

Background: The interactions between estrogen receptor β (ERβ1) and different coregulators are responsible for the distinct functions of ERβ1.
Results: Tip60 enhances ERβ1 transactivation at the AP-1 site but inhibits it at ERE sites.
Conclusion: Tip60 is either a coactivator or a corepressor for ERβ1 in a regulatory element-dependent manner.
Significance: Tip60 is the first multifaceted coregulator of the transcriptional activity of ERβ1 that has been identified.
Keywords: Estrogen Receptor, Protein-Protein Interactions, Transcription, Transcription Enhancers, Transcription Repressor, AP-1, ERβ1, Acetyltransferase, Coregulator, Estrogen-response Element
Abstract
Estrogen receptor (ER) β1 and ERα have overlapping and distinct functions despite their common use of estradiol as the physiological ligand. These attributes are explained in part by their differential utilization of coregulators and ligands. Although Tip60 has been shown to interact with both receptors, its regulatory role in ERβ1 transactivation has not been defined. In this study, we found that Tip60 enhances transactivation of ERβ1 at the AP-1 site but suppresses its transcriptional activity at the estrogen-response element (ERE) site in an estradiol-independent manner. However, different estrogenic compounds can modify the Tip60 action. The corepressor activity of Tip60 at the ERE site is abolished by diarylpropionitrile, genistein, equol, and bisphenol A, whereas its coactivation at the AP-1 site is augmented by fulvestrant (ICI 182,780). GRIP1 is an important tethering mediator for ERs at the AP-1 site. We found that coexpression of GRIP1 synergizes the action of Tip60. Although Tip60 is a known acetyltransferase, it is unable to acetylate ERβ1, and its coregulatory functions are independent of its acetylation activity. In addition, we showed the co-occupancy of ERβ1 and Tip60 at ERE and AP-1 sites of ERβ1 target genes. Tip60 differentially regulates the endogenous expression of the target genes by modulating the binding of ERβ1 to the cis-regulatory regions. Thus, we have identified Tip60 as the first dual-function coregulator of ERβ1.
Introduction
Estrogen normally exerts its effects via two main receptor subtypes, estrogen receptor (ER)2 α and β (ERβ1) (1). These receptors function as transcription factors and regulate gene expression either by binding directly to estrogen-response elements (EREs) within the regulatory region of target genes (2, 3) or by interacting with other transcription factors, such as AP-1, NFκB, and Sp1 (4, 5). The activation of ERs is controlled by interplay between the binding of ligands and coregulators (coactivators and corepressors) (6). Most ER signaling pathways require ligand binding because ligands are able to induce the dimerization of ERs and conformational changes in receptors and thus to increase the potency of coactivator recruitment (7). However, studies of the ligand-independent regulation of ERβ1 by coregulators are limited to previous findings demonstrating this mode of action for SRC1 and GRIP1 (8, 9). Global transcriptional profiling also reveals that unliganded ERβ1 regulates a significant number of target genes (10, 11). These findings, taken together, have stimulated significant interest in the topic of ligand-independent action.
Coregulators regulate the activity of transcription factors through several mechanisms, including post-translational modification. Activities of ERs are regulated, for example, by acetylation, phosphorylation, and ubiquitination (12–14). A putative acetylation motif is present in many hormone receptors conserved among different species (13, 15), revealing that acetylation is a common regulatory mechanism of receptor activity. ERα is acetylated by p300 and SRC1 (16, 17), whereas its hormone sensitivity and transactivation are regulated by acetylation (17). Moreover, acetylation of ERα modulates or is modulated by other post-translational modifications, such as ubiquitination and phosphorylation (18, 19). However, acetylation of ERβ1 has not yet been reported. Alternatively, coregulators can act as scaffold proteins to allow tethering of ERs and associated proteins onto other transcription factors (4, 20). For example, AP-1 recruits CBP and p300, which bind to p160 coactivators. ERs then tether onto the transcriptional complex of AP-1 through the physical interaction with p160 coactivators (4, 20). In short, the diverse actions of a nuclear receptor such as ERβ1 could depend largely on its interacting coregulators.
Tip60 (lysine acetyltransferase 5 (KAT5)) is a well studied ERα coregulator. It belongs to the MYST (MOZ, Ybf2/Sas3, Sas2, and Tip60) family. Members of this family possess an acetyltransferase domain capable of acetylating histones and other proteins (21). Moreover, Tip60 functions as either a coactivator (22–26) or a corepressor (27, 28), depending on its interacting transcription factors. Tip60 enhances ERα transactivation at ERE sites in a ligand-dependent manner (29, 30) and thus increases the expression of certain ERα target genes (29, 31). A study also shows that Tip60 interacts with ERβ1 in the presence of estrogen (32). However, it remains unclear how Tip60 modulates ERβ1 function.
This study investigated the biological function of Tip60 on ERβ1 transactivation, particularly at the various cis-regulatory sequences and/or in the presence of different types of ligand. The dependence of histone acetyltransferase (HAT) domain activity in Tip60 was evaluated with a HAT domain mutant. Its interactions with other common coregulators such as SRC-1 and GRIP1 were determined. Moreover, the co-occupancy of ERβ1 and Tip60 at cis-regulatory elements of endogenous ERβ1 target genes and their differential regulation by Tip60 were evaluated. Here, we showed that Tip60 is a unique dual-function coregulator of ERβ1 in a cis-acting element-dependent manner.
EXPERIMENTAL PROCEDURES
Cell Culture Conditions
HEK293 and DU-145 cells were grown in Eagle's minimal essential medium supplemented with 10% fetal bovine serum, l-glutamine. PC-3 cells were grown in F-12K medium supplemented with 10% fetal bovine serum (ATCC, Manassas, VA). All cells were grown in 1% penicillin/streptomycin. The phenol red-free DMEM was supplemented with 10% charcoal-stripped fetal bovine serum (CSS) prior to the addition of ligands in experiments. Cells were grown at 37 °C and 5% CO2.
Transfection Reagents and Chemicals
Transient transfection of plasmids into HEK293 cells was performed using Lipofectamine 2000 (Invitrogen). Transient transfection of plasmids into PC-3 and DU-145 cells was performed using X-tremeGENE HP (Roche Applied Science). DharmaFECT 2 was used as the siRNA transfection reagent for PC-3 (Thermo Scientific Dharmacon, Florence, KY). Chemicals such as estradiol (E2), diarylpropionitrile (DPN), genistein (GEN), equol (EQ), daizein (DAI), apigenin (API), 4-OH-tamoxifen (TAM), raloxifene (RAL), bisphenol A (BPA), anacardic acid, trichostatin A (TSA), and nicotinamide were purchased from Sigma. ICI 182,780 (ICI) was a gift from Zeneca Pharmaceuticals (Cheshire, UK).
Plasmids, siRNAs, and Recombinant Protein
Full-length ERβ1 and ERα were subcloned into pGBKT7 vector, whereas Tip60 was cloned into pACT2 vector (Clontech). ERβ1 and Tip60 were also cloned into pcDNA-HisMax (Invitrogen) or subcloned into pENTR entry vector (Invitrogen) and then transferred into destination vector pDEST40 through gateway cloning (Invitrogen). In addition, full-length ERβ1 and ERα were subcloned into the pGBKT7 vector, whereas Tip60 was cloned into the pACT2 vector (Clontech). SRC-1 and GRIP-1, gifts from Dr. Nancy Weigel (Baylor College of Medicine, Houston), were cloned into pcDNA3.1. ONTARGETplus SMARTpool 4 siRNAs specific to Tip60 were used for gene knockdown. ONTARGETplus nontargeting siRNA was used as the negative control (Thermo Scientific Dharmacon). Recombinant ERβ1 protein was purchased from Thermo Scientific Pierce.
To generate different domain-deleted ERβ1 constructs, a c-Myc tag was first added by PCR to the N terminus of the full-length ERβ1 coding sequence, which was cloned into pDEST40. We generated different domain-deleted ERβ1 by performing PCR with different sets of primers (Table 1) and using ERβ1-pDEST40 as the template.
TABLE 1.
Primers used in the experiments of domain-deletion study of ERβ1 and site-directed mutagenesis of Tip60
F is forward, and R is reverse.
| Primers | Sequences |
|---|---|
| Domain deletion of ERβ1 | |
| ERβ1ΔAF-1-F | CACCATGGAGGAGCAGAAGCTGATCTCAGAGGAGGACCTGTGCGCTGTCTGCAGCGATTA |
| ERβ1ΔAF-1-R | TGGATCCTCACTGAGACTGTGGGTTCT |
| ERβ1ΔAF-1-DBD-F | CACCATGGAGGAGCAGAAGCTGATCTCAGAGGAGGACCTGGTGAAGTGTGGCTCCCGGAG |
| ERβ1ΔAF-1-DBD-R | TGGATCCTCACTGAGACTGTGGGTTCT |
| ERβ1ΔAF-1-HD-F | CACCATGGAGGAGCAGAAGCTGATCTCAGAGGAGGACCTGGAGTTGGTACACATGATCAG |
| ERβ1ΔAF-1-HD-R | TGGATCCTCACTGAGACTGTGGGTTCT |
| ERβ1ΔLBD-AF-2-F | CACCATGGAGGAGCAGAAGCTG |
| ERβ1ΔLBD-AF-2-R | TCAAAGCACGTGGGCATTCAGCA |
| ERβ1ΔAF-2-F | CACCATGGAGGAGCAGAAGCTG |
| ERβ1ΔAF-2-R | TCACTTGTCGGCCAACTTGGTCA |
| Site-directed mutagenesis of Tip60's HAT domain | |
| Tip60ΔHAT-F | GCCTCCCTACGAGCGCCGGGAATACGGCAAGC |
| Tip60ΔHAT-R | GCTTGCCGTATTCCCGGCGCTCGTAGGGAGGC |
Antibodies
Rabbit polyclonal anti-ERβ (H-150), goat polyclonal anti-Tip60 (N-17 and K-17), goat polyclonal anti-SRC-1 (C-20), rabbit polyclonal anti-GRIP-1 (M-343), mouse monoclonal anti-c-Myc (9E10), and control IgG were purchased from Santa Cruz Biotechnology (Dallas, TX). Mouse monoclonal anti-ERβ1 was purchased from AbD Serotec (Raleigh, NC). Rabbit polyclonal anti-acetyl-lysine and IgG XP isotype control were purchased from Cell Signaling Technology (Danvers, MA). EZview red anti-HA and anti-c-Myc affinity gel were purchased from Sigma.
Construction of ERβ1 Stably Expressed Cell Lines
Stably expressed cell lines were constructed according to the published data (33). Full-length ERβ1 or LacZ (negative control) was subcloned, respectively, into pLenti6 lentiviral vector by Multisite Gateway Cloning (Invitrogen) and transfected into 293FT for production of lentivirus. The titer of lentivirus was measured, and the multiplicity of infection of PC-3 cells was determined. Lentivirus-infected PC-3 cells were selected with blasticidin (10 μg/ml) for 3 weeks. Quantitative reverse transcription (RT)-PCR, Western blot, and β-galactosidase assay were performed to confirm the stable expression of ERβ5 or LacZ.
In Vitro Coimmunoprecipitation (Co-IP)
T7 promoter and HA tag were added to the N terminus of the coding sequence of Tip60 by PCR. pGBKT7 vector containing the full-length of ERβ1, ERα, and purified PCR product of Tip60 were, respectively, translated in vitro by the TnT T7-reticulocyte system (Promega, Fitchburg, WI) labeled with EasyTag EXPRESS 35S protein labeling mix (PerkinElmer Life Sciences). Tip60 (10 μl) and ERβ1 or ERα (each 10 μl) proteins were mixed at 4 °C for 1 h. Lysates were incubated with 20 μl of EZview red anti-HA affinity gel (Sigma) at 4 °C overnight with agitation. The samples were subjected to SDS-PAGE. The dried gel was exposed to x-ray film for 72 h, and an intensifying screen (Eastman Kodak) was used for signal enhancement. Films were scanned using the Odyssey Infrared Imaging System (LiCor Bioscience, Lincoln, NE).
Yeast Two-hybrid Assays
ERα- or ERβ1-pGBKT7 and Tip60-pACT2 were cotransformed into yeast strain Y187 through the polyethylene glycol/lithium acetate method with the use of the Yeastmaker yeast transformation system (Clontech). Procedures followed the manufacturer's protocol. The transformed yeast cells were grown on quadruple dropout (SD/−Ade−His−Leu−Trp) (QDO) agar with X-α-galactosidase until the appearance of blue colonies.
Ni-NTA Purification of His-tagged Proteins
HEK293 cells were transfected with ERβ1 and Tip60. After a 24-h transfection, medium was added with 10 nm E2. Cells were lysed in lysis buffer (50 mm NaH2PO4, 300 mm NaCl, 10 mm imidazole, 0.1% Tween 20) containing complete EDTA-free protease inhibitor mixture (Calbiochem) followed by sonication. About 1 mg of total lysate was incubated with 20 μl of Ni-NTA-agarose beads (Qiagen, Valencia, CA) at 4 °C overnight. Washing and elution procedures followed the manufacturer's protocol. The samples were subjected to Western blot analysis. IRDye secondary antibody was used to detect the protein bands, and the Odyssey Infrared Imaging System (LiCor Bioscience) was used to detect the signals.
Mammalian Co-IP
HEK293 cells transfected with plasmids or ERβ1 stably expressed PC-3 cells were used. Medium was added with or without 10 nm E2 as indicated. Cells were lysed in M-PER lysis buffer (Thermo Scientific Pierce) containing protease inhibitor mixture. Lysates were incubated with 2 μg of Tip60 or ERβ1 antibody at 4 °C overnight and then with protein G Dynabeads (Invitrogen) at room temperature for 1.5 h. The immunoprecipitates were subjected to Western blot analysis.
In the domain-deletion study, full-length and domain-deleted ERβ1 constructs were immunoprecipitated by EZview red anti-c-Myc affinity gel (Sigma). IgG XP isotype was used as negative control (Cell Signaling Technology).
Immunofluorescence Staining
HEK293 cells or ERβ1 stably expressed PC-3 cells were seeded on a round coverslip. HEK293 cells were transfected with ERβ1 and Tip60. Cells were fixed in 10% formalin and permeabilized with 1% Nonidet P-40. Normal chicken serum was used for blocking. Cells were incubated with rabbit ERβ (H150) and goat Tip60 (N-17) at room temperature for 1 h followed by incubation with different fluorescent-tagged secondary antibodies. DAPI (Sigma) was used for nuclear counterstaining. Prolong R Gold anti-fade reagent (Invitrogen) was used for signal enhancement. Fluorescent images were obtained with an Axiovert 200 M fluorescent microscope equipped with an AxioCam MRm camera and Axiovision 4.8 software (Carl Zeiss, Oberkochen, Germany).
Site-directed Mutagenesis
The acetylation-deficient mutant of Tip60, Tip60ΔHAT (Q377E/G380E), was generated with the use of the Stratagene QuikChange lightning site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) as described in the protocol. Primers for mutagenesis were designed through the QuikChange primer design program (Agilent Technologies) (Table 1). In brief, the mutant strand synthesis was done by PCR, and products were treated with the restriction endonuclease DpnI to digest the parental DNA. The mutated single-stranded DNA was converted to the duplex form in vivo through bacterial transformation. Plasmids were extracted and sequenced to confirm the mutations.
In Vitro and in Vivo Acetylation Assay
For the in vitro acetylation assay, HEK293 cells were transfected with either wild-type Tip60 (Tip60WT) or Tip60ΔHAT. Cells were treated with 3 μm TSA and 5 mm nicotinamide for 6 h. Recombinant Tip60 was purified on the Ni-NTA column as described above, and the lysis and wash buffers were added with 1 μm TSA and 5 mm nicotinamide, which are inhibitors of different deacetylase families. The Tip60-bound Ni-NTA column was resuspended in HAT buffer (50 mm Tris-HCl, pH 8, 10% glycerol, 100 μm EDTA, 1 mm DTT, 1 mm PMSF, 10 mm sodium butyrate, 5 mm nicotinamide) with 500 μm acetyl-CoA and 500 μg of recombinant ERβ1. The mixture was incubated at 30 °C for 1 h. Lysates were subjected to Western blot analysis.
For the in vivo acetylation assay, HEK293 cells were transfected with ERβ1, Tip60WT, or Tip60ΔHAT. Cells were treated with 3 μm TSA and 5 mm nicotinamide for 6 h. Immunoprecipitation was performed with ERβ1 or Tip60 antibody, and the lysis and wash buffers were added with 1 μm TSA and 5 mm nicotinamide. Lysates were subjected to Western blot analysis.
Luciferase Reporter Assay
Different luciferase reporter plasmids were used. The pt109-ERE3-Luc carrying 3× vitellogenin ERE was provided by Dr. Craig Jordan (Fox Chase Cancer Center, Philadelphia). The pAP-1-Luc was purchased from Clontech. The C3 ERE-Luc, c-Fos ERE-Luc, progesterone receptor (PR) ERE-Luc, and pS2 ERE-Luc reporters were gifts from Dr. Carolyn Klinge (University of Louisville, Louisville, KY). NFκB-Luc and pSp13-Luc were provided by Dr. Francis Chan (University of Massachusetts Medical School, Worcester, MA). HEK293 cells were seeded on 24-well plates at 2.8 × 105 in phenol red-free medium supplemented with 10% charcoal-stripped serum (CSS). Expression plasmids of ERβ1, GFP, or Tip60, together with luciferase reporter plasmids and β-galactosidase, were transiently transfected into cells. Different ligands, such as E2, DPN, GEN, EQ, DAI, API, TAM, RAL, ICI, and BPA were added to the medium after a 24-h transfection. Transactivation activities of ERβ1 were measured by using the Bright-Glo luciferase kit (Promega). Normalization of transfection efficiency was done by measuring β-galactosidase activity using the β-gal assay kit (Promega). Each independent experiment was carried out in technical triplicates.
Quantitative RT-PCR
Total RNA was extracted with TRIzol reagent (Invitrogen), and cDNA synthesis was done with SMART Moloney murine leukemia virus reverse transcriptase with poly(dT) primer following the manufacturer's protocols (Promega). Quantitative RT-PCR was performed with ABI7900 real time PCR system (Invitrogen). The sequences of primers used are summarized in Table 2.
TABLE 2.
Primers used in the experiments of quantitative RT-PCR and ChIP real time PCR
F is forward, and R is reverse.
| Primers | Sequences |
|---|---|
| Quantitative RT-PCR | |
| ERβ1-RT-F | TGGCTAACCTCCTGATGCTC |
| ERβ1-RT-R | TCCAGCAGCAGGTCATACAC |
| Tip60-RT-F | CGGAGGTGGGGGAGATAAT |
| Tip60-RT-R | ATGTCCTTCACGCTCAGGAT |
| CXCL12-RT-F | TTGACCCGAAGCTAAAGTGG |
| CXCL12-RT-R | TGGGCTCCTACTGTAAGGGTT |
| CyclinD2-RT-F | TGAGCTGCTGGCTAAGATCA |
| CyclinD2-RT-R | ACGTTGGTCCTGACGGTACT |
| GAPDH-RT-F | GAAGGTGAAGGTCGGAGTCA |
| GAPDH-RT-R | GACAAGCTTCCCGTTCTCAG |
| ChIP real time PCR | |
| CXCL12-ChIP-F | AGGCATCACAATGCAAATCA |
| CXCL12-ChIP-R | AGGCTGGTGAGATGCTGAGT |
| CyclinD2-ChIP-F | GTCTCTCCCCTTCCTCCTGG |
| CyclinD2-ChIP-R | GCCCTGACACGTGCTCTAA |
| ERβ5-ChIP-F | CCCTAAGGAGCTGCTCTGCTTG |
| ERβ5-ChIP-R | TATAAACCCCAGCAATTGAAA |
Chromatin Immunoprecipitation (ChIP) and Re-ChIP Assays
PC-3-ERβ1 cells were grown in CSS-containing medium supplemented with 10 nm E2. ChIP assays were performed as described previously (34), except for the use of magnetic beads (Dynabeads) for capturing antibodies (Invitrogen). In re-ChIP assays, DNA-containing magnetic beads were incubated in TE buffer with 10 mm dithiothreitol (DTT) to elute the immunoprecipitated DNA after the first ChIP assay. The second ChIP assay was performed with the purified DNA by the second antibody. The ChIP DNA was amplified by PCR with the ABI7900 real time PCR system. The sequences of primers used in the amplification are summarized in Table 2.
Statistical Analysis
The Student's t test of QuickCalcs (GraphPad Software, La Jolla, CA) was used for statistical analysis. p values calculated were two-sided, and values <0.05 were considered statistically significant.
RESULTS
ERβ1 Can Interact with Tip60 in Either the Absence or Presence of Estrogen
To show the physical binding between ERβ1 and Tip60, we performed in vitro coimmunoprecipitation. Tip60 translated in vitro was incubated with ERα or ERβ in the presence of E2 and immunoprecipitated with HA antibody. The translated Tip60 interacted with both ERα (Fig. 1A, lane 2) and ERβ1 (Fig. 1A, lane 6). To confirm the interactions in a cellular system, we cotransformed ERβ1, ERα, or empty vector with Tip60 into yeast cells. We were surprised to find that Tip60 interacted with ERβ1 in the absence or presence of E2, as indicated by the growth of blue yeast colonies (Fig. 1B, left panel). Consistent with previous findings (29, 32), ERα-Tip60 interaction occurred only in the presence of E2 (Fig. 1B, middle panel). To verify the interaction in a mammalian system, we transfected HEK293 cells with Tip60 or empty vector along with ERβ1, followed by immunoprecipitation (Fig. 1C). ERβ1 was coimmunoprecipitated with Tip60 in the absence and presence of E2 (Fig. 1C, lanes 1 and 3). Their interaction was verified by reciprocal coimmunoprecipitation using ERβ1-specific antiserum. Tip60 was coimmunoprecipitated only when cells overexpressed ERβ1 and Tip60 (Fig. 1D, lane 1). However, no Tip60 was coimmunoprecipitated when the cells overexpressed only ERβ1 (Fig. 1D, lane 2) or Tip60 alone (Fig. 1D, lane 3). The interaction was also confirmed in a cell line with a high endogenous level of Tip60. A prostate cancer cell line, PC-3, with ectopic expression of ERβ1 (PC-3-ERβ1) was used (35). Tip60 was coimmunoprecipitated with ERβ1 in the absence or presence of E2 (Fig. 1E, lanes 1 and 3).
FIGURE 1.

ERβ1 can interact with Tip60 in either the absence or presence of estrogen. A, Tip60 interacts with ERβ1 and ERα in vitro. ERβ1, ERα, and HA-tagged Tip60 were translated in vitro and labeled with [35S]methionine. The lysates were mixed and incubated with E2 and then immunoprecipitated (IP) with HA antibody. The immunoprecipitates were resolved by SDS-PAGE and analyzed by autoradiography. B, ERβ1 interacts with Tip60 in yeast cells independent of E2. ERβ1, ERα, or empty vector (pGBKT7) was transformed into yeast with Tip60. The transformed cells were grown on quadruple dropout agar (QDO) containing X-α-galactosidase and DMSO or E2 until the appearance of blue colonies. C, ERβ1 interacts with Tip60 in vivo. HEK293 cells were grown in CSS-containing medium and transfected with ERβ1 and His-tagged Tip60 before the addition of E2. Lysates were precipitated on an Ni-NTA column and immunoblotted (IB) with ERβ1 or Tip60 antibody. The samples were run on the same gel. D, ERβ1-Tip60 interaction was confirmed by reciprocal coimmunoprecipitation. Procedures were similar to those in C, except that lysates were immunoprecipitated with ERβ1 antibody. E, ERβ1 interacts with Tip60 in an E2-independent manner in a hormone-sensitive prostate cancer cell line, PC-3. ERβ1 stably expressed PC-3 cells (PC-3-ERβ1) were grown in CSS-containing medium before the addition of DMSO or E2. Lysates were immunoprecipitated by ERβ1 antibody and immunoblotted with ERβ1 or Tip60 antibody. F, ERβ1 colocalized with Tip60 with or without E2. HEK293 cells were grown in CSS-containing medium transfected with ERβ1 and Tip60 followed by the incubation of DMSO (vehicle) (upper panel) or E2 (lower panel). G, ERβ1 colocalized with Tip60 in PC-3. PC-3-ERβ1 cells were grown in full-serum containing medium. F and G, antibodies to ERβ1 and Tip60 were used for immunostaining, and DAPI was used as the nuclear marker. The images in F and G were captured by a fluorescence microscope. Bar, 20 μm.
We further determined the presence of ERβ1 and Tip60 in the same subcellular compartments. ERβ1 (red) was shown to be colocalized with Tip60 (green) (Fig. 1F) in the nucleus of HEK293 cells in the absence or presence of E2. Colocalization of the two proteins also was observed in PC-3-ERβ1 (Fig. 1G). These data show that ERβ1 physically interacts with Tip60 inside the nucleus in either the absence or presence of E2.
Hinge Domain of ERβ1 Is Responsible for the Interaction with Tip60
We performed interaction analysis of different domains of ERβ1 with Tip60 to further characterize the interaction between ERβ1 and Tip60. Functional domains of ERβ1 include activation function 1 (AF-1), DNA-binding domain (DBD), hinge domain (HD), ligand-binding domain (LBD), and AF-2 domain. We constructed five domain-deleted ERβ1 mutants (ERβ1ΔAF-1, ERβ1ΔAF-1-DBD, ERβ1ΔAF-1-HD, ERβ1ΔLBD-AF-2, and ERβ1ΔAF-2) with the c-Myc tag at the N termini (Fig. 2A). Tip60 together with full-length ERβ1 or its domain-deleted mutants were transfected into HEK293 cells followed by immunoprecipitation. A considerable amount of Tip60 was pulled down simultaneously with the N-terminally deleted mutants ERβ1ΔAF-1 and ERβ1ΔAF-1-DBD (Fig. 2B, upper panel) and the C-terminally deleted mutants ERβ1ΔLBD-AF-2 and ERβ1ΔAF-2 (Fig. 2B, lower panel). However, no Tip60 was coimmunoprecipitated with the ERβ1ΔAF-1-HD construct (Fig. 2B, lower panel). The data show that the hinge domain of ERβ1 is responsible for interacting with Tip60.
FIGURE 2.
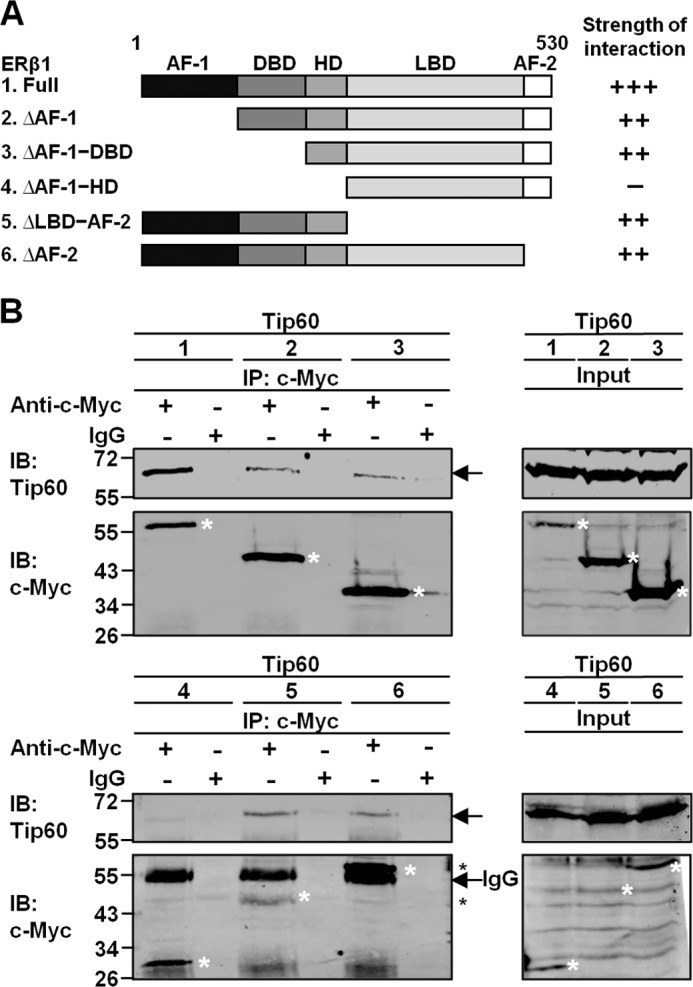
Hinge domain of ERβ1 is responsible for the interaction with Tip60. A, schematic diagram shows the domains of full-length ERβ1 and different domain-deleted constructs. The c-Myc tag was added to the N terminus of each construct. The strength of interaction between different ERβ1 constructs and Tip60 is represented by “+” and “−” signs. “+++” represents the strongest interaction, and “−” represents no interaction. AF-1, activation function 1; AF-2, activation function 2. B, HEK293 cells were grown in CSS-containing medium and transfected with Tip60 and different domain-deleted ERβ1 constructs. Lysates were immunoprecipitated (IP) with c-Myc antibody. Immunoglobulin IgG was used as the negative control. The immunoprecipitates were immunoblotted (IB) with c-Myc or Tip60 antibody. Asterisks denote the positions of ERβ1 and its mutants.
Tip60 Differentially Regulates ERβ1 Transactivation at ERE and AP-1 Sites
ERβ1 is a transcription factor controlling gene expression by either directly binding to consensus DNA sequences or tethering on other transcription factors (2, 5, 36). We were interested in investigating whether Tip60 enhances ERβ1 transactivation and whether the effect is dependent on a cis-regulatory element.
We therefore transfected Tip60, ERβ1, and different luciferase reporter plasmids into HEK293 cells. Tip60 reduced ERβ1 transactivation at the vitellogenin-ERE site in the absence or presence of E2 (Fig. 3A). Moreover, we verified its inhibitory effect at ERE sequences of different ERβ1-target genes. Tip60 inhibited ERβ1 transactivation at C3-ERE (Fig. 3B) and c-Fos-ERE sites (Fig. 3C) in the absence or presence of E2 and also at pS2-ERE (Fig. 3D) and PR-ERE sites (Fig. 3E) in the absence of E2. To determine its mode of regulatory action, we showed that the inhibitory effect of Tip60 on ERβ1 transactivation was concentration-dependent (Fig. 3F). Tip60 decreased constitutive and E2-induced transactivation, and the fold change also was similar in the absence and presence of E2 (Fig. 3F). Apart from directly binding to DNA sequences, ERβ1 can interact with coregulators to tether onto other transcription factors to activate the transcription. Tip60 enhanced ERβ1 transactivation at the AP-1-response element (Fig. 3G). Tip60 increased ERβ1 transactivation more significantly in the absence of E2 than in the presence of E2. The transcriptional regulation by Tip60 required ERβ1 expression because cells transfected with only Tip60 showed very little luciferase activity (data not shown). In contrast, no regulatory effect of Tip60 was observed at NFκB- and Sp1-binding sites (Fig. 3, H and I). Differential regulation of ERβ1 transactivation at vitellogenin ERE and AP-1 sites was also observed in the different prostate cancer cell lines PC-3 (Fig. 3, J and K) and DU-145 (Fig. 3, L and M). These data suggest that Tip60 enhances ERβ1 transactivation at the AP-1 site but reduces the transactivation at different ERE sites.
FIGURE 3.

Tip60 differentially regulates ERβ1 transactivation at ERE and AP-1 sites but has minimal effect on other transcription factor-binding sites. A–E, Tip60 reduces ERβ1 transactivation at various ERE sites. ERβ1 was transfected with GFP or Tip60 together with pCMV-β-gal and vitellogenin ERE (A), C3 ERE (B), c-Fos ERE (C), pS2 ERE (D), or progesterone receptor ERE (E) reporter plasmids into HEK293 cells grown in CSS-containing medium. F, inhibition of ERβ1 transactivation by Tip60 is concentration-dependent. ERβ1 was transfected with different amounts of GFP and Tip60 together with pCMV-β-gal and vitellogenin ERE reporter plasmid. Different ratios of plasmids of Tip60 to GFP were transfected. G–I, Tip60 enhances ERβ1 transactivation at AP-1 sites but has minimal effect on other transcription factor-binding sites. ERβ1 was transfected with GFP or Tip60 together with pCMV-β-gal and reporter plasmids containing the binding site of AP-1 (G), NFκB (H), or Sp1 (I) into HEK293 cells grown in CSS-containing medium. J–M, Tip60 reduces ERβ1 transactivation at ERE site but increases its transactivation at AP-1 site in different PCa cell lines. ERβ1 was transfected with GFP or Tip60 together with pCMV-β-gal and reporter plasmids containing vitellogenin ERE (J and L) or AP-1-binding site (K and M) into PC-3 or DU-145 cells grown in CSS-containing medium. After the transfection, HEK293, PC-3, and DU-145 cells were added with DMSO or E2. Relative luciferase activity was determined and normalized with the β-gal activity. Results were the average of three independent experiments. All data are represented as mean ± S.D. The statistical significance of the difference in luciferase activity between the overexpression of GFP and Tip60 in the presence of DMSO or E2 is shown as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Various Ligands Modulate the Regulatory Effects by Tip60 on ERβ1 Transactivation
Because we found that the regulatory effect of Tip60 at AP-1 site was reduced by E2, we sought to determine whether various ligands could influence its regulation. This was especially relevant because transcriptional activity of ERβ1 responds differently depending on ligands and binding sites (36). We tested five categories of chemicals, estrogens (E2 and DPN), phytoestrogens (GEN, EQ, DAI, and API), selective estrogen receptor modulators (SERMs) (RAL and TAM), antiestrogen (ICI), and an endocrine disruptor (BPA). As with previous findings (8, 36, 37), ERβ1 transactivation at the ERE site was enhanced in the presence of estrogens or phytoestrogens but was suppressed in the presence of TAM, RAL, and ICI (Fig. 4A). In stark contrast, SERMs and antiestrogen stimulated the transactivation at the AP-1 site, whereas estrogens and phytoestrogens inhibited ERβ1 transcriptional activity (Fig. 4B). Moreover, we examined the regulatory effect by Tip60 in the presence of these ligands. The transcriptional inhibition by Tip60 persisted at the ERE site in response to all of the ligands except DPN, GEN, EQ, and BPA (Fig. 4A). In contrast, all the estrogens and phytoestrogens except apigenin down-regulated the enhancement of ERβ1 transactivation by Tip60 at the AP-1 site (Fig. 4B). SERMs could not further up-regulate the effect of Tip60, whereas ICI was the only ligand that increased Tip60 enhancement over that of the control (Fig. 4B). Hence, we suggest that various ligands differentially modulate the regulatory effects by Tip60 at ERE and AP-1 sites.
FIGURE 4.
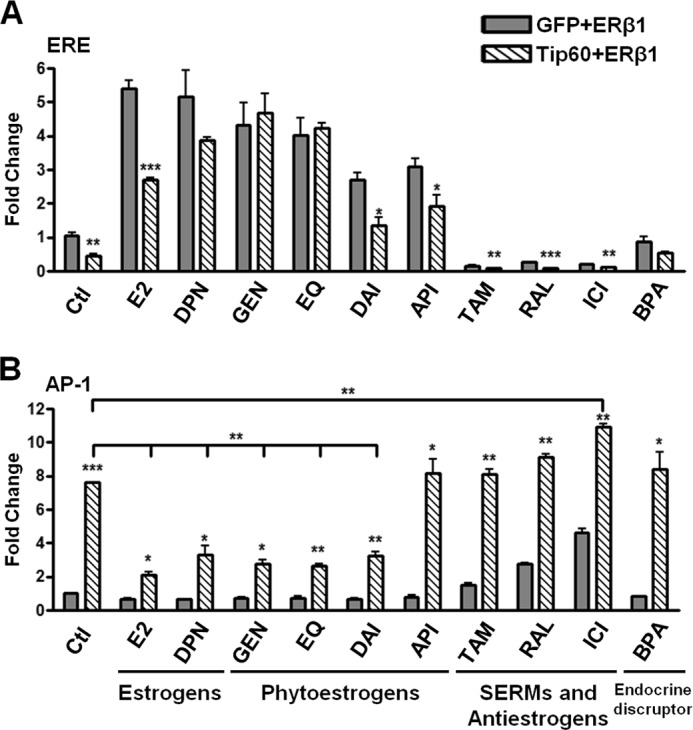
Various ligands modulate Tip60-mediated regulatory effects on ERβ1 transactivation. A and B, ERβ1 was transfected with GFP or Tip60 together with pCMV-β-gal and vitellogenin ERE (A) or AP-1 reporter plasmids (B) into HEK293 cells grown in CSS-containing medium. Various ligands, namely E2 (10 nm), DPN (10 nm), GEN (1 μm), EQ (1 μm), DAI (1 μm), API (100 nm), TAM (1 μm), RAL (1 μm), ICI (1 μm), and BPA (10 nm), were added, respectively, or DMSO was used as the control after transfection for 24 h. Relative luciferase activity was determined as above. The results are the average of at least two independent experiments. All data are represented as mean ± S.D. The statistical significance of the difference in luciferase activity between the overexpression of GFP and Tip60 in the presence of each ligand is shown as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
ERβ1 Cannot Be Acetylated by Tip60 and Preferentially Interacts with Unacetylated Tip60
Tip60 was found to acetylate different transcription factors, such as androgen receptor, p53, c-Myc, and ataxia telangiectasia mutated (ATM) (24–26, 38). To examine whether ERβ1 can be acetylated by Tip60, we performed an in vitro acetylation assay. The structural domains of the Tip60 wild-type (Tip60WT) and the mutation sites of its HAT-defective mutant (Tip60ΔHAT) (Q377E/G380E) are shown in Fig. 5A. His-tagged Tip60WT and Tip60ΔHAT proteins were purified on Ni-NTA columns. Recombinant ERβ1 protein and purified Tip60 were incubated with acetyl-CoA. Consistent with the finding by another group (36), we found that Tip60WT, but not Tip60ΔHAT, was able to auto-acetylate in vitro (Fig. 5B, lanes 1 and 3). However, ERβ1 could not be acetylated by either Tip60WT or Tip60ΔHAT as shown by the absence of signal when pan-acetyl-lysine antibody was used (Fig. 5B, lanes 3 and 4).
FIGURE 5.

ERβ1 cannot be acetylated by Tip60 and preferentially interacts with unacetylated Tip60. A, schematic diagram shows the structural domains of Tip60 and the substitution of amino acids on the HAT-defective mutant (Q377E/G380E) (Tip60ΔHAT). B, ERβ1 is not acetylated by Tip60 in vitro. His-tagged wild-type of Tip60 (Tip60WT) or Tip60ΔHAT was transfected, respectively, into HEK293 cells, and Tip60 proteins were purified on an Ni-NTA column. Recombinant ERβ1 protein and Tip60 were incubated in HAT buffer containing acetyl-CoA. The immunoprecipitates (IP) were immunoblotted (IB) with acetyl-lysine, ERβ1, or Tip60 antibody. Asterisk denotes the nonspecific band that appeared when the blot was immunoblotted with pan-acetyl-lysine antibody. C, ERβ1 is not acetylated by Tip60 in vivo and preferentially interacts with unacetylated Tip60. Tip60WT or HAT was transfected with ERβ1 into HEK293 cells. Lysates were immunoprecipitated with either ERβ1 (left panel) or Tip60 (middle panel) antibody. Immunoglobulin IgG was used as the negative control. The immunoprecipitates were immunoblotted with acetyl-lysine, ERβ1, or Tip60 antibody.
Next, we verified the results in vivo. Either Tip60WT or Tip60ΔHAT was expressed simultaneously with ERβ1. The cells were incubated with TSA and nicotinamide to maximize the level of acetylation. Immunoprecipitation was performed with either ERβ1 or Tip60 antibody to isolate different populations of protein complex. Tip60WT, but not Tip60ΔHAT, was able to auto-acetylate in vivo, as shown in the input lysate (Fig. 5C, right panel). Immunoprecipitation was first performed with ERβ1 antibody to isolate ERβ1 complexes that may or may not contain Tip60. Although Tip60 was coimmunoprecipitated with ERβ1, no acetylation of ERβ1 or Tip60 was detected (Fig. 5C, left panel). Similarly, Tip60 antibody was then used in the pulldown assay to isolate two populations of Tip60 complexes, including the one with or without ERβ1. As expected, Tip60 and ERβ1 were isolated simultaneously. Although there was no acetylation of ERβ1, auto-acetylation of Tip60WT was detected in the immunoprecipitate (Fig. 5C, middle panel), and its unacetylated form may interact preferentially with ERβ1. To conclude, ERβ1 is not acetylated by Tip60 in vitro or in vivo and may preferentially interact with the unacetylated form of Tip60.
HAT Activity of Tip60 Is Not Involved in the Regulation of ERβ1 Transactivation at AP-1 and ERE Sites
Acetylation of androgen receptor (AR) by Tip60 is essential for up-regulating the transactivation of AR at AR-response elements (24). The inability of Tip60 to acetylate ERβ1 infers that its HAT activity may not be important for regulating ERβ1 activity. Luciferase reporter assays were performed to determine ERβ1 activity with the overexpression of Tip60WT and Tip60ΔHAT. Western blot analysis showed that their expression was similar (Fig. 6A). At the vitellogenin-ERE site, the two Tip60 proteins were equally effective in reducing ERβ1 transactivation (Fig. 6B). However, Tip60ΔHAT enhanced ERβ1 transactivation to a greater extent than Tip60WT did at the AP-1 site (Fig. 6C). To further determine the significance of the HAT activity of Tip60 to the transcriptional activity of ERβ1, we used a HAT inhibitor, anacardic acid, which inhibits Tip60-dependent acetylation (39). Similar to the results shown in Fig. 6, B and C, enhancement of ERβ1 transactivation by Tip60 was up-regulated at the AP-1 site in the presence of anacardic acid (Fig. 6E), but no change was observed at the ERE site (Fig. 6D). The results suggest that HAT activity of Tip60 is not required to regulate ERβ1 transactivation at ERE or AP-1 site.
FIGURE 6.
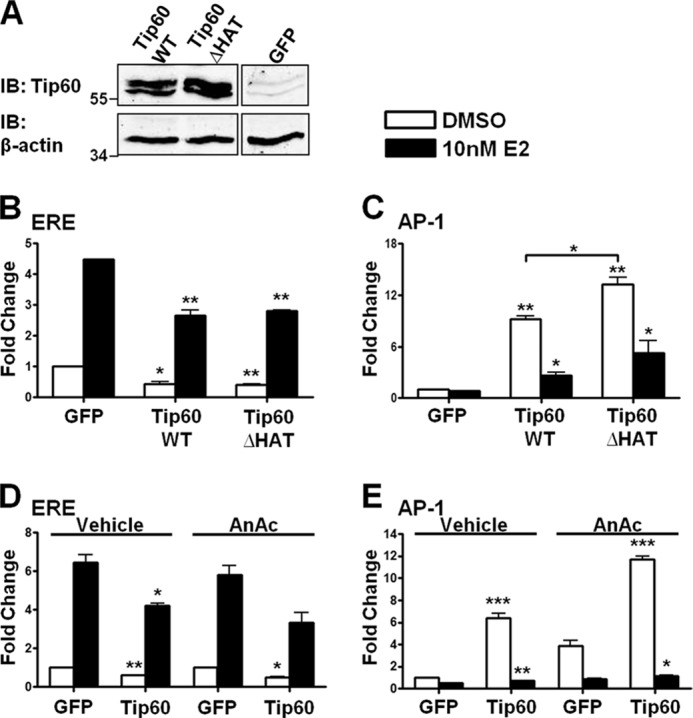
HAT activity of Tip60 is not necessary for regulation of the ERβ1 transactivation at AP-1 and ERE sites. A, expression of Tip60ΔHAT was similar to that of Tip60WT. Lysates were extracted and immunoblotted (IB) with Tip60 antibody. β-Actin was used as the loading control. B and C, HAT activity of Tip60 is not necessary for the regulation of ERβ1 transactivation at AP-1 and ERE sites. GFP, Tip60WT, or Tip60ΔHAT was transfected, respectively, with ERβ1, pCMV-β-gal (B), AP-1(C), or vitellogenin-ERE reporter plasmids into HEK293 cells before the addition of E2. D and E, GFP or Tip60 was transfected, respectively, with ERβ1, pCMV-β-gal (D), AP-1 (E), or vitellogenin-ERE reporters into HEK293 cells. After the transfection, DMSO or E2 together with ethanol (vehicle) or anacardic acid (AnAc) was added as indicated. B–D, relative luciferase activity was determined as in Fig. 3. Results are the average of three independent experiments. Data are represented as mean ± S.D. The statistical significance of the difference in luciferase activity between the overexpression of GFP and Tip60 in the presence of DMSO or E2 is shown as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Tip60 Interacts with GRIP1 to Enhance ERβ1 Transactivation at the AP-1 Site Synergistically
The p160 SRC family consists of three homologous members, SRC1, GRIP1, and SRC3 (40–42). Of these, SRC1 and GRIP1 are coactivators of ERs at the AP-1 and ERE sites (4, 43). Because Tip60 enhanced ERβ1 transactivation at the AP-1 site but diminished transactivation at different ERE sites, we investigated whether Tip60 has any combinatorial effect with p160 coactivators. We overexpressed different combinations of Tip60, SRC1, and GRIP1 together with ERβ1 and determined the regulation of ERβ1 transactivation by these proteins at the ERE and AP-1 sites. SRC1 enhanced ERβ1 transactivation in the absence of E2, whereas Tip60 and GRIP1 reduced ERβ1 transactivation in the absence or presence of E2. The effect of inhibition persisted when Tip60 and GRIP1 were overexpressed simultaneously (Fig. 7A). At the AP-1 site, all three coregulators were able to enhance ERβ1 transactivation with or without E2 (Fig. 7B). In the absence of E2, coexpression of Tip60 and GRIP1 had the strongest stimulatory effect on the transactivation. Interestingly, overexpression of SRC1 abolished the synergistic effects of Tip60 and GRIP1 (Fig. 7B). To further investigate the synergistic effect of Tip60 and GRIP1 on ERβ1 transactivation at the AP-1 site, we performed luciferase assays with different ratios of GRIP1 and Tip60 plasmids. Consistent with the results in Fig. 7B, coexpression of GRIP1 and Tip60 resulted in a greater enhancement of ERβ1 transactivation than expression of GRIP1 alone, whereas a 1:1 ratio of GRIP1 and Tip60 plasmids resulted in the greatest enhancement at the AP-1 site (Fig. 7C). Next, an immunoprecipitation experiment was used to determine whether ERβ1, Tip60, and the two p160 coactivators are involved in a transcriptional complex. Tip60 interacted with ERβ1, GRIP1, and SRC1 (Fig. 7D). To conclude, ERβ1, Tip60, GRIP1, and SRC1 are able to form a multiprotein complex, whereas Tip60 and GRIP1 synergistically enhance ERβ1 transactivation at the AP-1 site.
FIGURE 7.

Tip60 interacts with GRIP1 to enhance ERβ1 transactivation at the AP-1 site synergistically. A and B, Tip60 and GRIP1 exert a synergistic effect on ERβ1 transactivation at the AP-1 site. Different combinations of GFP, Tip60, GRIP1, and SRC1 were transfected with ERβ1, pCMV-β-gal, vitellogenin-ERE (A) or AP-1 reporter plasmids (B) as indicated. After the transfection, DMSO or 10 nm E2 was added as indicated. C, synergistic effect of Tip60 and GRIP1 on the ERβ1 transactivation at the AP-1 site is concentration-dependent. GFP or different ratios of plasmids of Tip60 to GRIP1 were transfected. DMSO was added after the transfection. Relative luciferase activity was determined as in Fig. 3. Results are the average of three independent experiments. Data are presented as mean ± S.D. The statistical significance of the difference in luciferase activity between overexpressing Tip60, GRIP1, and GFP is shown as *, p < 0.05; **, p < 0.01; ***, p < 0.001. D, Tip60 forms a multiprotein complex with p160 coactivators and ERβ1. HEK293 cells were transfected with Tip60, ERβ1, SRC1, and GRIP1 and grown in CSS-containing medium. Lysates were immunoprecipitated (IP) with Tip60 antibody. Immunoglobulin IgG was used as the negative control. The immunoprecipitates were immunoblotted (IB) with Tip60, ERβ1, GRIP1, or SRC1 antibody as indicated.
Tip60 Differentially Regulates ERβ1 Target Genes by Modulating ERβ1 Binding to the cis-Regulatory Regions Possessing the ERE or AP-1 Site
In our study, Tip60 either enhanced or reduced ERβ1 transactivation at the AP-1 or ERE site. To investigate whether ERβ1 target genes are differentially regulated by Tip60, we determined their gene expressions in ERβ1 or LacZ stably expressed PC-3 cells (PC-3-ERβ1/PC-3-LacZ) after the knockdown of Tip60. The ectopic expression of ERβ1 and the efficiency of Tip60 knockdown were confirmed by quantitative RT-PCR (Fig. 8, A and B) and Western blotting (data not shown). We found that the expressions of CXCL12 and cyclin D2 were drastically increased in PC-3-ERβ1 compared with the control (PC-3-LacZ) (Fig. 8, C and D). Moreover, their expressions were differentially regulated with the knockdown of Tip60 in PC-3-ERβ1 cells. Expression of CXCL12 was further up-regulated (Fig. 8C), whereas that of cyclin D2 was down-regulated after Tip60 depletion (Fig. 8D). The cis-regulatory sequence of the CXCL12 gene was found to have an ERE site (44), and sequence analysis revealed a predicted AP-1-binding site at the upstream region of the cyclin D2 gene (data not shown). In ChIP assays, ERβ1 and Tip60 were significantly recruited to the respective investigated regions (Fig. 8, E and F). Moreover, the co-occupancy of ERβ1 and Tip60 on the respective cis-regulatory regions of CXCL12 and cyclin D2 was confirmed in the re-ChIP assay (Fig. 8G). Similar results were observed in the reciprocal re-ChIP assay (data not shown). Next, we investigated the molecular mechanism of differential regulation of ERβ1 target genes by Tip60. Upon the depletion of Tip60, the recruitment of ERβ1 to the cis-regulatory region of CXCL12 was significantly enhanced, whereas the recruitment of ERβ1 to the investigated region of cyclin D2 was decreased (Fig. 8H). Collectively, our results showed that Tip60 differentially regulates the expression of ERβ1 target genes by modulating the binding of ERβ1 to their respective cis-regulatory regions.
FIGURE 8.

Tip60 differentially regulates ERβ1 target genes possessing ERE or AP-1 sites at their cis-regulatory regions in PC-3 cells. A and B, expression of ERβ1 and Tip60 in ERβ1 and LacZ stably expressed PC-3 cells upon the knockdown of Tip60 was determined. PC-3-LacZ/-ERβ1 cells were grown in CSS-containing medium and transfected with nontargeting control siRNA (siNT) or siRNAs specific to Tip60 (siTip). E2 was added after 24 h. Expression of ERβ1 (A) and Tip60 (B) was determined by quantitative RT-PCR. Human GAPDH was used as the housekeeping gene. C and D, Tip60 differentially regulates ERβ1 target genes. PC-3-LacZ/-ERβ1 cells were treated as described in A and B. Expression of CXCL12 (C) and cyclin D2 (D) was determined by quantitative RT-PCR. The results are the average of three independent experiments. All data are represented as mean ± S.D. The statistical significance of the difference in gene expression between different treatments is shown as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001. E–G, ERβ1 and Tip60 are both recruited to the cis-regulatory regions of CXCL12 and cyclin D2. PC-3-ERβ1 cells were grown in CSS-containing medium added with E2. ChIP assays were performed with ERβ1 (E) or Tip60 antibody (F). G, re-ChIP assay was performed with Tip60 antibody followed by the second immunoprecipitation with ERβ1 antibody. The ChIP DNA was amplified by real time PCR for the target regions containing an ERE site of CXCL12 or an AP-1 site of cyclin D2. The genomic region of ERβ isoform 5 (ERβ5) containing neither an ERE nor an AP-1 site was used as the negative control. The fold enrichment of recruitment of ERβ1 and/or Tip60 at the target regions is relative to respective IgG controls. The results are the average of two independent experiments. All data are represented as mean ± S.D. The statistical significance of the difference in the recruitment between ERβ1 (and/or Tip60) and IgG is shown as follows: *, p < 0.05. H, Tip60 differentially regulates the recruitment of ERβ1 to the cis-regulatory regions of CXCL12 and cyclin D2. PC-3-ERβ1 cells were grown in CSS-containing medium added with E2 and transfected with siRNAs (siNT or siTip) for 48 h. ChIP assays were performed with ERβ1 antibody. The procedures of the amplification of ChIP DNA were similar to those described in E–G. The results are the average of two independent experiments. Data are represented as mean ± S.D. The statistical significance of the difference in the ERβ1 recruitment with or without the knockdown of Tip60 is shown as follows: *, p < 0.05.
DISCUSSION
Estrogen signaling is mediated primarily by ERα and ERβ1, whereas ERβ1 is able to activate a distinct set of target genes and also to antagonize ERα transactivation (45–49). Although ERs share many common coregulators, the differential interaction between the coregulatory proteins and ERs may be responsible for their distinct functions (8). In this study, Tip60 was found to interact with ERβ1 in the absence or the presence of E2. Tip60 either enhances or inhibits ERβ1 transactivation, depending on the cis-regulatory sites. Moreover, Tip60 and GRIP1 enhance the transactivation at the AP-1 site synergistically. We also showed that ERβ1 is not acetylated by Tip60 and thus that the regulation of ERβ1 activity by Tip60 is independent of its HAT activity. In addition, Tip60 is able to differentially control the endogenous expression of ERβ1 target genes possessing the ERE or AP-1 site by modulating ERβ1 binding to the respective cis-regulatory regions. On the basis of these data, we suggest that ERβ1 transactivation is differentially regulated by Tip60 in a regulatory element-dependent manner.
Tip60 is an interacting partner of some hormone receptors, including ERs, AR, and PR. Their interactions were shown to require the presence of respective agonists (32). In this study, we found that the binding of Tip60 to ERβ1 does not require ligands and that the strength of the interaction is similar in the absence or presence of E2. The discrepancy between our finding and that from another group may be due to our use of different ERβ1 sequences and interaction assays. Gaughan et al. (32) used a construct containing only LBD of ERβ1 in the mammalian two-hybrid assay. We used the full-length ERβ1, which is more biologically relevant in terms of protein folding, to show the interaction in yeast two-hybrid assays, in vitro and in vivo coimmunoprecipitation, and subcellular localization studies in different cell lines. It is not uncommon for ligand-independent interactions to occur between ERβ1 and coregulators. For example, phosphorylation of ERβ1 leads to ligand-independent recruitment of SRC1 (48), and GRIP1 is also recruited by unliganded ERβ1 (8, 10, 20). Both coactivators stimulated unliganded ERβ1 transactivation (8, 10). Our data suggest that Tip60 interacts with ERβ1 regardless of E2 presence.
The interaction of Tip60 with LBD of ERα in a ligand-dependent manner is well documented (29, 30, 32). The distinct mechanisms of recruiting Tip60 by ERα and ERβ1 imply that they may have different domains interacting with Tip60. We performed domain deletion of ERβ1 followed by immunoprecipitation to show that the hinge domain of ERβ1 is the interacting region. Although ERs interact with the most coactivators and corepressors at either or both N and C termini (50), they also bind to some coregulators at the hinge domain. L7/SPA interacts with the hinge domain of ERα and enhances transactivation of antagonist-occupied ERα at the ERE site (51). ERα also binds to PGC-1 at its hinge domain in a ligand-independent manner (52). Although the hinge domain of ERs is not as well characterized, it has been shown to affect protein degradation and activity of ERβ1 (53, 54), ERα tethered-mediated AP-1 transactivation (55), and the functional synergy between AF-1 and AF-2 of ERs (56). Because AF-1 and AF-2 domains are responsible for E2-independent and E2-dependent activation of the transactivation of ERs (50), we speculate that the atypical interaction interface between ERβ1 and Tip60 at the hinge domain may contribute to the unique regulation of ERβ1 activity by Tip60.
Tip60 functions as a coregulator of many transcription factors (57). Hence, we determined its role in the regulation of ERβ1 transactivation by the luciferase assay and used reporter constructs with different cis-regulatory sequences of the target genes of ERβ1. Tip60 reduced ERβ1 transactivation at different ERE sequences, such as vitellogenin-, C3-, c-Fos-, pS2- and PR-EREs. Moreover, the inhibitory action of ERβ1 transactivation by Tip60 is concentration-dependent but E2-independent. Our results imply that Tip60 can inhibit transcription of certain ERβ1-regulated genes possessing ERE sites. In contrast, Tip60 increased the expression of some estrogen-regulated ERα target genes containing EREs (29, 30). Because ERβ1 antagonizes ERα-dependent transcription through hetero-dimerization (50), Tip60 may be a key factor in determining the antagonism between ERs. ERβ1 also interacts with other transcription factors to mediate the transcription through tethering. We showed that Tip60 did not regulate ERβ1 transactivation at either the NFκB or the Sp1 site but that it drastically increased the transactivation at the AP-1 site. Moreover, the enhancement by Tip60 was more drastic in the absence of E2. It is not surprising for a coregulator to show dual regulation of the activity of transcription factors. GRIP1 acts as a coactivator of ERα at ERE and AP-1 sites (4, 8) but inhibits the activity of E2-bound ERα, which tethers on c-Jun and NFκB at TNFα promoter (58). In addition, GRIP1 is either a coactivator or a corepressor of glucocorticoid receptor in a hormone-response element-dependent manner (59). Our study not only shows that the regulation of ERβ1 transactivation by Tip60 occurs in an E2-independent manner but also provides evidence that it can enhance or inhibit the transactivation at the AP-1-response element or ERE, respectively.
The modulation by ligands of ERβ1 signaling at different response elements has been well documented (36). We extensively investigated the effects of various steroidal compounds on the transcriptional regulation by Tip60. Consistent with the previous findings (36, 60, 61), we found that estrogenic compounds (E2 and DPN) and phytoestrogens (GEN, EQ, DAI, and API) up-regulated ERβ1 transactivation at ERE, whereas SERMs (TAM and RAL) and antiestrogen (ICI) did the opposite. Surprisingly, DPN, GEN, and EQ abolished Tip60-mediated inhibition at the ERE site. Moreover, all estrogenic chemicals except apigenin significantly inhibited enhancement by Tip60 at the AP-1 site. The discrepancy may be due to differential conformational changes of ERβ1 through binding to different estrogenic chemicals (62, 63) and thus affect the formation of the ERβ1 transcriptional complex (62). Perhaps the binding of these compounds triggers the recruitment of other coactivators to counteract the Tip60-mediated inhibition (7). For example, GEN can recruit SRC1 isoforms to ERβ1 (64, 65). Moreover, all estrogenic chemicals except apigenin significantly inhibited the enhancement by Tip60 at the AP-1 site. Although Fujimoto et al. (37) suggested that estrogens and phytoestrogens do not exert any regulatory effect on ERβ1-mediated AP-1 transactivation, previous findings and this study have clearly shown that estrogens or phytoestrogens repress the transactivation at the AP-1 site (36, 66). It is tempting to speculate that these compounds reduce the potency of recruitment of coactivators, such as Tip60, by ERβ1 at AP-1 site. In contrast, ICI and SERMs were agonists of ERβ1-mediated AP-1 transactivation, but SERMs did not further up-regulate the enhancement by Tip60 as compared with the control. We suggest that SERMs cannot improve the potency of Tip60 recruitment by ERβ1. Another possible explanation may be that the binding of either Tip60 or SERMs causes a similar conformational change in ERβ1 that is favorable to tethering on the AP-1 site (67–69). Tip60 and SERMs are thus redundant to the enhancement of ERβ1 transactivation. To conclude, we showed that the differential regulation of ERβ1 transactivation by Tip60 at ERE and AP-1 sites is controlled through binding to different ligands.
Tip60 enhances the activities of certain transcription factors through acetylation (57). Thus, we sought to determine whether its regulation of ERβ1 activity is mediated through acetylation. We used different acetylation assays to illustrate that Tip60 is incapable of acetylating ERβ1. This is consistent with studies of other coregulators of ERβ1 that possess HAT activity, but none of them was found to acetylate ERβ1 (9, 13, 16, 45, 48). Moreover, acetylation of nuclear receptors is assumed to occur at a conserved motif “(K/R)XKK” (13), which is absent in ERβ1 (data not shown). These findings suggest that ERβ1 may not be post-translationally modified through acetylation.
In addition to acetylating its interacting partners, Tip60 can auto-acetylate to regulate its activity (71, 72). In our in vivo acetylation assays, acetylation of Tip60 was detected only in the immunoprecipitation that used Tip60 antibody but not ERβ1 antibody, revealing that those Tip60 proteins in the ERβ1-Tip60 complex are probably unacetylated. The result implies that ERβ1 may preferentially interact with unacetylated Tip60, perhaps because auto-acetylation modifies the structure of Tip60 (71). Our study verified that HAT activity of Tip60 does not increase ERβ1 transactivation. In contrast, Tip60ΔHAT did not reduce but enhanced ERβ1 activity at the AP-1 site. The result was confirmed with the use of anacardic acid, which inhibits the HAT activity of Tip60 (37). The observation may be explained by the increased amount of unacetylated Tip60 that binds to ERβ1. In fact, HAT activity of Tip60 is not essential for the regulation of the activity of some transcription factors, such as CREB, STAT3, and PGC-1α (27, 28, 73). Our data indicate that ERβ1 transactivation is not regulated through HAT activity of Tip60. Furthermore, the receptor appears to interact preferentially with unacetylated Tip60.
In this study, we found that ERβ1 activity was enhanced by Tip60 at the AP-1 site. The ERβ1-mediated transactivation requires the recruitment of CBP/p300 and p160 coactivators at the AP-1-response element (4), where ERβ1 interacts primarily with p160 coactivators (4, 43, 74, 75). These observations urged us to investigate whether Tip60 interacts with p160 coactivators to regulate ERβ1 transactivation. We found that Tip60 interacted with SRC1 and GRIP1, although it only enhanced ERβ1 activity synergistically with GRIP1 at the AP-1 site. Moreover, expression of different amounts of GRIP1 and Tip60 always resulted in a greater enhancement of ERβ1 transactivation compared with expression of GRIP1 alone, revealing that they simultaneously act as coactivators of ERβ1 at the AP-1 site. It is interesting that SRC1 was not synergistic with the other two coregulators, implying that it may have other mechanisms regulating ERβ1 transactivation. Because ERβ1 interacts with Tip60 at its hinge domain and GRIP1 binds to AF-1 and AF-2 domains of the receptor (43), we therefore hypothesize that Tip60 and GRIP1 cooperate to modify the conformation of ERβ1, permitting more efficient tethering on the AP-1 site.
In addition, we showed that Tip60 modulates ERβ1 regulation of endogenous gene expression in prostate cancer cells. In our search for ERβ1-regulated genes (10, 11), CXCL12 (76) and cyclin D2 (previously unknown) were the only two that we identified in this study that were regulated by both ERβ1 and Tip60. We found that upon the knockdown of Tip60, the expression of CXCL12 increased and that of cyclin D2 decreased. Interestingly, the promoter region of CXCL12 contains multiple EREs (44, 76, 77) and that of cyclin D2 harbors two AP-1 sites based on bioinformatics. In the ChIP and re-ChIP assays, ERβ1 and Tip60 were shown to co-occupy the investigated regions. Moreover, the depletion of Tip60 appeared to increase ERβ1 binding to the promoter of CXCL12 and decrease its recruitment to the promoter of cyclin D2. These results raise the possibility that Tip60 promotes the recruitment of ERβ1 to AP-1 site but reduces its ERE binding, a mechanism that likely contributes to the differential regulation of ERβ1-targeted gene expression.
In conclusion, we showed that Tip60 modulates ERβ1 action in a regulatory element-dependent manner as exemplified by its opposing roles on ERβ1 transactivation at the ERE and AP-1 sites. Furthermore, its coregulatory action on ERβ1 appears to be E2-independent at both cis-elements, unlike its action on ERα. Contrary to common belief, Tip60 action is not mediated by its HAT activity. Our data also suggest that the interaction between Tip60 and GRIP1 synergistically enhances ERβ1 tethering on the AP-1 site. Moreover, Tip60 can modulate the recruitment of ERβ1 to the promoters of CXCL12 and cyclin D2, harboring the ERE and AP-1 site, respectively. Collectively, these data put Tip60 into the category of a multifaceted coregulator in the ERβ1 context, similar to GRIP1 in the regulation of the activities of ERα and glucocorticoid receptor (4, 8, 58, 59).
Acknowledgments
We thank Drs. Carolyn Klinge, Craig Jordan, Francis Chan, and Nancy Weigel for providing different plasmids; Dr. Xiaoting Zhang for critical reading of the manuscript, and Nancy Voynow for professional editing of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA015776, R01DK061084, R01ES015584, U01ES019480, U01ES020988, and P30ES006096 from NIEHS (to S. M. H.). This work was also supported by Veterans Affairs Grant I01BX000675 (to S. M. H.) and Congressionally Directed Medical Research Program Department of Defense Grants PC094619 (to P. T.) and BC094017 (to M. T. L.).
- ER
- estrogen receptor
- AP-1
- activation protein 1
- Sp1
- specificity protein 1
- NFκB
- nuclear factor κ-light-chain-enhancer of activated B cell
- ERE
- estrogen-response element
- E2
- estradiol
- DPN
- diarylpropionitrile
- GEN
- genistein
- EQ
- equol
- DAI
- daizein
- API
- apigenin
- TAM
- 4-OH-tamoxifen
- RAL
- raloxifene
- BPA
- bisphenol A
- TSA
- trichostatin A
- ICI
- ICI 182,780
- Ni-NTA
- nickel-nitrilotriacetic acid
- HAT
- histone acetyltransferase
- CSS
- charcoal-stripped serum
- HD
- hinge domain
- LBD
- ligand-binding domain
- DBD
- DNA-binding domain
- PR
- progesterone receptor
- SRC
- steroid receptor coactivator
- AR
- androgen receptor
- SERM
- selective estrogen receptor modulator.
REFERENCES
- 1. Ho S. M., Lee M. T., Lam H. M., Leung Y. K. (2011) Estrogens and prostate cancer: etiology, mediators, prevention, and management. Endocrinol. Metab Clin. North Am. 40, 591–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carroll J. S., Brown M. (2006) Estrogen receptor target gene: an evolving concept. Mol. Endocrinol. 20, 1707–1714 [DOI] [PubMed] [Google Scholar]
- 3. Klinge C. M. (2001) Estrogen receptor interaction with estrogen-response elements. Nucleic Acids Res. 29, 2905–2919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kushner P. J., Agard D. A., Greene G. L., Scanlan T. S., Shiau A. K., Uht R. M., Webb P. (2000) Estrogen receptor pathways to AP-1. J. Steroid Biochem. Mol. Biol. 74, 311–317 [DOI] [PubMed] [Google Scholar]
- 5. Saville B., Wormke M., Wang F., Nguyen T., Enmark E., Kuiper G., Gustafsson J. A., Safe S. (2000) Ligand-, cell-, and estrogen receptor subtype (α/β)-dependent activation at GC-rich (Sp1) promoter elements. J. Biol. Chem. 275, 5379–5387 [DOI] [PubMed] [Google Scholar]
- 6. Heldring N., Pike A., Andersson S., Matthews J., Cheng G., Hartman J., Tujague M., Ström A., Treuter E., Warner M., Gustafsson J. A. (2007) Estrogen receptors: how do they signal and what are their targets. Physiol. Rev. 87, 905–931 [DOI] [PubMed] [Google Scholar]
- 7. Jeyakumar M., Carlson K. E., Gunther J. R., Katzenellenbogen J. A. (2011) Exploration of dimensions of estrogen potency: parsing ligand binding and coactivator binding affinities. J. Biol. Chem. 286, 12971–12982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klinge C. M., Jernigan S. C., Mattingly K. A., Risinger K. E., Zhang J. (2004) Estrogen-response element-dependent regulation of transcriptional activation of estrogen receptors α and β by coactivators and corepressors. J. Mol. Endocrinol. 33, 387–410 [DOI] [PubMed] [Google Scholar]
- 9. Wong C. W., Komm B., Cheskis B. J. (2001) Structure-function evaluation of ER α and β interplay with SRC family coactivators. ER selective ligands. Biochemistry 40, 6756–6765 [DOI] [PubMed] [Google Scholar]
- 10. Vivar O. I., Zhao X., Saunier E. F., Griffin C., Mayba O. S., Tagliaferri M., Cohen I., Speed T. P., Leitman D. C. (2010) Estrogen receptor β binds to and regulates three distinct classes of target genes. J. Biol. Chem. 285, 22059–22066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhao C., Gao H., Liu Y., Papoutsi Z., Jaffrey S., Gustafsson J. A., Dahlman-Wright K. (2010) Genome-wide mapping of estrogen receptor-β-binding regions reveals extensive cross-talk with transcription factor activator protein-1. Cancer Res. 70, 5174–5183 [DOI] [PubMed] [Google Scholar]
- 12. Lonard D. M., O'malley B. W. (2007) Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol. Cell 27, 691–700 [DOI] [PubMed] [Google Scholar]
- 13. Wang C., Tian L., Popov V. M., Pestell R. G. (2011) Acetylation and nuclear receptor action. J. Steroid Biochem. Mol. Biol. 123, 91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu R. C., Smith C. L., O'Malley B. W. (2005) Transcriptional regulation by steroid receptor coactivator phosphorylation. Endocr. Rev. 26, 393–399 [DOI] [PubMed] [Google Scholar]
- 15. Fu M., Wang C., Zhang X., Pestell R. G. (2004) Acetylation of nuclear receptors in cellular growth and apoptosis. Biochem. Pharmacol. 68, 1199–1208 [DOI] [PubMed] [Google Scholar]
- 16. Kim M. Y., Woo E. M., Chong Y. T., Homenko D. R., Kraus W. L. (2006) Acetylation of estrogen receptor α by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol. Endocrinol. 20, 1479–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang C., Fu M., Angeletti R. H., Siconolfi-Baez L., Reutens A. T., Albanese C., Lisanti M. P., Katzenellenbogen B. S., Kato S., Hopp T., Fuqua S. A., Lopez G. N., Kushner P. J., Pestell R. G. (2001) Direct acetylation of the estrogen receptor α hinge region by p300 regulates transactivation and hormone sensitivity. J. Biol. Chem. 276, 18375–18383 [DOI] [PubMed] [Google Scholar]
- 18. Cui Y., Zhang M., Pestell R., Curran E. M., Welshons W. V., Fuqua S. A. (2004) Phosphorylation of estrogen receptor α blocks its acetylation and regulates estrogen sensitivity. Cancer Res. 64, 9199–9208 [DOI] [PubMed] [Google Scholar]
- 19. Ma Y., Fan S., Hu C., Meng Q., Fuqua S. A., Pestell R. G., Tomita Y. A., Rosen E. M. (2010) BRCA1 regulates acetylation and ubiquitination of estrogen receptor-α. Mol. Endocrinol. 24, 76–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Webb P., Nguyen P., Valentine C., Lopez G. N., Kwok G. R., McInerney E., Katzenellenbogen B. S., Enmark E., Gustafsson J. A., Nilsson S., Kushner P. J. (1999) The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol. Endocrinol. 13, 1672–1685 [DOI] [PubMed] [Google Scholar]
- 21. Utley R. T., Côté J. (2003) The MYST family of histone acetyltransferases. Curr. Top. Microbiol. Immunol. 274, 203–236 [DOI] [PubMed] [Google Scholar]
- 22. Baek S. H., Ohgi K. A., Rose D. W., Koo E. H., Glass C. K., Rosenfeld M. G. (2002) Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-κB and β-amyloid precursor protein. Cell 110, 55–67 [DOI] [PubMed] [Google Scholar]
- 23. Brady M. E., Ozanne D. M., Gaughan L., Waite I., Cook S., Neal D. E., Robson C. N. (1999) Tip60 is a nuclear hormone receptor coactivator. J. Biol. Chem. 274, 17599–17604 [DOI] [PubMed] [Google Scholar]
- 24. Gaughan L., Logan I. R., Cook S., Neal D. E., Robson C. N. (2002) Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J. Biol. Chem. 277, 25904–25913 [DOI] [PubMed] [Google Scholar]
- 25. Legube G., Linares L. K., Tyteca S., Caron C., Scheffner M., Chevillard-Briet M., Trouche D. (2004) Role of the histone acetyl transferase Tip60 in the p53 pathway. J. Biol. Chem. 279, 44825–44833 [DOI] [PubMed] [Google Scholar]
- 26. Patel J. H., Du Y., Ard P. G., Phillips C., Carella B., Chen C. J., Rakowski C., Chatterjee C., Lieberman P. M., Lane W. S., Blobel G. A., McMahon S. B. (2004) The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 24, 10826–10834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gavaravarapu S., Kamine J. (2000) Tip60 inhibits activation of CREB protein by protein kinase A. Biochem. Biophys. Res. Commun. 269, 758–766 [DOI] [PubMed] [Google Scholar]
- 28. Xiao H., Chung J., Kao H. Y., Yang Y. C. (2003) Tip60 is a co-repressor for STAT3. J. Biol. Chem. 278, 11197–11204 [DOI] [PubMed] [Google Scholar]
- 29. Jeong K. W., Kim K., Situ A. J., Ulmer T. S., An W., Stallcup M. R. (2011) Recognition of enhancer element-specific histone methylation by TIP60 in transcriptional activation. Nat. Struct. Mol. Biol. 18, 1358–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nakajima A., Maruyama S., Bohgaki M., Miyajima N., Tsukiyama T., Sakuragi N., Hatakeyama S. (2007) Ligand-dependent transcription of estrogen receptor α is mediated by the ubiquitin ligase EFP. Biochem. Biophys. Res. Commun. 357, 245–251 [DOI] [PubMed] [Google Scholar]
- 31. Won Jeong K., Chodankar R., Purcell D. J., Bittencourt D., Stallcup M. R. (2012) Gene-specific patterns of coregulator requirements by estrogen receptor-α in breast cancer cells. Mol. Endocrinol. 26, 955–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gaughan L., Brady M. E., Cook S., Neal D. E., Robson C. N. (2001) Tip60 is a co-activator specific for class I nuclear hormone receptors. J. Biol. Chem. 276, 46841–46848 [DOI] [PubMed] [Google Scholar]
- 33. Leung Y. K., Lam H. M., Wu S., Song D., Levin L., Cheng L., Wu C. L., Ho S. M. (2010) Estrogen receptor β2 and β5 are associated with poor prognosis in prostate cancer and promote cancer cell migration and invasion. Endocr. Relat. Cancer 17, 675–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leung Y. K., Gao Y., Lau K. M., Zhang X., Ho S. M. (2006) ICI 182,780-regulated gene expression in DU145 prostate cancer cells is mediated by estrogen receptor-β/NFκB crosstalk. Neoplasia 8, 242–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shiota M., Yokomizo A., Masubuchi D., Tada Y., Inokuchi J., Eto M., Uchiumi T., Fujimoto N., Naito S. (2010) Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate 70, 540–554 [DOI] [PubMed] [Google Scholar]
- 36. Paech K., Webb P., Kuiper G. G., Nilsson S., Gustafsson J., Kushner P. J., Scanlan T. S. (1997) Differential ligand activation of estrogen receptors ERα and ERβ at AP1 sites. Science 277, 1508–1510 [DOI] [PubMed] [Google Scholar]
- 37. Fujimoto N., Honda H., Kitamura S. (2004) Effects of environmental estrogenic chemicals on AP1-mediated transcription with estrogen receptors α and β. J. Steroid Biochem. Mol. Biol. 88, 53–59 [DOI] [PubMed] [Google Scholar]
- 38. Sun Y., Jiang X., Chen S., Fernandes N., Price B. D. (2005) A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. U.S.A. 102, 13182–13187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun Y., Jiang X., Chen S., Price B. D. (2006) Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS Lett. 580, 4353–4356 [DOI] [PubMed] [Google Scholar]
- 40. Lonard D. M., O'Malley B. W. (2005) Expanding functional diversity of the coactivators. Trends Biochem. Sci. 30, 126–132 [DOI] [PubMed] [Google Scholar]
- 41. Puigserver P., Adelmant G., Wu Z., Fan M., Xu J., O'Malley B., Spiegelman B. M. (1999) Activation of PPARγ coactivator-1 through transcription factor docking. Science 286, 1368–1371 [DOI] [PubMed] [Google Scholar]
- 42. Xu J., Wu R. C., O'Malley B. W. (2009) Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 9, 615–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Webb P., Nguyen P., Shinsako J., Anderson C., Feng W., Nguyen M. P., Chen D., Huang S. M., Subramanian S., McKinerney E., Katzenellenbogen B. S., Stallcup M. R., Kushner P. J. (1998) Estrogen receptor activation function 1 works by binding p160 coactivator proteins. Mol. Endocrinol. 12, 1605–1618 [DOI] [PubMed] [Google Scholar]
- 44. Holmes K. A., Song J. S., Liu X. S., Brown M., Carroll J. S. (2008) Nkx3-1 and LEF-1 function as transcriptional inhibitors of estrogen receptor activity. Cancer Res. 68, 7380–7385 [DOI] [PubMed] [Google Scholar]
- 45. Chang E. C., Charn T. H., Park S. H., Helferich W. G., Komm B., Katzenellenbogen J. A., Katzenellenbogen B. S. (2008) Estrogen receptors α and β as determinants of gene expression: influence of ligand, dose, and chromatin binding. Mol. Endocrinol. 22, 1032–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu Y., Gao H., Marstrand T. T., Ström A., Valen E., Sandelin A., Gustafsson J. A., Dahlman-Wright K. (2008) The genome landscape of ERα- and ERβ-binding DNA regions. Proc. Natl. Acad. Sci. U.S.A. 105, 2604–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matthews J., Wihlén B., Tujague M., Wan J., Ström A., Gustafsson J. A. (2006) Estrogen receptor (ER) β modulates ERα-mediated transcriptional activation by altering the recruitment of c-Fos and c-Jun to estrogen-responsive promoters. Mol. Endocrinol. 20, 534–543 [DOI] [PubMed] [Google Scholar]
- 48. Tremblay A., Tremblay G. B., Labrie F., Giguère V. (1999) Ligand-independent recruitment of SRC-1 to estrogen receptor β through phosphorylation of activation function AF-1. Mol. Cell 3, 513–519 [DOI] [PubMed] [Google Scholar]
- 49. Williams C., Edvardsson K., Lewandowski S. A., Ström A., Gustafsson J. A. (2008) A genome-wide study of the repressive effects of estrogen receptor β on estrogen receptor α signaling in breast cancer cells. Oncogene 27, 1019–1032 [DOI] [PubMed] [Google Scholar]
- 50. Zhao C., Dahlman-Wright K., Gustafsson J. A. (2008) Estrogen receptor β: an overview and update. Nucl. Recept. Signal. 6, e003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jackson T. A., Richer J. K., Bain D. L., Takimoto G. S., Tung L., Horwitz K. B. (1997) The partial agonist activity of antagonist-occupied steroid receptors is controlled by a novel hinge domain-binding coactivator L7/SPA and the corepressors N-CoR or SMRT. Mol. Endocrinol. 11, 693–705 [DOI] [PubMed] [Google Scholar]
- 52. Tcherepanova I., Puigserver P., Norris J. D., Spiegelman B. M., McDonnell D. P. (2000) Modulation of estrogen receptor-α transcriptional activity by the coactivator PGC-1. J. Biol. Chem. 275, 16302–16308 [DOI] [PubMed] [Google Scholar]
- 53. Sanchez M., Sauvé K., Picard N., Tremblay A. (2007) The hormonal response of estrogen receptor β is decreased by the phosphatidylinositol 3-kinase/Akt pathway via a phosphorylation-dependent release of CREB-binding protein. J. Biol. Chem. 282, 4830–4840 [DOI] [PubMed] [Google Scholar]
- 54. Sanchez M., Picard N., Sauvé K., Tremblay A. (2013) Coordinate regulation of estrogen receptor β degradation by Mdm2 and CREB-binding protein in response to growth signals. Oncogene 32, 117–126 [DOI] [PubMed] [Google Scholar]
- 55. Burns K. A., Li Y., Arao Y., Petrovich R. M., Korach K. S. (2011) Selective mutations in estrogen receptor α D-domain alters nuclear translocation and non-estrogen-response element gene regulatory mechanisms. J. Biol. Chem. 286, 12640–12649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zwart W., de Leeuw R., Rondaij M., Neefjes J., Mancini M. A., Michalides R. (2010) The hinge region of the human estrogen receptor determines functional synergy between AF-1 and AF-2 in the quantitative response to estradiol and tamoxifen. J. Cell Sci. 123, 1253–1261 [DOI] [PubMed] [Google Scholar]
- 57. Sapountzi V., Logan I. R., Robson C. N. (2006) Cellular functions of TIP60. Int. J. Biochem. Cell Biol. 38, 1496–1509 [DOI] [PubMed] [Google Scholar]
- 58. Cvoro A., Tzagarakis-Foster C., Tatomer D., Paruthiyil S., Fox M. S., Leitman D. C. (2006) Distinct roles of unliganded and liganded estrogen receptors in transcriptional repression. Mol. Cell 21, 555–564 [DOI] [PubMed] [Google Scholar]
- 59. Rogatsky I., Luecke H. F., Leitman D. C., Yamamoto K. R. (2002) Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc. Natl. Acad. Sci. U.S.A. 99, 16701–16706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Harris D. M., Besselink E., Henning S. M., Go V. L., Heber D. (2005) Phytoestrogens induce differential estrogen receptor α- or β-mediated responses in transfected breast cancer cells. Exp. Biol. Med. 230, 558–568 [DOI] [PubMed] [Google Scholar]
- 61. Kuiper G. G., Lemmen J. G., Carlsson B., Corton J. C., Safe S. H., van der Saag P. T., van der Burg B., Gustafsson J. A. (1998) Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor β. Endocrinology 139, 4252–4263 [DOI] [PubMed] [Google Scholar]
- 62. Nikov G. N., Hopkins N. E., Boue S., Alworth W. L. (2000) Interactions of dietary estrogens with human estrogen receptors and the effect on estrogen receptor-estrogen-response element complex formation. Environ. Health Perspect. 108, 867–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Paige L. A., Christensen D. J., Grøn H., Norris J. D., Gottlin E. B., Padilla K. M., Chang C. Y., Ballas L. M., Hamilton P. T., McDonnell D. P., Fowlkes D. M. (1999) Estrogen receptor (ER) modulators each induce distinct conformational changes in ER α and ER β. Proc. Natl. Acad. Sci. U.S.A. 96, 3999–4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mueller S. O., Simon S., Chae K., Metzler M., Korach K. S. (2004) Phytoestrogens and their human metabolites show distinct agonistic and antagonistic properties on estrogen receptor α (ERα) and ERβ in human cells. Toxicol. Sci. 80, 14–25 [DOI] [PubMed] [Google Scholar]
- 65. Routledge E. J., White R., Parker M. G., Sumpter J. P. (2000) Differential effects of xenoestrogens on coactivator recruitment by estrogen receptor (ER) α and ERβ. J. Biol. Chem. 275, 35986–35993 [DOI] [PubMed] [Google Scholar]
- 66. Björnström L., Sjöberg M. (2004) Estrogen receptor-dependent activation of AP-1 via non-genomic signalling. Nucl. Recept. 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pike A. C., Brzozowski A. M., Hubbard R. E., Bonn T., Thorsell A. G., Engström O., Ljunggren J., Gustafsson J. A., Carlquist M. (1999) Structure of the ligand-binding domain of oestrogen receptor β in the presence of a partial agonist and a full antagonist. EMBO J. 18, 4608–4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pike A. C., Brzozowski A. M., Walton J., Hubbard R. E., Thorsell A. G., Li Y. L., Gustafsson J. A., Carlquist M. (2001) Structural insights into the mode of action of a pure antiestrogen. Structure 9, 145–153 [DOI] [PubMed] [Google Scholar]
- 69. Wang Y., Chirgadze N. Y., Briggs S. L., Khan S., Jensen E. V., Burris T. P. (2006) A second binding site for hydroxytamoxifen within the coactivator-binding groove of estrogen receptor β. Proc. Natl. Acad. Sci. U.S.A. 103, 9908–9911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Deleted in proof
- 71. Wang J., Chen J. (2010) SIRT1 regulates autoacetylation and histone acetyltransferase activity of TIP60. J. Biol. Chem. 285, 11458–11464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yang C., Wu J., Zheng Y. G. (2012) Function of the active site lysine autoacetylation in Tip60 catalysis. PLoS One 7, e32886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lerin C., Rodgers J. T., Kalume D. E., Kim S. H., Pandey A., Puigserver P. (2006) GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1α. Cell Metab. 3, 429–438 [DOI] [PubMed] [Google Scholar]
- 74. Kamei Y., Xu L., Heinzel T., Torchia J., Kurokawa R., Gloss B., Lin S. C., Heyman R. A., Rose D. W., Glass C. K., Rosenfeld M. G. (1996) A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 85, 403–414 [DOI] [PubMed] [Google Scholar]
- 75. Lee S. K., Kim H. J., Na S. Y., Kim T. S., Choi H. S., Im S. Y., Lee J. W. (1998) Steroid receptor coactivator-1 coactivates activating protein-1-mediated transactivations through interaction with the c-Jun and c-Fos subunits. J. Biol. Chem. 273, 16651–16654 [DOI] [PubMed] [Google Scholar]
- 76. Sauvé K., Lepage J., Sanchez M., Heveker N., Tremblay A. (2009) Positive feedback activation of estrogen receptors by the CXCL12-CXCR4 pathway. Cancer Res. 69, 5793–5800 [DOI] [PubMed] [Google Scholar]
- 77. Zhu Y., Sullivan L. L., Nair S. S., Williams C. C., Pandey A. K., Marrero L., Vadlamudi R. K., Jones F. E. (2006) Coregulation of estrogen receptor by ERBB4/HER4 establishes a growth-promoting autocrine signal in breast tumor cells. Cancer Res. 66, 7991–7998 [DOI] [PubMed] [Google Scholar]