

Background: Glycosaminoglycan (GAG)-chemokine dimer interactions regulate neutrophil trafficking, but the molecular basis underlying their interactions is not well understood.
Results: NMR studies of murine CXCL1 indicate that heparin spans the dimer interface and enhances its structural integrity and stability.
Conclusion: Heparin binding modulates multiple structural properties of the chemokine dimer.
Significance: This study provides novel structural insights into how chemokine dimers orchestrate neutrophil recruitment.
Keywords: Chemokines, Glycobiology, Glycosaminoglycan, Heparin, Neutrophil, NMR
Abstract
Glycosaminoglycan (GAG)-bound and soluble chemokine gradients in the vasculature and extracellular matrix mediate neutrophil recruitment to the site of microbial infection and sterile injury in the host tissue. However, the molecular principles by which chemokine-GAG interactions orchestrate these gradients are poorly understood. This, in part, can be directly attributed to the complex interrelationship between the chemokine monomer-dimer equilibrium and binding geometry and affinities that are also intimately linked to GAG length. To address some of this missing knowledge, we have characterized the structural basis of heparin binding to the murine CXCL1 dimer. CXCL1 is a neutrophil-activating chemokine and exists as both monomers and dimers (Kd = 36 μm). To avoid interference from monomer-GAG interactions, we designed a trapped dimer (dCXCL1) by introducing a disulfide bridge across the dimer interface. We characterized the binding of GAG heparin octasaccharide to dCXCL1 using solution NMR spectroscopy. Our studies show that octasaccharide binds orthogonally to the interhelical axis and spans the dimer interface and that heparin binding enhances the structural integrity of the C-terminal helical residues and stability of the dimer. We generated a quadruple mutant (H20A/K22A/K62A/K66A) on the basis of the binding data and observed that this mutant failed to bind heparin octasaccharide, validating our structural model. We propose that the stability enhancement of dimers upon GAG binding regulates in vivo neutrophil trafficking by increasing the lifetime of “active” chemokines, and that this structural knowledge could be exploited for designing inhibitors that disrupt chemokine-GAG interactions and neutrophil homing to the target tissue.
Introduction
A fundamental aspect of the innate immune response is the rapid chemokine-mediated recruitment of circulating neutrophils in response to infection and sterile tissue injury (1). There is now compelling evidence that chemokines, via their interactions with cell surface and free glycosaminoglycans on the endothelial and epithelial cells and the extracellular matrix, regulate every step of the recruitment process (1–3). It is believed that these interactions regulate chemokine monomer and dimer levels and influence the steepness and duration of the chemotactic and haptotactic gradients, whereby they play a critical role in orchestrating neutrophil homing to the target tissue (3–5).
Glycosaminoglycans (GAGs)2, such as heparin and heparan sulfate (HS), are long, linear, sulfated polysaccharides covalently attached to cell membrane proteins to form proteoglycans (6, 7). They have a phenomenal structural diversity because of differences in the positioning of the sulfate groups along the polysaccharide chain (8, 9). GAGs interact with a wide range of proteins, including cytokines, bone morphogenic proteins, growth factors, coagulation enzymes, and chemokines, to regulate diverse physiological processes (10–14).
Chemokines (chemotactic cytokines) are small (8- to 10-kDa) proteins that mediate leukocyte recruitment by activating G protein-coupled receptors (3, 15–17). Humans express ∼50 chemokines that are divided into four subclasses, depending on the position of N-terminal cysteine residues (CC, CXC, CX3C, and C). Chemokines share the fundamental property of oligomerization and GAG binding (14–16, 18). Although all chemokines share a conserved three-dimensional tertiary structure, they can dimerize using different regions of the protein, resulting in distinctly different quaternary structures (19–23).
Neutrophil-activating chemokines (NACs), a subgroup of CXC chemokines characterized by the highly conserved ELR motif, recruit neutrophils by differentially activating CXCR1/CXCR2 receptors (24–27). NACs reversibly exist as monomers, dimers, and/or tetramers, and recent in vivo studies using disulfide-trapped dimers have shown that dimers are highly efficient in recruiting neutrophils. As dimers have a similar or lower receptor activity, the higher recruitment may be due to GAG interactions (5, 28). However, the molecular mechanisms by which GAG interactions mediate recruitment are not known because there is a lack of experimental data on the structural architecture of NAC dimer-GAG complexes. Structural studies of GAG-chemokine interactions are highly challenging because of limitations such as the inherent heterogeneity of the GAGs, the dynamic oligomerization behavior of the chemokines, and the aggregation/precipitation of the complexes with physiologically relevant GAGs at high concentrations used in structural studies (15, 16, 21, 29–31).
To address this missing knowledge, we have chosen murine CXCL1 (also known as keratinocyte-derived chemokine), the homolog of human CXCL1 (melanoma growth-stimulatory activity), and have characterized the structural basis of heparin binding using NMR spectroscopy. We circumvent the phenomenon of dynamic oligomerization by designing a trapped non-dissociating dimer and the precipitation issues of the GAG-chemokine complexes by performing NMR structural studies at low micromolar protein concentrations. Moreover, nothing is known regarding the structural properties of murine CXCL1, although CXCL1-mediated neutrophil recruitment and the role of CXCL1-CXCR2 axis in health and disease have been studied extensively in animal models, including in KO mice and various bacterial and tissue injury models (32, 33). As WT CXCL1 exists in equilibrium between monomers and dimers, we engineered a disulfide-linked CXCL1-trapped dimer (dCXCL1) and characterized its structure and dynamics in both free and heparin octasaccharide-bound forms using multidimensional NMR spectroscopy. Our data indicate that the heparin octasaccharide binds perpendicularly to the interhelical axis and spans the dimer interface and that heparin binding restricts the mobility and enhances the stability of the dimer. To our knowledge, this is the first experimental evidence of the structural basis of chemokine-GAG interactions of a NAC dimer.
EXPERIMENTAL PROCEDURES
Construction of Murine CXCL1 and Its Mutants
A murine CXCL1 (GenBanktm accession no. AAB03376.1) cDNA fragment was codon-optimized for Escherichia coli expression, and the gene was synthesized using custom gene synthesis from Genscript. The gene was amplified by PCR and inserted into the pET 32Xa-LIC vector using a ligation-independent cloning method. The mutant protein genes were generated using the QuikChange site-directed mutagenesis protocol (Stratagene). All plasmid constructs were verified by DNA sequencing. Disulfide-trapped dimeric CXCL1 was constructed by introducing a K28C mutation on a WT-CXCL1 background. The quadruple mutant (H20A/K22A/K62A/K66A), labeled as dCXCL1-M4, was generated on the dCXCL1 background by performing iterative cycles of mutagenesis.
Protein Expression and Purification of CXCL1 Variants
Transformed E. coli BL21 (DE3) cells were cultured in LB medium or isotopically enriched 13C/15N-labeled minimal medium (containing 15NH4Cl and 13C glucose as the sole nitrogen and carbon sources) with 100 μg/ml ampicillin. Cells were cultured at 37 °C until an A600 of 0.5–0.6 was reached and subsequently induced by addition of 0.1 mm isopropyl 1-thio-β-d-galactopyranoside for protein expression. The cells were grown at 25 °C at 200 rpm in the shaker for another 12–16 h before harvesting.
The harvested cells (stored at −80 °C) were thawed on ice, resuspended in lysis buffer (pH 8.0) containing 50 mm Tris, 150 mm NaCl, 5 mm benzamidine, and 0.5 mm freshly added phenylmethylsulfonyl fluoride. The cells were treated with lysozyme on ice for 40 min and then lysed by sonication. The cell lysate was separated into supernatant and pellet by high-speed centrifugation. The lysate supernatant was applied to a nickel-nitrilotriacetic acid column that was equilibrated with lysis buffer, and the fusion protein was eluted using lysis buffer containing 400 mm imidazole. The fusion protein was dialyzed against a buffer containing 50 mm Tris, 50 mm NaCl, and 5 mm CaCl2 (pH 7.4) and digested using Factor Xa. The cleavage products were passed through a nickel-nitrilotriacetic acid column and the unbound CXCL1 was collected, concentrated, and further purified through HPLC using a gradient of water and acetonitrile. The purity and molecular weight of the proteins were confirmed using SDS-PAGE and MALDI-TOF.
Analytical Ultracentrifugation Analysis
Sedimentation equilibrium studies were performed using a Beckman-Coulter Optima XL-A analytical ultracentrifuge equipped with absorbance optics and a Ti-60a titanium rotor. 15, 30, and 40 μm protein samples in 50 mm phosphate, 50 mm NaCl (pH 6.0) were centrifuged at 25,000, 35,000, and 45,000 rpm at 25 °C. The protein absorbance was measured at 232 nm, and the dissociation constant was calculated from the global data analysis using Hetero-Analysis software version 1.1.33 (J. L. Cole and J. W. Larry, University of Connecticut).
Chemotaxis
Chemotactic activities of WT CXCL1 and dCXCL1, at 10 and 100 nm, were measured using calcein-AM-labeled HL60 cells stably transfected with the CXCR2 receptor using a modified Boyden chamber 96-well plate assay as discussed previously (34).
NMR Data Acquisition and Processing
15N- and 15N/13C-labeled CXCL1 variants and their heparin octasaccharide complexes were prepared in 50 mm sodium phosphate buffer (pH 6.0) containing 1 mm sodium azide and 5% D2O. The heparin oligosaccharides used for NMR experiments were purchased from Iduron (United Kingdom). According to the manufacturer's specifications, these oligosaccharides contain uronic acid (ΔUA) at the non-reducing end and have a C4-C5 double bond as a result of the heparinase endolytic action. The main disaccharide unit in these oligosaccharides is IdoA,2S-GlcNS,6S (∼75%) and show some variation in sulfation.
The NMR experiments were carried out using a triple-channel Bruker Avance III 800 MHz (equipped with a TXI cryoprobe) and 600 MHz (with QCI cryoprobe) equipped with pulse-shaping and pulse field gradient capabilities. For resonance assignments, a series of standard three-dimensional experiments (35), such as 15N-edited NOESY, 15N-edited TOCSY, HNCO, HNCA, HN(CO)CA, CBCANH, and CBCA(CO)NH, were recorded at 35 °C for free dCXCL1 (100 μm in dimer units) and at 25 °C for the dCXCL1-heparin octasaccharide complex (30 μm). 15N-edited NOESY was recorded for dCXCL1 at both 25 °C and 35 °C. The 1H and 15N carrier frequencies were set at 4.7 and 119.5 ppm, respectively. The 13C carrier frequency was set at 54 ppm for HNCA/HN(CO)CA, at 42 ppm for HNCACB/CBCA(CO)NH, and 174 ppm for HNCO. The mixing times for 15N-edited NOESY and TOCSY experiments were 150 and 80 ms, respectively. The chemical shifts were calibrated relative to 2,2-dimethyl-2-silapentane-5-sulfonate.
The octasaccharide titration experiments were performed at 25 °C. The dissociation constant (Kd) for the dCXCL1-octasaccharide complex was calculated as described previously (36). Chemical shift perturbation (CSP) was calculated using the following equation:
 |
The spectra were processed with NMRPipe (37) and analyzed using Bruker Topspin 3.2 or NMRView (38).
Steady-state Heteronuclear 1H-15N NOE
Steady-state heteronuclear (1H-15N) NOE experiments were recorded for the free and heparin octasaccharide-bound dCXCL1 on a 600-MHz Bruker spectrometer in an interleaved fashion with a proton saturation time of 2.5 s and a relaxation delay of 2.5 s. 200 complex increments and 256 scans/fid were used for signal-averaging the data. Steady-state 1H-15N NOEs were calculated as a ratio of intensities of the peaks with and without proton saturation. The errors in the NOEs were obtained as described by Farrow et al. (39).
Hydrogen Exchange Measurements
For native-state hydrogen exchange studies, dCXCL1 and the dCXCL1-heparin octasaccharide complex were prepared in 50 mm sodium phosphate (pH 6.0) and then lyophilized. Native-state hydrogen exchange was initiated by dissolving the protein samples in D2O. The samples were loaded on a pretuned and shimmed NMR spectrometer at 25 °C. The time from the addition of D2O to the start of the HSQC spectrum was 10 min (dead time). Each spectrum was recorded with 24 scans and consisted of 80 complex increments in the indirect 15N dimension (total experiment time, ∼ 90 min).
Structural Model of the Free dCXCL1
CXCL1 dimer Cα, Cβ, CO, Hα, NH, 15N chemical shifts were used as input to generate the monomer fold of the CXCL1 dimer using the CS23D 2.0 (40). The structure of the CXCL1 dimer was obtained from the NMR-derived CXCL1 monomeric fold by defining the dimer symmetry elements. The intermolecular disulfide linkage (Cys-28-Cys-28′) across the dimer interface was defined, and the CXCL1 dimer was subjected to constrained energy minimization using AMBER 12 suite (41). Only the residues Leu-27, Cys-28, and Val-29 were allowed to relax. This relieved any steric contacts because of the substitution and resulted in proper geometry for the disulfide bond. Finally, the structure was subjected to multiple cycles of free energy minimization. The quality of the energy-minimized structure was evaluated using PROCHECK (42).
Structural Model of dCXCL1-Heparin Octasaccharide Complex Structure
Molecular docking of the heparin oligosaccharide with dCXCL1 was carried out using HADDOCK 2.1 (43). Docking was carried out using the dCXCL1 structure obtained as discussed above, and the octasaccharide was generated from the NMR structure of heparin (PDB code 1HPN) (44). CSP, native-state hydrogen exchange, and mutational data were used as ambiguous interaction restraints to drive the docking process. Active and passive residues were defined on the basis of the experimental data to generate ambiguous interaction restraints. For the octasaccharide, parameters were generated using the PRODRG server, and necessary modifications were made in the parameter file and included into the HADDOCK 2.1 program suite. The docking was performed in a 1:1 protein:octasaccharide ratio, as determined from isothermal calorimetry and NMR diffusion experiments. A total of 1000 structures were generated during the initial rigid-body docking. The best 200 structures in terms of intermolecular energies were further subjected to semiflexible simulated annealing. The clusters were scored using the “HADDOCK score,” which is a weighted sum of a combination of energy terms.
RESULTS
WT-CXCL1 Exists in Equilibrium between Monomers and Dimers
In general, chemokines share the property of reversibly existing as monomers, dimers, and higher-order oligomers (15, 16, 18). The sedimentation equilibrium studies of murine CXCL1 indicate that it exists in equilibrium between monomers and dimers (Kd 36 ± 4 μm) (Fig. 1A). NMR studies also showed evidence of monomer-dimer equilibrium (Fig. 1B). The total number of observed resonances (∼120) is approximately twice what can be expected from a monomer. Together, these studies establish that, under the experimental conditions, murine CXCL1 exists in equilibrium between monomers and dimers.
FIGURE 1.

Sedimentation equilibrium analysis of wild-type CXCL1 in 50 mm phosphate and 50 mm NaCl (pH 6. 0) (A). The concentrations calculated in fringe displacement units (mg/ml) are plotted against the radius. Residuals of the corresponding least square fit are random, indicating the goodness of the fit. B, 1H-15N HSQC spectrum of WT-CXCL1 (120 μm) at pH 6.0 at 25 °C. The spectra show approximately twice the number of peaks than what is expected for a single species, indicating the presence of both the CXCL1 monomer and dimer.
Design and Characterization of the Disulfide-trapped CXCL1 Dimer
Unraveling the structural features of monomer/dimer mixtures is challenging because the very process of monomer-dimer equilibrium interferes in the study of one or the other form. Recent functional data from our laboratory have shown that the human CXCL1 dimer is a potent agonist for its receptor (34). Therefore, we engineered a dimeric variant of murine CXCL1 using the disulfide trapping strategy by substituting a Cys residue for Lys-28, which is at the 2-fold symmetry axis (Fig. 2, A and B). A similar strategy has been used in the past to successfully engineer a trapped dimer for human CXCL8 and CXCL1 (27, 34).
FIGURE 2.

Sequence alignment of two murine and seven human CXC neutrophil-activating chemokines (A). The murine chemokines are represented with their common names (mKC for CXCL1 and mMIP2 for CXCL2). The functional ELR sequence motif is shown in green. The conserved cysteine residues and their respective disulfide linkages are shown in red. The positively charged residues His-20, Lys-22, Lys-62, and Lys-66 (numbered with respect to mKC) are shown in blue. Residues Lys-28 in mKC, Arg-26 in CXCL8, and Asn-27 in CXCL1 represent the 2-fold symmetry and act as an anchor point for introducing the intermolecular disulfide bond (asterisk). B, schematic showing the design strategy for connecting two monomeric CXCL1 units using the K28C mutation. C, SDS-PAGE analysis of murine CXCL1 variants. The formation of intermolecular disulfide bond is evident for the CXCL1 dimer (dCXCL1) from the band at 16 kDa. M, protein marker; R, reduced; NR, non-reduced. D, HNCACB strips of all the cysteine residues. The Cβ shifts indicate that all form disulfides. E, the chemotactic activity of the WT and trapped dimer at 10 and 100 nm concentrations was measured using a Boyden chamber-type assay. The data were collected in quadruplicate, and the results are expressed as mean ± S.D. and are representative of three independent experiments.
The formation of the interdisulfide bond was confirmed from the non-reducing SDS-PAGE (Fig. 2C) and doubling of mass of dCXCL1 from MALDI-TOF spectrometry (data not shown). Furthermore, the oxidation state of Cys-28 and other Cys residues was confirmed from their characteristic Cβ chemical shifts (> 40 ppm for oxidized cysteines) (45) (Fig. 2D). These data collectively demonstrate that the designed disulfide trap facilitates the formation of a non-dissociating dimer. The activity of the trapped CXCL1 dimer was characterized using an in vitro chemotaxis assay (Fig. 2E). The trapped dimer was as active as the WT, indicating that the murine CXCL1 dimer is a potent agonist for CXCR2 receptor function.
The HSQC spectrum of the disulfide-linked dimer showed a single set of resonances with excellent dispersion, indicating a well folded and symmetric protein dimer (Fig. 3A). The backbone and side chain resonances were assigned using a set of conventional triple-resonance experiments (see “Experimental Procedures”). The backbone NH assignments for the majority of the residues are labeled in Fig. 3A. The secondary structure prediction analysis using chemical shifts indicates that the monomeric subunit consists of an extended N-loop followed by a turn of a helix (most likely a 310 helix, as evidenced by dihedral angles), three β-strands, and a C-terminal α-helix. Although the dimer interface β-strand amides showed several intra- and intermolecular backbone and side chain NOEs in the 15N-edited NOESY experiment, no NOEs were observed between the β-strands and the C-terminal helix (Fig. 3B). Similar intermolecular NOEs between the β1 and β′1 strands are also observed for human CXCL8- and CXCL1-trapped dimers (27, 34).
FIGURE 3.

1H-15N HSQC spectrum of dCXCL1 at 35 °C (A). Backbone N-H resonance assignments are shown for all the resolved peaks in the spectrum. The peaks enclosed in the rectangles correspond to the Asn/Gln side chain NH2 resonances. B, two-dimensional strip plots from the 15N-edited three-dimensional NOESY spectrum. Representative two-dimensional strips are shown for Lys-27, Val-29, Val-64, Gln-65, and Lys-66 amides. The characteristic interstrand intermolecular NOEs between the β1 and β′1 strand residues are highlighted as prime (′), and the proton type is shown in italics.
The absence of NOEs from the C-terminal helix could be a direct consequence of its inherent structural dynamics (see below). In the absence of the NOEs, it is challenging to pin down the position of the helix on the strands and obtain an overall topology of dCXCL1. Chemical shift threading is a relatively powerful and alternate approach to generate accurate three-dimensional structural folds without the aid of NOEs (40). In chemical shift threading, the protein sequence and experimentally observed chemical shifts are threaded simultaneously to render three-dimensional structures. This method is proven to yield high-quality structures when structures of homologous proteins are known. Therefore, we generated the structural fold of dCXCL1 using chemical shift threading. The stereochemical quality assessed using PROCHECK showed a well folded protein with no major violations.
The structural fold of the dCXCL1 and its monomeric subunit are shown in Fig. 4. The intermolecular NOEs observed across the dimer interface β-strand residues are in good agreement with the back-calculated distances from the structural model. Further, the dCXCL1 fold agrees with the known dimeric structures of NACs (Fig. 5). The different structural elements and functional motifs are labeled in Fig. 4A. The β sheet and α-helical faces for the dCXCL1 are shown in Fig. 4B. Analysis of the surface electrostatics indicates that the β sheet face is negatively charged and that the helical face is positively charged (Fig. 4C). A structural comparison between dCXCL1 and murine MIP2/CXCL2 and human CXCL1 and human CXCL8 reveal that the length of the C-terminal helix is shorter in murine chemokines, compared with their human counterparts, by at least one turn (Fig. 5). These observations suggest that the structural and dynamic characteristics of C-terminal helix most likely regulate the structural stability and function of chemokines, as discussed below.
FIGURE 4.

Structural features of the murine CXCL1 dimer. A, tertiary fold of a single monomeric subunit of the dCXCL1 dimer. The structural elements, including disulfide linkages and functionally important N loop and ELR motif regions, are highlighted. B, the individual monomers in the dimer are shown in cyan and pink. The Cys-28-Cys-28′ intermolecular disulfide bond is shown in yellow, and other disulfides are not shown for clarity. C, electrostatic surface representation of dCXCL1. All structures were generated using PYMOL. Blue, positively charged residues; red, negatively charged residues; white, hydrophobic residues).
FIGURE 5.
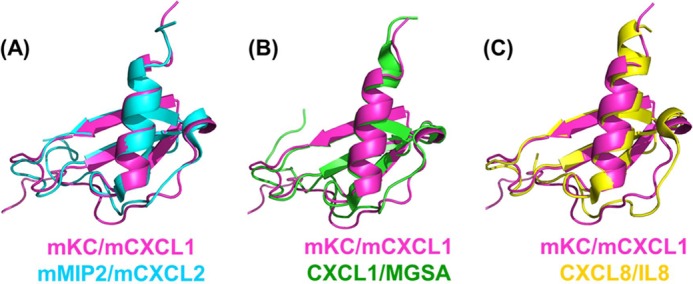
Structural comparison of the monomeric subunit of dimeric murine CXCL1/mKC with other ELR-CXC chemokines. A, murine CXCL2/MIP2 (PDB code 3N52). B, human CXCL1/melanoma growth-stimulatory activity (PDB code 1MGS). C, human CXCL8/IL-8 (PDB code 3IL8).
Heparin Binds Orthogonally to the Interhelical Axis of the CXCL1 Dimer
The availability of high-field spectrometers equipped with sensitive cryoprobes have allowed us to use solution NMR experiments to obtain residue-level structural information of chemokine-GAG complexes at low protein concentrations (∼30 μm). NMR-derived backbone amide and 15N chemical shift changes upon ligand binding are sensitive probes to map the binding surface on the protein structure. We have used this chemical shift perturbation approach to map the binding site of heparin octasaccharide on dCXCL1 (Fig. 6A). Perturbation of only a subset of residues, such as Gly-18, Lys-22, Ser-26, and Gly-70 in the depicted spectral region suggests that the binding process is specific. A representative binding profile of octasaccharide binding is shown in Fig. 6B. The calculated apparent dissociation constant for a 1:1 complex is 34 ± 6 μm. Fig. 6C shows the CSP map of dCXCL1 upon complete saturation (dCXCL1:octasaccharide molar ratio = 1:11). The perturbations are centered on the N loop, 310 helix, 40s loop, β3 sheet, 50s loop, and C-terminal helix. The perturbed residues superimpose with the positively charged patches (Figs. 4C and 6D), indicating that electrostatic interactions are the major contributors for heparin binding.
FIGURE 6.

A section of the 1H-15N HSQC spectrum of the free dCXCL1 (blue) and in the presence of heparin octasaccharide (red) (A). The arrow indicates the direction of binding-induced chemical shift changes. B, representative binding profile for calculating the binding constant of dCXCL1-octasaccharide (Octa) interactions. C, CSP map of dCXCL1-octasaccharide interactions. The horizontal line at 0.1 ppm represents the cutoff for a residue to be considered perturbed. The sequence-specific secondary structural elements are shown on the top of the CSP map with arrows for β sheets and cylinders for helices. D, molecular plot of the dCXCL1 residues that are significantly perturbed on octasaccharide binding. The perturbed residues are shown in red.
Using our NMR experimental data as distance constraints, we generated the structure of the dCXCL1-octasaccharide complex using the HADDOCK 2.1 program. The 10 lowest energy dCXCL1-octasaccharide structures from the best cluster, on the basis of HADDOCK energy, are shown in Fig. 7A. The structure shows that the octasaccharide binds orthogonally to the C-terminal helices and proximal to the 310 helix (Fig. 7, B and C). Analysis of the structure shows that four basic charge residues from each monomer, His-20, Lys-22, Lys-62, and Lys-66 act as “hot spots” for heparin binding through electrostatic interactions with the negatively charged sulfate groups of the octasaccharide (Fig. 7, B and C).
FIGURE 7.

The 10 lowest energy-minimized structures of the dCXCL1-heparin octasaccharide complex calculated using HADDOCK 2.1. (A). B, magnification highlighting the electrostatic interactions between the positively charged residues (blue) of dCXCL1 and the negatively charged sulfate groups (yellow) of heparin octasaccharide (backbone in green). C, schematic of the electrostatic model of the dCXCL1-glycosaminoglycan complex. GAG is shown in red. Positively charged residues that are perturbed upon GAG binding are shown in blue. Other residues that are perturbed are shown in yellow. D, electrostatic surface representation of the quadruple mutant dCXCL1-M4. Blue, positively charged residues; red, negatively charged residues; white, hydrophobic residues. E, overlay of a section of the 1H-15N HSQC spectrum of dCXCL1-M4. Blue, dCXCL1-M4; red, dCXCL1-M4 + heparin octasaccharide (ratio ∼1:11).
To validate that the hot spot residues mediate GAG binding, we generated a quadruple dCXCL1 mutant (referred as dCXCL1-M4) lacking these hot spot residues (H20A/K22A/K62A/K66A). The dCXCL1-M4 mutant has the same structural features and retains the interdisulfide trap as in dCXCL1 (data not shown). Loss of the hot spot residues, as expected, resulted in complete loss of the basic patch on the helical face (Figs. 4C and 7D). NMR HSQC titration experiments of dCXCL1-M4 with heparin octasaccharide (molar ratio ∼ 1:11) showed negligible chemical shift changes, indicating that dCXCL1-M4 does not bind the octasaccharide (Fig. 7E). This clearly demonstrates that H20A/K22A/K62A/K66A residues predominantly mediate GAG binding in the dimer structure.
Heparin-induced Structural Integrity of dCXCL1
Structural analysis using the NOESY data indicated that the C-terminal helices are not tightly packed against the β strand (Fig. 3B). Considering that heparin octasaccharide binds to the C-terminal helical face of the dimer, we further explored the role of dynamics and stabilities by measuring the steady-state heteronuclear NOE and native state hydrogen exchange of dCXCL1 in both the free and octasaccharide-bound forms.
1H-15N steady-state heteronuclear NOE measurements are sensitive to picosecond to nanosecond (ps-ns) timescale motions, and low positive (< 0.6) or negative heteronuclear NOE values signify the presence of ps-ns motions. The residue-wise plots of the heteronuclear NOEs (Fig. 8A) indicated that the ps-ns time scale motions of free dCXCL1 varied substantially among structural elements. In addition to the N- and C termini, N loop, 310 helix, 50s loop, and C-terminal helical residues were also highly dynamic (Fig. 8A). These ps-ns dynamics observed in the free dimer were quenched in the octasaccharide-bound form, indicating that the dimer becomes more structured upon octasaccharide binding (Fig. 8A). Both N loop/310 helix and C-terminal helix residues show substantial changes on binding, and, in particular, the entire C-terminal helix residues (Val-60-Leu-68) attain uniform ps-ns motions similar to those observed for the structured β strand residues (Fig. 8A).
FIGURE 8.

1H-15N heteronuclear steady-state NOE relaxation measurements of dCXCL1 (blue) and dCXCL1-octasaccharide complex (red). B, 1H-15N HSQC spectra of free dCXCL1 (blue) and the dCXCL1-octasaccharide complex (red) after initiating hydrogen exchange with 100% D2O at pH 6.0 and 25 °C (experiment time, 90 min).
Native-state hydrogen/deuterium exchange monitored by NMR spectroscopy is a powerful tool for studying residue-wise stabilities/free-energies of proteins. The exchange rates of backbone amide protons depend on their accessibility to the solvent deuterons, which, in turn, correlate to the stability of secondary structural elements and tertiary/quaternary structures. The initial native-state hydrogen exchange spectra of both forms are shown in Fig. 8B. In free dCXCL1 (Fig. 8B, blue), only residues corresponding to the antiparallel β sheets were protected, whereas, in the GAG-bound form, 50s loop and C-terminal helix residues (red) are also protected, indicating that these residues are stabilized upon GAG binding.
DISCUSSION
Knowledge of the structures of both free and GAG-bound chemokines is essential to describe the molecular mechanisms underlying chemokine-GAG interactions and how these interactions mediate in vivo function. Although a wealth of structural information for chemokines is available, structures of chemokine-GAG complexes have been hard to come by. Two x-ray structures of chemokine-disaccharide are known, and most NMR structural studies also report only disaccharide binding, which provides only limited insights (46, 47). However, NMR studies have had better success with longer GAGs, but the extent of the structural insights varies among different chemokines and seems to be critically dependent on experimental conditions (21, 31, 48). Recent studies by Handel and co-workers (21) on CCL27 and Volkman and co-workers (31) on CXCL12 using different oligosaccharides, protein concentrations, protein:GAG ratios, and variants (monomer and dimer) have shown that chemokine structural plasticity, oligomerization properties, GAG length, and binding-induced oligomerization/aggregation/precipitation are intimately coupled. These studies also highlight that designed chemokine monomer/dimer variants that show impaired protein-protein interactions and/or are structurally more homogeneous enable NMR studies by minimizing GAG-binding-induced oligomerization and aggregation.
For NACs, NMR studies of disaccharide binding to human CXCL8 and murine CXCL2 have identified GAG binding residues. Models of GAG binding geometry have been proposed on the basis of NMR, mutagenesis, and biochemical data (29, 49, 50). Interestingly, in the case of murine CXCL2, attempts to determine NMR and crystal structures using longer oligosaccharides were unsuccessful, although we could determine the binding of an octasaccharide to murine CXCL1. However, the terminal helical residues are unstructured in both proteins (Fig. 5). Lack of correlation between two highly related chemokines, such as murine CXCL1 and CXCL2, highlight the challenges and the complex interrelationship between chemokine structural properties and GAG binding. In the case of human CXCL8, one study used a hexasaccharide but could obtain only limited data because increasing GAG concentrations led to precipitation (29, 50). However, we observed that human CXCL8 can bind longer GAGs but only at low protein concentrations.3
Multiple computational docking studies have been carried out for the human CXCL8 dimer, but they report different GAG binding geometries. In model I (known as the horseshoe model), sulfated domains of GAG bind parallel to the helices and are linked by a non-sulfated domain (51, 52). In model II, GAG binds perpendicular to the two helices spanning the dimer (53, 54). In model III, GAG binds parallel and lies between the two helices (50). Our NMR-derived structural model of the murine CXCL1 dimer-octasaccharide complex shows that GAG binds perpendicularly to the C-terminal helices, is consistent with model II, and completely rules out model III. In model III, GAG is nestled between the helices and does not interact with the 310 helix, which contradicts our data and also the binding and functional data for human CXCL8 and murine CXCL2 (28, 29, 49). Our data are also inconsistent with model I but cannot completely rule it out and require structural studies using heparan sulfate GAGs containing both sulfated (NS) and non-sulfated (NA) domains. Our structure also reveals that the octasaccharide cannot simultaneously interact with 310 helical residues of both the monomeric subunits of the dimer, indicating that a longer GAG is essential (Fig. 7A) (54). Because endogenous GAGs are much longer (> 40 saccharide units), their binding to the dimer will involve interactions with the 310 helix of both monomers. A theoretical distance calculation shows that a minimum length of 12–14 saccharide units is essential to cover the entire length of the dimeric surface (54). However, our attempts to experimentally verify the minimum length were unsuccessful because of precipitation (data not shown).
We propose, on the basis of this study, that the free chemokine dimer and GAG are conformationally plastic and exist as a dynamic ensemble (Fig. 8) and that chemokine structural plasticity confers another layer of regulatory mechanism in recognition of GAGs and defining the chemotactic and haptotactic gradients. Conformational dynamics of GAGs has also been shown to be essential for inducing conformational changes, oligomerization, and aggregation of a number of proteins that mediate diverse physiological functions (55–59). Such a dynamic regulation of a macromolecular recognition process could be a highly efficient way of programming the binding interactions because the associated energetic penalties are much less compared with a traditional path of new domain/structure formation (60).
Recent animal model studies have shown that Streptococcus pyogenes cell envelope proteinase (SpyCEP) inactivates CXCL1 and other NACs by cleaving the C-terminal helix (at Gln-61/Lys-62 in murine CXCL1) (61). Our studies show that the GAG binding site and the protease cleavage site on the dimer overlap, indicating that only the free soluble chemokine would be susceptible to proteolytic cleavage (Figs. 2A and 7C). It has also been shown that GAG binding increases the lifetime of CXCL12 by protecting it from enzymatic cleavage (31, 62). These observations suggest that NAC monomer-dimer equilibrium and GAG binding interactions and affinities must be highly regulated and that any disruption, such as proteolytic cleavage, impairs dimerization and GAG binding, which, in turn, disrupts chemokine gradient and neutrophil recruitment, enabling microbes to evade the host immune response.
We analyzed the structural and sequence features of NAC family members to shed light on their GAG-binding characteristics. The NAC dimer structures consistently reveal an interhelical distance of ∼12- to 14 Å between GAG-binding residues (Fig. 7, B and C) (19, 20, 23, 63–65). Sequences of nine NACs indicate that three of the four hot spot residues, corresponding to Lys-22, Lys-62, and Lys-66 in murine CXCL1, are absolutely conserved (Fig. 2A). The two basic residues in the C-terminal helix (Lys-62 and Lys-66) are separated by three amino acids with the signature BXXXB motif (B = Lys/Arg), and two basic residues in the N loop/310 helix are separated by an amino acid (HXK) of which His is less conserved and Lys is absolutely conserved (Fig. 2A). In addition to the conserved hot spots, NAC sequences show positively charged residues on the C-terminal helix and the 40s loop that are less conserved (Figs. 2A and 4C). We propose that the three hot spot residues and the interhelical distance dictate the geometry of the chemokine dimer on GAGs and that the His residue of the HXK motif and the other less conserved residues fine-tune the binding in a chemokine-specific manner (Fig. 7, B and C). In addition, dynamic characteristics of the individual chemokines also most likely fine-tune the binding. Structural studies of other NAC-GAG complexes, and also in different oligomeric states (such as monomers and tetramers), are necessary to better describe the molecular principles and mechanisms that govern the NAC-GAG interactions and their role in function.
Our studies also shed some light on whether GAG-bound chemokines can activate the receptor. NMR studies of human CXCL8 and CXCL1 binding to CXCR1 and CXCR2 N-terminal domain peptides have shown that the His and Lys of the HXK motif are also involved in receptor binding (34, 36). Therefore, GAG binding will occlude these residues and prevent access for receptor binding and activation, suggesting that GAG-bound NACs cannot bind or can only bind with impaired affinity compared with the soluble chemokines.
In summary, NMR methodologies, along with our design strategy of a disulfide-trapped dimer, have allowed us to delineate the structural basis of heparin binding to the CXCL1 dimer at low micromolar protein concentrations. Our data show that GAGs bind orthogonally to the interhelical axis and enhance the structural integrity and stability of the dimer. We propose that this enhanced stability and integrity of GAG-bound dimers regulate in vivo neutrophil trafficking by multiple mechanisms, including increasing the lifetime of “active” chemokines and by minimizing the proteolytic cleavage, which, in turn, fine-tunes the kinetics, flux, and duration of neutrophil recruitment into the target tissue during inflammation/infection. Knowledge of the structures could also be useful in the development of targeted chemokine/GAG-based therapeutics/inhibitors for regulating neutrophil function.
Acknowledgments
We thank Dr. Ann Richmond of Vanderbilt University for the HL60-CXCR2 cells; Dr. Luis Holthauzen for analytical ultracentrifugation and MALDI experiments; Dr. Amit Dutta, Renling Xu, Curtis Nutter, and Anna Troshkina for assistance; Dr. Tianzhi Wang for help with NMR instruments; Dr. Mohan Sepuru for critical comments; and the John S. Dunn Gulf Course Consortium for Magnetic Resonance for access to the 800-MHz spectrometer.
This work was supported, in whole or in part, by National Institutes of Health Grant P01 HL107152 (to K. R.).
P. R. B. Joseph and K. Rajarathnam, unpublished observations.
- GAG
- glycosaminoglycan
- HS
- heparan sulfate
- NAC
- neutrophil activating chemokine
- CSP
- chemical shift perturbation
- HSQC
- heteronuclear single-quantum coherence
- ps-ns
- picosecond to nanosecond
- mKC
- murine keratinocyte-derived chemokine
- CXC
- CXC ligand
- dCXCL1
- dimeric CXC ligand 1
- CXCR
- CXC chemokine receptor.
REFERENCES
- 1. Kolaczkowska E., Kubes P. (2013) Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13, 159–175 [DOI] [PubMed] [Google Scholar]
- 2. Wang L., Fuster M., Sriramarao P., Esko J. D. (2005) Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 6, 902–910 [DOI] [PubMed] [Google Scholar]
- 3. Blanchet X., Langer M., Weber C., Koenen R. R., von Hundelshausen P. (2012) Touch of chemokines. Front. Immunol. 3, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parish C. R. (2006) The role of heparan sulphate in inflammation. Nat. Rev. Immunol. 6, 633–643 [DOI] [PubMed] [Google Scholar]
- 5. Das S. T., Rajagopalan L., Guerrero-Plata A., Sai J., Richmond A., Garofalo R. P., Rajarathnam K. (2010) Monomeric and dimeric CXCL8 are both essential for in vivo neutrophil recruitment. PLoS ONE 5, e11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li L., Ly M., Linhardt R. J. (2012) Proteoglycan sequence. Mol. Biosyst. 8, 1613–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Prabhakar V., Capila I., Sasisekharan R. (2009) The structural elucidation of glycosaminoglycans. Methods Mol. Biol. 534, 147–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Esko J. D., Lindahl U. (2001) Molecular diversity of heparan sulfate. J. Clin. Invest. 108, 169–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shriver Z., Capila I., Venkataraman G., Sasisekharan R. (2012) Heparin and heparan sulfate. Analyzing structure and microheterogeneity. Handb. Exp. Pharmacol. 207, 159–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Capila I., Linhardt R. J. (2002) Heparin-protein interactions. Angew. Chem. Int. Ed. Engl. 41, 391–412 [DOI] [PubMed] [Google Scholar]
- 11. Bernfield M., Götte M., Park P. W., Reizes O., Fitzgerald M. L., Lincecum J., Zako M. (1999) Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 68, 729–777 [DOI] [PubMed] [Google Scholar]
- 12. Esko J. D., Selleck S. B. (2002) Order out of chaos. Assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 71, 435–471 [DOI] [PubMed] [Google Scholar]
- 13. Hileman R. E., Fromm J. R., Weiler J. M., Linhardt R. J. (1998) Glycosaminoglycan-protein interactions. Definition of consensus sites in glycosaminoglycan binding proteins. BioEssays 20, 156–167 [DOI] [PubMed] [Google Scholar]
- 14. Lortat-Jacob H. (2009) The molecular basis and functional implications of chemokine interactions with heparan sulphate. Curr. Opin. Struct. Biol. 19, 543–548 [DOI] [PubMed] [Google Scholar]
- 15. Lau E. K., Allen S., Hsu A. R., Handel T. M. (2004) Chemokine-receptor interactions. GPCRs, glycosaminoglycans and viral chemokine binding proteins. Adv. Protein Chem. 68, 351–391 [DOI] [PubMed] [Google Scholar]
- 16. Salanga C. L., Handel T. M. (2011) Chemokine oligomerization and interactions with receptors and glycosaminoglycans. The role of structural dynamics in function. Exp. Cell Res. 317, 590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fernandez E. J., Lolis E. (2002) Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 42, 469–499 [DOI] [PubMed] [Google Scholar]
- 18. Rajarathnam K., Kay C. M., Dewald B., Wolf M., Baggiolini M., Clark-Lewis I., Sykes B. D. (1997) Neutrophil-activating peptide-2 and melanoma growth-stimulatory activity are functional as monomers for neutrophil activation. J. Biol. Chem. 272, 1725–1729 [DOI] [PubMed] [Google Scholar]
- 19. Clore G. M., Appella E., Yamada M., Matsushima K., Gronenborn A. M. (1990) Three-dimensional structure of interleukin 8 in solution. Biochemistry 29, 1689–1696 [DOI] [PubMed] [Google Scholar]
- 20. Fairbrother W. J., Reilly D., Colby T. J., Hesselgesser J., Horuk R. (1994) The solution structure of melanoma growth stimulating activity. J. Mol. Biol. 242, 252–270 [DOI] [PubMed] [Google Scholar]
- 21. Jansma A. L., Kirkpatrick J. P., Hsu A. R., Handel T. M., Nietlispach D. (2010) NMR analysis of the structure, dynamics, and unique oligomerization properties of the chemokine CCL27. J. Biol. Chem. 285, 14424–14437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lodi P. J., Garrett D. S., Kuszewski J., Tsang M. L., Weatherbee J. A., Leonard W. J., Gronenborn A. M., Clore G. M. (1994) High-resolution solution structure of the β chemokine hMIP-1 β by multidimensional NMR. Science 263, 1762–1767 [DOI] [PubMed] [Google Scholar]
- 23. Malkowski M. G., Wu J. Y., Lazar J. B., Johnson P. H., Edwards B. F. (1995) The crystal structure of recombinant human neutrophil-activating peptide-2 (M6L) at 1.9-A resolution. J. Biol. Chem. 270, 7077–7087 [DOI] [PubMed] [Google Scholar]
- 24. Nasser M. W., Raghuwanshi S. K., Grant D. J., Jala V. R., Rajarathnam K., Richardson R. M. (2009) Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J. Immunol. 183, 3425–3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murphy P. M. (1997) Neutrophil receptors for interleukin-8 and related CXC chemokines. Semin. Hematol. 34, 311–318 [PubMed] [Google Scholar]
- 26. Stillie R., Farooq S. M., Gordon J. R., Stadnyk A. W. (2009) The functional significance behind expressing two IL-8 receptor types on PMN. J. Leukoc. Biol. 86, 529–543 [DOI] [PubMed] [Google Scholar]
- 27. Rajarathnam K., Prado G. N., Fernando H., Clark-Lewis I., Navarro J. (2006) Probing receptor binding activity of interleukin-8 dimer using a disulfide trap. Biochemistry 45, 7882–7888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gangavarapu P., Rajagopalan L., Kolli D., Guerrero-Plata A., Garofalo R. P., Rajarathnam K. (2012) The monomer-dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J. Leukocyte Biol. 91, 259–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rajasekaran D., Keeler C., Syed M. A., Jones M. C., Harrison J. K., Wu D., Bhandari V., Hodsdon M. E., Lolis E. J. (2012) A model of GAG/MIP-2/CXCR2 interfaces and its functional effects. Biochemistry 51, 5642–5654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoogewerf A. J., Kuschert G. S., Proudfoot A. E., Borlat F., Clark-Lewis I., Power C. A., Wells T. N. (1997) Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 36, 13570–13578 [DOI] [PubMed] [Google Scholar]
- 31. Ziarek J. J., Veldkamp C. T., Zhang F., Murray N. J., Kartz G. A., Liang X., Su J., Baker J. E., Linhardt R. J., Volkman B. F. (2013) Heparin oligosaccharides inhibit chemokine (CXC motif) ligand 12 (CXCL12) cardioprotection by binding orthogonal to the dimerization interface, promoting oligomerization, and competing with the chemokine (CXC motif) receptor 4 (CXCR4) N terminus. J. Biol. Chem. 288, 737–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Devalaraja R. M., Nanney L. B., Du J., Qian Q., Yu Y., Devalaraja M. N., Richmond A. (2000) Delayed wound healing in CXCR2 knockout mice. J. Invest. Dermatol. 115, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jang J. E., Hod E. A., Spitalnik S. L., Frenette P. S. (2011) CXCL1 and its receptor, CXCR2, mediate murine sickle cell vaso-occlusion during hemolytic transfusion reactions. J. Clin. Invest. 121, 1397–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ravindran A., Sawant K. V., Sarmiento J., Navarro J., Rajarathnam K. (2013) Chemokine CXCL1 dimer is a potent agonist for the CXCR2 receptor. J. Biol. Chem. 288, 12244–12252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Permi P. (2004) Coherence transfer in proteins. Prog. Nucl. Magn. Reson. Spectrosc. 44, 97–137 [Google Scholar]
- 36. Ravindran A., Joseph P. R., Rajarathnam K. (2009) Structural basis for differential binding of the interleukin-8 monomer and dimer to the CXCR1 N-domain. Role of coupled interactions and dynamics. Biochemistry 48, 8795–8805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 38. Johnson B. A., Blevins R. A. (1994) NMR View. A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 39. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., Kay L. E. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 [DOI] [PubMed] [Google Scholar]
- 40. Wishart D. S., Arndt D., Berjanskii M., Tang P., Zhou J., Lin G. (2008) CS23D. A web server for rapid protein structure generation using NMR chemical shifts and sequence data. Nucleic Acids Res. 36, W496–W502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Case D. A., Cheatham T. E., 3rd, Darden T., Gohlke H., Luo R., Merz K. M., Jr., Onufriev A., Simmerling C., Wang B., Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR. Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 43. de Vries S. J., van Dijk A. D., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., Bonvin A. M. (2007) HADDOCK versus HADDOCK. New features and performance of HADDOCK2.0 on the CAPRI targets. Proteins 69, 726–733 [DOI] [PubMed] [Google Scholar]
- 44. Mulloy B., Forster M. J., Jones C., Davies D. B. (1993) N.M.R., and molecular-modelling studies of the solution conformation of heparin. Biochem. J. 293, 849–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sharma D., Rajarathnam K. (2000) 13C NMR chemical shifts can predict disulfide bond formation. J. Biomol. NMR 18, 165–171 [DOI] [PubMed] [Google Scholar]
- 46. Murphy J. W., Cho Y., Sachpatzidis A., Fan C., Hodsdon M. E., Lolis E. (2007) Structural and functional basis of CXCL12 (stromal cell-derived factor-1 α) binding to heparin. J. Biol. Chem. 282, 10018–10027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shaw J. P., Johnson Z., Borlat F., Zwahlen C., Kungl A., Roulin K., Harrenga A., Wells T. N., Proudfoot A. E. (2004) The X-ray structure of RANTES. Heparin-derived disaccharides allow the rational design of chemokine inhibitors. Structure 12, 2081–2093 [DOI] [PubMed] [Google Scholar]
- 48. Laguri C., Sapay N., Simorre J. P., Brutscher B., Imberty A., Gans P., Lortat-Jacob H. (2011) 13C-labeled heparan sulfate analogue as a tool to study protein/heparan sulfate interactions by NMR spectroscopy. Application to the CXCL12α chemokine. J. Am. Chem. Soc. 133, 9642–9645 [DOI] [PubMed] [Google Scholar]
- 49. Kuschert G. S., Hoogewerf A. J., Proudfoot A. E., Chung C. W., Cooke R. M., Hubbard R. E., Wells T. N., Sanderson P. N. (1998) Identification of a glycosaminoglycan binding surface on human interleukin-8. Biochemistry 37, 11193–11201 [DOI] [PubMed] [Google Scholar]
- 50. Pichert A., Samsonov S. A., Theisgen S., Thomas L., Baumann L., Schiller J., Beck-Sickinger A. G., Huster D., Pisabarro M. T. (2012) Characterization of the interaction of interleukin-8 with hyaluronan, chondroitin sulfate, dermatan sulfate and their sulfated derivatives by spectroscopy and molecular modeling. Glycobiology 22, 134–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gandhi N. S., Mancera R. L. (2011) Molecular dynamics simulations of CXCL-8 and its interactions with a receptor peptide, heparin fragments, and sulfated linked cyclitols. J. Chem. Inf. Model. 51, 335–358 [DOI] [PubMed] [Google Scholar]
- 52. Spillmann D., Witt D., Lindahl U. (1998) Defining the interleukin-8-binding domain of heparan sulfate. J. Biol. Chem. 273, 15487–15493 [DOI] [PubMed] [Google Scholar]
- 53. Lortat-Jacob H., Grosdidier A., Imberty A. (2002) Structural diversity of heparan sulfate binding domains in chemokines. Proc. Natl. Acad. Sci. U.S.A. 99, 1229–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bitomsky W., Wade R. C. (1999) Docking of glycosaminoglycans to heparin-binding proteins. Validation for aFGF, bFGF, and antithrombin and applications to IL-8. J. Am. Chem. Soc. 121, 3004–3013 [Google Scholar]
- 55. Jasnin M. (2012) Use of neutrons reveals the dynamics of cell surface glycosaminoglycans. Methods Mol. Biol. 836, 161–169 [DOI] [PubMed] [Google Scholar]
- 56. Bazar E., Jelinek R. (2010) Divergent heparin-induced fibrillation pathways of a prion amyloidogenic determinant. ChemBioChem. 11, 1997–2002 [DOI] [PubMed] [Google Scholar]
- 57. Schedin-Weiss S., Richard B., Olson S. T. (2010) Kinetic evidence that allosteric activation of antithrombin by heparin is mediated by two sequential conformational changes. Arch. Biochem. Biophys. 504, 169–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Laguri C., Sadir R., Rueda P., Baleux F., Gans P., Arenzana-Seisdedos F., Lortat-Jacob H. (2007) The novel CXCL12gamma isoform encodes an unstructured cationic domain which regulates bioactivity and interaction with both glycosaminoglycans and CXCR4. PLoS ONE 2, e1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McCornack M. A., Boren D. M., LiWang P. J. (2004) Glycosaminoglycan disaccharide alters the dimer dissociation constant of the chemokine MIP-1 β. Biochemistry 43, 10090–10101 [DOI] [PubMed] [Google Scholar]
- 60. Tzeng S. R., Kalodimos C. G. (2012) Protein activity regulation by conformational entropy. Nature 488, 236–240 [DOI] [PubMed] [Google Scholar]
- 61. Kurupati P., Turner C. E., Tziona I., Lawrenson R. A., Alam F. M., Nohadani M., Stamp G. W., Zinkernagel A. S., Nizet V., Edwards R. J., Sriskandan S. (2010) Chemokine-cleaving Streptococcus pyogenes protease SpyCEP is necessary and sufficient for bacterial dissemination within soft tissues and the respiratory tract. Mol. Microbiol. 76, 1387–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sadir R., Imberty A., Baleux F., Lortat-Jacob H. (2004) Heparan sulfate/heparin oligosaccharides protect stromal cell-derived factor-1 (SDF-1)/CXCL12 against proteolysis induced by CD26/dipeptidyl peptidase IV. J. Biol. Chem. 279, 43854–43860 [DOI] [PubMed] [Google Scholar]
- 63. Kim K. S., Clark-Lewis I., Sykes B. D. (1994) Solution structure of GRO/melanoma growth stimulatory activity determined by 1H NMR spectroscopy. J. Biol. Chem. 269, 32909–32915 [PubMed] [Google Scholar]
- 64. Qian Y. Q., Johanson K. O., McDevitt P. (1999) Nuclear magnetic resonance solution structure of truncated human GROβ [5–73] and its structural comparison with CXC chemokine family members GROα and IL-8. J. Mol. Biol. 294, 1065–1072 [DOI] [PubMed] [Google Scholar]
- 65. Shao W., Jerva L. F., West J., Lolis E., Schweitzer B. I. (1998) Solution structure of murine macrophage inflammatory protein-2. Biochemistry 37, 8303–8313 [DOI] [PubMed] [Google Scholar]