

Background: Chemokines play a prominent role in inflammatory diseases.
Results: Nanobodies targeting chemokines display high affinity and potently neutralize chemokine-induced receptor binding and signaling.
Conclusion: Neutralizing Nanobodies targeting chemokines effectively inhibit chemokine function.
Significance: Nanobodies directed against inflammatory and homeostatic chemokines form a promising new class of potent and specific inhibitors of chemokine function, to be used for research and therapeutic purposes.
Keywords: Antibodies, Chemokines, Chemotaxis, G Protein-coupled Receptors (GPCR), Radioreceptor Assays, Nanobodies
Abstract
Chemokine receptors and their ligands play a prominent role in immune regulation but many have also been implicated in inflammatory diseases such as multiple sclerosis, rheumatoid arthritis, allograft rejection after transplantation, and also in cancer metastasis. Most approaches to therapeutically target the chemokine system involve targeting of chemokine receptors with low molecular weight antagonists. Here we describe the selection and characterization of an unprecedented large and diverse panel of neutralizing Nanobodies (single domain camelid antibodies fragment) directed against several chemokines. We show that the Nanobodies directed against CCL2 (MCP-1), CCL5 (RANTES), CXCL11 (I-TAC), and CXCL12 (SDF-1α) bind the chemokines with high affinity (at nanomolar concentration), thereby blocking receptor binding, inhibiting chemokine-induced receptor activation as well as chemotaxis. Together, we show that neutralizing Nanobodies can be selected efficiently for effective and specific therapeutic treatment against a wide range of immune and inflammatory diseases.
Introduction
Chemokines (chemotactic cytokines) are small peptides of ∼8–10 kDa that along with their receptors, belonging to the family of G protein-coupled receptors (GPCRs),5 form the chemokine system (1). Chemokines are important mediators of inflammation and are involved in a variety of immune and inflammatory diseases. Based on the location of conserved cysteine residues in their primary sequence, chemokines are divided into four subfamilies and named accordingly: CXC, CC, XC, and CX3C (1). Chemokines are often classified as inducible (inflammatory) or constitutive (homeostatic) chemokines. Inflammatory chemokines are expressed and released after injury or during infection, and are involved in the attraction of leukocytes, a process called chemotaxis. In contrast, constitutive chemokines are expressed in the absence of damage, and control developmental cell trafficking and homeostatic leukocyte homing during immune surveillance (2). Antagonizing the chemokine receptor interaction is considered to be beneficial in inflammatory disorders. Experimental therapies range from low molecular weight receptor antagonists, antibodies directed against chemokine or chemokine receptors, interference with chemokine-glycosaminoglycan interactions, and viral chemokine-binding proteins (3–6).
In this study, we explored a novel approach to target the chemokine system, using so-called Nanobodies. Nanobodies (NB) are therapeutic proteins based on the smallest functional fragments of heavy chain antibodies, naturally occurring in Camelidae. The camelid family (Llama and dromedary) have the striking characteristic of having conventional (composed of heavy and light chains) as well as heavy chain antibodies (composed of heavy chains only) (7, 8). To compensate for the reduced antigen binding interface (usually formed by both the heavy and light chain), heavy chain antibodies have evolved toward longer complementary determining regions (CDRs) in the variable domain. As a result, and combined with their small size (∼12–15 kDa) Nanobodies can recognize uncommon and cryptic epitopes, hidden or shielded from the much larger conventional antibodies, and bind into cavities or active sites (9, 10). Because of their compact single domain structure, Nanobodies are more stable at extreme pH and temperatures (11–14). Some Nanobodies have been shown to survive the harsh conditions of the stomach and remain biologically active in the gut due to a better resistance to proteases (15). In contrast to conventional antibodies, Nanobodies are encoded by single genes and are efficiently produced in almost all prokaryotic and eukaryotic hosts including bacteria and yeast (16). Furthermore, through engineering methods Nanobodies can be formatted to increase their half-life varying from 30 min to 3 weeks (16, 17), increasing their therapeutic range from acute to chronic indications.
Targets, such as GPCRs proven to be difficult to be targeted therapeutically by monoclonal antibodies were effectively targeted by Nanobodies. Earlier, we have identified Nanobodies formatted as a bivalent molecule binding and antagonizing CXCR4 function, showing high specificity, affinity, and inverse agonistic potency (18). Several Nanobodies, such as ALX-0081, a bivalent Nanobody targeting the von Willebrand factor, are currently in clinical trials, demonstrating the clinical potential of Nanobodies (see www.ablynx.com).
In this study, we generated and characterized Nanobodies against the inflammatory chemokines CCL2 (MCP-1), CCL5 (RANTES), and CXCL11 (I-TAC), as well as the homeostatic chemokine CXCL12 (SDF-1α). These chemokines are involved in a wide range of disorders: CCL2 binds the chemokine receptor CCR2, implicated in multiple sclerosis (19), rheumatoid arthritis (20), diabetic nephropathy (21), atherosclerosis (22), and cancer (23). CCL5 binds CCR1, CCR3, CCR5, thought to be involved in inflammatory diseases, transplant rejection, and cancer (1, 3). The chemokine CXCL11 binds CXCR3, thought to be involved in multiple sclerosis (24), rheumatoid arthritis (25), atherosclerosis (26), and inflammatory skin diseases such as psoriasis and to play a role in transplant rejection (27) and metastasis of cancer cells (28). CXCL11 and CXCL12 both bind to the recently de-orphanized receptor CXCR7, highly expressed on tumor cells (29). CXCL12 binds to CXCR4, which is implicated in metastasis (3–5, 30). In this study we show that the Nanobodies generated bind their target chemokines with high affinity and potently neutralize chemokine-induced signaling.
EXPERIMENTAL PROCEDURES
Materials
Dulbecco's modified Eagle's medium (DMEM), RPMI 1640 with l-glutamine, and 25 mm HEPES and trypsin were purchased from PAA Laboratories GmbH (Pasching, Austria), non-essential amino acids, sodium pyruvate, 2-mercaptoethanol, glutamine, penicillin, and streptomycin were obtained from Cambrex Bio Science (Verviers, Belgium), fetal bovine serum (FBS) was purchased from Integro B.V. (Dieren, The Netherlands), certified FBS was from Invitrogen. myo-[2-3H]Inositol (10–20 Ci/mmol) was from GE Healthcare. 125I-CXCL11 was obtained from GE Healthcare or PerkinElmer Life Sciences, 125I-CCL2, 125I-CCL5, and 125I-CXCL12 were from PerkinElmer Life Sciences. Unlabeled chemokines were obtained from PeproTech (Rocky Hill, NJ).
Llama Immunization and Phage Display Library Construction
Two llamas were immunized according to standard protocols with 6 biweekly intramuscular injection of a chemokine mixture. The mixture was a mixture of carrier-free human recombinant protein acquired from R&D Systems and included CCL2-Mucin stalk chimera (catalog number 979-MC/CF), CCL3/MIP1α (catalog number 270-LD/CF), CCL5/RANTES-Mucin stalk chimera (catalog number 978-RN/CF), CXCL11/I-TAC (catalog number 672-IT/CF), CXCL12/SDF1α (catalog number 350-NS/CF), and adjuvant (incomplete Freund's adjuvant). Blood was collected at 4 and 8 days after the last injection. Peripheral blood mononuclear cells were prepared from blood samples using Ficoll-Hypaque according to the manufacturer's instructions. Next, total RNA was extracted from these cells and used as starting material for RT-PCR to amplify Nanobody-encoding gene fragments. These fragments were cloned into phagemid vector pAX50 (31). Phage was prepared according to standard methods and stored at 4 °C for further use, making phage library 100 and 101.
Phage Display Selections
To identify Nanobodies recognizing the chemokines, phage libraries 100 and 101 were used for selections on the biotinylated chemokines. The biotinylated chemokines were immobilized independently at 5, 0.5, or 0 μg/ml (control) on Nunc Maxisorp ELISA plates previously coated with Neutravidine (5 μg/ml). For CCL2-mucin-like stalk chimera and CCL5-mucin-like stalk chimera, non-relevant mucin stalk protein was added during the selection procedure to ensure that chemokine-specific phage are selected (and excluding mucin stalk specific phages). After 2 h incubation in PBS containing 4% nonfat milk, the non-bound phages were washed away by 20× PBST (PBS + 0.2% Tween 20). Bound phages were eluted from the chemokines using triethylamine. Output of the first round of selections (R1) were analyzed for enrichment factor (phage present in eluate relative to controls). Based on these parameters the best selections were chosen for further analysis. Individual colonies were picked and grown in 96-deep well plates (1 ml volume) and Nanobody expression was induced by addition of 1 mm isopropyl 1-thio-β-d-galactopyranoside. Periplasmic extracts containing soluble Nanobodies were extracted by a freeze/thaw cycle. Bacteria were pelleted and frozen at −20 °C for 4 h to overnight and then thawed at room temperature and resuspended in 100 μl of PBS. After at least 30 min shaking at room temperature and centrifugation, the supernatant containing significant amounts of Nanobodies was collected (is the periplasmic fraction). For large scale production of Nanobodies, the Nanobody-encoding cDNA was recloned in the pAX51 expression vector (pAX50-derived vector without gene3). After overnight induction of a 400-ml bacterial culture with 1 mm isopropyl 1-thio-β-d-galactopyranoside, the bacteria were pelleted and lysed by freeze-thawing in PBS. The Nanobodies were purified using their C-terminal His tag and TALON beads (Clontech Laboratories, Mountain View, CA) according to the manufacturer's recommendation and dialyzed against PBS. Purity was confirmed on SDS-PAGE and Coomassie staining.
Nanobody ELISA
To determine binding specificity to the chemokines, the clones were tested in an ELISA binding assay. In short, 2 μg/ml of chemokines (for CCL2) was immobilized directly on Polysorp microtiter plates (Nunc) or 0.5 μg/ml of biotinylated chemokines (for CCL3, mucin-CCL5 and CXCL12) were captured on Neutravidine-coated (2 μg/ml) Maxisorp microtiter plates (Nunc). Free binding sites were blocked using 4% Marvel in PBS. Next, 10 μl of periplasmic extract containing Nanobody of the different clones in 100 μl of 2% Marvel PBST were allowed to bind to the immobilized chemokine. After incubation and extensive washing, Nanobody binding was revealed using a mouse anti-Myc secondary antibody, which after a wash step was detected with HRP-conjugated goat anti-mouse antibodies. Binding specificity was determined based on OD values compared with controls having received no Nanobody. Alternatively, to enhance detection, mouse anti-Myc antibody was coated on maxisorp microtiter plates (Nunc) and free binding sites were blocked using 4% Marvel in PBS. Next, 10 μl of periplasm was added to capture the Nanobodies present in the periplasmic extract. After washing, biotinylated CXCL11 was added and detected after washing using streptavidin-HRP.
DNA Constructs
The cDNA of human CXCR3 inserted in pcDNA3 (32) was amplified by PCR and inserted into pcDEF3 (a gift from Dr. Langer (33)). The cDNA encoding the chimeric G protein Gαqi5 (pcDNA1-HA-mGαqi5) was a gift from Dr. Conklin (34). The cDNA of CCR1 was a gift from Dr. C. P. Tensen and subcloned into pcDEF3. pcDNA3.1-CCR2 and pcDNA3.1-CXCR4 were obtained from UMR cDNA Resource Center.
Cell Culture and Transfection
HEK293T cells were grown at 5% CO2 and 37 °C in DMEM supplemented with 10% FBS, penicillin, and streptomycin. HEK293T cells were transfected with 2.5 μg of plasmid encoding a chemokine receptor supplemented with 2.5 μg of pcDNA1-HA-mGαqi5 (for PLC activation experiments) or 2.5 μg of pcDEF3 using linear polyethyleneimine (Mr 25,000; Polysciences Inc., Warrington, PA). Briefly, a total of 5 μg of DNA was diluted in 250 μl of 150 mm NaCl. Subsequently 30 μg of polyethyleneimine in 250 μl of 150 mm NaCl was added to the DNA solution and incubated for 10 min at RT. The mixture was added to adherent HEK293T cells in 100-mm tissue culture dishes. The following day, cells were trypsinized, resuspended into culture medium, and plated in poly-l-lysine (Sigma)-coated assay plates. Mouse fibroblast NIH-3T3 cells stably expressing CXCR7 were cultured in DMEM supplemented with 10% calf serum.
Murine pre-B lymphoma L1.2 cells were grown in RPMI 1640 medium with l-glutamine and 25 mm HEPES, supplemented with 10% heat-inactivated certified FBS, penicillin, streptomycin, glutamine, non-essential amino acids, 2-mercaptoethanol, and sodium pyruvate. L1.2 cells were transfected with 1 μg of pcDEF3-CXCR3 per million cells using a Bio-Rad Gene Pulser II (330 V and 975 microfarads) and grown in culture medium supplemented with 10 mm sodium butyrate. Untransfected L1.2 cells were grown without sodium butyrate.
Chemokine Binding
Transfected HEK293T cells were seeded in poly-l-lysine-coated 48-well plates 24 h after transfection. NIH3T3 cells stably expressing CXCR7 were seeded in poly-l-lysine-coated 96-well plates. The next day, Nanobodies were incubated with radiolabeled chemokine (∼50 pm) in binding buffer (50 mm HEPES, pH 7.4, 1 mm CaCl2, 5 mm MgCl2, 0.5% BSA) with or without 100 mm NaCl for 1 h at RT while shaking. For screening experiments, Nanobodies from the library were incubated at a 10 times dilution with radiolabeled chemokine. Next, the solutions were transferred to 48-well plates containing the transfected HEK293T cells and incubated for 3–4 h at 4 °C. Subsequently, cells were washed three times with ice-cold binding buffer supplemented with 0.5 m NaCl. Subsequently, cells were lysed and counted in a Wallac Compugamma counter.
Phospholipase C Activation
Twenty-four h after transfection, HEK293T cells were seeded in poly-l-lysine-coated 24-well plates and labeled overnight in Earle's inositol-free minimal essential medium (Invitrogen) supplemented with 10% FBS, penicillin, and streptomycin and myo-[2-3H]inositol (2 μCi/ml). The next day, increasing concentrations of Nanobodies were incubated with chemokine (5 nm) in inositol phosphate assay buffer (20 mm HEPES, pH 7.4, 140 mm NaCl, 5 mm KCl, 1 mm MgSO4, 1 mm CaCl2, 10 mm glucose, and 0.05% (w/v) BSA) supplemented with 10 mm LiCl for 1 h at RT while shaking. Cells were washed with inositol phosphate assay buffer and incubated for 2 h at 5% CO2 at 37 °C with the Nanobody/chemokine solutions. The incubation was stopped by aspiration of the solutions and addition of ice-cold 10 mm formic acid. After incubation for 90 min at 4 °C, [3H]inositol phosphate were isolated by anion-exchange chromatography (Dowex AG1-X8 columns, Bio-Rad) and counted by liquid scintillation.
Chemotaxis
Migration experiments were performed with untransfected L1.2 cells (for CXCR4-mediated transfection) or 24 h after transfection of L1.2 cells with pcDEF3-CXCR3. Increasing concentrations of Nanobodies were incubated with chemokine (1 nm) in RPMI 1640 with l-glutamine and 25 mm HEPES supplemented with 0.1% BSA for 1 h at RT while shaking. For chemokine dose-response curves, chemokine dilutions were made in the same medium. Next, chemotaxis of L1.2 cells was determined using 5 μm pore ChemoTx 96-well plates (Neuroprobe Inc., MD). Briefly, ChemoTx plates were blocked for 30 min using RPMI 1640 with glutamax-I and 25 mm HEPES supplemented with 1% (w/v) BSA. After removal of the blocking medium, Nanobody/chemokine solutions were added to the wells. L1.2 cells were added on top of the membrane and incubated for 4 h in a humidified chamber at 5% CO2 at 37 °C. Subsequently, the cells that migrated into the wells were transferred to white 96-well plates and incubated with Calcein-AM (1 μg/ml) (Alexis Biochemicals, Lausen, Switzerland) for 30 min at 37 °C. Next, flurorescense (485/535 nm) was measured using a Wallac Victor2 and compared with a standard curve made with L1.2 cells.
Data Analysis
Nonlinear regression analysis of the data and calculation of pIC50 values was performed using Prism 4.03.
RESULTS
Generation and Selection of Nanobodies from Immune Libraries
To obtain Nanobodies with high binding affinities, active immunization of 2 llamas was performed with a mixture of recombinant human chemokines (see “Experimental Procedures” for details). To establish the method, we selected chemokines from different chemokine families based on their therapeutic relevance (see Introduction). The chemokines used were CCL2, CCL3, CCL5, CXCL11, and CXCL12. The Nanobodies were cloned in-frame to gene3 in the phagemid vector pAX50. For the selection of specific Nanobodies, the chemokines were biotinylated and captured on Nunc Maxisorp ELISA plates previously coated with Neutravidine. This approach was chosen to prevent the possible denaturation of the chemokines when coated directly onto the plate. Phage selection was done as described previously (31, 35). To test the binding of the monoclonal Nanobodies selected after a single round of selection, Nanobodies were produced as periplasmic fraction of the isopropyl 1-thio-β-d-galactopyranoside-induced bacterial clones and tested in ELISA. The success rate using this method (2–100%) (Table 1) shows a high hit rate for 3 of 5 targets using this approach. Yet, also for CCL5 and CXCL12 high affinity binders were obtained. All Nanobodies tested were specific for their target chemokine and were not binding to other chemokines (data not shown). In view of the large diversity found, we decided to focus on Nanobodies targeting CCL2, CCL5, and particularly CXCL11 and CXCL12.
TABLE 1.
Positive clones identified by Nanobody ELISA according to the selections type and elution using libraries 100 and 101
Depicted are the number of positive clones (out of 48 clones) and representative percentage of positive clones.
| Chemokine | Elution | Library 100 (% positive) | Library 101 (% positive) |
|---|---|---|---|
| CCL2 | TEAa | 15 (32%) | 38 (81%) |
| CCL3 | TEA | 20 (67%) | 39 (83%) |
| CCL5 | TEA | 1 (2%) | 6 (13%) |
| CXCL11 | TEA | 40 (100%) | 42 (87%) |
| CXCL12 | TEA | 3 (6%) | 0 |
a TEA, triethylamine.
Functional Nanobody Screening
Nanobodies were also tested for their neutralizing activity, i.e. their ability to inhibit interaction of the chemokines with their respective chemokine receptor. To develop a high-throughput strategy, Nanobodies were again tested as periplasmic fractions. Anti-chemokine Nanobodies were preincubated with the corresponding radiolabeled chemokine for 1 h, after which the ability of the radiolabeled chemokine to bind their respective receptor expressed in HEK293T cells was determined. Fig. 1A shows an example of the screening results for Nanobodies directed against CXCL11. A commercially available anti-CXCL11 antibody was used as a positive control to demonstrate blocking of binding of 125I-CXCL11 to CXCR3-expressing HEK293T cells. In most cases, the ELISA-positive Nanobodies inhibited binding of 125I-CXCL11 to CXCR3, whereas control samples containing PBS had no effect on binding. We observed that several Nanobodies not only inhibited specific binding of 125I-CXCL11 to CXCR3, but also reduced nonspecific binding of 125I-CXCL11, thereby almost completely blocking all radioligand binding to the cells.
FIGURE 1.
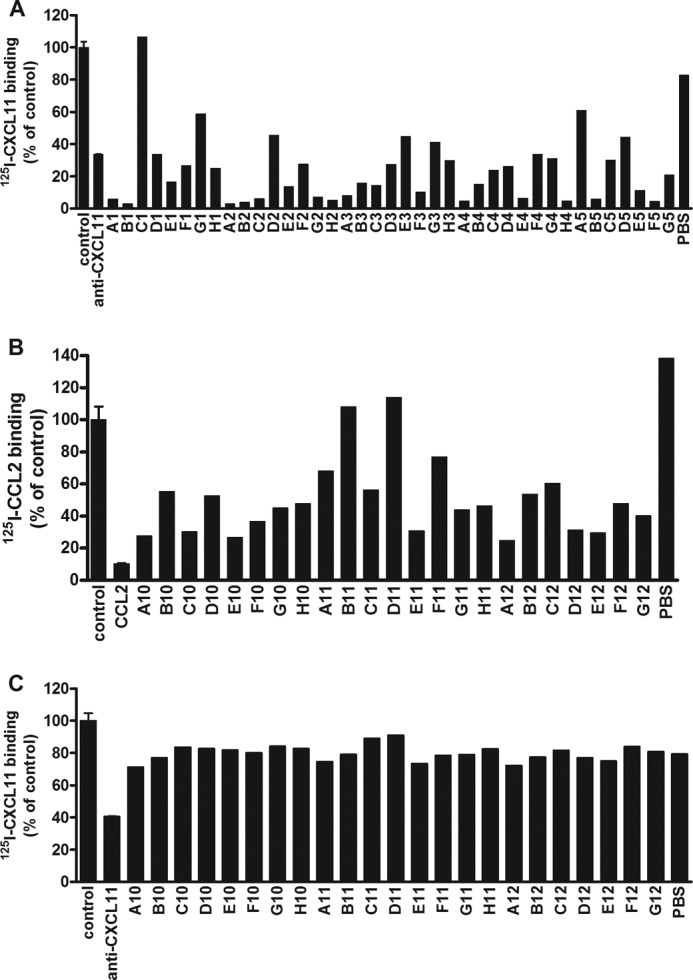
Screening and specificity of Nanobody libraries. A, screening of Nanobodies directed against CXCL11. Periplasm of anti-CXCL11 Nanobody-producing bacteria was diluted (1:10), incubated with 125I-CXCL11 (50 pm), and used for a binding experiment on CXCR3-expressing HEK293T. As positive control an anti-CXCL11 antibody (2 μg/ml) was used. B, screening of Nanobodies directed against CCL2. Periplasm of anti-CCL2 Nanobody-producing bacteria was diluted (1:10), incubated with 125I-CCL2 (50 pm), and used for a binding experiment on HCMV-US28-expressing HEK293T cells. As positive control unlabeled CCL2 was used. C, specificity of Nanobodies directed against CCL2. Periplasm of anti-CCL2 Nanobody-producing bacteria was diluted (1:10), incubated with 125I-CXCL11 (50 pm) and used for a binding experiment on CXCR3-expressing HEK293T. Experiments were performed in triplicate for the controls and single concentrations for the Nanobodies.
As a functional screening of anti-CCL2 Nanobodies (as periplasmic fraction), inhibition of the binding of 125I-CCL2 to the viral chemokine receptor US28, encoded by human cytomegalovirus (HCMV) was assessed (Fig. 1B). HCMV-US28 is expressed at higher levels in transiently transfected HEK293T cells than the human receptor for CCL2, i.e. CCR2, and therefore better qualified for screening purposes. Again, most binding Nanobodies identified by ELISA screening also inhibited binding to HCMV-US28. Similarly, Nanobodies directed against CCL5 were screened for competition of 125I-CCL5 binding to CCR1-expressing HEK293T cells, and a single clone of anti-CXCL12 Nanobody was tested for competition of 125I-CXCL12 binding to CXCR4-expressing HEK293T cells (data not shown), again demonstrating the presence of antagonistic Nanobodies for both chemokines.
The specificity of the anti-CCL2 Nanobodies was tested against CXCL11. As expected, the Nanobodies against CCL2 were not able to prevent the binding of 125I-CXCL11 to CXCR3 (Fig. 1C). Similarly, none of the other tested mismatching combinations displayed nonspecific effects (several anti-CCL2 Nanobodies were tested with 125I-CXCL12 and 125I-CCL5 as well; anti-CCL5 Nanobodies were used in combination with 125I-CCL2, 125I-CXCL11, and 125I-CXCL12; the anti-CXCL12 Nanobody was probed with 125I-CCL5 and 125I-CXCL11, data not shown).
Nanobodies Diversity
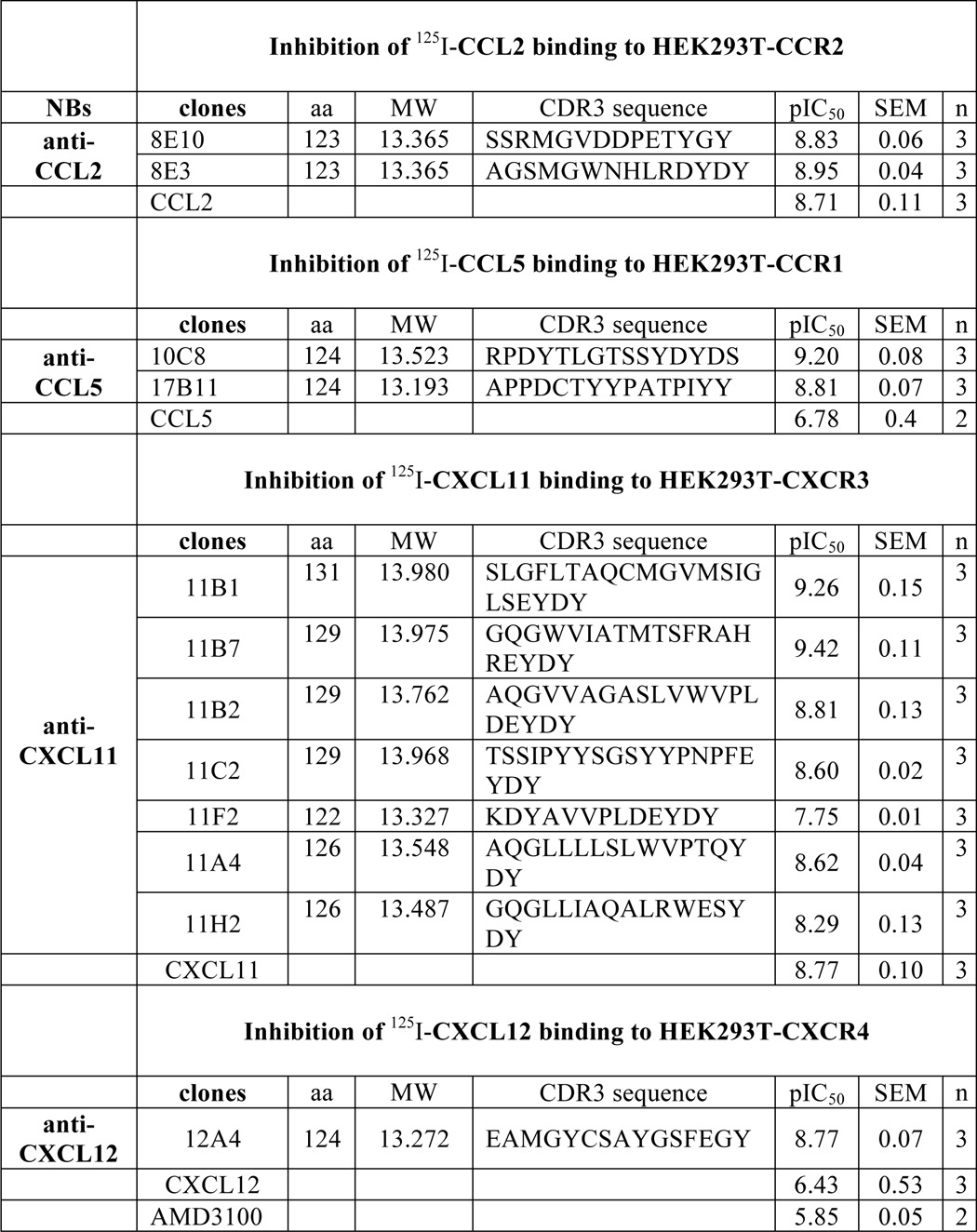
Large numbers of Nanobodies were selected based on the ELISA results and their neutralizing activities using the periplasm fraction, and were sequenced. A surprisingly large diversity of Nanobodies was found against several chemokines. As an example, out of 44 sequenced clones, positive against CXCL11, 31 different Nanobody sequences were found (1 sequence was found 10 times, 1 sequence was found 4 times, 1 sequence was found 2 times, and the rest were unique sequences). When those 31 Nanobody sequences were analyzed for homology, 18 different families (clones with identical CDR3 length and having related CDR3 sequence), among which half were represented by single clones (CDRs are too different to be grouped with any other Nanobodies). Differences between families are most apparent in the CDR3 regions, known to be highly variable and implicated in antigen recognition. All Nanobodies further characterized, with distinct amino acid sequences in the CDR3 region as depicted in Table 2, were derived from different families. The high diversity shows the power of such approach combining active immunization and phage display selection. This is especially remarkable for such a relatively small antigen such as the chemokines.
TABLE 2.
Potencies of different Nanobodies in binding assays
Nanobodies were incubated with approximately 50 pm radiolabeled chemokine. Length (number of amino acids (aa), molecular mass (kDa) and amino acid sequences of the CDR3 region of the Nanobodies (NB) are depicted. All selected Nanobodies are derived from different families.
Inhibition of Chemokine Binding
After analysis of the sequenced Nanobodies, a member of each family identified as well as several unique clones were selected for further analysis. Large quantities of each Nanobody were produced and purified (see “Experimental Procedures”). The potency of the purified Nanobodies was determined in radiolabeled ligand binding experiments, using increasing concentrations of the anti-chemokine Nanobodies. As can be seen in Fig. 2A, several anti-CXCL11 Nanobodies dose dependently inhibited the ability of 125I-CXCL11 to bind CXCR3. The Nanobodies covered a wide range of potencies, with pIC50 of 9.4 to 7.7 (IC50 ranging from 0.4 to 20 nm) (Table 2). The pIC50 value of unlabeled CXCL11 was 8.8 (IC50 2 nm).
FIGURE 2.

Inhibition of chemokine binding. A, inhibition of CXCL11 binding to CXCR3. Purified Nanobodies were incubated at the indicated concentrations with 125I-CXCL11 (50 pm). Subsequently, binding was analyzed on CXCR3-expressing HEK293T cells. The Nanobodies inhibited 125I-CXCL11 binding with the following pIC50 ± S.E. values: 11B7 (○), 9.4 ± 0.1 (n = 3); 11B1 (●), 9.3 ± 0.1 (n = 4); 11B2 (■), 8.8 ± 0.1 (n = 3); 11A4 (□), 8.6 ± 0.0 (n = 3); 11H2 (▴), 8.3 ± 0.1 (n = 3); 11F2 (▵), 7.7 ± 0.0 (n = 3). Unlabeled CXCL11 (♢, dashed line) inhibited 125I-CXCL11 binding with pIC50 ± S.E. of 8.8 ± 0.1. B, inhibition of CXCL12 binding to CXCR4. Purified Nanobody 12A4 was incubated at the indicated concentrations with 125I-CXCL12 (50 pm). Subsequently, binding was analyzed on CXCR4-expressing HEK293T cells. The Nanobody 12A4 inhibited 125I-CXCL12 binding with a pIC50 ± S.E. value of 8.8 ± 0.1 (n = 3). C, inhibition of CCL2 binding to CCR2. Purified Nanobodies were incubated at the indicated concentrations with 125I-CCL2 (50 pm). Subsequently, binding was performed on CCR2-expressing HEK293T cells. The Nanobodies inhibited 125I-CCL2 binding with the following pIC50 ± S.E. values: 8E3 (●), 9.0 ± 0.0 (n = 3); 8E10 (○), 8.8 ± 0.1 (n = 3). D, inhibition of CCL5 binding to CCR1. Purified Nanobodies were incubated at the indicated concentrations with 125I-CCL5 (50 pm). Subsequently, binding was performed on CCR1-expressing HEK293T cells. The Nanobodies inhibited 125I-CCL5 binding with the following pIC50 ± S.E. values: 17B11 (●), 8.8 ± 0.1 (n = 3); 10C8 (○), 9.2 ± 0.1 (n = 3). Experiments were performed in duplicate and repeated the indicated amount of times. E, inhibition of CXCL12 and CXCL11 binding to CXCR7. CXCL12 (100 nm) and CXCL11 (100 nm) effectively displace 125I-CXCL12 (50 pm). Purified Nanobody 12A4 (1 μm) was incubated with 125I-CXCL12 (50 pm) and CXCL11 (100 nm) with CXCL11 Nanobodies 11B1, 11B7, and 11B2 (1 μm) and binding was analyzed on CXCR7-expressing NIH-3T3 cells. Purified anti-CXCL12 Nanobody 12A4 prevented binding of 125I-CXCL12 to CXCR7 and preincubation of CXCL11 with purified anti-CXCL11 Nanobodies 11B1, 11B7, and 11B2 neutralized CXCL11 allowing 125I-CXCL12 to bind to CXCR7 (n = 4).
Similar experiments were performed using the anti-CXCL12 Nanobody, 12A4, resulting in a pIC50 of 8.8 (IC50 2 nm) (Fig. 2B). Dose-inhibition curves for anti-CCL2 Nanobodies were generated using 125I-CCL2 at the human receptor CCR2, instead of HCMV-US28, which was used in the initial screen (Fig. 2C). Dose-inhibition curves for anti-CCL5 Nanobodies were generated using 125I-CCL5 and CCR1-expressing HEK293T cells (Fig. 2D). The anti-CCL2 and anti-CCL5 Nanobodies all inhibited chemokine binding at nanomolar concentration (Table 2).
Because CXCR7 is known to bind both CXCL11 and CXCL12 (29, 36), we determined whether preincubation of anti-CXCL12 and anti-CXCL11 Nanobodies affect CXCL12 and CXCL11 binding, respectively, to CXCR7-expressing NIH-3T3 cells. As can be seen in Fig. 2E anti-CXCL12 Nanobodies 12A4 prevented binding of 125I-CXCL12 to CXCR7 (Fig. 2E). CXCL11 fully displaced 125I-CXCL12, whereas preincubation of CXCL11 with anti-CXCL11 Nanobodies 11B1, 11B7, and 11B2 did not inhibit binding of 125I-CXCL12 to CXCR7 (Fig. 2E). These data show that anti-CXCL11 and CXCL12 Nanobodies also neutralized chemokine binding to CXCR7.
Inhibition of Chemokine Receptor Activation
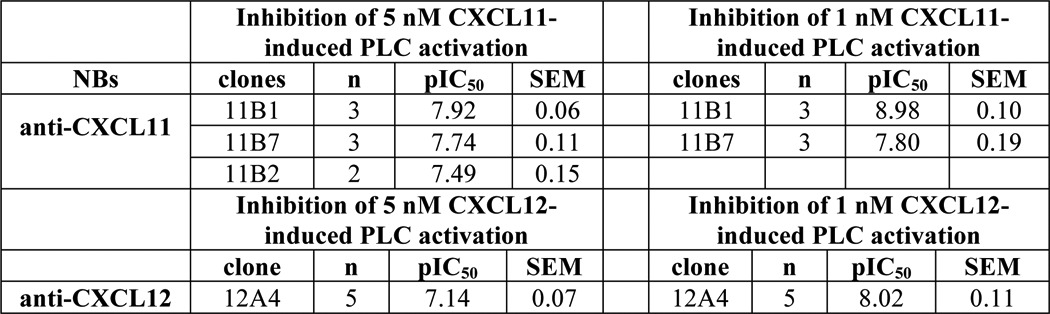
Next, we determined the ability of the Nanobodies to inhibit CXCL11- and CXCL12-induced receptor activation. Chemokine receptors were co-transfected with the chimeric G protein Gαqi5 (34) in HEK293T cells to measure activation of phospholipase C (PLC). Increasing concentrations of Nanobodies were incubated for 1 h with chemokine (5 nm), after which chemokine-mediated PLC activation was measured. The anti-CXCL11 Nanobodies 11B1 and 11B7 inhibited CXCR3 activation with pIC50 of 7.9 and 7.7 respectively, corresponding with IC50 values between 10 and 20 nm (Fig. 3A). Similarly, the anti-CXCL12 Nanobody 12A4 inhibited CXCL12-mediated CXCR4 activation with a pIC50 of 7.1 (IC50 of 80 nm) (Fig. 3B, Table 3).
FIGURE 3.
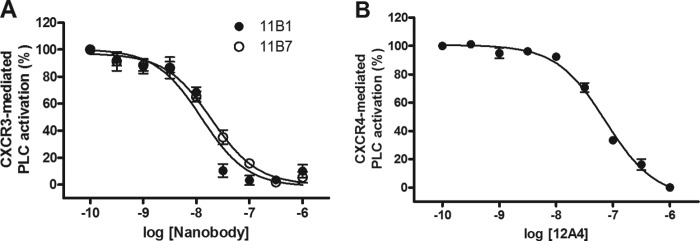
Inhibition of chemokine receptor activation. A, inhibition of CXCR3 activation. Purified Nanobodies were incubated at the indicated concentrations with CXCL11 (5 nm). Subsequently, CXCR3-mediated PLC activation was determined in HEK293T cells co-expressing CXCR3 and Gαqi5. The Nanobodies inhibited CXCL11-induced signaling with the following pIC50 ± S.E. values: 11B1 (●), 7.9 ± 0.1 (n = 3); 11B7 (○), 7.7 ± 0.1 (n = 3). B, inhibition of CXCR4 activation. Purified 12A4 Nanobody was incubated at the indicated concentrations with CXCL12 (5 nm). Subsequently, CXCR4-mediated PLC activation was determined in HEK293T cells co-expressing CXCR4 and Gαqi5. Nanobody 12A4 inhibited CXCL12-induced signaling with a pIC50 ± S.E. value of 7.1 ± 0.1 (n = 5).
TABLE 3.
Inhibition of Nanobodies (NBs) in functional assays
Inhibition of Chemotaxis
One of the major downstream effects of chemokine receptor activation is cellular migration. We determined the ability of the Nanobodies to inhibit chemokine-induced migration of L1.2 cells, a murine pre-B lymphoma cell line. CXCR3-transfected L1.2 cells migrated to increasing concentrations of CXCL11, resulting in a typical bell-shaped curve characteristic for chemotaxis assays (Fig. 4A). L1.2 cells transfected with an empty vector did not migrate toward CXCL11 (data not shown). Anti-CXCL11 Nanobodies were incubated with CXCL11 (1 nm) for 1 h and chemotaxis of CXCR3-expressing L1.2 cells was determined. The Nanobodies 11B1 and 11B7 inhibited CXCR3-mediated migration with pIC50 of 9.0 and 7.8, respectively, corresponding with IC50 values between 1 and 16 nm (Fig. 4B, Table 3). Subsequently, migration of L1.2 cells, which endogenously express CXCR4, to increasing concentrations of CXCL12 was determined (Fig. 4C). Addition of the CXCR4 antagonist AMD3100 (10 μm) inhibited CXCL12-induced (1 nm) migration, indicating that migration was mediated by CXCR4 (Fig. 4C). As can be seen in Fig. 4D, the anti-CXCL12 Nanobody 12A4 inhibited CXCR4-mediated migration with a pIC50 of 7.9 (corresponding with an IC50 of 13 nm) (Table 3).
FIGURE 4.

Inhibition of chemotaxis. A, migration of CXCR3-expressing L1.2 cells. A migration assay with increasing concentrations of CXCL11 was performed using L1.2 cells transfected with cDNA encoding CXCR3. Data are shown as percentage of migrated cells and obtained in three experiments. B, inhibition of CXCL11-induced chemotaxis. Purified Nanobodies were preincubated at the indicated concentrations with CXCL11 (1 nm). Subsequently, CXCL11-induced migration of CXCR3-expressing L1.2 cells was determined. The Nanobodies inhibited CXCL11-induced chemotaxis with the following pIC50 ± S.E. values: 11B1 (●), 9.0 ± 0.1 (n = 4); 11B7 (○), 7.8 ± 0.2 (n = 4). C, migration of L1.2 cells. A migration assay with increasing concentrations of CXCL12 was performed using L1.2 cells. The CXCR4 antagonist AMD3100 (10 μm) inhibited migration toward CXCL12 (1 nm). Data are shown as percentage of migrated cells and was obtained in three experiments. D, inhibition of CXCL12-induced chemotaxis. Purified 12A4 Nanobody was incubated at the indicated concentrations with CXCL12 (1 nm) for 1 h at RT while shaking. Subsequently, CXCL12-induced migration of L1.2 cells was determined. The 12A4 Nanobody inhibited CXCL12-induced chemotaxis with a pIC50 ± S.E. value of 7.9 ± 0.1 (n = 5). Experiments were performed in triplicate.
DISCUSSION
Chemokines and their cognate GPCRs are important mediators of the inflammatory response (1). Consequently, they are also involved in many inflammatory diseases, (auto-)immune diseases, and cancer. In general, GPCRs are readily targeted with low molecular weight antagonists, exemplified by the notion that GPCRs are targeted by more than 30% of clinically marketed drugs (37). However, despite the existence of about 20 chemokine receptors, there are currently only two drugs on the market that target chemokine receptor, i.e. the HIV entry inhibitor Maraviroc, which binds to CCR5 (38) and CXCR4 antagonist AMD3100 (Mozobil). Recently, the anti-CCR4-ADCC antibody has been approved for adult T cell leukemia lymphoma treatment for use in Japan (39), showing that it is also feasible to therapeutically target chemokine receptors with biologicals. The low success rate of chemokine receptor antagonists reaching the market may be due to their relatively recent discovery, the first chemokine receptor being reported in 1991. Yet, the complexity and apparent promiscuity of the chemokine system with numerous peptide chemokines exerting their effects by binding to a wide range of receptors may also account for this. When a small molecule compound inhibits a chemokine receptor, some chemokine ligands may still bind and signal via another chemokine receptor. Blocking chemokine action or removal of chemokines by chemokine-binding proteins to lower immune response is largely exploited by pathogens and parasites that infect mammals including humans (40, 41). Therefore, targeting the chemokine instead of targeting the receptor can be considered as an interesting alternative.
A small molecule compound from the family of chalcones was shown to bind CXCL12 (42). The chalcone inhibited chemotaxis of human peripheral blood lymphocytes with an affinity of about 1 μm and was effective in vivo in a mouse model of allergic eosinophilic airway inflammation. In addition, targeting sulfotyrosine binding sites on chemokines, as shown for CXCL12 and of importance for CXCR4 binding, appears effective (43). Targeting chemokines with antibodies in animal models has also proven successful in a number of cases (44). Anti-CCL2 antibody treatment during relapsing experimental autoimmune encephalomyelitis, a mouse model for multiple sclerosis, decreased clinical severity of relapsing disease and reduced CNS macrophage accumulation during relapsing EAE (45). Using a tumor cell-mediated angiogenesis assay, it was shown that an anti-CCL2 antibody has anti-angiogenic properties in vivo (46) and mediate prostate cancer growth through macrophage infiltration (47, 48). The anti-CCL2 antibody is now in phase II clinical study for prostate cancer. Anti-CCL5 delayed rejection time in an experimental rat model of cardiac allograft rejection (49). An anti-CCL5 antibody and has reached phase I clinical study. Finally, several antibodies targeted against CXCL10 are in phase 2 clinical trial for rheumatoid arthritis, ulcerative colitis, Crohn disease, or primary biliary cirhosis (for review, see Klareenbeek et al. (6)). In a mouse model of allergic airway disease, antibodies against CXCL12 resulted in a reduction in lung allergic inflammation and airway hyper-responsiveness (50).
Here, we report on the generation and characterization of Nanobodies, a relatively novel class of highly effective biologicals, directed against CCL2, CCL5, CXCL11, and CXCL12. The Nanobodies prevented binding of the chemokines to their receptors with nanomolar affinities, thereby effectively preventing the chemokine to signal via its respective receptor, neutralizing chemokine receptor activation and chemotaxis. Nanobodies targeting CXCL11 and CXCL12, e.g. prevented binding of these chemokines to, respectively, CXCR3 and CXCR4 as well as CXCR7, known to bind both chemokines as well (29, 36). This further illustrates the broad neutralizing capacity of Nanobodies in targeting chemokines.
For some chemokines, e.g. CXCL11, various families of Nanobodies were identified, whereas for others, e.g. CXCL12 and CCL5, only a limited number of clones were obtained. The difference in diversity and the low immunogenicity may be explained by a technical reason (biotinylation of the chemokine can cause loss of epitope recognition) or may result from high sequence conservation between the llama and human chemokines (CXCL12 95%, CCL5 85%, and CXCL11 81% identity between the two species).
Interestingly, the anti-CXCL11 Nanobodies 11B7 and 11B1 inhibited 125I-CXCL11 binding with a 3–4-fold lower IC50 value than unlabeled CXCL11, indicating the high neutralizing activity of the Nanobodies. Moreover, most Nanobodies inhibited binding of radiolabeled chemokines to their cognate receptors as well as dramatically lowered nonspecific binding of the radioligand to the cells. The nonspecific binding most likely entails binding to glycosaminoglycans, which is of importance for a large number of chemokines (51). In a physiological setting this would be equivalent to complete removal of the Nanobody-bound chemokine from circulation, also taking into account the small molecular weight of the complex of chemokine-Nanobody, which would allow rapid clearance of the bound chemokine. Functional receptor activation, measured as PLC activation and chemotaxis was inhibited by the Nanobodies against CXCL12 or CXCL11 with affinities between 1 and 80 nm. This in vitro experimental setup is relevant because we already showed that blocking the CXCL12/CXCR4 axis effectively abrogates the mobilization of CD34-positive stem cells in cynomolgus monkeys (18).
Nanobodies will specifically bind with high affinity to the chemokine and ensure that the chemokine is unavailable for receptor interaction and/or glycosaminoglycan interaction and are highly selective, whereas a small molecule shows possibly more off-target effects. In addition, small molecule compounds acting on CXCR4 such as AMD3100 and ALX40-4C have been reported to be partial agonists and T140 was found to have inverse agonistic properties (52). Because CXCR4 is involved in metastasis of breast cancer cells (30), especially compounds with partial agonistic effects may have unwanted effects. Likewise, we have shown that a series of CXCR3 antagonists behave as inverse agonists (53). Although the physiological consequences of these intrinsic activities of small molecule antagonists are still unknown, it may be safer to target the chemokines instead of their receptors. The potential of targeting chemokines was also recently demonstrated in vitro and in vivo by the Evasins. Evasins are natural chemokine-binding proteins from bloodsucking parasites such as ticks that allow immune response evasion. Evasins are even smaller than Nanobodies but do not show a strict chemokine specificity (54, 55). During inflammatory conditions neutralizing several chemokines at once may be of added value.
All together, the Nanobodies presented here, directed against inflammatory and homeostatic chemokines, form a promising new class of specific and powerful inhibitors of chemokine function, which can be used for research and therapeutic purposes.
Acknowledgment
We thank Francis Descamp (Ablynx, NV) for critically reviewing the manuscript.
This work was supported by Ablynx N.V.
- GPCR
- G protein-coupled receptor
- HCMV
- human cytomegalovirus
- I-TAC
- interferon-inducible T cell α chemoattractant
- MCP-1
- monocyte chemotactic protein-1
- PLC
- phospholipase C
- RANTES
- regulated upon activation, normal T-cell expressed, and secreted
- SDF-1α
- stromal cell derived factor 1 alpha
- TEA
- triethylamine
- CDR
- complementarity determining region
- NB
- Nanobodies.
REFERENCES
- 1. Murphy P. M., Baggiolini M., Charo I. F., Hébert C. A., Horuk R., Matsushima K., Miller L. H., Oppenheim J. J., Power C. A. (2000) International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol. Rev. 52, 145–176 [PubMed] [Google Scholar]
- 2. Comerford I., Nibbs R. J. (2005) Post-translational control of chemokines. A role for decoy receptors? Immunology Letters 96, 163–174 [DOI] [PubMed] [Google Scholar]
- 3. Allen S. J., Crown S. E., Handel T. M. (2007) Chemokine. Receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 25, 787–820 [DOI] [PubMed] [Google Scholar]
- 4. Jensen K. K., Chen S. C., Hipkin R. W., Wiekowski M. T., Schwarz M. A., Chou C. C., Simas J. P., Alcami A., Lira S. A. (2003) Disruption of CCL21-induced chemotaxis in vitro and in vivo by M3, a chemokine-binding protein encoded by murine gammaherpesvirus 68. J. Virol. 77, 624–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trinker M., Kungl A. (2012) Targeting chemokine-glycan interactions. The CellJammer® technology platform. Drug Discovery Today Technol. 9, e253-e259 [DOI] [PubMed] [Google Scholar]
- 6. Klarenbeek A., Maussang D., Blanchetot C., Saunders M., van der woning S., Smit M. J., de Haard H., Hoffmann M. (2012) Targeting chemokines and chemokine receptors with antibodies. Drug Discovery Today Technol. 9, e237-e244 [DOI] [PubMed] [Google Scholar]
- 7. Hamers-Casterman C., Atarhouch T., Muyldermans S., Robinson G., Hamers C., Songa E. B., Bendahman N., Hamers R. (1993) Naturally occurring antibodies devoid of light chains. Nature 363, 446–448 [DOI] [PubMed] [Google Scholar]
- 8. Nguyen V. K., Desmyter A., Muyldermans S. (2001) Functional heavy-chain antibodies in Camelidae. Adv. Immunol. 79, 261–296 [DOI] [PubMed] [Google Scholar]
- 9. Stijlemans B., Conrath K., Cortez-Retamozo V., Van Xong H., Wyns L., Senter P., Revets H., De Baetselier P., Muyldermans S., Magez S. (2004) Efficient targeting of conserved cryptic epitopes of infectious agents by single domain antibodies. African trypanosomes as paradigm. J. Biol. Chem. 279, 1256–1261 [DOI] [PubMed] [Google Scholar]
- 10. De Genst E., Silence K., Decanniere K., Conrath K., Loris R., Kinne J., Muyldermans S., Wyns L. (2006) Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. U.S.A. 103, 4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van der Linden R. H., Frenken L. G., de Geus B., Harmsen M. M., Ruuls R. C., Stok W., de Ron L., Wilson S., Davis P., Verrips C. T. (1999) Comparison of physical chemical properties of llama VHH antibody fragments and mouse monoclonal antibodies. Biochim. Biophys. Acta 1431, 37–46 [DOI] [PubMed] [Google Scholar]
- 12. Dumoulin M., Conrath K., Van Meirhaeghe A., Meersman F., Heremans K., Frenken L. G., Muyldermans S., Wyns L., Matagne A. (2002) Single-domain antibody fragments with high conformational stability. Protein Sci. 11, 500–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dolk E., van der Vaart M., Lutje Hulsik D., Vriend G., de Haard H., Spinelli S., Cambillau C., Frenken L., Verrips T. (2005) Isolation of llama antibody fragments for prevention of dandruff by phage display in shampoo. Appl. Environ. Microbiol. 71, 442–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dolk E., van Vliet C., Perez J. M., Vriend G., Darbon H., Ferrat G., Cambillau C., Frenken L. G., Verrips T. (2005) Induced refolding of a temperature denatured llama heavy-chain antibody fragment by its antigen. Proteins 59, 555–564 [DOI] [PubMed] [Google Scholar]
- 15. van der Vaart J. M., Pant N., Wolvers D., Bezemer S., Hermans P. W., Bellamy K., Sarker S. A., van der Logt C. P., Svensson L., Verrips C. T., Hammarstrom L., van Klinken B. J. (2006) Reduction in morbidity of rotavirus induced diarrhoea in mice by yeast produced monovalent llama-derived antibody fragments. Vaccine 24, 4130–4137 [DOI] [PubMed] [Google Scholar]
- 16. Harmsen M. M., De Haard H. J. (2007) Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 77, 13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saerens D., Ghassabeh G. H., Muyldermans S. (2008) Single-domain antibodies as building blocks for novel therapeutics. Curr. Opin. Pharmacol. 8, 600–608 [DOI] [PubMed] [Google Scholar]
- 18. Jähnichen S., Blanchetot C., Maussang D., Gonzalez-Pajuelo M., Chow K. Y., Bosch L., De Vrieze S., Serruys B., Ulrichts H., Vandevelde W., Saunders M., De Haard H. J., Schols D., Leurs R., Vanlandschoot P., Verrips T., Smit M. J. (2010) CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc. Natl. Acad. Sci. U.S.A. 107, 20565–20570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mahad D. J., Ransohoff R. M. (2003) The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin. Immunol. 15, 23–32 [DOI] [PubMed] [Google Scholar]
- 20. Koch A. E., Kunkel S. L., Harlow L. A., Johnson B., Evanoff H. L., Haines G. K., Burdick M. D., Pope R. M., Strieter R. M. (1992) Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J. Clin. Invest. 90, 772–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tesch G. H. (2008) MCP-1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am. J. Physiol. Renal Physiol. 294, F697–701 [DOI] [PubMed] [Google Scholar]
- 22. Charo I. F., Taubman M. B. (2004) Chemokines in the pathogenesis of vascular disease. Circ. Res. 95, 858–866 [DOI] [PubMed] [Google Scholar]
- 23. Craig M. J., Loberg R. D. (2006) CCL2 (monocyte chemoattractant protein-1) in cancer bone metastases. Cancer Metastasis. Rev. 25, 611–619 [DOI] [PubMed] [Google Scholar]
- 24. Sørensen T. L., Tani M., Jensen J., Pierce V., Lucchinetti C., Folcik V. A., Qin S., Rottman J., Sellebjerg F., Strieter R. M., Frederiksen J. L., Ransohoff R. M. (1999) Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J. Clin. Invest. 103, 807–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Qin S., Rottman J. B., Myers P., Kassam N., Weinblatt M., Loetscher M., Koch A. E., Moser B., Mackay C. R. (1998) The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J. Clin. Invest. 101, 746–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mach F., Sauty A., Iarossi A. S., Sukhova G. K., Neote K., Libby P., Luster A. D. (1999) Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J. Clin. Invest. 104, 1041–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hancock W. W., Lu B., Gao W., Csizmadia V., Faia K., King J. A., Smiley S. T., Ling M., Gerard N. P., Gerard C. (2000) Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J. Exp. Med. 192, 1515–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kawada K., Sonoshita M., Sakashita H., Takabayashi A., Yamaoka Y., Manabe T., Inaba K., Minato N., Oshima M., Taketo M. M. (2004) Pivotal role of CXCR3 in melanoma cell metastasis to lymph nodes. Cancer Res. 64, 4010–4017 [DOI] [PubMed] [Google Scholar]
- 29. Burns J. M., Summers B. C., Wang Y., Melikian A., Berahovich R., Miao Z., Penfold M. E., Sunshine M. J., Littman D. R., Kuo C. J., Wei K., McMaster B. E., Wright K., Howard M. C., Schall T. J. (2006) A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 203, 2201–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Müller A., Homey B., Soto H., Ge N., Catron D., Buchanan M. E., McClanahan T., Murphy E., Yuan W., Wagner S. N., Barrera J. L., Mohar A., Verástegui E., Zlotnik A. (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56 [DOI] [PubMed] [Google Scholar]
- 31. Roovers R. C., Laeremans T., Huang L., De Taeye S., Verkleij A. J., Revets H., de Haard H. J., van Bergen en Henegouwen P. M. (2007) Efficient inhibition of EGFR signaling and of tumour growth by antagonistic anti-EFGR Nanobodies. Cancer Immunol. Immunother. 56, 303–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Loetscher M., Gerber B., Loetscher P., Jones S. A., Piali L., Clark-Lewis I., Baggiolini M., Moser B. (1996) Chemokine receptor specific for IP10 and mig. Structure, function, and expression in activated T-lymphocytes. J. Exp. Med. 184, 963–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goldman L. A., Cutrone E. C., Kotenko S. V., Krause C. D., Langer J. A. (1996) Modifications of vectors pEF-BOS, pcDNA1, and pcDNA3 result in improved convenience and expression. BioTechniques 21, 1013–1015 [DOI] [PubMed] [Google Scholar]
- 34. Coward P., Chan S. D., Wada H. G., Humphries G. M., Conklin B. R. (1999) Chimeric G proteins allow a high-throughput signaling assay of Gi-coupled receptors. Anal. Biochem. 270, 242–248 [DOI] [PubMed] [Google Scholar]
- 35. Verheesen P., Roussis A., de Haard H. J., Groot A. J., Stam J. C., den Dunnen J. T., Frants R. R., Verkleij A. J., Theo Verrips C., van der Maarel S. M. (2006) Reliable and controllable antibody fragment selections from Camelid non-immune libraries for target validation. Biochim. Biophys. Acta 1764, 1307–1319 [DOI] [PubMed] [Google Scholar]
- 36. Balabanian K., Lagane B., Infantino S., Chow K. Y., Harriague J., Moepps B., Arenzana-Seisdedos F., Thelen M., Bachelerie F. (2005) The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J. Biol. Chem. 280, 35760–35766 [DOI] [PubMed] [Google Scholar]
- 37. Wise A., Gearing K., Rees S. (2002) Target validation of G-protein coupled receptors. Drug Discovery Today 7, 235–246 [DOI] [PubMed] [Google Scholar]
- 38. Esté J. A., Telenti A. (2007) HIV entry inhibitors. Lancet 370, 81–88 [DOI] [PubMed] [Google Scholar]
- 39. Ishida T., Joh T., Uike N., Yamamoto K., Utsunomiya A., Yoshida S., Saburi Y., Miyamoto T., Takemoto S., Suzushima H., Tsukasaki K., Nosaka K., Fujiwara H., Ishitsuka K., Inagaki H., Ogura M., Akinaga S., Tomonaga M., Tobinai K., Ueda R. (2012) Defucosylated anti-CCR4 monoclonal antibody (KW-0761) for relapsed adult T-cell leukemia-lymphoma. A multicenter phase II study. J. Clin. Oncol. 30, 837–842 [DOI] [PubMed] [Google Scholar]
- 40. Alcami A. (2003) Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol 3, 36–50 [DOI] [PubMed] [Google Scholar]
- 41. Galzi J. L., Haas M., Frossard N., Hibert M. (2012) Why and how to find neutraligands targeting chemokines? Drug Discovery Today Technol. 9, e245–e251 [DOI] [PubMed] [Google Scholar]
- 42. Hachet-Haas M., Balabanian K., Rohmer F., Pons F., Franchet C., Lecat S., Chow K. Y., Dagher R., Gizzi P., Didier B., Lagane B., Kellenberger E., Bonnet D., Baleux F., Haiech J., Parmentier M., Frossard N., Arenzana-Seisdedos F., Hibert M., Galzi J. L. (2008) Small neutralizing molecules to inhibit actions of the chemokine CXCL12. J. Biol. Chem. 283, 23189–23199 [DOI] [PubMed] [Google Scholar]
- 43. Veldkamp C. T., Ziarek J. J., Peterson F. C., Chen Y., Volkman B. F. (2010) Targeting SDF-1/CXCL12 with a ligand that prevents activation of CXCR4 through structure-based drug design. J. Am. Chem. Soc. 132, 7242–7243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johnson Z., Schwarz M., Power C. A., Wells T. N., Proudfoot A. E. (2005) Multi-faceted strategies to combat disease by interference with the chemokine system. Trends Immunol. 26, 268–274 [DOI] [PubMed] [Google Scholar]
- 45. Kennedy K. J., Strieter R. M., Kunkel S. L., Lukacs N. W., Karpus W. J. (1998) Acute and relapsing experimental autoimmune encephalomyelitis are regulated by differential expression of the CC chemokines macrophage inflammatory protein-1α and monocyte chemotactic protein-1. J. Neuroimmunol. 92, 98–108 [DOI] [PubMed] [Google Scholar]
- 46. Tsui P., Das A., Whitaker B., Tornetta M., Stowell N., Kesavan P., Kaiser E., Lacy E. R., Yan L., Snyder L. A., Sweet R. (2007) Generation, characterization and biological activity of CCL2 (MCP-1/JE) and CCL12 (MCP-5) specific antibodies. Hum. Antibodies 16, 117–125 [PubMed] [Google Scholar]
- 47. Loberg R. D., Ying C., Craig M., Yan L., Snyder L. A., Pienta K. J. (2007) CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia 9, 556–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Loberg R. D., Ying C., Craig M., Day L. L., Sargent E., Neeley C., Wojno K., Snyder L. A., Yan L., Pienta K. J. (2007) Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 67, 9417–9424 [DOI] [PubMed] [Google Scholar]
- 49. Mulligan M. S., McDuffie J. E., Shanley T. P., Guo R. F., Vidya Sarma J., Warner R. L., Ward P. A. (2000) Role of RANTES in experimental cardiac allograft rejection. Exp. Mol. Pathol. 69, 167–174 [DOI] [PubMed] [Google Scholar]
- 50. Gonzalo J. A., Lloyd C. M., Peled A., Delaney T., Coyle A. J., Gutierrez-Ramos J. C. (2000) Critical involvement of the chemotactic axis CXCR4/stromal cell-derived factor-1α in the inflammatory component of allergic airway disease. J. Immunol. 165, 499–508 [DOI] [PubMed] [Google Scholar]
- 51. Proudfoot A. E. (2006) The biological relevance of chemokine-proteoglycan interactions. Biochem. Soc. Trans. 34, 422–426 [DOI] [PubMed] [Google Scholar]
- 52. Zhang W. B., Navenot J. M., Haribabu B., Tamamura H., Hiramatu K., Omagari A., Pei G., Manfredi J. P., Fujii N., Broach J. R., Peiper S. C. (2002) A point mutation that confers constitutive activity to CXCR4 reveals that T140 is an inverse agonist and that AMD3100 and ALX40–4C are weak partial agonists. J. Biol. Chem. 277, 24515–24521 [DOI] [PubMed] [Google Scholar]
- 53. Verzijl D., Storelli S., Scholten D. J., Bosch L., Reinhart T. A., Streblow D. N., Tensen C. P., Fitzsimons C. P., Zaman G. J., Pease J. E., de Esch I. J., Smit M. J., Leurs R. (2008) Noncompetitive antagonism and inverse agonism as mechanism of action of nonpeptidergic antagonists at primate and rodent CXCR3 chemokine receptors. J. Pharmacol. Exp. Ther. 325, 544–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Frauenschuh A., Power C. A., Déruaz M., Ferreira B. R., Silva J. S., Teixeira M. M., Dias J. M., Martin T., Wells T. N., Proudfoot A. E. (2007) Molecular cloning and characterization of a highly selective chemokine-binding protein from the tick Rhipicephalus sanguineus. J. Biol. Chem. 282, 27250–27258 [DOI] [PubMed] [Google Scholar]
- 55. Déruaz M., Frauenschuh A., Alessandri A. L., Dias J. M., Coelho F. M., Russo R. C., Ferreira B. R., Graham G. J., Shaw J. P., Wells T. N., Teixeira M. M., Power C. A., Proudfoot A. E. (2008) Ticks produce highly selective chemokine binding proteins with antiinflammatory activity. J. Exp. Med. 205, 2019–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]