

Background: P450 BM3 is a high activity enzyme with biotechnological potential.
Results: Mutations perturbing P450 BM3's conformational state and active site facilitate human P450-like oxidation of the drug omeprazole.
Conclusion: Conformational destabilization enables P450 BM3 to explore novel conformations and accept diverse substrates.
Significance: “Gatekeeper” mutations that decrease the energetic barrier for transition to the substrate-bound state can reconfigure P450 BM3 specificity and reactivity.
Keywords: Calorimetry, Crystal Structure, Cytochrome P450, Drug Metabolism, Enzyme Catalysis, Hydroxylase, Site-directed Mutagenesis, P450 BM3, Conformational Destabilization, Omeprazole
Abstract
Cytochrome P450 monooxygenases (P450s) have enormous potential in the production of oxychemicals, due to their unparalleled regio- and stereoselectivity. The Bacillus megaterium P450 BM3 enzyme is a key model system, with several mutants (many distant from the active site) reported to alter substrate selectivity. It has the highest reported monooxygenase activity of the P450 enzymes, and this catalytic efficiency has inspired protein engineering to enable its exploitation for biotechnologically relevant oxidations with structurally diverse substrates. However, a structural rationale is lacking to explain how these mutations have such effects in the absence of direct change to the active site architecture. Here, we provide the first crystal structures of BM3 mutants in complex with a human drug substrate, the proton pump inhibitor omeprazole. Supported by solution data, these structures reveal how mutation alters the conformational landscape and decreases the free energy barrier for transition to the substrate-bound state. Our data point to the importance of such “gatekeeper” mutations in enabling major changes in substrate recognition. We further demonstrate that these mutants catalyze the same 5-hydroxylation reaction as performed by human CYP2C19, the major human omeprazole-metabolizing P450 enzyme.
Introduction
The cytochrome P450 monooxygenases (P450s)2 are hemoproteins that catalyze a huge range of biochemical transformations, including key reactions involved in the biosynthesis of steroids, antibiotics, and signaling lipids (1, 2). Recent years have seen intensive efforts to exploit the P450s' ability to catalyze regio- and stereoselective oxidations of substrates to make biotechnologically useful products. Examples include the rational engineering of the Pseudomonas putida camphor hydroxylase P450cam (CYP101A1) for improved binding and oxidation of environmentally recalcitrant polychlorinated benzenes, and the use of directed evolution for the conversion of specificity of the Bacillus megaterium P450 BM3 (CYP102A1, BM3) from long chain fatty acids toward short chain hydrocarbons (3, 4). BM3 has proven to be a particularly versatile and popular model system for use in engineering studies, benefiting from the fact that it is a natural fusion of a P450 to its mammalian-like diflavin reductase redox partner (enabling efficient electron transfer that underpins its high monooxygenase activity (5, 6)) and that the structure of its P450 domain and roles of many active site amino acids are well understood (7–10).
Recent work to diversify BM3's substrate selectivity and reactivity has produced variants that catalyze olefin cyclopropanation by carbene transfer and oxidation of e.g. testosterone, polycyclic aromatic hydrocarbons, and pharmaceuticals (11–14). In the latter case, an aim is to engineer BM3 to generate high levels of metabolites typical of those formed by human P450s. Approaches to engineering BM3 have included directed evolution, chimeragenesis (with homologs from Bacillus subtilis) and CASTing, as well as structure-led mutagenesis guided by x-ray crystal structures of BM3's P450 (heme) domain in substrate-free (SF), fatty acid-bound (SB), and various mutant forms (7, 8, 15, 16). Many studies identified common residues that help facilitate substrate diversification. These include Phe-87, Ala-82, Val-78, and Arg-47, the first three of which are internal residues, whereas Arg-47 interacts with the fatty acid carboxylate at the protein surface (6, 10). Phe-87 interacts with the ω-end of fatty acids and prevents oxidation at this position. Phe-87 mutations alter regioselectivity of fatty acid oxidation, with positions of hydroxylation of lauric acid moving from ω-1/ω-2/ω-3 (WT BM3) toward ω-5 in F87A, F87S, and F87G point mutants, and even further from the ω-position in a F87A/V78A double mutant (6, 17). The F87A mutation also activated BM3 in hydroxylation of testosterone (12).
Although alterations that influence local structure in the BM3 active site cavity clearly have potential to alter the binding position of fatty acids and to enable docking of novel substrates, other BM3 mutations were shown to have more profound effects on the P450's conformational landscape (18, 19). One of the more common activity-altering BM3 mutants (A82F) shows greatly enhanced affinity for fatty acid substrates. Although the crystal structure of palmitate-bound A82F heme domain was solved in the SB-type conformation (20), this did not provide a clear rationale for its much improved affinity for fatty acids. Also, despite several studies involving mutagenesis of BM3 to improve its capacity to oxidize human pharmaceuticals, there are no structural data to provide insight into how improved mutants enable binding of the new substrates or to guide subsequent engineering to further enhance binding and desired activities.
In this study we characterize the structural and biochemical/catalytic properties of the A82F, F87V, and F87V/A82F mutants of BM3, demonstrating a novel activity in oxidation of the widely used gastric proton pump inhibitor (PPI) omeprazole (Fig. 1) and generating products typical of those formed by the major human metabolizing P450 enzyme CYP2C19 (21). Structural data for substrate-free and omeprazole-bound forms of the A82F and double mutant BM3 heme domains provide clear evidence for the gatekeeper nature of the A82F mutation, which produces a major structural rearrangement of the BM3 heme domain that leads to novel molecular selectivity. These first structural data for BM3 in complex with a human drug substrate highlight how combinations of conformational effector mutations (e.g. A82F) with secondary mutations that make local structural changes to alter binding in the heme vicinity (e.g. F87A/V) can be combined to cause dramatic changes in P450 substrate selectivity for biotechnological applications.
FIGURE 1.

Structure of omeprazole. The chemical structure of the proton pump inhibitor OMP is shown. The pyridine ring shows the accepted numbering (hydroxylation occurs at the 5-methyl position). Also shown is the characteristic MS fragmentation position that gives the methoxybenzimidazole and 4-methoxy-3,5-dimethylpyridin-2-yl (pyridinyl) fragments. Hydroxylation on the 5-methyl group is performed by engineered variants of P450 BM3 described in this study. 5-Hydroxylation is also the primary reaction catalyzed by the major human OMP-metabolizing enzyme CYP2C19. Omeprazole is chiral around the central sulfur atom. As a drug preparation, omeprazole is a racemate of two isomers.
EXPERIMENTAL PROCEDURES
Generation, Expression, and Purification of WT and Mutant P450 BM3 Proteins
Mutants of the intact P450 BM3 and its heme domain were generated by oligonucleotide-directed mutagenesis. Intact WT P450 BM3 in pET15b was used for mutagenesis to create A82F, F87V, and F87V/A82F (DM) mutants. Mutations (positions underlined) were generated using the QuikChange Lightning site-directed mutagenesis kit (Stratagene-Agilent UK). Primers used were as follows: A82F, 5′-CTTAAATTTGTACGTGATTTTTTCGGAGACGGGTTA-3′; F87V, 5′-TTGCAGGAGACGGGTTAGTTACAAGCTGGACGCATG-3′; F87V in A82F background, 5′-TTTTCGGAGACGGGTTAGTTACAAGCTGGACGCATG-3′ (and their reverse complements). These intact BM3 enzymes were expressed as N-terminal hexahistidine-tagged enzymes either using the pET15b (F87V, DM) constructs directly or following cloning of the WT and A82F genes into pET14b using NdeI/BamHI sites. WT and mutant heme domain genes were generated using the relevant pET14b/15b constructs. To generate the heme domain constructs, a stop codon pair (underlined) was introduced after residue 473 by PCR using the same mutagenesis kit, and with primers StopF 5′-CAGTCTGCTAAAAAAGTACGCAAATAGTAGGAAAACGCTCATAATACGCCGCTG-3′ and StopR 5′-CAGCGGCGTATTATGAGCGTTTTCCTACTATTTGCGTACTTTTTTAGCAGACTG-3′. The heme domain genes (amino acids 1–473) were transferred as NdeI/BamHI fragments to pET20b to enable heme domain production in the absence of an N-terminal His tag for improved crystallization. Genes were sequenced to ensure the presence of desired mutation(s) and the absence of other mutations. The WT and A82F intact BM3 and the WT and all mutant P450 BM3 heme domain proteins were expressed in BL21-Gold (DE3) Escherichia coli cells (Stratagene-Agilent UK) using TB medium with cell growth at 37 °C and with agitation at 200 rpm in an orbital incubator. The F87V and DM intact BM3 proteins were grown in autoinduction TB medium (Melford Ltd., Ipswich, UK). Typically, 4-liter bacterial cultures were used for protein production with cell growth for 24–36 h.
Following cell growth, bacterial cells were recovered by centrifugation at 4 °C (6000 × g, 10 min) and resuspended in ice-cold buffer B (50 mm KPi, 250 mm NaCl, 10% (v/v) glycerol, pH 7.0) containing protease inhibitors (EDTA-free CompleteTM tablets, Roche Applied Science). Protease inhibitors were maintained in all subsequent buffers used for protein purification. Cells were lysed by sonication on ice using a Bandelin Sonopuls sonicator (40% power, 50 pulses for 5 s with 25 s between pulses). The supernatant containing soluble intact BM3 and heme domain proteins was separated from cell debris by centrifugation (20,000 × g, 40 min, 4 °C), and ammonium sulfate was added to 30% saturation on ice with slow stirring for ∼4 h. Centrifugation (20,000 × g, 40 min, 4 °C) was done to fractionate soluble BM3 and heme domain proteins from insoluble material. Intact BM3 WT and mutant proteins in the supernatant were purified using His tag affinity by mixing with nickel-iminodiacetic acid (Ni-IDA) resin (Qiagen, UK) overnight at 4 °C in buffer B with 5 mm imidazole, prior to elution with 200 mm imidazole in buffer B. Isolated proteins were dialyzed into buffer A (50 mm Tris, 1 mm EDTA, pH 7.2) and further purified by size exclusion chromatography using a Sephacryl S-200 column (GE Healthcare, 26 × 60 cm on an AKTA purifier system). BM3 fractions were checked for purity by SDS-PAGE, concentrated by ultrafiltration (Vivaspin, Vivaproducts), and frozen in buffer A plus 50% glycerol at −80 °C. For the nontagged BM3 heme domains, supernatants subsequent to the 30% ammonium sulfate fractionation were subjected to a second ammonium sulfate fractionation step at 60% salt saturation. The pellet was resuspended in buffer A and extensively dialyzed into the same buffer to desalt, prior to loading onto a Q-Sepharose anion exchange column (16 × 10 cm, on an AKTA) and eluting using a gradient of 0–500 mm KCl in buffer A. Heme domain-containing fractions were desalted by using a desalting column (GE Healthcare, 26 × 10 cm on an AKTA) into 25 mm KPi, pH 7.0, and then loaded onto a hydroxyapatite column (Bio-Rad, 16 × 11 cm) and eluted in a linear gradient of 25–500 mm KPi, pH 7.0 (200 ml). Pure heme domain fractions were concentrated by ultrafiltration (Vivaspin) and used immediately for crystallography or flash-frozen in liquid nitrogen and stored at −80 °C. Because of the enhanced affinity for fatty acids in BM3 and heme domain proteins carrying the A82F mutation, all enzymes were passed through a Lipidex 1000 column (PerkinElmer Life Sciences) in 25 mm KPi, pH 7.0, to remove any fatty acid bound during purification and prior to use for crystallization (heme domains) or for binding or turnover studies (heme domain and intact BM3 enzymes).
Quantification of P450 BM3 Enzymes and Determination of Their Substrate Affinity and Steady-state Kinetic Properties
Concentrations of the low spin forms of the WT and mutant forms of intact P450 BM3 and its heme domain were determined using extinction coefficients of ϵ = 105 and 95 mm−1 cm−1, respectively, at the Soret maximum (418 or 419 nm), as described previously (6). Thiolate coordination of the heme iron in all samples was established by formation of the Fe(II)CO complex at ∼450 nm by bubbling of sodium dithionite-reduced WT and mutant BM3 and heme domain samples (∼2–4 μm) with carbon monoxide gas, as described by Omura and Sato (22). All samples showed near complete conversion to the P450 form, with negligible formation of an ∼420-nm peak relating to the thiol-coordinated P420 form (23, 24).
Dissociation constants (Kd values) for binding of the substrates N-palmitoylglycine (NPG) and omeprazole (OMP) to WT and mutant BM3 heme domains were determined by UV-visible absorption titrations using ∼1–4 μm protein in 100 mm KPi, pH 7.0, at 25 °C (assay buffer) in 1-cm path length quartz cuvettes and as in our previous studies (6, 16, 25). Spectra were recorded for substrate-free enzymes and following addition of ligands during substrate titrations (typically 800 to 250 nm). Titrations were recorded until no further spectral changes were observed in the P450s. Difference spectra were generated by subtraction of the spectrum for ligand-free protein from spectra recorded after each addition of substrate. Maxima and minima in difference spectra were identified (using the same wavelength pair in each titration), and the overall absorbance changes Δ(Apeak minus Atrough) were plotted versus [substrate]. Data were fitted using either a standard (Michaelis-Menten) hyperbolic function or (for tight binding substrates where the Kd value is ≤5× the P450 concentration) by using the Morrison equation (as described previously) to determine Kd values (26, 27). UV-visible spectroscopy was carried out on a Cary 50 UV-visible spectrometer (Cary-Agilent, UK), with data analysis and fitting done using Origin Pro software (OriginLab).
Steady-state kinetic parameters for WT and F87V, A82F, and DM mutants of intact BM3 were analyzed on a Cary 50 UV-visible spectrophotometer using substrate (OMP and NPG)-dependent NADPH oxidation at 340 nm (Δϵ340 = 6210 m−1 cm−1) across a range of substrate concentrations and using pure BM3 enzymes (25–125 nm) in a total of 1 ml of assay buffer at 25 °C. NADPH was maintained at a saturating concentration (200 μm). Data points were collected in at least triplicate. Rate constants for substrate-dependent NADPH oxidation were plotted versus [substrate], and data were fitted using the Michaelis-Menten function (using Origin Pro) to obtain kcat and Km parameters in each case. Vmax values for WT and mutant BM3 enzymes with 5-OH OMP were measured similarly using a saturating concentration of the metabolite.
Omeprazole and 5-OH Omeprazole Turnover and Analysis by LC-MS
Turnover reactions for oxidation of OMP were carried out in deep well blocks at 37 °C with shaking for 30 min. Reaction mixtures contained purified WT or mutant (F87V, A82F, or DM BM3) enzymes (0.1 μm), substrate (10 μm), and NADPH regeneration system (glucose 6-phosphate 7.76 mm, NADP+ 0.6 mm, and glucose-6-phosphate dehydrogenase, 0.75 units/ml) in turnover buffer (50 mm KPi, 5 mm CaCl2, pH 7.4) in a final volume of 500 μl. Following completion of the reaction, protein was precipitated by addition of an equal volume of acetonitrile containing 1 μg/ml fluconazole by shaking the mixed samples at 800 rpm for 10 min. The precipitated protein was filtered through protein precipitation plates (Phenomenex, Macclesfield, UK) into mass spectrometry vials (FluidX, Nether Alderley, UK) and clarified by centrifugation (4000 × g, 25 min, 10 °C). Analysis was carried out on a Thermo Exactive LC-MS with a CTC PAL autosampler (Thermo Scientific, UK) with a Kinetex 2.6U XB-C18 100A column (Phenomenex). A gradient of 0.1% formic acid to acetonitrile was used to resolve products. Reaction of the DM BM3 enzyme with 5-OH OMP and subsequent product analysis were done in the same way as for the OMP turnovers.
Omeprazole Turnover and Analysis by NMR
Turnover reactions with OMP were carried out in a 100-ml flask at 37 °C with shaking of reagents at 100 rpm for 30 min. Reaction mixtures contained purified WT or mutant (F87V, A82F, or DM intact BM3) enzymes (1 μm), substrate (100 μm), NADPH regeneration system (glucose 6-phosphate 7.76 mm, NADP+ 0.6 mm, and glucose-6-phosphate dehydrogenase 0.75 units/ml) in 60 ml of assay buffer (100 mm KPi, pH 7.0). Products were extracted using Strata-X SPE columns (Phenomenex), dried under vacuum, and eluted in CDCl3. Analysis was carried out on a Bruker Avance 400 MHz NMR (Bruker, Coventry, UK). 1H spectra were collected at 400 MHz and 13C spectra at 101 MHz. Spectra were base-line corrected and referenced to tetramethylsilane standard by the residual nondeuterated solvent in the sample. δ values are in ppm;, J values are in Hz. Full assignments were made by COSY, HMBC, and HMQC methods. Signal splittings were recorded as singlet (s), doublet (d), doublet of doublets (dd), αβ system (AB), and multiplet (m). Processing was carried out using MestReNova Lite (Mestrelab Research, Santiago de Compostela, Spain) and an NMR processor (Advanced Chemistry Development, Inc., Toronto, Canada).
Examination of Hemoprotein Stability by Differential Scanning Calorimetry
DSC was carried out on a Microcal VP-DSC instrument. Data analysis was done using Microcal Origin software. The parameters used were as follows: 20–80 °C temperature gradient, 90 °C/h scan rate, 10-min prescan thermostat. Background scans were carried out with degassed assay buffer and saturated with OMP or NPG for the substrate-bound samples. Protein samples were prepared in assay buffer by extensive buffer exchange and dialysis. All samples were run using 20 μm protein and saturating substrate. Once two overlapping base-line scans were achieved, a degassed protein sample was run.
Crystallization of P450 BM3 Heme Domains and Determination of Protein Structures
Crystallography was performed using the sitting drop method using a seeding protocol at 4 °C. Crystals obtained during initial screens for each mutant were used to create microcrystal screen stocks, and consecutive screens (Molecular Dimensions) were made with drops that consisted of 150 nl of WT or mutant heme domain proteins (230 μm), 50 μl of seed stock, and 200 nl of well solution using a Mosquito liquid handling robot (TTP LabTech Ltd., Melbourn, UK). For the OMP mutant heme domain complex structures, proteins were saturated with racemic OMP ligand prior to crystallization. Ligands were titrated into heme domain samples until no further change in heme iron spin state (toward high spin) was observed. Thereafter, samples were concentrated by ultrafiltration in the presence of saturating ligand. Micro seeding was also used to produce diffraction quality crystals. Crystals were obtained under a range of conditions and flash-cooled in liquid nitrogen prior to data collection. The mother liquor was supplemented with 10% PEG 200 where an additional cryo-protectant was required. Data were collected at Diamond synchrotron beamlines and reduced and scaled using XDS (28). Structures were solved by molecular replacement with previously solved BM3 heme domain structures (PDB 1JPZ) using PHASER (29). Structures were refined using Refmac5 (29) and Coot (30). PDB codes for the new heme domain structures are as follows: A82F, 4KF0; F87V/A82F (DM, imidazole-bound), 4KF2; A82F (OMP-bound), 4KEW; and DM (OMP-bound), 4KEY.
Oligonucleotide primers were from Eurofins MWG Operon (Ebersberg, Germany). Omeprazole was from Cypex Ltd. (Dundee, Scotland, UK). 5-OH omeprazole was from Santa Cruz Biotechnology, Inc. (Dallas). The bacterial growth medium (Terrific Broth) was from Melford Ltd (Ipswich, UK). Unless otherwise stated, other chemicals were from Sigma and of the highest purity available.
RESULTS
Characterization of Omeprazole Binding Properties of BM3 Mutants
Preceding studies identified Ala-82 and Phe-87 as important in controlling molecular selectivity and regioselectivity of BM3 substrate oxidation (16, 20, 31). WT, F87V, A82F, and the F87V/A82F DM forms of intact P450 BM3 and of its heme domain (residues 1–473) were expressed and purified as described under “Experimental Procedures.” The BM3 ferric heme iron undergoes a shift from low spin toward high spin on binding substrates that displace its 6th ligand (a water molecule), accompanied by a shift of Soret absorption maximum from ∼418 nm to ∼392 nm (32). Binding of the substrate NPG was done for WT, F87V, A82F, and the DM BM3 proteins. WT and all mutants bound NPG tightly, with Kd values <1 μm (Table 1).
TABLE 1.
Substrate binding and turnover data for BM3 enzymes
Table shows data for the binding of substrates (NPG and OMP) to WT and A82F, F87V, and DM P450 BM3 enzymes (Kd values from optical titrations) and for the kinetics of substrate-dependent NADPH oxidation (kcat and Km values). All data points were collected in triplicate; errors are S.E. NA indicates that no evidence of binding of OMP to WT BM3 was found in optical titrations.
| Protein | Substrate | Kd | kcat | Km |
|---|---|---|---|---|
| μm | min−1 | μm | ||
| WT | NPG | 0.082 ± 0.011 | 4770 ± 160 | 13.9 ± 2.7 |
| A82F | NPG | 0.297 ± 0.069 | 5130 ± 570 | 26.3 ± 5.3 |
| F87V | NPG | 0.204 ± 0.045 | 4970 ± 240 | 14.9 ± 2.8 |
| DM | NPG | 0.004 ± 0.003 | 4050 ± 250 | 1.91 ± 0.31 |
| WT | OMP | NA | 238 ± 12 | 124 ± 20 |
| A82F | OMP | 1.67 ± 0.05 | 1460 ± 30 | 18.7 ± 0.8 |
| F87V | OMP | 49.0 ± 2.7 | 2180 ± 20 | 38.7 ± 4.6 |
| DM | OMP | 0.212 ± 0.014 | 1500 ± 45 | 1.93 ± 0.22 |
The A82F mutant has particularly high affinity for lipids and was purified from E. coli with fatty acid bound, as was the DM. Separation of the bound lipid by Lipidex chromatography was done prior to analysis. Binding studies with the PPI omeprazole (OMP) revealed no evidence for its association with WT BM3, but partial spin-state conversion was observed for F87V BM3, indicative of the ability of the drug to access the active site (Kd = 49 μm). Much tighter binding and more extensive high spin heme accumulation was observed with both A82F and the DM (Kd values = 1.67 and 0.21 μm, respectively), indicating that the mutations had synergistic effects in enhancing OMP affinity, with A82F having the major role (Fig. 2A and Table 1).
FIGURE 2.

Binding and oxidation of omeprazole by P450 BM3 mutants. A, binding titration for F87V/A82F (DM) intact P450 BM3 (1 μm) with omeprazole. Main panel, plot of the induced heme Soret absorption change (ΔA389–ΔA419) versus [OMP] with data fitted to yield a Kd = 0.212 ± 0.014 μm. Inset, selected OMP-induced absorption difference spectra from titration at OMP concentrations: 0.05 μm (green), 0.15 μm (blue), 0.30 μm (magenta), 0.40 μm (purple), and 1.0 μm (red). B, turnover data for OMP with WT (black column), A82F (red), F87V (blue), and F87V/A82F (DM, orange) P450 BM3 enzymes. Assays were done for 30 min. Products (5-OH OMP and 5-COOH OMP) are shown as a percentage of the initial OMP concentration used in the assay, with data corrected for an internal standard (fluconazole) and for enzyme-independent degradation of OMP substrate.
Steady-state Kinetics of P450 BM3 Mutants with Omeprazole
Steady-state kinetic analysis was done for WT and each of the intact BM3 mutants by measuring NADPH oxidation following the addition of various concentrations of NPG or OMP (24). Table 1 details high kcat values for all BM3 enzymes with NPG (4050–5130 min−1), with DM BM3 having the lowest Km value (1.91 μm). With OMP, some substrate-stimulated NADPH oxidation was seen for WT BM3 (kcat = 238 min−1), but kinetics were substantially improved in all the mutants (e.g. 1500 min−1 for DM), and apparent OMP affinity for the DM was ∼65-fold greater than for WT BM3 (Km values of 1.9 μm versus 124 μm), resulting in a >400-fold improvement in catalytic efficiency (kcat/Km ratio) for the DM over WT BM3.
Oxidation of Omeprazole by WT and Mutant P450 BM3 Enzymes
To validate novel omeprazole oxidase activity in mutant BM3 enzymes, in vitro turnover studies were done as described in detail under “Experimental Procedures,” using LC-MS and NMR to characterize products formed. WT BM3 exhibited very small amounts of oxidation of OMP (<1% of the starting material), but the low amounts obtained precluded determination of position(s) of oxidation. However, each of the three mutants extensively oxidized the drug.
LC-MS revealed a 362.1162 atomic mass unit species in all the mutant turnover reactions, corresponding to introduction of an oxygen atom into OMP (Fig. 3). A natural fragmentation of the OMP molecule occurs in the MS, with bond breakage between the sulfur and the methoxybenzimidazole moiety (Fig. 1). A +16 increase in mass of the larger fragment indicated that oxidation occurs on this portion of OMP. Further fragmentation of the oxidized molecule showed that the position of oxidation was likely on one of the two 4-methoxy-3,5-dimethylpyridin-2-yl (hereafter termed pyridinyl) methyl groups (Fig. 3). This was confirmed by 1H NMR spectroscopy after larger scale reactions of mutant enzymes with OMP, and the absolute position of oxidation was confirmed using two-dimensional NMR, showing that a specific hydroxylation occurred on the pyridinyl 5-methyl group (forming 5-OH OMP) (supplemental Figs. S1–S4). This same reaction is also catalyzed by CYP2C19, the major metabolizing P450 for OMP and other PPIs in humans (33).
FIGURE 3.

LC-MS analysis of products derived from omeprazole oxidation by the P450 BM3 F87V/A82F (DM) double mutant enzyme. The figures show data from LC-MS studies of OMP before and after its enzymatic turnover by the P450 BM3 DM (F87V/A82F) enzyme. These data demonstrate hydroxylation and subsequent oxidation of OMP, and also the fragmentation of the OMP (and its oxidized products) that occurs during MS analysis. A (retention time = 5.39 min) shows data for OMP prior to addition of enzyme and initiation of its oxidation by the BM3 DM enzyme. Peaks at m/z 346.1212 and 198.0581 (circled) correspond to the fragmentation of OMP at the sulfone group (between the sulfur and the methoxybenzimidazole moiety), with the smaller species representing the sulfur-containing fragment. B (retention time = 5.26 min) is following an enzymatic reaction for 30 min. The m/z peaks at 362.1162 and 214.0530 (circled) are for the 5-OH OMP and its hydroxylated fragment. C, (retention time = 5.32 min) is following an enzymatic reaction for 30 min. The m/z peaks at 376.0955 and 228.0323 (circled) are for the 5-COOH OMP and its carboxylated fragment.
In OMP turnovers done over 30 min, the total oxidative turnover of OMP was greatest for the DM (∼55%), followed by the F87V (∼50%) and the A82F (∼20%) mutants (Fig. 2B). LC-MS demonstrates the formation of a considerable amount of a +32 species (∼10%) in the case of the DM, which was shown to be the carboxylic acid product at the pyridinyl 5-methyl group (5-COOH OMP), resulting from further P450-mediated oxidation at the same position. By comparing the amount of NADPH oxidized with the quantity of OMP oxidized over the first 2 min of reactions, enzymatic coupling was estimated as 68% for both the A82F and DM BM3 enzymes and 39% for the F87V variant. These data suggest that structural changes induced by the A82F mutation are the major determinant of the productive binding mode for OMP, and thus the coupling efficiency.
Consistent with the model of further BM3 mutant-catalyzed oxidation of the primary product, 5-OH OMP showed tight binding to both A82F and the DM (Kd values of 15.2 ± 0.9 and 2.62 ± 0.19 μm, respectively) (Table 2 and Fig. 4), indicating that oxidized products do bind the enzyme. The further oxidation of 5-OH OMP to 5-COOH OMP was confirmed by LC-MS studies with the BM3 DM enzyme (data not shown). Vmax values determined for 5-OH OMP-stimulated NADPH oxidation in the BM3 mutants were 1050 ± 50 min−1 (A82F), 645 ± 30 min−1 (F87V), and 785 ± 35 min−1 (DM), but NADPH oxidation was not stimulated significantly above background in the WT BM3 (Table 2). A time course for the oxidation of OMP into 5-OH OMP and 5-COOH OMP products by the BM3 DM enzyme is shown in Fig. 5. Approximately 30% of the 5-OH OMP is further oxidized to 5-COOH OMP by the DM BM3 over 30 min.
TABLE 2.
Binding and kinetics of oxidation of 5-OH OMP by WT and mutant P450 BM3 enzymes
The table shows Kd values (where determinable) for the binding of the oxidized product 5-OH OMP to WT and mutant intact P450 BM3 enzymes. The Kd values were derived from optical titrations. Also shown are Vmax values for 5-OH OMP-dependent NADPH oxidation catalyzed by WT, F87V, A82F, and DM BM3 enzymes. All data points were collected in triplicate; errors are S.E. 5-OH OMP is the primary metabolite of OMP generated by human CYP2C19 and by the BM3 mutants (most efficiently by the F87V/A82F DM). Data were collected as described under “Experimental Procedures.” ND indicates not determinable.
| Protein | Substrate | Kd | Vmax |
|---|---|---|---|
| μm | min−1 | ||
| WT | 5-OH OMP | ND | ND |
| A82F | 5-OH OMP | 15.2 ± 0.9 | 1050 ± 50 |
| F87V | 5-OH OMP | ND | 645 ± 30 |
| DM | 5-OH OMP | 2.62 ± 0.19 | 785 ± 35 |
FIGURE 4.

Optical binding titration for the BM3 DM heme domain with 5-OH OMP. A shows UV-visible binding spectra for a titration of intact DM BM3 (∼1.0 μm, red spectrum) and following addition of 2 μm (blue), 6 μm (magenta), 16 μm (orange), and 40 μm (black) 5-OH OMP. The Soret band shifts from 418 to 393 nm on binding 5-OH OMP. The inset shows difference spectra obtained by subtraction of the substrate-free spectrum from each of the shown 5-OH-bound spectra (color coding remains same). B shows a plot of induced Soret absorbance change (ΔA389 nm − ΔA421 nm) versus the relevant [5-OH OMP], with data fitted using the Morrison equation to give a Kd value of 2.62 ± 0.19 μm (26). 5-OH OMP binds both A82F and DM heme domains to induce a substrate-like shift in heme iron spin state equilibrium toward the high spin state. Full Kd and Vmax data for 5-OH OMP binding/turnover with WT and mutant BM3 enzymes are given in Table 2.
FIGURE 5.

Time course of substrate oxidation and product formation in the reaction of the P450 BM3 DM enzyme with omeprazole. OMP substrate is shown in black squares, and the products 5-OH OMP and 5-COOH OMP are shown in open circles and open triangles, respectively. Reactions were done as described under “Experimental Procedures.” The reactions reach completion in ∼10–15 min, with most substrate oxidation (and 5-OH OMP formation) occurring in the first 2.5 min.
Structural Analysis of Omeprazole-binding P450 BM3 Mutants
The effect of the F87V mutation in enabling OMP binding may be explained using available structural information for the BM3 heme domain, as space vacated by the F87V substitution directly above the heme plane is likely to allow binding of the bulky substrate (6). The marked effects of the A82F mutation, however, are not intuitive given the peripheral position of Ala-82 to the substrate binding pocket.
To gain detailed understanding of the effects of the A82F mutation in promoting OMP binding, both the A82F and the F87V/A82F double mutant were co-crystallized with OMP, and structures solved by molecular replacement using the NPG-bound heme domain structure (PDB code 1JPZ) (34). Crystal structure PDB codes are 4KEW for the OMP-bound A82F heme domain, and 4KEY for the OMP-bound DM heme domain. In both cases, the global conformation of the heme domain is remarkably similar to previously determined fatty-acid bound structures (0.3 Å over 450 Cα atoms) (Fig. 6A, Table 3) (8, 34).
FIGURE 6.

Structures of P450 BM3 enzymes and their omeprazole-binding sites. A, comparison of WT and mutant BM3 heme domain structures. The FG-helices are in color, and the remainder of the protein structures is depicted in grayscale. The A82F mutation is shown in spheres (where present) and substrate molecules are shown in atom-colored spheres. The heme is shown as red sticks. Panel 1, F87V/A82F (DM) P450 BM3 mutant heme domain in complex with OMP. Panel 2, DM heme domain in the ligand-free form. Panel 3, WT heme domain complex with NPG (PDB code 1JPZ) (34). Panel 4, WT heme domain in the ligand-free form (PDB code 1BU7) (57). B, mode of binding of OMP is shown for the DM (left panel) and A82F (right panel) mutant BM3 heme domain active sites. Because of weak electron density, the labile sulfone oxygen is omitted from the models shown. Key residues contacting the ligand are shown as sticks, and water molecules hydrogen bonding to the OMP are in red. Right panel, OMP from the A82F heme domain structure is overlaid with that from the DM heme domain. Right panel, Phe-82 residues are shown in green for the DM and in cyan for the A82F mutant. The DM Val-87 is in green, and the A82F Phe-87 is in cyan. The distance between the P450 heme iron and the OMP 5-methyl group is 3.9 Å in the A82F heme domain and 4.1 Å in the DM heme domain.
TABLE 3.
Data reduction and final structural refinement statistics for P450 BM3 mutants and their OMP-substrate complexes
| A82F (4KF0), space group P21 | DM-imidazole (4KF2), space group P21 | A82F-OMP (4KEW), space group P212121 | DM-OMP (4KEY), space group P212121 | |
|---|---|---|---|---|
| Cell parameters | a = 59.1 Å, b = 147.2 Å, c = 64.0 Å, β = 97.5° | a = 59.3 Å, b = 151.7 Å, c = 60.8 Å, β = 95.9° | a = 59.4 Å, b = 129.5 Å, c = 145.1 Å | a = 59.3 Å, b = 130.7 Å, c = 146.0 Å |
| Resolution | 64 to 1.45 Å (1.5 to 1.45 Å) | 47 to 1.82 Å (1.86 to 1.82) | 65 to 1.89 (1.94 to 1.89) | 55 to 2.05 (2.1 to 2.05) |
| Rmerge | 7.8% (35.5%) | 6.8% (47.2%) | 8.4% (43.8%) | 8.5% (39.2%) |
| I/σI | 16.2 (2.1) | 14.4 (1.9) | 11.8 (3.2) | 11.4 (3.0) |
| R/Rfree | 14.5/17.8% (28.1/32.3%) | 18.2/22.2% (27.6/31.3%) | 18.8/23.6% (28.5/33.6%) | 18.7/23.5% (24.2/29.2%) |
| Average B | 16.8 Å2 | 22.2 Å2 | 19.7 Å2 | 30.5 Å2 |
| Root mean square deviation bonds/angles | 0.025 Å/2.04° | 0.024 Å/1.91° | 0.022 Å/1.88° | 0.022 Å/1.79° |
| Crystallization conditions | 25% PEG2000MME, 0.2 m MgCl2, pH 6.5 (0.1 m sodium cacodylate) | 15% PEG20K, 15% PEG550MME 0.06 m MgCl2, pH 6.5 (0.1 m imidazole/MES) | 15% PEG4K, 0.2 m MgCl2, pH 6.5 (0.1 m sodium cacodylate) | 15% PEG4K, 0.2 m MgCl2, pH 6.5 (0.1 m sodium cacodylate) |
The PPI ligand is clearly identified from the electron density occupying the substrate binding channel for the OMP complex. In both cases, electron density corresponding to the oxygen atom of the central sulfinyl groups is weak, and it is possible both stereoisomers are present from the racemic mixture. OMP was also shown to be relatively labile, with loss of oxygen from the sulfone noted in aqueous solution (Fig. 6B) (35). We thus modeled OMP without the sulfone oxygen. The pyridinyl moiety is placed in a near-perpendicular orientation directly above the heme plane. The close distance of the heme to the OMP 5-methyl group, in particular, leads to displacement of the water 6th ligand, and this conformation is consistent with the observed 5-OH OMP product. The heme iron to OMP 5-methyl group distances are 3.9 and 4.1 Å in the A82F and DM heme domains, respectively. A single direct hydrogen bond is made between the backbone carbonyl of Leu-437 and one of the methoxybenzimidazole nitrogens. Water molecules are present in close proximity to the other OMP polar groups, and these also mediate a network of hydrogen bonds between protein and ligand. Specific interactions are made via a bridging water molecule between (i) the Ala-74 backbone nitrogen and the methoxy oxygen of the methoxybenzimidazole and (ii) the hydroxyl oxygen of Ser-72 and the second methoxybenzimidazole nitrogen. Although there are no substantial differences in the orientation of the methoxybenzimidazole group for both mutants, close contacts between Phe-87 and the pyridinyl moiety in the A82F mutant structure lead to small reorientation of the substrate in comparison with the DM structure. In the latter, the reduced bulk of the Val-87 appears to provide sufficient space for the substrate to adopt a less strained conformation. This likely explains the difference in affinity observed between the A82F and DM mutants. Other protein-OMP interactions in the A82F and DM complex structures involve hydrophobic interactions between residues near both the pyridinyl (e.g. Thr-438, Ile-263, and Ala-328) and methoxybenzimidazole (e.g. Val-26 and Leu-188) ends of the OMP substrate (Fig. 7).
FIGURE 7.

Interactions of omeprazole in the active site of the A82F BM3 heme domain. The diagram shows the binding site of OMP (without the labile sulfinyl oxygen) in the A82F mutant BM3 heme domain. For OMP, carbon atoms are shown in black, oxygens in red, sulfur in yellow, nitrogens in blue, and the oxygens of water molecules in cyan. Bonds in the OMP substrate are shown in purple, and bonds in selected amino acids are in brown. Hydrogen bonds are shown (with their lengths) as green dashed lines. Amino acids making hydrophobic interactions with the OMP are shown as red arcs with radiating lines. OMP atoms involved in these hydrophobic interactions are shown with radiating red lines. A direct hydrogen bond interaction is made between the backbone carbonyl of Leu-437 and one of the OMP benzimidazole group nitrogens (NE1). A further bridging hydrogen bond occurs from the Ser-72 hydroxyl group through a water molecule (water 781) to the other benzimidazole nitrogen (NV). A final bridging hydrogen bond interaction occurs between the backbone nitrogen of Ala-74 and the benzimidazole methoxy oxygen (O3) via another water molecule (water 761). A number of hydrophobic protein-OMP interactions are seen. These include interactions with Leu-188 at the benzimidazole methoxy group (C4) and with Ala-328 at the pyridinyl 5-methyl group (C1). The diagram was produced using Ligplot+ using the structure of the A82F heme domain-omeprazole complex solved in this study (PDB code 4KEW) (58).
A clear rationale for the marked difference in OMP affinity between A82F containing mutants and the WT BM3 is not provided by the mutant-OMP structures. Fig. 8A shows the active sites in detail for the overlaid A82F OMP-bound BM3 heme domain and the NPG-bound WT heme domain. The additional bulk of the Phe-82 side chain only interacts edge-on with the pyridinyl moiety of OMP, and the only significant change in the positions of nearby key residues occurs for Phe-87. The Phe-87 side chain is seen in different conformations in the A82F/OMP structure, compared with the WT BM3/NPG heme domain. Fig. 8B shows an alternative view of the active site for the DM/OMP-bound heme domain and the WT/NPG-bound P450, looking along the I-helix, with the F/G-helices highlighted. This reinforces the strong structural similarity between the WT and DM mutant substrate-bound structures. All these structures occupy the SB conformation. However, the ligand-free A82F and DM heme domains also have the SB conformation (Fig. 6A). However, the WT BM3 heme domain occupies a distinct conformation when SF, and thus the A82F mutation is key to shifting the equilibrium between SF and SB conformations. Previous studies also showed that the structural state of the heme domain can be significantly affected by single mutations at key positions (36).
FIGURE 8.

Stereoviews of structural overlays of substrate-bound forms of the BM3 A82F-containing mutant heme domains with WT BM3. A shows a stereoview of the A82F-OMP heme domain active site (in red) with that of the WT-NPG structure (PDB 1JPZ in blue) (34). Key amino acids are shown in lines, and the bound ligands are shown in atom-colored sticks (OMP with magenta carbons; NPG with light blue carbons). Besides the nature of the ligand itself, and the obvious difference of the A82F mutation, there are very few differences between the structures, and these are mainly limited to Phe-87 occupying multiple conformations in the A82F-OMP structure. B shows an alternative view of the BM3 double mutant (DM, F87V/A82F) OMP-bound heme domain structure overlaid with the NPG substrate-bound structure of the WT BM3 heme domain. The F/G-helix region is colored in red for the BM3 DM and in blue for the substrate-bound WT BM3. Key amino acid residues and the respective ligands are shown as sticks. As also seen for A, surprisingly little change can be observed in the DM protein structure compared with WT BM3, despite the distinct nature of the ligand and the introduction of two mutations.
We also determined the crystal structures of the ligand-free and imidazole-bound A82F (PDB code 4KF0) and DM (4KF2) heme domains. In the latter case, imidazole present in the crystallization buffer ligated to the heme iron. These structures share a similar conformation that is again distinct from previously observed WT BM3 heme domain SF structures (Figs. 6A and 9). These data again point to the A82F substitution producing dramatic changes in the structure and behavior of ligand-free A82F-containing mutants. The most significant changes occur in the positioning of the FG-helices, which adopt a conformation that places the FG-loop further away from the protein core, leading to increased mobility. This altered position is a consequence of a reorganization of the hydrophobic contact between the FG-helices and the I-helix. In the WT SF conformation, the N-terminal region of the I-helix adopts a slightly different orientation compared with the SB conformation. In the case of the A82F-containing mutants, this motion leads to a close contact between Ile-263 and Phe-82 (Fig. 9). This results in a minor reorientation of the I-helix and the Ile-263 side chain, which directly affects the position of the G-helix residue Met-177. The repositioning of the G-helix is accompanied by reorientation of hydrophobic residues on the B- and F-helices.
FIGURE 9.

Structural overlay of the omeprazole-bound A82F mutant with the WT BM3 heme domain. A stereoview is shown for a structural overlay of the A82F BM3 heme domain with the WT heme domain (PDB 1BU7). Color coding is as in Fig. 6, with F/G-helices in green for the substrate-free A82F mutant and in yellow for the substrate-free WT BM3 heme domain.
Although the F87V mutant enables binding of OMP detectable by heme absorbance shift, the Kd value for OMP binding to F87V BM3 heme domain is ∼30- and 230-fold weaker than that observed for the A82F and DM heme domains, respectively. The improved ligand binding properties of the A82F mutants can be understood in view of the large changes introduced by the A82F mutation in the ligand-free but not ligand-bound structures. The shift in the BM3 conformational equilibrium from SF to SB (as induced by ligand binding) is dependent on the free energy difference between both conformations and the free energy associated with ligand binding. The A82F-containing mutant crystal structures suggest that the mutation leads to a substantially altered free energy difference between both conformations. The fact OMP only appears to bind with measurable affinity to A82F-containing mutants suggests that the A82F mutation destabilizes the substrate-free conformation.
To confirm this hypothesis, we performed DSC analysis of the substrate-free and OMP-bound forms of WT and all mutant heme domains, and we compared data with NPG-bound forms. The results showed that both F87V and A82F mutations diminished thermal stability of the BM3 heme domain. The WT heme domain has two unfolding transitions (Tm values) at 65.7 °C (Tm1, major) and 59.0 °C (Tm2, minor). The Tm values are not altered significantly by OMP, but NPG stabilizes the P450 (Tm1 = 70.4 °C). Consistent with our model, the A82F Tm values are 59.7 and 49.7 °C, substantially lower than WT BM3. F87V was also destabilized, albeit to a lesser extent, with the minor transition no longer seen (Tm1 = 61.3 °C). The DM showed greatest destabilization (Tm = 50.9 °C and 58.0 °C). Binding of OMP to A82F-containing mutants resulted in single unfolding transitions, with negligible change in Tm1 for A82F (59.6 °C) and an ∼2 °C stabilization for the DM (59.9 °C). A similar effect was seen for F87V heme domain (Tm = 63.1 °C) (supplemental Table S1). Thus, A82F has the major effect on stability of BM3, but F87V also destabilizes the protein, and there is an additive effect on combining the mutations (Fig. 10 and supplemental Fig. S5 and Table S1).
FIGURE 10.
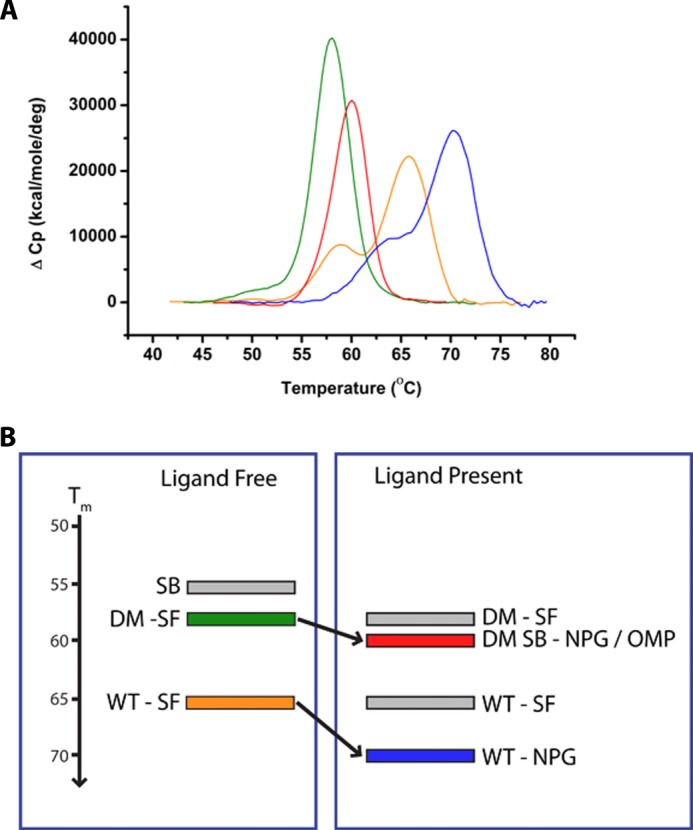
Conformational equilibria and the relationship with structural stability in P450 BM3. A, DSC data for the WT and DM P450 BM3 heme domains in substrate-free, OMP-, and NPG-bound forms. B, schematic overview of the conformational equilibria proposed for the BM3 WT and DM mutant heme domains. Individual conformational states (as represented by crystal structures) are depicted as gray-shaded rectangles when largely unpopulated and color rectangles (color-coded to match A) when significantly populated. The y axis indicates the relative Tm values for the unfolding of these proteins.
DISCUSSION
The development of efficient biocatalysts that generate human drug metabolites has become an area of great interest. Human P450s are responsible for most phase I xenobiotic metabolism, producing numerous oxidized and other metabolites from human drugs (37). For omeprazole, the principal metabolic pathway is 5-hydroxylation catalyzed by CYP2C19, the product of which can then be converted to the sulfone by CYP3A4. Oxidized metabolites of human drugs are required for pharmaceutical compliance, for metabolite safety testing, and for molecular interaction studies as required by Food and Drug Administration guidelines. Use of human P450s to produce small quantities of specific metabolites is a possibility, but caveats include slow reaction rates, requirement for a separate redox partner, and enzyme instability. Synthetic chemistry to make oxidized metabolites is an alternative, but controlling regioselectivity of oxidation, maintaining stability of compounds, and requirements for several steps giving low yield are major issues. An attractive alternative is to engineer high activity microbial P450s for specific oxidation of drugs.
A system of choice is P450 BM3, due to its catalytically self-sufficient nature, high turnover rates, and availability of excellent structural data to guide protein engineering. Crystal structures of substrate-free and fatty acid-bound forms of the wild-type BM3 heme domain drove early protein engineering studies on BM3 (7, 8). This enabled identification of residues important in binding the substrate carboxylate at the mouth of the active site (Arg-47 and Tyr-51), as well as amino acids crucial for regulating heme iron redox potential (Phe-393) and the coupling of electron transfer to substrate oxidation (Thr-268) (6, 38–40). Parallel studies documented its fast rates of fatty acid substrate oxidation, resulting from rapid electron transfer from NADPH through its fused cytochrome P450 reductase partner (5, 41), and more recent studies have confirmed that the enzyme is functional as a dimer (42, 43) and that electron transfer to drive catalysis occurs between the NADPH-cytochrome P450 reductase module of one monomer and the heme domain of the other in the BM3 dimer (44).
The application of random mutagenesis, recombination, and directed evolution approaches (pioneered by Arnold and co-workers) brought a new dimension to research on BM3, demonstrating that radical changes in its substrate selectivity could be engineered. A key development was the production of the 139-3 BM3 mutant (containing 11 mutations), which enabled the hydroxylation of a range of alkanes from octane through to propane, including cyclohexane (45). However, only one mutation (V78A) occurred at a position known to be in contact with fatty acid from preceding structural data (8). Although mutations at Ala-82 and Phe-87 were not present in the 139-3 mutant, the addition of an A82L mutation to 139-3 resulted in increased coupling of NADPH oxidation to the hydroxylation of propane and octane, indicating improvements to the productive binding of these substrates. A related engineered BM3 variant (named 9/10A and containing 13 mutations, 8 of which are shared with 139-3) was inactive in ethane hydroxylation but was shown to develop ethane hydroxylase activity and to improve propane hydroxylation when an A82S mutation was incorporated, together with either two or four further mutations (46). A related propane hydroxylase variant (35E11) containing 16 mutations (mostly accumulated from the 139-3 and 9/10A progenitors) also has the A82S mutation, although propane hydroxylation rates and coupling were improved on introduction of further groups of mutations that included L188P (47). The later structural data for the 139-3 mutant heme domain in complex with NPG provided insights into structural changes that could promote binding and oxidation of short chain alkanes, while retaining a conformational state (SB) similar to the WT-NPG complex structure (34). In particular, active site mutation V78A enlarged a hydrophobic pocket close to the heme, whereas A184V enabled interactions with Leu-437 across the active site channel, likely enabling van der Waals contacts with the terminal carbon of octane and protecting the substrate from solvent (48). Predictive modeling based on the 139-3 structure indicated that A82S and other mutations altered substrate channel organization but that the destabilizing L188P mutation was a major determinant in promoting efficient propane oxidation, and likely acted by inducing structural disruption at the end of the F-helix, favoring a conformational change toward a catalytically productive form (48). Given the conservative nature of the A82S mutation, it is unclear whether this mutation alone might favor the SB conformation (as does the A82F mutation in our work). However, the SB structural conformation might be expected in any case for the NPG-bound enzyme.
The fact that the structurally destabilizing L188P mutation has profound effects on BM3 binding/turnover of propane has parallels in recent studies by Wong and co-workers, in which (i) a substrate-free I401P mutant (the mutated residue being adjacent to the heme proximal ligand Cys-400) was crystallized in a SB-like conformation, and (ii) an A330P mutant in an inter-helical β-sheet part of the P450 caused structural disruption that reshaped the active site cavity. For both these mutants, large increases in catalytic activity with non-natural substrates such as toluene, propylbenzene, and 3-methylpentane were observed (18, 49).
Although some structural data are available for BM3 variants promoting short chain/alkane binding and oxidation, there is also much interest in the application of BM3 mutants for the production of metabolites of human drugs. BM3 variants have been shown to oxidize drugs such as diclofenac, ibuprofen, and acetaminophen, but no structural data were obtained (50, 51). In this study, we present the first structure of a BM3 P450 bound to a human drug substrate (OMP), and we show that a single active site mutation (A82F) is sufficient to facilitate tight binding of this PPI drug, enabling the same oxidative reaction (hydroxylation at the pyridinyl 5-methyl group) as catalyzed by the main human metabolizing P450 CYP2C19. OMP binding is further enhanced by introduction of the F87V mutation in the immediate vicinity of the heme, avoiding close contacts between Phe-87 and the OMP. Despite the obvious differences between the natural fatty acid substrates and OMP, the conformation of the OMP-bound mutants is nearly identical to previously determined WT fatty acid complexes. This suggests BM3 active site architecture is largely determined by the overall protein conformation (SB versus SF) rather than the exact nature of the ligand. Studies of human urinary metabolites of OMP showed that the 5-OH OMP and 5-COOH OMP were the major derivatives detected, with the former predominant (35, 52). The relatively tight binding of the primary metabolite (5-OH OMP) to both A82F and (particularly) the DM BM3 reinforces that the oxidized derivative retains the ability to bind in a catalytically competent mode to A82F-containing mutants, consistent with its further oxidation to 5-COOH OMP by the DM BM3 (Fig. 4 and Table 2). The BM3 DM's ability to catalyze successive OMP oxidations to generate 5-COOH OMP points to the possibility that CYP2C19 and/or other human cytochrome P450s might further oxidize 5-OH OMP to 5-COOH OMP.
Early structural studies of BM3's heme domain identified Phe-87 as a key active site residue, mutations of which altered binding and regioselectivity of fatty acid oxidation (6, 10, 17), and both Phe-87 and Ala-82 mutations are frequently contained in BM3 mutants generated by directed evolution and other strategies for altered substrate specificity (13, 46). Other studies on Phe-87 mutants have further highlighted its importance in controlling access to the heme center and in altering reactivity. Notable recent studies have highlighted that an F87V BM3 mutant catalyzes hydroxylation of both testosterone and progesterone. In the case of testosterone, 2β-hydroxytestosterone and 16β-hydroxytestosterone were formed in roughly equal amounts by F87V BM3. However, triple mutants that also contained the A82F mutation (F87V, A82F, and V78L/V78T/V78I) showed 3–4-fold greater catalytic efficiency and produced the 16β-hydroxytestosterone product at ∼90%, suggesting an important influence of the A82F mutation on steroid binding mode (12). Other work has pointed to the importance of Phe-87 in tolerance to solvents used for substrate solubilization. Crystal structure data showed that DMSO was able to compete with the heme's 6th ligand water molecule for coordination to the iron in the F87A heme domain mutant but not in the WT heme domain at the same DMSO concentration (53).
Although the influence of Phe-87 mutations on accessibility to the heme center is easily rationalized from crystal structure data, the mechanism by which Ala-82 mutations diversify BM3 substrate selectivity has been relatively poorly understood. Huang et al. (20) solved the crystal structure of a palmitate-bound A82F heme domain, after purifying the mutant from E. coli in the lipid-bound form. The structure was similar to that of the NPG-bound WT heme domain (34). Our studies of the substrate-free A82F structure reveal that the mutation induces a change in the protein conformational landscape, leading to a distinct and previously unobserved position of the FG-helices. These structural changes have profound consequences for substrate binding in the A82F mutants and point to a role for this residue as a gatekeeper for preferred substrate access as a consequence of its regulation of the conformational state and/or dynamics of structural change in the P450. Thus, although the mutation is removed from the immediate vicinity of the heme iron, it induces structural reconfiguration that enables the binding of OMP and evidently that of other molecules. Our DSC studies show that the A82F mutation (in particular) alters the thermodynamic stability of BM3, decreasing the Tm for protein unfolding. This suggests that its new conformational “flexibility” is a key facet underpinning its diversification of substrate selectivity, at least in part due to removal of inherent substrate bias toward long chain fatty acids by decreasing the free energy barrier corresponding to the transition to the SB state (Fig. 10B). These data are consistent with conclusions drawn by Bloom et al. (54) with respect to the capacity of protein-destabilizing mutations to impart novel substrate selectivity/reactivity on an enzyme.
In conclusion, our data point to new lessons to be learned from the outcomes of previous studies of BM3 (and other enzymes) in that amino acid changes that affect the dynamics of proteins in unpredictable ways may lead to mutations “distant” from the active site (or otherwise appearing not to impact significantly on substrate binding/affinity) having profound effects on enzyme catalysis and substrate specificity. The gatekeeper hypothesis thus points to Ala-82 as a crucial target residue in research to enable further engineering of BM3 for diverse functions. Our findings show that, contrary to previous approaches focusing on increasing enzyme stability as a path to biotechnologically relevant enzymes (55, 56), enzyme conformational destabilization is key to reducing the thermodynamic barrier to substrate binding and therefore to altered enzymatic activities, which could enable rapid identification of P450 (and other enzyme) variants with biotechnologically important activities.
Acknowledgments
We acknowledge the following University of Manchester staff: Dr. Colin Levy for assistance with synchrotron x-ray data collection, Dr. Tom Jowitt for assistance with DSC studies, and Dr. Robert Šardzík for helpful discussions on NMR analysis.
This work was supported by United Kingdom Biotechnology and Biological Sciences Research Council Research Grant BB/F00883X1 (to A. W. M. and D. L. supporting A. E. M.) and Industrial CASE Studentship BB/G01698/1 from Cypex Ltd. (to A. W. M. and M. W. V. supporting C. F. B.).

This article contains supplemental Results, Figs. S1–S5, and Table S1.
The atomic coordinates and structure factors (codes 4KEW, 4KEY, 4KF0, and 4KF2) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- P450
- cytochrome P450 monooxygenase
- BM3
- flavocytochrome P450 BM3
- DM
- BM3 F87V/A82F double mutant
- DSC
- differential scanning calorimetry
- NPG
- N-palmitoylglycine
- OMP
- omeprazole
- PPI
- proton pump inhibitor
- SB
- substrate bound
- SF
- substrate free
- 5-OH OMP
- 5-hydroxy omeprazole
- 5-COOH OMP
- 5-carboxy omeprazole
- PDB
- Protein Data Bank.
REFERENCES
- 1. Guengerich F. P. (2005) in Cytochrome P450: Structure, Mechanism and Biochemistry (Ortiz de Montellano P. R., ed) 3rd Ed., pp. 377–530, Kluwer Academic/Plenum Press, New York [Google Scholar]
- 2. Munro A. W., Girvan H. M., McLean K. J. (2007) Variations on a (t)heme–novel mechanisms, redox partners, and catalytic functions in the cytochrome P450 superfamily. Nat. Prod. Rep. 24, 585–609 [DOI] [PubMed] [Google Scholar]
- 3. Xu F., Bell S. G., Rao Z., Wong L. L. (2007) Structure-activity correlations in pentachlorobenzene oxidation by engineered cytochrome P450cam. Protein Eng. Des. Sel. 20, 473–480 [DOI] [PubMed] [Google Scholar]
- 4. Peters M. W., Meinhold P., Glieder A., Arnold F. H. (2003) Regio- and enantioselective alkane hydroxylation with engineered cytochromes P450 BM-3. J. Am. Chem. Soc. 125, 13442–13450 [DOI] [PubMed] [Google Scholar]
- 5. Munro A. W., Daff S., Coggins J. R., Lindsay J. G., Chapman S. K. (1996) Probing electron transfer in flavocytochrome P-450 BM3 and its component domains. Eur. J. Biochem. 239, 403–409 [DOI] [PubMed] [Google Scholar]
- 6. Noble M. A., Miles C. S., Chapman S. K., Lysek D. A., MacKay A. C., Reid G. A., Hanzlik R. P., Munro A. W. (1999) Roles of key active-site residues in flavocytochrome P450 BM3. Biochem. J. 339, 371–379 [PMC free article] [PubMed] [Google Scholar]
- 7. Ravichandran K. G., Boddupalli S. S., Hasermann C. A., Peterson J. A., Deisenhofer J. (1993) Crystal structure of hemoprotein domain of P450BM-3, a prototype for microsomal P450s. Science 261, 731–736 [DOI] [PubMed] [Google Scholar]
- 8. Li H., Poulos T. L. (1997) The structure of the cytochrome P450BM-3 haem domain complexed with the fatty acid substrate, palmitoleic acid. Nat. Struct. Biol. 4, 140–146 [DOI] [PubMed] [Google Scholar]
- 9. Munro A. W., Leys D. G., McLean K. J., Marshall K. R., Ost T. W., Daff S., Miles C. S., Chapman S. K., Lysek D. A., Moser C. C., Page C. C., Dutton P. L. (2002) P450 BM3: The very model of a modern flavocytochrome. Trends Biochem. Sci. 27, 250–257 [DOI] [PubMed] [Google Scholar]
- 10. Whitehouse C. J., Bell S. G., Wong L. L. (2012) P450BM3 (CYP102A1): connecting the dots. Chem. Soc. Rev. 41, 1218–1260 [DOI] [PubMed] [Google Scholar]
- 11. Coelho P. S., Brustad E. M., Kannan A., Arnold F. H. (2013) Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science 339, 307–310 [DOI] [PubMed] [Google Scholar]
- 12. Kille S., Zilly F. E., Acevedo J. P., Reetz M. T. (2011) Regio- and stereoselectivity of P450-catalysed hydroxylation of steroids controlled by laboratory evolution. Nat. Chem. 3, 738–743 [DOI] [PubMed] [Google Scholar]
- 13. Carmichael A. B., Wong L. L. (2001) Protein engineering of Bacillus megaterium CYP102. The oxidation of polycyclic aromatic hydrocarbons. Eur. J. Biochem. 268, 3117–3125 [DOI] [PubMed] [Google Scholar]
- 14. Sawayama A. M., Chen M. M., Kulanthaivel P., Kuo M. S., Hemmerle H., Arnold F. H. (2009) A panel of cytochrome P450 BM3 variants to produce drug metabolites and diversify lead compounds. Chemistry 15, 11723–11729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reetz M. T. (2011) Laboratory evolution of stereoselective enzymes: a prolific source of catalysts for asymmetric reactions. Angew. Chem. Int. Ed. Engl. 50, 138–174 [DOI] [PubMed] [Google Scholar]
- 16. Ost T. W., Miles C. S., Murdoch J., Cheung Y., Reid G. A., Chapman S. K., Munro A. W. (2000) Rational re-design of the substrate binding site of flavocytochrome P450 BM3. FEBS Lett. 486, 173–177 [DOI] [PubMed] [Google Scholar]
- 17. Dietrich M., Do T. A., Schmid R. D., Pleiss J., Urlacher V. B. (2009) Altering the regioselectivity of the subterminal fatty acid hydroxylase P450 BM-3 towardγ and δ positions. J. Biotechnol. 139, 115–117 [DOI] [PubMed] [Google Scholar]
- 18. Whitehouse C. J., Yang W., Yorke J. A., Rowlatt B. C., Strong A. J., Blanford C. F., Bell S. G., Bartlam M., Wong L. L., Rao Z. (2010) Structural basis for the properties of two single-site proline mutants of CYP102A1 (P450BM3). ChemBioChem 11, 2549–2556 [DOI] [PubMed] [Google Scholar]
- 19. Joyce M. G., Girvan H. M., Munro A. W., Leys D. (2004) A single mutation in cytochrome P450 BM3 induces the conformational rearrangement seen upon substrate binding in the wild-type enzyme. J. Biol. Chem. 279, 23287–23293 [DOI] [PubMed] [Google Scholar]
- 20. Huang W.-C., Westlake A. C., Maréchal J. D., Joyce M. G., Moody P. C., Roberts G. C. (2007) Filling a hole in cytochrome P450 BM3 improves substrate binding and catalytic efficiency. J. Mol. Biol. 373, 633–651 [DOI] [PubMed] [Google Scholar]
- 21. Ibeanu G. C., Ghanayem B. I., Linko P., Li L., Pederson L. G., Goldstein J. A. (1996) Identification of residues 99, 220, and 221 of human cytochrome P450 2C19 as key determinants of omeprazole activity. J. Biol. Chem. 271, 12496–12501 [DOI] [PubMed] [Google Scholar]
- 22. Omura T., Sato R. (1964) The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J. Biol. Chem. 239, 2379–2385 [PubMed] [Google Scholar]
- 23. Perera R., Sono M., Sigman J. A., Pfister T. D., Lu Y., Dawson J. H. (2003) Neutral thiol as a proximal ligand to ferrous heme iron: implications for heme proteins that lose cysteine thiolate on reduction. Proc. Natl. Acad. Sci. U.S.A. 100, 3641–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dunford A. J., McLean K. J., Sabri M., Seward H. E., Heyes D. J., Scrutton N. S., Munro A. W. (2007) Rapid P450 heme iron by laser photoexcitation of Mycobacterium tuberculosis CYP121 and CYP51B1. Analysis of CO complexation reactions and reversibility of the P450/P420 equilibrium. J. Biol. Chem. 282, 24816–24824 [DOI] [PubMed] [Google Scholar]
- 25. Driscoll M. D., McLean K. J., Levy C., Mast N., Pikuleva I. A., Lafite P., Rigby S. E., Leys D., Munro A. W. (2010) Structural and biochemical characterization of Mycobacterium tuberculosis CYP142: Evidence for multiple cholesterol 27-hydroxylase activities in a human pathogen. J. Biol. Chem. 285, 38270–38382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morrison J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 27. Bui S. H., McLean K. J., Cheesman M. R., Bradley J. M., Rigby S. E., Levy C. W., Leys D., Munro A. W. (2012) Unusual spectroscopic and ligand binding properties of the cytochrome P450-flavodoxin fusion enzyme XplA. J. Biol. Chem. 287, 19699–19714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kabsch W. (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 29. Collaborative Computation Project No. 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 30. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 31. Venkataraman H., Beer S. B., Bergen L. A., Essen Nv, Geerke D. P., Vermeulen N. P., Commandeur J. N. (2012) A single active site mutation inverts stereoselectivity of 16-hydroxylation of testosterone catalyzed by engineered cytochrome P450 BM3. ChemBioChem 13, 520–523 [DOI] [PubMed] [Google Scholar]
- 32. Miles J. S., Munro A. W., Rospendowski B. N., Smith W. E., McKnight J., Thomson A. J. (1992) Domains of the catalytically self-sufficient cytochrome P-450 BM-3. Genetic construction, overexpression, purification and spectroscopic characterization. Biochem. J. 288, 503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Desta Z., Zhao X., Shin J. G., Flockhart D. A. (2002) Clinical significance of the P4502C19 genetic polymorphism. Clin. Pharmacokinet. 41, 913–958 [DOI] [PubMed] [Google Scholar]
- 34. Haines D. C., Tomchick D. R., Machius M., Peterson J. A. (2001) Pivotal role of water in the mechanism of P450BM-3. Biochemistry 40, 13456–13465 [DOI] [PubMed] [Google Scholar]
- 35. Renberg L., Simonsson R., Hoffmann K.-J. (1989) Identification of two main urinary metabolites of [14C]omeprazole in humans. Drug Metab. Dispos. 17, 69–76 [PubMed] [Google Scholar]
- 36. Girvan H. M., Seward H. E., Toogood H. S., Cheesman M. R., Leys D., Munro A. W. (2007) Structural and spectroscopic characterization of P450 BM3 mutants with unprecedented P450 heme iron ligand sets. New heme ligation states influence conformational equilibria in P450 BM3. J. Biol. Chem. 282, 564–572 [DOI] [PubMed] [Google Scholar]
- 37. Guengerich F. P. (2008) Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol. 21, 70–83 [DOI] [PubMed] [Google Scholar]
- 38. Li H., Poulos T. L. (1999) Fatty acid metabolism, conformational change, and electron transfer in cytochrome P450 BM-3. Biochim. Biophys. Acta 1441, 141–149 [DOI] [PubMed] [Google Scholar]
- 39. Ost T. W., Miles C. S., Munro A. W., Murdoch J., Reid G. A., Chapman S. K. (2001) Phenylalanine 393 exerts thermodynamic control over the heme of flavocytochrome P450 BM3. Biochemistry 40, 13421–13429 [DOI] [PubMed] [Google Scholar]
- 40. Yeom H., Sligar S. G., Li H., Poulos T. L., Fulco A. J. (1995) The role of Thr268 in oxygen activation of cytochrome P450 BM-3. Biochemistry 34, 14733–14740 [DOI] [PubMed] [Google Scholar]
- 41. Narhi L. O., Fulco A. J. (1986) Characterization of a catalytically self-sufficient 119,000-dalton cytochrome P-450 monooxygenase induced by barbiturates in Bacillus megaterium. J. Biol. Chem. 261, 7160–7169 [PubMed] [Google Scholar]
- 42. Neeli R., Girvan H. M., Lawrence A., Warren M. J., Leys D., Scrutton N. S., Munro A. W. (2005) The dimeric form of flavocytochrome P450 BM3 is catalytically functional as a fatty acid hydroxylase. FEBS Lett. 579, 5582–5588 [DOI] [PubMed] [Google Scholar]
- 43. Kitazume T., Haines D. C., Estabrook R. W., Chen B., Peterson J. A. (2007) Obligatory intramolecular electron-transfer from FAD to FMN in dimeric P450BM-3. Biochemistry 46, 11892–11901 [DOI] [PubMed] [Google Scholar]
- 44. Girvan H. M., Dunford A. J., Neeli R., Ekanem I. S., Waltham T. N., Joyce M. G., Leys D., Curtis R. A., Williams P., Fisher K., Voice M. W., Munro A. W. (2011) Flavocytochrome P450 BM3 mutant W1046A is a NADH-dependent fatty acid hydroxylase: implications for the mechanism of electron transfer in the P450 BM3 dimer. Arch. Biochem. Biophys. 507, 75–85 [DOI] [PubMed] [Google Scholar]
- 45. Glieder A., Farinas E. T., Arnold F. H. (2002) Laboratory evolution of a soluble, self-sufficient, highly active alkane hydroxylase. Nat. Biotechnol. 20, 1135–1139 [DOI] [PubMed] [Google Scholar]
- 46. Meinhold P., Peters M. W., Chen M. M., Takahashi K., Arnold F. H. (2005) Direct conversion of ethane to ethanol by engineered cytochrome P450 BM3. ChemBioChem 6, 1765–1768 [DOI] [PubMed] [Google Scholar]
- 47. Fasan R., Chen M. M., Crook N. C., Arnold F. H. (2007) Engineered alkane-hydroxylating cytochrome P450BM3 exhibiting native-like catalytic properties. Angew. Chem. Int. Ed. Engl. 46, 8414–8418 [DOI] [PubMed] [Google Scholar]
- 48. Fasan R., Meharenna Y. T., Snow C. D., Poulos T. L., Arnold F. H. (2008) evolutionary history of a specialized P450 propane monooxygenase. J. Mol. Biol. 383, 1069–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Whitehouse C. J., Bell S. G., Yang W., Yorke J. A., Blanford C. F., Strong A. J., Morse E. J., Bartlam M., Rao Z., Wong L.-L. (2009) A highly active single-mutation variant of P450BM3 (CYP102A1). ChemBioChem 10, 1654–1656 [DOI] [PubMed] [Google Scholar]
- 50. Tsotsou G. E., Sideri A., Goyal A., Di Nardo G., Gilardi G. (2012) Identification of mutant Asp251Gly/Gln307His of cytochrome P450 BM3 for the generation of metabolites of diclofenac, ibuprofen, and tolbutamide. Chemistry 18, 3582–3588 [DOI] [PubMed] [Google Scholar]
- 51. Damsten M. C., van Vugt-Lussenburg B. M., Zeldenthuis T., de Vlieger J. S., Commandeur J. N., Vermeulen N. P. (2008) Application of drug metabolising mutants of cytochrome P450 BM3 (CYP102A1) as biocatalysts for the generation of reactive metabolites. Chem. Biol. Interact. 171, 96–107 [DOI] [PubMed] [Google Scholar]
- 52. Abelö A., Andersson T. B., Antonsson M., Naudot A. K., Skånberg I., Weidolf L. (2000) Stereoselective metabolism of omeprazole by human cytochrome P450 enzymes. Drug Metab. Dispos. 28, 966–972 [PubMed] [Google Scholar]
- 53. Kuper J., Tee K. L., Wilmanns M., Roccatano D., Schwaneberg U., Wong T. S. (2012) The role of active-site Phe-87 in modulating the organic co-solvent tolerance of cytochrome P450 BM3 monooxygenase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 1013–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bloom J. D., Labthavikul S. T., Otey C. R., Arnold F. H. (2006) Protein stability promotes evolvability. Proc. Natl. Acad. Sci. U.S.A. 103, 5869–5874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Romero P. A., Krause A., Arnold F. H. (2013) Navigating the protein fitness landscape with Gaussian processes. Proc. Natl. Acad. Sci. U.S.A. 110, E193–E201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li Y., Drummond D. A., Sawayama A. M., Snow C. D., Bloom J. D. (2007) A diverse family of thermostable cytochrome P450s created by recombination of stabilizing fragments. Nat. Biotechnol. 25, 1051–1056 [DOI] [PubMed] [Google Scholar]
- 57. Sevrioukova I. F., Li H., Zhang H., Peterson J. A., Poulos T. L. (1999) Structure of a cytochrome P450 redox partner electron transfer complex. Proc. Natl. Acad. Sci. U.S.A. 96, 1863–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Laskowski R. A., Swindells M. B. (2011) Ligplot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 [DOI] [PubMed] [Google Scholar]