 2.4 Hz, J2 = 6 Hz, 1H), 6.91 (d, J = 8 Hz, 1H), 7.06 (d, J = 1.6 Hz, 2H), 7.32 – 7.43 (m,3H), 7.52 – 7.60 (m, 3H), 7.85 – 7.89 (m, 2H), 8.34 (d, J = 12 Hz, 2H), 9.87 (d, J = 1.6 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 19.1, 76.2, 107.3, 110.4, 111.0, 111.1, 113.7, 117.5, 119.5, 120.5, 121.5, 122.3, 123.6, 126.0, 126.0, 126.1, 127.0, 128.1, 134.1, 134.9, 142.0, 147.2, 152.6, 159.0, 170.5; ESI-HRMS for C24H20N3O3 (M+H)+ calcd. 398.1505, found 398.1509.
2-(naphthalene-1-yloxy)-N-(2-(pyrimidin-2-yl)benzo[d]oxazol-5-yl)propionamide (67)
63%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.83 (d, J = 6.8 Hz, 3H), 5.04 (q, J = 6.8 Hz, 1H), 6.93 (d, J = 7.6 Hz, 1H), 7.39 (t, J = 8.4 Hz, 1H), 7.61 – 7.53 (m, 4H), 7.88 – 7.86 (m, 1H), 8.09 (s, 1H), 8.34 (d, J = 7.2 Hz, 1H), 8.44 (s, 1H), 8.99 (d, J = 4.8 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 19.1, 76.4, 107.5, 111.7, 111.7, 113.1, 120.5, 121.5, 122.2, 122.4, 125.9, 126.0, 126.1, 127.0, 128.1, 134.8, 134.9, 142.2, 148.4, 152.7, 155.2, 158.2, 170.7; ESI-HRMS for C24H19N4O3 (M+H)+ calcd. 411.1457, found 411.1460.
(S)-N-(7-chloro-2-(pyridin-4-yl)benzo[d]oxazol-5-yl)-2-(2,3-dichlorophenoxy)propionamide (68)
68%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.73 (d, J = 6.8 Hz, 3H), 4.87 (q, J = 6.8 Hz, 1H), 6.90 (dd, J1 = 6.4 Hz, J2 = 3.2 Hz, 1H), 7.18 – 7.23 (m, 2H), 7.75 (d, J = 2 Hz, 1H), 7.97 (d, J = 1.6 Hz, 1H), 8.08 (d, J = 5.2 Hz, 2H), 8.82 (d, J = 8 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ 18.5, 77.0, 110.4, 113.3, 116.5, 119.2, 121.3, 124.5, 128.1, 133.7, 134.6, 135.3, 143.2, 144.8, 150.9, 153.6, 162.1, 162.6, 169.3; ESI-HRMS for C21H15N3O3Cl3 (M+H)+ calcd. 462.0179 found 462.0182, Chiral purity (% ee >99, tR= 4.52).
2-methyl-3-phenyl-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (69)
75%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.29 (d, J = 6 Hz, 3H), 2.63 (q, J = 6.4 Hz, 1H), 2.77 (dd, J1 = 16 Hz, J2 = 5.6 Hz, 1H), 3.02 (dd, J1 = 16 Hz, J2 = 9.2 Hz, 1H), 7.17 – 7.25 (m, 5H), 7.34 – 7.44 (m, 3H), 7.75 (s, 1H), 7.99 (d, J = 4 Hz, 2H), 8.75 (s, 2H); 13C NMR (CDCl3, 100 MHz) δ 18.0, 40.9, 44.9, 110.9, 112.5, 120.0, 121.2, 126.7, 128.8, 129.1, 134.4, 135.3, 139.8, 142.1, 147.8, 150.8, 174.3; ESI-HRMS for C22H20N3O2 (M+H)+ calcd. 358.1556 found 358.1556.
2-phenyl-N-(2-(pyridine-4-yl) benzo[d]oxazol-5-yl)propionamide (70)
77%; 1H NMR (CDCl3, 400 MHz) δ 1.61 (d, J = 7.2 Hz, 3H), 3.74 (q, J = 7.2 Hz, 1H), 7.17 (s, 1H), 7.23 (s, 1H), 7.29 – 7.32 (m, 1H), 7.37 – 7.41 (m, 4H), 7.47 (d, J = 8.8 Hz, 1H), 7.91 (d, J = 1.6 Hz, 1H), 8.01 (dd, J1 = 1.6 Hz, J2 = 1.2 Hz, 2H), 8.77 (d, J = 5.6 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 18.8, 48.3, 110.9, 112.1, 119.4, 121.1, 127.92, 127.94, 129.4, 134.3, 135.4, 140.9, 142.3, 147.8, 150.9, 161.6, 172.5; ESI-HRMS for C21H18N3O2 (M+H)+ calcd. 344.1399, found 344.1400.
(S)-2-phenyl-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (71)
77%; 1H NMR (CDCl3, 400 MHz) δ 1.52 (d, J = 7.2 Hz, 3H), 3.67 (q, J = 7.2 Hz, 1H), 7.15 (t, J = 8.4Hz, 1H), 7.21 – 7.35 (m, 5H), 7.69 (s, 1H), 7.83 (s, 1H), 7.91 (d, J = 5.6 Hz, 2H), 8.68 (d, J = 5.2 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 18.9, 31.1, 42.7, 48.0, 110.8, 112.2, 119.7, 120.5, 121.1, 127.7, 127.8, 128.1, 129.3, 134.3, 135.6, 141.1, 142.1, 147.7, 148.4, 149.1, 150.8, 161.4, 172.9; ESI-HRMS for C21H18N3O2 (M+H)+ calcd. 344.1399, found 344.1397, Chiral purity (% ee >99, tR = 8.07 min).
(R)-2-phenyl-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (72)
78%; white solid; 1H NMR (CDCl3, 400 MHz) δ 1.59 (d, J = 7.2 Hz, 3H), 3.76 (q, J = 6.8 Hz, 1H), 7.21 – 7.29 (m, 1H), 7.32 – 7.41 (m, 6H), 7.90 (s, 1H), 7.97 (d, J = 4.8 Hz, 3H), 8.74 (s, 2H); 13C NMR (CDCl3, 100 MHz) δ 18.9, 48.0, 110.8, 112.3, 119.8, 121.2, 127.7, 127.8, 129.2, 134.3, 135.6, 141.1, 142.1, 147.7, 150.7, 161.4, 172.9; ESI-HRMS for C21H18N3O2 (M+H)+ calcd. 344.1399, found 344.1393, Chiral purity (% ee 99, tR=11.29 min).
2-(2,3-dichlorophenylamino)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (73)
71%; 1H NMR (CDCl3, 400 MHz) δ 1.72 (d, J = 6.8 Hz, 3H), 3.95 (dq, J1 = 7.2 Hz, J2 = 3.2 Hz, 1H), 4.79 (d, J = 2.8 Hz, 1H), 6.56 (d, J = 8.4 Hz, 1H), 6.96 (d, J = 8 Hz, 1H), 7.09 (t, J = 8 Hz, 1H), 7.46 (dd, J1 = 9.2 Hz, J2 = 2 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 8.06 (d, J = 4 Hz, 2H), 8.09 (d, J = 1.6 Hz, 1H), 8.56 (s, 1H), 8.8 (bs, 2H); 13C NMR (CDCl3, 100 MHz) δ 20.0, 56.4, 110.8, 111.1, 112.4, 118.6, 119.6, 120.9, 121.2, 128.4, 133.5, 134.3, 134.7, 142.4, 144.1, 148.0, 150.9, 161.7, 171.7; ESI-HRMS for C21H17N4O2Cl2 (M+H)+ calcd. 427.0729 found 427.0724.
Preparation of (S)-N-methyl-2-(naphthalen-1-yloxy)-N-(2-(thiazol-5-yl)benzo[d]oxazol-5-yl)propanamide (9, R1 = 5-thiazole, R2 = 1-naphthyl, R3= (S)-Me, Y = O, X = H)
A solution of 63 (30 mg, 0.072 mmol) in 2 mL anhydrous THF was added drop-wise to a suspension of sodium hydride (60% in mineral oil, 1.89 mg, 0.079 mmol) in anhydrous THF (3 mL) under a nitrogen atmosphere at 0 °C. The resulting mixture was stirred for 30 min at room temperature. The mixture was cooled to 0 °C and then MeI (11.2 mg, 0.079 mmol) was added. The reaction mixture was stirred for 1 h at room temperature. The reaction mixture was quenched with a few drops of water and extracted with DCM. The organic layers were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by silica gel column chromatography using ethyl acetate/n-hexane (40:60) to yield 9 (15 mg, 48%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 1.62 (d, J = 6.4 Hz, 3H), 3.34 (s, 3H), 4.92 (q, J = 6.4 Hz, 1H), 6.49 (d, J = 7.6 Hz, 1H), 7.03 (d, J = 8.4 Hz, 1H), 7.19 – 7.39 (m, 6H), 7.69 (d, J = 8 Hz, 1H), 7.90 (d, J = 8.4 Hz, 1H), 8.61 (s, 1H), 8.61 (s, 1H), 8.98 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 18.7, 39.0, 72.6, 105.9, 111.4, 119.0, 121.3, 122.3, 125.2, 125.29, 125.3, 125.5, 126.0, 126.5, 127.4, 134.7, 140.0, 142.6, 146.0, 149.7, 153.0, 156.9, 158.4, 171.3; ESI-HRMS for C24H20N3O3S (M+H)+ calcd. 430.1225, found 430.1222, Chiral purity (% ee >99, tR=12.2 min.
Preparation of (S)-2-((2,3-dichlorophenyl)amino)propionoic acid (11)
A mixture of 10 (163 mg, 1.82 mmol), 2,3-dichloro iodobenzene (500 mg, 1.83 mmol), Cs2CO3 (1.19 gr, 3.65 mmol), and CuI (69.7 mg, 0.36 mmol) in DMF (3 mL) under a nitrogen atmosphere was heated at 90 °C for 48 h. The mixture was allowed to cool to room temperature and then diluted with water and the pH was adjusted to 3 to 5 by the addition of concentrated HCl. The mixture was extracted with DCM. The organic layers were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by silica gel flash column chromatography using ethyl acetate/n-hexane (50:50) to afforded 11 (240 mg, 57%) as a brown solid. 1H NMR (CDCl3, 400 MHz) δ 1.60 (d, J = 7.2 Hz, 3H), 4.17 (q, J = 7.2 Hz, 1H), 6.48 (d, J = 8 Hz, 1H), 6.85 (d, J = 8 Hz, 1H), 7.05 (t, J = 8.4 Hz, 1H).
Synthesis of (S)-2-(2,3-dichlorophenylamino)-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl) propionamide (15a)
The general procedure for 8 was followed using 11 and 4 to produce 15a; 70%; brown solid; 1H NMR (CDCl3, 400 MHz) δ 1.72 (d, J = 6.8 Hz, 3H), 3.95 (dq, J1 = 7.2 Hz, J2 = 3.2 Hz, 1H), 4.79 (d, J = 2.8 Hz, 1H), 6.56 (d, J = 8.0 Hz, 1H), 6.96 (d, J = 8 Hz, 1H), 7.09 (t, J = 8 Hz, 1H), 7.47 (d, J = 8.4 Hz, 1H), 7.54 (d, J = 8.8 Hz, 1H), 8.08 (bd, 3H), 8.56 (s, 1H), 8.8 (bs, 2H); 13C NMR (CDCl3, 100 MHz) δ 19.9, 56.35, 110.7, 111.0, 112.5, 118.5, 119.7, 120.7, 121.2, 128.3, 133.5, 134.3, 134.8, 142.3, 144.1, 148.0, 150.8, 161.6, 171.9; ESI-HRMS for C21H17N4O2Cl2 (M+H)+ calcd. 427.0729 found 427.0731, Chiral purity (% ee 90.5, tR = 20.4 min).
Preparation of (S)-methyl 2-(naphthalen-1-ylamino)propanoate (13)
Into a dry round bottom flask equipped with a stir bar was added L-alanine methyl ester hydrochloride (500 mg, 3.58 mmol), 1-naphthalene boronic acid (1000 mg, 5.81 mmol), Cu(OAc)2 (715 mg, 3.93 mmol), and 4 Å molecular sieves (1.34 g). The flask was sealed with a septum, evacuated and back filled with oxygen. Triethylamine (0.92 mL) and dry DCM (30 mL) were added. The reaction mixture was stirred at room temperature for 48 h. The reaction mixture was quenched with 13 mL 2M NH3 in methanol. The volatiles were removed in vacuo and the resulting crude oil was purified by silica gel flash chromatography using ethyl acetate/n-hexane (10:90) to give 13 (280 mg, 34%) as a brown viscous oil. 1H NMR (CDCl3, 400 MHz) δ 1.27 (t, J = 6.8 Hz, 3H), 1.61 (d, J = 6.8 H, 3H), 4.23 (q, J = 6.8 Hz, 2H), 4.31 (q, J = 6.4 Hz, 1H), 4.98 (bs, NH), 6.55 (d, J = 7.2 Hz, 1H), 7.27–7.35 (m, 2H), 7.45–7.49 (m, 2H), 7.78–7.81 (m, 1H), 7.90–7.93 (m, 1H).
Preparation of N-1-naphthalenyl L-alanine (14)
Ester 13 (40 mg, 0.174 mmol) was dissolved in methanol (1mL) and 1M NaOH in aqueous solution (0.18 mmol, 1.1 eq) was added drop-wise. The reaction mixture was stirred at room temperature for 12 h and then concentrated. The residue was dissolved in DCM and then extracted with 10% aqueous Na2CO3 solution. The aqueous layer was acidified with 1M HCl. The precipitate was collected and washed with DCM. The solid was purified by silica gel flash column chromatography using ethyl acetate/n-hexane (40:70) to give 14 (25 mg, 67%) as a brown solid. 1H NMR (CDCl3, 400 MHz) δ 1.60 (d, J = 6.8 Hz, 3H), 4.24 (q, J = 6.8 Hz, 1H), 6.49 (d, J1 = 6 Hz, J2 = 1.6 Hz, 1H), 7.18–7.25 (m, 2H), 7.39–7.41 (m, 2H), 7.72–7.81 (m, 2H).
Synthesis of (S)-2-(naphthalen-1-ylamino)-N-[2-(pyridin-4-yl)benzo[d]oxazol-5-yl]propionamide (15b)
The general procedure for 8 was followed using 14 and 4 to produce 15b (60%) as a brown solid. 1H NMR (CDCl3, 400 MHz) δ 1.79 (d, J = 6.8 Hz, 3H), 4.13 (q, J = 6.8 Hz, 1H), 4.69 (bs, 1H), 6.66 (d, J = 7.2 Hz, 1H), 7.56- 7.32 (m, 6H), 7.87 (d, J = 7.2 Hz, 1H), 7.95 (d, J = 8 Hz, 1H), 8.08 (bs, 3H), 8.79 (bs, 3H); 13C NMR (CDCl3, 100 MHz) δ 20.2, 56.4, 106.9, 110.9, 112.4, 119.6, 12.7, 120.3, 121.1, 123.6, 125.7, 126.3, 126.6, 129.2, 134.3, 134.4, 135.0, 141.4, 142.3, 147.9, 150.9, 161.6, 172.4; ESI-HRMS for C25H20N4O2 (M+H)+ calcd. 409.1586 found 409.1668, Chiral purity (% ee 97, tR = 36.4 min).
Preparation of 2-(2,3-dichlorophenyl)acetyl chloride (17)
2,3-Dichlorophenylacetic acid (16, 500 mg, 2.43 mmol) was dissolved in thionyl chloride (4 mL) under a nitrogen atmosphere at 0 °C. The reaction mixture was heated at 90 °C for 2 h. The excess thionyl chloride was removed in vacuo to afford 17 as a colorless liquid. This material was used without further purification.
Preparation of (R)-4-benzyl-3-(2-(2,3-dichlorophenyl)acetyl)oxazolidin-2-one (19)
(R)-4-benzyloxazolidin-2-one (18, 212.6 mg, 1.19 mmol) was dissolved in anhydrous THF (8 mL) under a nitrogen atmosphere. The reaction mixture was cooled to −78 °C and then a 2.5M solution of n-butyl lithium in hexanes (0.9 mL, 1.2 mmol) was added drop-wise. After 1 h, 17 (492 mg, 2.4 mmol) was added. The reaction mixture was stirred for 15 min −78 °C. Then the reaction mixture was allowed to warm to 0 °C and stirred for 30 min. The reaction mixture was then quenched with saturated aqueous NH4Cl solution. The solvent was removed in vacuo, and then the mixture was extracted with DCM. The organic layer was washed with brine, dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by silica gel column chromatography using EtOAc/n-hexane (20:80) to give 19 (350 mg, 80%) as a brown semi-solid. 1H NMR (CDCl3, 400 MHz) δ 2.77 (t, J = 12 Hz, 1H), 3.29 (d, J = 12.8 Hz, 1H), 4.23 (m,2H), 4.35 (d, J = 18.4 Hz, 1H), 4.47 (d, J = 18.4 Hz, 1H), 4.67 (m,1H), 7.16–7.30 (m, 7H), 7.40 (dd, J1 = 4 Hz, J2 = 2.4 Hz, 1H).
Preparation of (4R)-4-benzyl-3-(2-(2,3-dichlorophenyl)propanoyl)oxazolidin-2-one (20)
To a solution of 19 (250 mg, 0.686 mmol) in anhydrous THF (10 mL) was added sodium bis(trimethylsilyl)amide (0.61 mL, 0.617 mmol) at −78 °C under a nitrogen atmosphere. After 1 h, methyl iodide (0.192 mL, 3 mmol) was slowly added. The reaction mixture was stirred for 2 h at −78 °C and then allowed to warm to room temperature over 5 h. Reaction mixture was quenched with saturated aqueous NH4Cl. The mixture was diluted with DCM. The organic layer was washed sequentially with water, saturated sodium sulfite and brine. The organic phase was dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by silica gel chromatography eluting with a linear gradient ranging from 5 to 20% ethyl acetate in n-hexane to provide 20 (200 mg, 77%) as a white foam. 1H NMR (CDCl3, 400 MHz) δ 1.56 (d, J = 6.8 Hz, 3H), 2.79 (t, J = 12.0 Hz, 1H), 3.28 (d, J = 13.6 Hz, 1H), 4.09–4.16 (m, 2H), 4.66 (bs, 1H), 5.37 (q, J = 6.8 Hz,1 H), 7.19–7.37 (m, 8H).
Preparation of (R)-2-(2,3-dichlorophenyl)propionic acid (21)
To a solution of 20 (160 mg, 0.42 mmol) in THF (5 mL) and water at 0 °C, was added drop-wise a solution of lithium peroxide [prepared by adding 30% hydrogen peroxide (2.9mL, 2.10 mmol) to lithium hydroxide (17.6 mg, 0.41 mmol) in water (0.679 mL)]. The reaction mixture was stirred for 0 °C for 1 h, and then quenched with saturated aqueous Na2SO3 (1.28 mL). The solvent was removed in vacuo. The residue was diluted with water and then extracted with DCM (2 times). The aqueous layer was acidified with concentrated HCl and then extracted with EtOAc (2 times). The EtOAc extracts were combined, washed with brine, dried over anhydrous MgSO4, and concentrated. The residue was purified by silica gel column chromatography using EtOAc/n-hexane (40:60) to afforded 21 (80 mg, 87%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 1.51 (d, J = 7.2 Hz, 3H), 4.27 (q, J = 6.8 Hz, 1H), 7.16–7.24 (m, 2H), 7.37(d, J = 7.6 Hz, 1H).
Synthesis of (R)-2-(2,3-dichlorophenyl-N-(2-(pyridine-4-yl)benzo[d]oxazol-5-yl)propionamide (22)
The general procedure for 8 using 21 and 4 yielded 22 as a white solid (70%); 1H NMR (CDCl3, 400 MHz) δ 1.61 (d, J = 7.2 Hz, 3H), 4.31 (q, J = 6.8 Hz, 1H), 7.26 (t, J = 8 Hz, 1H), 7.42 – 7.54 (m, 4H), 7.98 (s, 1H), 8.05 (d, J = 4.4 Hz, 2H), 8.80 (d, J = 4 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 17.8, 45.0, 111.0, 112.4, 119.7, 121.2, 126.8, 128.2, 129.7, 132.0, 133.7, 134.3, 135.4, 140.9, 142.3, 147.9, 150.8, 161.6, 171.3. ESI-HRMS for C21H15Cl2N3O2 (M+H)+ calcd. 412.0620, found 412.0611, Chiral purity (% ee 83, tR = 1.58 min).
Synthesis of 5-nitro-2-(pyridine-4-yl)-1H-benzo[d]imidazole (24)
4-Nitro-1,2-phenylenediamine (23, 500 mg, 3.26 mmol) and 4-pyridine carboxaldehyde (419 mg, 3.91 mmol) were dissolved in DMF (10 mL). Disodium metabisulfite (742 mg, 3.91 mmol) was added. The reaction mixture was heated at 120 °C for 24 h under a nitrogen atmosphere. After allowing the mixture to cool to room temperature, volatiles were removed under reduced pressure. The reaction mixture was then diluted with water and extracted from DCM. The organic layer was dried on anhydrous MgSO4, filtered and concentrated. The residue was purified by silica gel column chromatography using MeOH/CHCl3 (5:95) to give 24 (480 mg, 61%) as a red solid. 1H NMR (CDCl3, 400 MHz) 8.7 (d, J = 9.2 Hz, 2H), 8.1 (d, J = 2.4 Hz, 1H), 8.03 (d, J = 8.2 Hz, 2H), 7.92 (dd, J1 = 8.0 Hz, J2 = 2.4 Hz, 1H), 7.82 (s, 1H).
Synthesis of 2-(pyridin-4-yl)-1H-benzo[d]imidazole-5-amine (25)
Substrate 24 was reduced by hydrogenation using the general procedure described for 4 to yield 25 (64%). 1H NMR (CDCl3, 400 MHz) 8.50 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 8.2 Hz, 2H), 6.70 (dd, J1 = 8 Hz, J2 = 2Hz, 1H), 6.20–6.17 (dd, J1= 8 Hz, J2 = 2 Hz, 1H), 6.10 (s, 1H), 4.30 (s, 2H).
Preparation of 2-phenoxy-N-[2-pyridin-4-yl)-1H-benzo[d]imidazole-5-yl]propionamide (26)
To a solution of 25 (50 mg, 0.237 mmol) in anhydrous THF (6 mL) was added 2-phenoxypropionyl chloride (52.5 mg, 0.284 mmol), TEA (36.2 mg, 0.355 mmol), 4-DMAP (2.89 mg, 0.023 mmol) at 0 °C. The reaction mixture was stirred for 30 min at room temperature and quenched with sodium bicarbonate solution. The reaction mixture was diluted with DCM and washed with sodium bicarbonate solution followed by brine solution. The organic layer was dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by silica gel column chromatography using MeOH/CHCl3 (10:90) to furnish 26 (60 mg, 70%) as a brown solid. 1H NMR (CDCl3, 400 MHz) δ 1.68 (d, J = 6.8 Hz, 3H), 4.85 (q, J = 6.4 Hz, 1H), 6.95 – 7.05 (m, 4H), 7.26 – 7.32 (m, 2H), 7.52 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 5.2 Hz, 2H), 8.08 (s, 1H), 8.57 (d, J = 5.2 Hz, 2H), 8.65 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 18.9, 75.5, 115.9, 117.72, 117.74, 120.8, 122.8, 130.1, 132.8, 137.6, 150.0, 150.3, 156.7, 171.5; ESI-HRMS for C21H16N3O3Cl2 (M+H)+ calcd. 428.0569 found 428.0578.
Synthesis of (but-3-yn-2-yloxy)benzene (28)
Phenol (27, 422.9 mg, 4.49 mmol) and but-3-yn-2-ol (300 mg, 4.28 mmol) were dissolved in anhydrous THF (10 mL) under a nitrogen atmosphere at 0 °C. Then Ph3P (1.12 g, 4.26 mmol) was added portion-wise. The reaction mixture was stirred for 10 min and then DEAD (894.6 mg, 5.14 mmol) was slowly added. The resulting solution was heated at 70 °C for 20 h. The reaction mixture was allowed to cool to room temperature and then water was added. The mixture was extracted with DCM. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated. The residue was purified by silica gel flash chromatography using ethyl acetate/n-hexane (5:95) providing 28 (450 mg, 69%) as a solid. 1H NMR (CDCl3, 400 MHz) δ 1.67 (d, J = 6.8 Hz, 3H), 2.47 (d, J = 2 Hz, 1H), 4.88 (q, J = 2 Hz, 1H), 6.97 – 7.03 (m, 3H), 7.28 – 7.32 (m, 2H).
Preparation of 5-azido-2-(thiazol-5-yl)benzo[d]oxazole (29)
A solution of 2-(thiazol-5-yl)benzo[d]oxazol-5-amine (4, R1 = 5-thiazole, X = H, 50 mg, 0.23 mmol) dissolved in 2 mL concentrated HCl:H2O (1:1) was cooled at −5 °C. Then a solution of sodium nitrite (31.7 mg, 0.459 mmol) dissolved in water (15 mL) was slowly added. The reaction mixture was stirred for 60 min and then neutralized with sodium acetate (37.7 mg, 0.459 mmol). Next, a solution of NaN3 (29.9 mg, 0.49 mmol) in water (0.5 mL) was slowly added over a 30 min period maintaining a temperature between 0 – 5 °C. After additional stirring for 30 min, the solution was allowed to warm to room temperature and then it was extracted with ethyl acetate. The organic layer was dried over anhydrous MgSO4, filtered and concentrated to yield 29 (50 mg, 89%) as a solid, which was used without further purification.
Preparation of 5-(4-(1-phenoxyethyl)-1H-1,2,3-triazol-1-yl)-2-(thiazol-5-yl)benzo[d]oxazole (30)
A mixture of 29 (37 mg, 0.15 mmol) and 28 (20 mg, 0.13 mmol) were dissolved in anhydrous acetonitrile (3 mL) under a nitrogen atmosphere. Then DIPEA (53 mg, 0.40 mmol) was added and the mixture stirred at room temperature for 10 min. Next, CuI (51.7 mg, 0.27 mmol) was added portionwise and then the reaction mixture was stirred for 40 min. The mixture was quenched with aqueous saturated NH4Cl, diluted with water and extracted with DCM. The organic layer was dried over anhydrous MgSO4, filtered and concentrated. The residue was purified by silica gel column chromatography using ethyl acetate/n-hexane (50:50) to furnish 30 (43 mg, 84%) as a solid. 1H NMR (CDCl3, 400 MHz) δ 1.77 (d, J = 6.4 Hz, 3H), 5.69 (q, J = 6.4 Hz, 1H), 6.90 – 6.97 (m, 3H), 7.22 – 7.24 (m, 2H), 7.66 (d, J = 8.8 Hz, 1H), 7.74 (dd, J1 = 8 Hz, J2 = 2 Hz, 1H), 7.90 (s, 1H), 7.99 (d, J = 2 Hz, 1H), 8.66 (s, 1H), 8.98 (s, 1H); 13C NMR (Pyridine-d5, 100 MHz) δ 22.1, 69.5, 112.3, 112.5, 116.6, 119.1, 121.7, 121.8, 125.8, 130.3, 135.3, 143.2, 146.9, 150.5, 151.1, 158.5, 159.0, 159.3; ESI-HRMS for C20H16N5O2S (M+H)+ calcd. 390.1025, found 390.1031.
Preparation of N-(3-nitrophenyl)isonicotinamide (32)
The general procedure for 8 was followed using 3-nitroaniline (31) and isonicotinic acid to afforded 32 (68%) as a yellow solid. 1H NMR (DMSO-d6, 400 MHz) δ 7.67 – 7.71 (m, 1H), 7.89 – 7.91 (m, 2H), 8.01 (dd, J1 = 8.4 Hz, J2 = 1.6Hz, 1H), 8.19 (dd, J1 = 8 Hz, J2= 1.6 Hz, 1H), 8.79–8.83 (m, 3H), 10.9 (NH, s, 1H).
Preparation of N-(3-aminophenyl)isonicotinamide (33)
N-(3-nitrophenyl)isonicotinamide (32, 0.823 mmol) in 4 mL ethanol was heated to 70 °C. Then SnCl2•H2O (4.2 mmol) was added in portions and refluxed until starting material disappeared (~ 4 h). The reaction mixture was cooled and quenched with saturated sodium bicarbonate solution, and extracted with ethyl acetate (3 × 10 mL). The organic layer was washed with saturated sodium bicarbonate, brine, dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to give a residue, which was used without further purification.
Preparation of (S)-N-(3-(2-(2,3-dichlorophenoxy)propanamido)phenyl)isonicotinamide (34)
The general procedure for 8 was followed using 33 and 7 (R2 = 2,3-di-ClPh, Y = O, R3 = (S)-Me) to afford 34 (63%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 1.67 (d, J = 6.8 Hz, 3H), 4.79 (q, J = 6.8 Hz, 1H), 6.84 – 6.88 (m, 1H), 7.15 – 7.23 (m, 3H), 7.31 (t, J = 8 Hz, 1H), 7.59 (d, J = 8 Hz, 1H), 7.68 (d, J = 4.4 Hz, 2H), 8.02 (s, 1H), 8.24 (s, 1H), 8.68 (s, 1H), 8.75 (s, 2H); 13C NMR (CDCl3, 100 MHz) δ 18.5, 77.0, 111.9, 113.3, 116.5, 117.0, 121.2, 123.0, 124.4, 128.0, 130.0, 134.5, 137.8, 138.3, 142.1, 150.8, 153.7, 164.0, 169.5; ESI-HRMS for C21H18N3O3Cl2 (M+H)+ calcd. 430.0725 found 430.0725, Chiral purity (% ee >99, tR=10.54).
Synthesis of N-(1-phenylethyl)2-pyridin-4-yl)benzo[d]oxazole-5-carboxamide (36)
The general procedure for 8 was followed using 35 and α-methyl benzylamine to give 36 (71%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 1.57 (d, J = 6.8 Hz, 3H), 5.29 (q, J = 6.8 Hz, 1H), 6.41 (d, J = 7.2 Hz, 1H), 7.19–7.25 (m, 1H), 7.29–7.36 (m, 4H), 7.58 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 4.8 Hz, 2H), 8.13 (s, 1H), 8.77 (bs, 2H); 13C NMR (CDCl3, 100 MHz) δ 21.9, 49.7, 111.2, 119.6, 121.3, 126.0, 126.4, 127.8, 129.0, 132.4, 134.0, 142.0, 143.1, 151.0, 152.8, 162.1, 166.1; ESI-HRMS for C21H18N3O2 (M+H)+ calcd. 344.1399 found 344.1403.
Preparation of 6-nitro-2-(pyridin-4-yl)benzo[d]oxazole (38a)
The general procedure for 3 was followed using 2-amino-5-nitrophenol (37) and 4-pyridyl carboxaldehyde to give 38a (75%) as a yellow solid. 1H NMR (DMSO-d6, 400 MHz) 6.99–7.05 (m, 1H), 8.13 (d, J= 5.2 Hz, 2H), 8.36 (d, J= 8.4 Hz, 1H), 8.89 (d, J = 7.4 Hz, 2H), 8.81 (s, 1H).
Preparation of 2-(pyridin-4-yl)benzo[d]oxazol-6-amine (39a)
The general procedure for 4 was followed using 38a to obtained 39a (85%) as a yellow solid. 1H NMR (DMSO-d6, 400 MHz) 5.61 (s, 2H), 6.66 (dd, J1 = 8 Hz, J2 = 2 Hz, 1H), 6.79 (d, J = 1.6 Hz, 1H), 7.44 (d, J = 8.8 Hz, 1H), 7.92 (d, J = 6 Hz, 2H), 8.71 (d, J = 6 Hz, 2H).
(S)-2-(2,3-dichlorophenoxy)-N-(2-(pyridin-4-yl)benzo[d]oxazol-6-yl)propionamide (40a)
The general procedure for 8 was followed using 7 (R2 = 2,3-di-ClPh, Y=O, R3=(S)-Me) and 39a to afford 40a (76%) as a white solid. 1H NMR (CDCl3, 400 MHz) δ 1.71 (d, J = 6.8 Hz, 3H), 4.85 (q, J = 6.6 Hz, 1H), 6.88 (dd, J1 = 7.2 Hz, J2 = 2.8 Hz, 1H), 7.12 – 7.18 (m, 2H), 7.24 (dd, J1 = 8.8 Hz, J2 = 2 Hz, 2H), 7.69 (d, J = 8.4 Hz, 1H), 7.99 (d, J = 5.6 Hz, 2H), 8.33 (s, 1H), 8.75 (d, J = 5.2 Hz, 2H), 8.91 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 18.3, 76.7, 102.6, 113.0, 117.4, 120.5, 120.7, 122.6, 124.1, 127.8, 134.0, 134.2, 135.6, 138.3, 150.6, 151.1, 153.4, 160.8, 169.0; ESI-HRMS for C21H16N3O3Cl2 (M+H)+ calcd. 428.0569, found 428.0567, Chiral purity (% ee >99, tR=21.0).
(S)-2-(2,3-dichlorophenoxy)-N-(2-(thiazol-5-yl)benzo[d]oxazol-6-yl)propionamide (40b)
A similar sequence of reactions that was used to prepare 40a was also used to generate 40b as a white solid. 1H NMR (CDCl3, 400 MHz) δ 1.73 (d, J = 6.8 Hz, 3H), 4.88 (q, J = 6.4 Hz, 1H), 6.89 – 6.92 (m, 1H), 7.17 – 7.19 (m, 1H), 7.23- 7.26 (m, 2H), 7.66 (d, J = 8.8 Hz, 1H), 8.29 (s, 1H), 8.61 (s, 1H), 8.85 (s, 1H), 8.94(s, 1H); 13C NMR (CDCl3, 100 MHz) δ 18.5, 76.9, 102.8, 113.2, 117.5, 120.1, 122.9, 124.3, 125.7, 128.1, 134.5, 135.4, 138.6, 142.4, 151.0, 153.6, 153.68, 156.2, 157.3, 169.2; ESI-HRMS for C19H14N3O3SCl2 (M+H)+ calcd. 434.0133, found 434.0139, Chiral purity (% ee >99, tR= 22.5).
Gene cloning, protein expression, purification and crystallization
The coding sequence of CpIMPDH enzyme was amplified by PCR from CpIMPDH-90-134-pET28a plasmid10b using primers compatible with the ligation independent cloning vector pMCSG7.21 The gene was cloned into pMCSG7 using a modified ligation-independent cloning protocol.22 The recombinant construct produced fusion proteins with an N-terminal His6-tag and a TEV protease recognition site (ENLYFQ↓S). The fusion protein was expressed in an E. coli strain BL21(DE3) harboring pMAGIC plasmid encoding one rare E. coli Arg tRNAs (covering codons AGG/AGA) in the presence of 100 μg/mL ampicillin and 30 μg/mL kanamycin. The cells were grown in enriched M9 media at 37 °C followed by an overnight induction with 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) at 18 °C. Cells were harvested, resuspended in lysis buffer (50 mM HEPES pH 8.0, 500 mM KCl, 20 mM imidazole, 10 mM 2-mercaptoethanol, 5% glycerol) and stored at –80° C.
CpIMPDH protein was purified according to a standard protocol.22 The protocol included cell lysis by sonication, Ni2+-affinity chromatography on an ÄKTAxpress system (GE Healthcare Life Sciences) followed by His6-tag cleavage using recombinant TEV protease23 and an additional Ni2+-affinity chromatography performed to remove the protease, the uncut protein, and the affinity tag. In the final step, the protein was dialyzed against crystallization buffer containing 20 mM HEPES pH 8.0, 150 mM KCl and 1 mM TCEP, concentrated, flash frozen, and stored in liquid nitrogen. Crystallizations were set up with the help of a Mosquito liquid dispenser (TTP LabTech) using the sitting-drop vapor-diffusion method in 96-well CrystalQuick plates (Greiner Bio-One). For each condition, 0.4 μl protein solution and 0.4 μl crystallization formulation were mixed and equilibrated against a 135 μl reservoir. Five crystallization screens were used. Crystals with IMP and 54 were obtained from a drop solution containing 6 mg/ml protein, 3 mM IMP and 0.5 mM 54. Diffraction quality crystals appeared at 18 °C in 0.1 M succinic acid pH 7.0 and 15% (w/v) PEG 3350. The crystals were mounted on CryoLoops (Hampton Research) and flash-cooled in liquid nitrogen. The cryoprotectant consisted of 20% ethylene glycol.
Data collection and Structure determination
Diffraction data were collected at 100 K at the 19-ID beamline of the Structural Biology Center at the Advanced Photon Source, Argonne National Laboratory.24 The single wavelength data at 0.97923 Å (12.661406 keV) up to 2.1 Å were collected from a single crystal of CpIMPDH complexed with IMP and 54. The crystal was exposed for 5 s per 1.0° rotation of ω with the crystal to detector distance of 300 mm. The data were recorded on a CCD detector Q315 from ADSC scanning 220°. The SBC-Collect program was used for all data collection and visualization. Data collection strategy, integration, and scaling were performed with the HKL3000 program package.25 A summary of the crystallographic data can be found in Table S1.
The structure was determined by molecular replacement using chain A of the structure of CpIMPDH (PDB ID 3FFS) as a search model with HKL3000 using the data to 3.0 Å.25 The initial model contained 4 copies of the search model and there was extra electron density for additional protein residues that were not part of the search model. The presence of IMP and 54 in the active site was apparent from the initial electron density map (Fo). Extensive manual model building with coot26 and the subsequent refinement using phenix.refine27 was performed against the full data set up to 2.1 Å until the structure converged to the R factor (Rwork) of 0.162, and Rfree of 0.210 with the r.m.s. bond distances of 0.007 and the r.m.s. bond angles of 1.104°. The asymmetric unit contains four protein chains, A, B, C and D comprised of residues 1–92 and 135–392 (chains A and C) and 2–92 and 135–392 (chains B and D). The final model also includes four IMP molecules, four 54 molecules, five ethylene glycol molecules and 486 ordered water molecules. Several residues including several N- and C-terminal residues and those introduced as a cloning artifact (SNA)28 are missing due to disorder. In addition, residues 312–325 of chain A, 309–235 of chain B, 311–325 of chains C and D located on the surface of the tetramer were disordered and not modeled. The stereochemistry of the structure was checked with PROCHECK29 and the Ramachandran plot. Atomic coordinates and experimental structure factors of the structure have been deposited in the PDB under the ID code 4IXH.
Inhibition of recombinant CpIMPDH
Recombinant CpIMPDH, purified from E. coli,12 was assessed by monitoring the production of NADH by fluorescence at varying inhibitor concentrations (25 pM – 5 YM). IMPDH was incubated with inhibitor for 5 min at room temperature prior to addition of substrates. The following conditions were used: 50 mM Tris-HCl, pH 8.0, 100 mM KCl, 3 mM EDTA, 1 mM dithiothreitol (assay buffer) at 25 °C, 10 nM CpIMPDH, 300 μM NAD and 150 μM IMP. To characterize the non-specific binding of inhibitors, assays were also carried out in the presence of 0.05% BSA (fatty acid free). IC50 values were calculated for each inhibitor according to Equation 1 using the SigmaPlot program (SPSS, Inc.):
| (Eq. 1) |
where υi is initial velocity in the presence of inhibitor (I) and υo is the initial velocity in the absence of inhibitor. When the values of IC50 approach the concentration of enzyme, tight binding treatment is used:
| (Eq. 2) |
Inhibition at each inhibitor concentration was measured in quadruplicate and averaged; this value was used as υi. The IC50 values were determined three times; the average and standard deviations are reported. Mechanism of inhibition was determined by fitting to equations for competitive, noncompetitive/mixed and uncompetitive inhibition as previously described.16c
Supplementary Material
Figure 2.
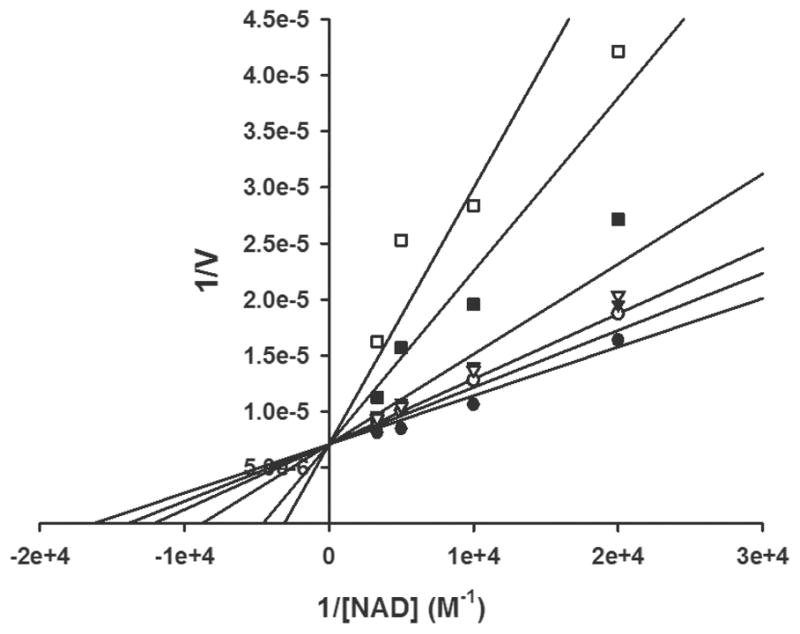
Inhibition of CpIMPDH by 68. Inhibitor concentrations: open squares, 630 nM; closed squares, 380 nM; closed triangles, 130 nM; open triangles, 50 nM; open circles, 25 nM; no inhibitor, closed circles.
Scheme 4.
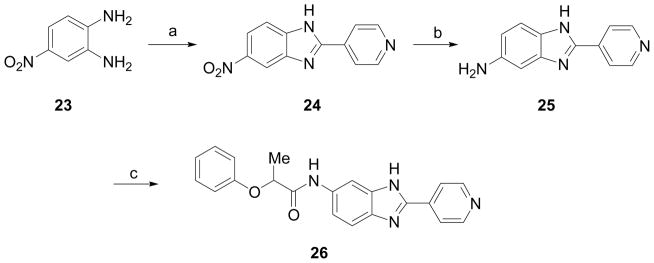
Synthesis of a benzimidazole derivative.
Reagents and conditions: (a) 4-PyCHO, Na2S2O5, DMF, 120 °C, 24 h, 61% (b) H2 (1 atm), 10% Pd-C, MeOH, rt, 6 h, 64% (c) PhOCH(CH3)COCl, TEA, cat. 4-DMAP, THF, 0 °C – rt, 30 min, 70%. A benzimidazole analogue of 1 was prepared using the method outlined in Scheme 4. 6-Nitro-2-(pyridin-4-yl)-1H-benzo[d]imidazole (24) was prepared by condensation of 4-pyridine carboxaldehyde with 4-nitro-1,2-phenylenediamine (23) in the presence of the oxidizing agent sodium metabisulphite.10d The product was reduced in the presence of hydrogen (1 atm) and 10% Pd-C to give 25, which was then coupled with 2-(phenoxy)propionoyl chloride in the presence of triethylamine and catalytic DMAP to give 26.
Scheme 7.
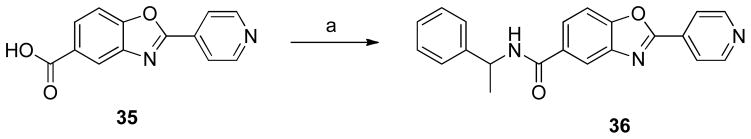
Synthesis of amide derivative 36.
Reagents and conditions: (a) α-methyl benzyl amine, EDCI•HCl, DMF, 0 °C - rt, 12 h, 71%. An analogue of 22 in which the amide group is inverted was prepared using the method outlined in Scheme 7. Amide 36 was generated by coupling 2-(pyridin-4-yl)benzo[d]oxazole-5-carboxylic acid (35) and α-methylbenzylamine in the presence of EDCI•HCl.
Acknowledgments
This work has been funded in whole or in part with funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services (U01AI075466 to LH, and contracts HHSN272200700058C and HHSN272201200026C to CSGID). GDC thanks the New England Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (NERCE/BEID) and the Harvard NeuroDiscovery Center for financial support. The authors would also like to thank Prof. Li Deng and Yongwei Wu (Department of Chemistry, Brandeis University) for use of a chiral HPLC instrument and Minyi Gu for help with gene cloning.
Abbreviations
- Cp
Cryptosporidium parvum
- CLint
intrinsic clearance
- BSA
bovine serum albumin
- DCM
dichloromethane
- DEAD
Diethylazodicarboxylate
- DIPEA
diisopropylethylamine
- DMF
N,N-dimethylformamide
- EDCI•HCl
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- IMP
inosine 5′-monophosphate
- IMPDH
inosine 5′-monophosphate dehydrogenase
- hIMPDH
human IMPDH
- HTS
high-throughput screening
- MPA
mycophenolic acid
- MR
molecular replacement
- N.D
not determined
- r.m.s
root-mean-square
- TEA
triethylamine
- Toxo
Toxoplasma
- XMP
xanthosine 5′-monophosphate
Footnotes
Supporting Information: Statistics for data collection and refinement of the x-ray crystal structure are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Carey CM, Lee H, Trevors JT. Biology, persistence and detection of Cryptosporidium parvum and Cryptosporidium hominis oocyst. Water Res. 2004;38:818–62. doi: 10.1016/j.watres.2003.10.012. [DOI] [PubMed] [Google Scholar]; (b) Fayer R. Cryptosporidium: a water-borne zoonotic parasite. Veterinary Parasitology Veterinary parasitology. 2004;126:37–56. doi: 10.1016/j.vetpar.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Guerrant RL, Oriá RB, Moore SR, Oriá MOB, Lima AAM. Malnutrition as an enteric infectious disease with long-term effects on child development. Nutrition Reviews. 2008;66:487–505. doi: 10.1111/j.1753-4887.2008.00082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snelling WJ, Xiao L, Ortega-Pierres G, Lowery CJ, Moore JE, Rao JR, Smyth S, Millar BC, Rooney PJ, Matsuda M, Kenny F, Xu J, Dooley JSG. Cryptosporidiosis in developing countries. 2007;1 [PubMed] [Google Scholar]
- 4.DuPont HL, Chappell CL, Sterling CR, Okhuysen PC, Rose JB, Jakubowski W. The infectivity of Cryptosporidium parvum in healthy volunteers. N Engl J Med. 1995;332:855–859. doi: 10.1056/NEJM199503303321304. [DOI] [PubMed] [Google Scholar]
- 5.Mead JR. Cryptosporidiosis and the challenges of chemotherapy. Drug resistance updates: reviews and commentaries in antimicrobial and anticancer chemotherapy. 2002;5:47–57. doi: 10.1016/s1368-7646(02)00011-0. [DOI] [PubMed] [Google Scholar]
- 6.(a) Striepen B, Pruijssers AJ, Huang J, Li C, Gubbels MJ, Umejiego NN, Hedstrom L, Kissinger JC. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc Natl Acad Sci USA. 2004;101:3154–3159. doi: 10.1073/pnas.0304686101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu P, Widmer G, Wang Y, Ozaki LS, Alves JM, Serrano MG, Puiu D, Manque P, Akiyoshi D, Mackey AJ, Pearson WR, Dear PH, Bankier AT, Peterson DL, Abrahamsen MS, Kapur V, Tzipori S, Buck GA. The genome of Cryptosporidium hominis. Nature. 2004;431:1107–1112. doi: 10.1038/nature02977. [DOI] [PubMed] [Google Scholar]; (c) Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, Buck GA, Xu P, Bankier AT, Dear PH, Konfortov BA, Spriggs HF, Iyer L, Anantharaman V, Aravind L, Kapur V. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science. 2004;304:441–445. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- 7.Hedstrom L. IMP Dehydrogenase: structure, mechanism and inhibition. Chem Rev. 2009;109:2903 – 2928. doi: 10.1021/cr900021w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Striepen B, White MW, Li C, Guerini MN, Malik SB, Logsdon JM, Jr, Liu C, Abrahamsen MS. Genetic complementation in apicomplexan parasites. Proc Natl Acad Sci USA. 2002;99:6304–6309. doi: 10.1073/pnas.092525699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Umejiego NN, Gollapalli D, Sharling L, Volftsun A, Lu J, Benjamin NN, Stroupe AH, Riera TV, Striepen B, Hedstrom L. Targeting a prokaryotic protein in a eukaryotic pathogen: identification of lead compounds against cryptosporidiosis. Chem Biol. 2008;15:70–77. doi: 10.1016/j.chembiol.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Maurya SK, Gollapalli DR, Kirubakaran S, Zhang M, Johnson CR, Benjamin NN, Hedstrom L, Cuny GD. Triazole inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J Med Chem. 2009;52:4623–4630. doi: 10.1021/jm900410u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) MacPherson IS, Kirubakaran S, Gorla SK, Riera TV, D’Aquino JA, Zhang M, Cuny GD, Hedstrom L. The structural basis of Cryptosporidium-specific IMP dehydrogenase inhibitor selectivity. J Am Chem Soc. 2010;132:1230–1231. doi: 10.1021/ja909947a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gorla SK, Johnson C, Khan J, Sun X, Sharling L, Striepen B, Hedstrom L. Targeting Prokaryotic Enzymes in the Eukaryotic Pathogen Cryptosporidium. In: Becker K, editor. Apicomplexan Parasites. Vol. 2. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2011. Chapter 14. [Google Scholar]; (d) Kirubakaran S, Gorla SK, Sharling L, Zhang M, Liu X, Ray SS, MacPherson IS, Striepen B, Hedstrom L, Cuny GD. Structure–activity relationship study of selective benzimidazole-based inhibitors of Cryptosporidium parvum IMPDH. Bioorg Med Chem Lett. 2012;22:1985–1988. doi: 10.1016/j.bmcl.2012.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gorla SK, Kavitha M, Zhang M, Liu X, Sharling L, Gollapalli DR, Striepen B, Hedstrom L, Cuny GD. Selective and potent urea inhibitors of Cryptosporidium parvum inosine monophosphate dehydrogenase. J Med Chem. 2012;55:7759–7771. doi: 10.1021/jm3007917. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Johnson CR, Gorla SK, Kavitha M, Zhang M, Liu X, Striepen B, Mead JR, Cuny GD, Hedstrom L. Phthalazinone inhibitors of inosine-5′-monophosphate dehydrogenase from Cryptosporidium parvum. Bioorg Med Chem Lett. 2013;23:1004–1007. doi: 10.1016/j.bmcl.2012.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kawashita Y, Nakamichi N, Kawabata H, Hayashi M. Direct and practical synthesis of 2-arylbenzoxazoles promoted by activated carbon. Org Lett. 2003;5:3713–3715. doi: 10.1021/ol035393w. [DOI] [PubMed] [Google Scholar]
- 12.Bui M, Conlon P, Erlanson DA, Fan J, Guan B, Hopkins BT, Ishchenko A, Jenkins TJ, Kumaravel G, Marcotte D, Powell N, Scott D, Taveras A, Wang D, Zhong M. Preparation of 4-(3-aminopiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidine and 4-(3-aminopiperidin-1-yl)pyrimidine derivatives as Bruton’s tyrosine kinase (Btk kinase) inhibitors. WO 2011029043 2011
- 13.Petit M, Lapierre AJB, Curran DP. Relaying asymmetry of transient atropisomers of o-iodoanilides by radical cyclizations. J Am Chem Soc. 2005;127:14994–14995. doi: 10.1021/ja055666d. [DOI] [PubMed] [Google Scholar]
- 14.Mori K. Synthesis of (R)-ar-turmerone and its conversion to (R)-ar-himachalene, a pheromone component of the flea beetle: (R)-ar-himachalene is dextrorotatory in hexane, while levorotatory in chloroform. Tetrahedron: Asymmetry. 2005;16:685–692. [Google Scholar]
- 15.(a) Sharpless KB, Manetsch R. In situ click chemistry: a powerful means for lead discovery. Exp Opin Drug Discovery. 2006;1:525–538. doi: 10.1517/17460441.1.6.525. [DOI] [PubMed] [Google Scholar]; (b) Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]; (c) Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem, Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 16.(a) Farazi T, Leichman J, Harris T, Cahoon M, Hedstrom L. Isolation and characterization of mycophenolic acid resistant mutants of inosine 5′monophosphate dehydrogenase. J Biol Chem. 1997;272:961–965. doi: 10.1074/jbc.272.2.961. [DOI] [PubMed] [Google Scholar]; (b) Mortimer SE, Hedstrom L. Autosomal dominant retinitis pigmentosa mutations in inosine 5′-monophosphate dehydrogenase type I disrupt nucleic acid binding. Biochem J. 2005;390(Pt 1):41–47. doi: 10.1042/BJ20042051. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Umejiego NN, Li C, Riera T, Hedstrom L, Striepen B. Cryptosporidium parvum IMP dehydrogenase: Identification of functional, structural and dynamic properties that can be exploited for drug design. J Biol Chem. 2004;279:40320–40327. doi: 10.1074/jbc.M407121200. [DOI] [PubMed] [Google Scholar]
- 17.(a) Merrikin DJ, Briant J, Rolinson GN. Effect of protein binding on antibiotic activity in vivo. J Antimicrob Chemother. 1983;11:233–238. doi: 10.1093/jac/11.3.233. [DOI] [PubMed] [Google Scholar]; (b) Craig WA, Ebert SC. Protein binding and its significance in antibacterial therapy. Infect Dis Clin North Am. 1989;3:407–414. [PubMed] [Google Scholar]; (c) Smith DA, Di L, Kerns EH. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov. 2010;9:929–939. doi: 10.1038/nrd3287. [DOI] [PubMed] [Google Scholar]
- 18.Sharling L, Liu X, Gollapalli DR, Maurya SK, Hedstrom L, Striepen B. A screening pipeline for antiparasitic agents targeting Cryptosporidium inosine monophosphate dehydrogenase. PLoS Negl Trop Dis. 2010;4:e794. doi: 10.1371/journal.pntd.0000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sintchak MD, Fleming MA, Futer O, Raybuck SA, Chambers SP, Caron PR, Murcko MA, Wilson KP. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophenolic acid. Cell. 1996;85:921 – 930. doi: 10.1016/s0092-8674(00)81275-1. [DOI] [PubMed] [Google Scholar]
- 20.Gubbels MJ, Striepen B. Studying the cell biology of apicomplexan parasites using fluorescent proteins. Microsc Microanal. 2004;10:568–579. doi: 10.1017/S1431927604040899. [DOI] [PubMed] [Google Scholar]
- 21.Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25:8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 22.Kim Y, Babnigg G, Jedrzejczak R, Eschenfeldt WH, Li H, Maltseva N, Hatzos-Skintges C, Gu M, Makowska-Grzyska M, Wu R, An H, Chhor G, Joachimiak A. High-throughput protein purification and quality assessment for crystallization. Methods. 2011;55:2–28. doi: 10.1016/j.ymeth.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blommel PG, Fox BG. A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr Purif. 2007;55:53–68. doi: 10.1016/j.pep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenbaum G, Alkire RW, Evans G, Rotella FJ, Lazarski K, Zhang RG, Ginell SL, Duke N, Naday I, Lazarz J, Molitsky MJ, Keefe L, Gonczy J, Rock L, Sanishvili R, Walsh MA, Westbrook E, Joachimiak A. The Structural Biology Center 19ID undulator beamline: Facility specifications and protein crystallographic results. J Synchrotron Radiat. 2006;13(Pt 1):30–45. doi: 10.1107/S0909049505036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: The integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 26.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr Section D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 27.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim Y, Dementieva I, Zhou M, Wu R, Lezondra L, Quartey P, Joachimiak G, Korolev O, Li H, Joachimiak A. Automation of protein purification for structural genomics. J Struct Funct Genomics. 2004;5:111–118. doi: 10.1023/B:JSFG.0000029206.07778.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.