

Background: Ligands that selectively target α6β2* nAChRs are needed.
Results: An analog of α-conotoxin PeIA was synthesized that was >15,000-fold more potent at inhibiting α6/α3β2β3 receptors than the closely related α3β2 subtype.
Conclusion: PeIA analogs can be synthesized that distinguish between α6/α3β2β3 and α3β2 receptors.
Significance: Selective ligands will facilitate the identification of α6β2* receptors in tissues that co-express α3β2* receptors.
Keywords: Electrophysiology, Neurotoxin, Neurotransmitter Receptors, Nicotinic Acetylcholine Receptors, Oocyte, alpha-Conotoxin, alpha3beta2, alpha6beta2
Abstract
The nicotinic acetylcholine receptor (nAChR) subtype α6β2* (the asterisk denotes the possible presence of additional subunits) has been identified as an important molecular target for the pharmacotherapy of Parkinson disease and nicotine dependence. The α6 subunit is closely related to the α3 subunit, and this presents a problem in designing ligands that discriminate between α6β2* and α3β2* nAChRs. We used positional scanning mutagenesis of α-conotoxin PeIA, which targets both α6β2* and α3β2*, in combination with mutagenesis of the α6 and α3 subunits, to gain molecular insights into the interaction of PeIA with heterologously expressed α6/α3β2β3 and α3β2 receptors. Mutagenesis of PeIA revealed that Asn11 was located in an important position that interacts with the α6 and α3 subunits. Substitution of Asn11 with a positively charged amino acid essentially abolished the activity of PeIA for α3β2 but not for α6/α3β2β3 receptors. These results were used to synthesize a PeIA analog that was >15,000-fold more potent on α6/α3β2β3 than α3β2 receptors. Analogs with an N11R substitution were then used to show a critical interaction between the 11th position of PeIA and Glu152 of the α6 subunit and Lys152 of the α3 subunit. The results of these studies provide molecular insights into designing ligands that selectively target α6β2* nAChRs.
Introduction
Nicotinic acetylcholine receptors are ligand-gated ion channels ubiquitously expressed throughout the nervous system. In vertebrates, there are nine α (α2–α10) and three β (β2–β4) subunits that assemble in various combinations to form pentameric channels (1). Some α subunits, including α7, α8, and α9, can form homopentamers, but all other α subunits, with the exception of α10 (which can form α9α10 receptors), require a β subunit for functional expression, such as those that contain α6 or α3 subunits. In the mammalian central nervous system, α6- and α3-containing nAChRs2 may be co-expressed in several regions, including the inferior and superior colliculi, optic nerve, ventrolateral geniculate, striatum, medial habenula, interpeduncular nucleus, and dorsal horn of the spinal cord (2–6). The α6β2* subtype has been shown to play a critical role in the modulation of striatal dopamine release (7–9) and has been implicated in diseases of the basal ganglia, including Parkinson disease (10, 11). More recently, α6β2* receptors have been shown to modulate dopaminergic activity in reward centers of the brain and are thus likely to be key contributors to the reinforcing properties of nicotine (12–15). The ability to pharmacologically distinguish between α6β2* and α3β2* in these areas has been hampered by the scarcity of ligands that can discriminate between these two subtypes. This lack of selective ligands is due in part to the high homology of the N-terminal, ligand-binding domains of α6 and α3 subunits; consequently, receptors that contain these subunits share similar pharmacological profiles.
Peptides identified in the venom of marine cone snails that target nAChRs are called α-conotoxins (α-Ctxs) (16, 17) and have proven to be highly valuable tools for distinguishing among the various nAChR subtypes. These peptides are usually 12–20 amino acids in length and are characterized by the presence of two disulfide bridges between cysteine residues that serve to keep the peptide in its biologically active configuration. Some of these α-Ctxs include the widely used MII as well as PnIA, OmIA, LtIA, and PeIA (18–23). Unfortunately, these peptides all target both α6- and α3-containing nAChRs with similar potencies and thus do not discriminate well between α6β2* and α3β2* nAChRs.
PeIA was originally described as a potent inhibitor of α9α10 nAChRs (23) and was subsequently discovered to also inhibit α6/α3β2β3 and α3β2 subtypes with equal potencies (24). However, PeIA is much less active on the closely related α6β4 and α3β4 subtypes and has very little activity on all other nAChR subtypes. In contrast, MII has substantial affinity for the α6β4 subtype, whereas PnIA inhibits the α7 subtype (18, 20). For these reasons, we chose PeIA as a starting point to develop ligands that selectively target α6β2* versus α3β2* nAChRs.
In this report, we used positional scanning mutagenesis of PeIA to identify residues that confer potency for α6/α3β2β3 (the α6/α3 subunit is a chimera where the extracellular ligand binding domain of α6 is spliced with the transmembrane domain of α3) and α3β2 nAChRs. The results show that a single amino acid substitution in PeIA is sufficient to abolish activity for the α3β2 subtype and confer selectivity for α6-containing nAChRs. Through successive substitutions of several amino acids, we generated a ligand that was >15,000-fold more potent at inhibiting the α6/α3β2β3 over the α3β2 subtype.
EXPERIMENTAL PROCEDURES
Materials and Methodologies
Rat α3, α4, α6, and α7 nAChR subunit clones were provided by S. Heinemann (Salk Institute, San Diego, CA), and C. Luetje (University of Miami, Miami, FL) provided the β2, β3, and β4 subunits in the high expressing pGEMHE vector. Construction of the α6/α3 subunit chimera has been described previously and consists of an α3 subunit where the first 237 amino acids of the ligand binding domain were replaced with the corresponding α6 amino acids (18). This chimera was used because injection of non-chimeric α6 with β2 produces few functional receptors (25–27). However, injection of β2 and β3 cRNA in conjunction with the α6/α3 chimera produces sufficient numbers of receptors for electrophysiological measurement. The β3 subunit is not believed to form part of the canonical agonist binding site and serves a structural role in the pentamer (28). However, most native α6β2* receptors likely contain the β3 subunit because genetic deletion of β3 substantially reduces levels of α6β2* nAChR expression (29, 30). Point mutations in the α6/α3 and α3 subunits were made by PCR as described previously (31). Acetylcholine chloride (ACh) and bovine serum albumin were obtained from Sigma-Aldrich. HEPES was purchased from Research Organics (Cleveland, OH).
Peptide Synthesis
Solid phase Fmoc peptide chemistry was used to generate the α-Ctx peptides either as described previously (21) or with an AAPPTec Apex 396 automated peptide synthesizer (Louisville, KY). The peptides were initially constructed on a preloaded Fmoc-Rink Amide MBHA resin (substitution: 0.4 mmol/g−1; Peptides International Inc.). All standard amino acids were purchased from AAPPTec except for N-α-Fmoc-O-t-butyl-l-trans-4-hydroxyproline (O, Hyp), which was purchased from EMD Millipore (Billerica, MA). Side-chain protection for the following amino acids was as follows: Arg, 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl; Lys, tert-butyloxycarbonyl Hyp; His and Asn, trityl; Ser, tert-butyl. Cys residues were orthogonally protected by trityl for Cys1 and Cys3 and acetamidomethyl for Cys2 and Cys4. The peptides were synthesized at 50 μmol scale. Coupling activation was achieved with 1 eq of 0.4 m benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate and 2 eq of 2 m N,N-diisopropylethyl amine in N-methyl-2-pyrrolidone as the solvent. For each coupling reaction, a 10-fold excess of amino acid was used, and the reaction was carried out for 60 min. Fmoc deprotection was performed for 20 min with 20% (v/v) piperidine in dimethylformamide. The peptides were cleaved from the resin using Reagent K, trifluoroacetic acid/phenol/ethanedithiol/thioanisol/H2O (9:0.75:0.25:0.5:0.5 by volume), and a two-step oxidation protocol was used to selectively fold the peptides into the correct disulfide configuration. Briefly, the first disulfide bridge was closed using 20 mm potassium ferricyanide and 0.1 m Tris-HCl, pH 7.5. The solution was allowed to react for 45 min, and the monocyclic peptide was purified by reverse-phase HPLC. Simultaneous removal of the acetamidomethyl groups and closure of the second disulfide bridge was accomplished by iodine oxidation. The monocyclic peptide in HPLC eluent was dripped into an equal volume of 10 mm iodine in H2O/trifluoroacetic acid/acetonitrile (78:3:25 by volume) and allowed to react for 10 min. The reaction was terminated by the addition of ascorbic acid diluted 20-fold with 0.1% (v/v) trifluoroacetic acid, and the bicyclic product was purified by reverse-phase HPLC. The masses of the peptides were verified by matrix-assisted laser desorption ionization time-of-flight mass spectrometry at the Salk Institute for Biological Studies (San Diego, CA) under the direction of Dr. J. Rivier and are provided in supplemental Table 1.
Two-electrode Voltage Clamp Electrophysiology of Xenopus laevis Oocytes
Detailed methods for conducting electrophysiological experiments of nAChRs heterologously expressed in X. laevis oocytes have been described previously (32). Briefly, stage IV-V oocytes were injected at a 1:1 ratio with cRNA encoding cloned rat nAChR subunits α3, α4, α6/α3, α7, β2, β3, and β4 and used 1–5 days after injection. The oocytes were clamped at a holding potential of −70 mV and continuously gravity-perfused with standard ND96 solution buffered with HEPES and stimulated with 1-s pulses of ACh once every minute. The concentrations of ACh used were 100 μm for all receptor subtypes except α7, where 300 μm ACh was used. These concentrations are submaximal and within ∼3-fold of the EC50 for all subtypes. The ACh concentrations and brief exposure time were chosen to avoid open channel block and long term desensitization of the receptors. The solution changes were controlled through a series of three-way solenoid valves interfaced with a personal computer via a CoolDrive valve driver (Neptune Research & Development, West Caldwell, NJ) and LabVIEW software (National Instruments, Austin, TX). The ACh-gated currents (IACh) were acquired using an Oocyte OC-725 series voltage clamp amplifier (Warner Instruments, Hamden, CT), filtered through a 5-Hz low pass Bessel filter (model F1B1; Frequency Devices, Ottawa, IL), and digitized at 50 Hz using a National Instruments USB-6009 digital to analog converter. The toxins were dissolved in ND96 and either perfusion-applied (for concentrations of ≤1 μm) or applied in a static bath for 5 min (for concentrations of ≥10 μm).
Data Analysis
Concentration-response curves for inhibition of IACh were generated by fitting the data to the Hill equation, % response = 100/(1 + ([toxin]/IC50)nH), using GraphPad Prism software (La Jolla, CA).
Sequence Alignments
Sequence alignments and pairwise analysis of rat α3, α6, β2, and β3 subunits were performed using MacVector version 10.5.1 (MacVector Inc., Cary, NC).
Homology Binding Modeling
The 2.4 Å resolution crystal structure of the Aplysia californica acetylcholine-binding protein (AChBP) complexed with α-Ctx PnIA(A10L,D14K) (33) was used to model the binding of PeIA analogs to rat α6 and α3 subunits. The coordinates of the complex were rendered using PyMOL (version 1.2r3pre, Schrödinger, LLC), and changes in residues of the AChBP and PnIA(A10L,D14K) were accomplished using the mutagenesis function. The numbering system used to identify residues of the AChBP follow the alignment of rat α3 and α6 subunits (Fig. 7).
FIGURE 7.

Sequence alignment of rat nAChR subunits. A, sequence alignment of the N-terminal ligand binding domains of α6 and α3. A pairwise comparison of the sequences of the two subunits identified 137 (67%) identities and 32 (15%) similarities at the amino acid level. The arrows indicate the three amino acids of the α6 and α3 ligand binding domains that were examined in this study using mutagenesis. B, the first 210 amino acid pairs of the β2 and β3 subunits contained 81 (38%) identities and 22 (21%) similarities. The asterisks in B identify residues of the β2 subunit that have been shown previously to be important for α-Ctx binding (19, 47).
RESULTS
Alanine Scanning Mutagenesis Reveals Amino Acids Critical for PeIA Activity
PeIA is a 16-amino acid peptide whose sequence was determined from a cDNA library of Conus pergrandis. It is a potent antagonist of rat α3β2, α6/α3β2β3, and α9α10 nAChRs (23, 24). This peptide shares a similar amino acid sequence with other 4/7 framework α-Ctxs that target α6/α3β2β3 and α3β2 nAChRs (Fig. 1). We used positional scanning mutagenesis to identify residues of PeIA that were important for its binding to α6/α3β2β3 and α3β2 nAChRs. The Ala-substituted analogs were tested on cloned rat nAChRs expressed in X. laevis oocytes, and their activities were compared with that of native PeIA. Substitution of His5 or Pro6 in the first Cys loop with Ala or Hyp, respectively, significantly reduced the potency of PeIA for both receptor subtypes, whereas substitution of Ser4 with Ala had no effect (Fig. 2, A and B). In the second Cys loop, a slight increase in potency for both subtypes was observed with an Ala substitution of Ser9, whereas H12A significantly reduced potency (Fig. 2, C and D). Alanine substitution of Glu14 or Leu15 slightly reduced the potency for α3β2 nAChRs, whereas of these, only the L15A substitution resulted in decreased potency for α6/α3β2β3 nAChRs (Fig. 2, C and D). Substitutions of Asn11 with Ala or of Pro13 with either Ala or Hyp had little effect on the potency for either subtype. A summary of the changes in potencies of the Ala- and Hyp-substituted analogs is shown in Table 1.
FIGURE 1.
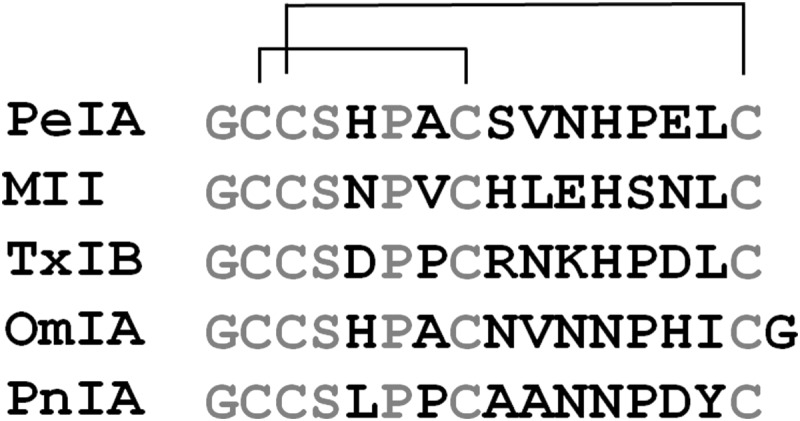
Sequence alignment of α-Ctxs PeIA, MII, TxIB, OmIA, and PnIA. Amino acids highlighted in boldface type are non-conserved among the five species.
FIGURE 2.

Effect of Ala or Hyp substitutions of non-cysteine amino acids on the potency of α-Ctx PeIA for rat α6/α3β2β3 and α3β2 nAChRs. Xenopus oocytes expressing α6/α3β2β3 or α3β2 nAChRs were subjected to TEVC as described under “Experimental Procedures,” and the IC50 values for inhibition of the IACh by each PeIA analog were determined. A and B, concentration-response analysis for inhibition of α3β2 and α6/α3β2β3 nAChRs by PeIA analogs with Ala or Hyp substitutions in the first Cys loop. C and D, concentration-response analysis for inhibition of α3β2 and α6/α3β2β3 nAChRs by PeIA analogs with Ala or Hyp substitutions in the second Cys loop. The IC50 values are summarized in Table 1. Error bars, S.E. from 4–5 oocytes for each experimental determination.
TABLE 1.
Potencies of Ala- and Hyp-substituted PeIA peptides on rat nAChRs expressed in Xenopus oocytes
| Peptide | α3β2 |
α6/α3β2β3 |
||||
|---|---|---|---|---|---|---|
| IC50 | 95% C.I. | IC50 ratio relative to PeIAa | IC50 | 95% C.I. | IC50 ratio relative to PeIAa | |
| PeIA | 19.2 nm | 17.2–21.5 | 0 | 17.2 nm | 15.5–19.0 | 0 |
| PeIA(S4A) | 35.9 nm | 32.2–40.0 | 0.3 | 12.7 nm | 10.6–15.1 | −0.1 |
| PeIA(H5A) | 25.9 μm | 14.1–47.8 | 3.1 | 1.11 μm | 0.330–3.76 | 1.8 |
| PeIA(P6A) | 11.1 μm | 7.4–16.6 | 2.8 | 340 nm | 121–895 | 1.3 |
| PeIA(P6O) | 25.1 μm | 15.6–40.4 | 3.1 | 2.22 μm | 0.292–16.7 | 2.1 |
| PeIA(S9A) | 5.91 nm | 4.99–7.00 | −0.5 | 7.20 nm | 5.89–8.80 | −0.4 |
| PeIA(V10A) | 2.23 nmb | 1.96–2.54 | −0.9 | 2.38 nmb | 2.01–2.69 | −0.9 |
| PeIA(N11A) | 45.9 nm | 24.1–87.4 | 0.4 | 29.6 nm | 25.2–34.7 | 0.2 |
| PeIA(H12A) | 51.5 μm | 31.1–85.1 | 3.4 | 7.27 μm | 2.11–24.9 | 2.6 |
| PeIA(P13A) | 63.0 nm | 51.7–76.7 | 0.5 | 13.3 nm | 10.9–16.2 | −0.1 |
| PeIA(P13O) | 27.9 nm | 24.2–32.1 | 0.2 | 9.70 nm | 6.82–13.8 | −0.2 |
| PeIA(E14A) | 98.9 nm | 66.7–147 | 0.7 | 12.5 nm | 9.39–16.6 | −0.1 |
| PeIA(L15A) | 139 nm | 114–172 | 0.9 | 82.5 nm | 63.5–107 | 0.7 |
a Indicates the log change in the ratio of IC50 values relative to PeIA. Negative values indicate an increase in potency, whereas positive values indicate a decrease in potency.
b From Hone et al. (24).
Positional Scanning Mutagenesis Using Amino Acids Found in Other 4/7 α-Ctxs Identifies Residues Critical for Discrimination between α6/α3β2β3 and α3β2 nAChRs
Substitution of the non-cysteine residues with Ala had little or no effect on the ability of PeIA to discriminate between α6/α3β2β3 and α3β2 subtypes. Therefore, we examined whether substitutions with amino acids found in other α-Ctxs might identify positions that are important for discriminating between these two subtypes. We started by substituting amino acids from MII into the sequence of PeIA. Because MII is ∼10-fold more potent at inhibiting α6/α3β2β3 over α3β2, one or more of the residues that differ between MII and PeIA must account for this difference in potency. We substituted H5N, A7V, V10L, N11E, and P13S, and of these, only the V10L substitution increased the potency of PeIA for α3β2, whereas A7V decreased potency (Fig. 3A and Table 2). Very little change, if any, was observed for the N11E or P13S substitutions (Fig. 3A and Table 2). For α6/α3β2β3 nAChRs, V10L and N11E substitutions decreased potency, whereas A7V increased potency (Fig. 3B and Table 2). The A7V mutation increased the potency for α6/α3β2β3 but decreased potency for α3β2 and produced a ∼12-fold selectivity for α6/α3β2β3 over the α3β2 subtype. The P13S mutation had no effect on the potency for α6/α3β2β3 nAChRs (Fig. 3B and Table 2).
FIGURE 3.

Effect of non-cysteine amino acid substitutions on the potency of α-Ctx PeIA for rat α6/α3β2β3 and α3β2 nAChRs. Xenopus oocytes expressing α6/α3β2β3 or α3β2 nAChRs were subjected to TEVC as described under “Experimental Procedures,” and the IC50 values for inhibition of the IACh by each PeIA analog were determined. A and B, concentration-response analysis for inhibition of α3β2 and α6/α3β2β3 nAChRs by PeIA analogs with substitutions from MII. C and D, positional scanning of residues 9, 10, and 11 of PeIA with positively charged amino acids and the concentration-response curves for inhibition of α3β2 and α6/α3β2β3 nAChRs by the resulting analogs. The IC50 values are summarized in Table 2. Error bars, S.E. from 4–5 oocytes for each experimental determination.
TABLE 2.
Potencies of PeIA peptides with select substitutions on rat nAChRs expressed in Xenopus oocytes
| Peptide | α3β2 |
α6/α3β2β3 |
||||
|---|---|---|---|---|---|---|
| IC50 | 95% C.I. | IC50 ratio relative to PeIAa | IC50 | 95% C.I. | IC50 ratio relative to PeIAa | |
| PeIA | 19.2 nm | 17.2–21.5 | 0 | 17.2 nm | 15.1–19.5 | 0 |
| PeIA(H5N) | 26.4 nm | 22.9–30.4 | 0.1 | 2.69 nm | 2.36–3.07 | −0.8 |
| PeIA(A7V) | 85.6 nm | 66.9–105 | 0.6 | 6.86 nm | 6.43–7.31 | −0.4 |
| PeIA(S9H) | 713 pmb | 480–1,060 | −1.4 | 991 pmb | 932–1054 | −1.2 |
| PeIA(S9R) | 4.15 nm | 3.55–4.86 | −0.7 | 2.29 nm | 1.67–3.14 | −0.9 |
| PeIA(V10R) | 45.8 μm | 33.8–62.0 | 3.4 | 562 nm | 374–843 | 1.5 |
| PeIA(V10L) | 3.81 nm | 2.970–4.89 | −0.7 | 18.8 nm | 16.0–22–1 | 0.04 |
| PeIA(N11E) | 37.6 nm | 34.1–41.4 | 0.3 | 114 nm | 92.7–140 | 1.2 |
| PeIA(N11R) | 30.9 μm | 22.8–42.1 | 3.2 | 46.5 nm | 35.0–61.2 | 0.4 |
| PeIA(N11K) | 45.4 μm | 30.6–67.2 | 3.4 | 40.8 nm | 30.1–55.2 | 0.4 |
| PeIA(P13S) | 29.2 nm | 26.2–32.4 | 0.2 | 17.7 nm | 16.0–19.4 | 0 |
| PeIA(E14N) | 149 nmb | 122–182 | 0.9 | 22.0 nmb | 19.6–24.6 | 0.1 |
a Indicates the log change in the ratio of IC50 values relative to PeIA. Negative values indicate an increase in potency, whereas positive values indicate a decrease in potency.
b From Hone et al. (24).
A study by Pucci et al. (34) suggested that a positively charged amino acid in the second Cys loop might be an important determinant favoring binding to the α6 subunit while disfavoring binding to the α3 subunit. The sequence of α-Ctx TxIB was recently determined from a cDNA library of Conus textile (35). Remarkably, TxIB and PeIA only differ by 6 amino acids, yet TxIB is exclusively selective for α6/α3β2β3 nAChRs. We note that TxIB has an Arg in the 9th position and a Lys in the 11th position instead of the Ser and Glu, respectively, found in PeIA (Fig. 1). Therefore, we substituted Ser9, Val10, and Glu11 one at a time with Arg and evaluated the resulting analogs for changes in potency for α6/α3β2β3 and α3β2 nAChRs. Substitution of Ser9 with Arg increased the potency for both subtypes, whereas substitution of Val10 with Arg significantly reduced the potency for both subtypes (Fig. 3 (C and D) and Table 2). The N11R substitution had the effect of essentially abolishing the activity of PeIA for the α3β2 subtype (Fig. 3C and Table 2) while minimally affecting the potency for α6/α3β2β3 receptors (Fig. 3D and Table 2). Similarly, the N11K substitution dramatically increased the IC50 for inhibition of α3β2 nAChRs by more than 2,300-fold (Fig. 3C and Table 2).
Combined Substitutions of Amino Acids in PeIA Increase the Selectivity of the Peptide for α6/α3β2β3 versus α3β2 nAChRs
The PeIA analog PeIA(S9H,V10A) was previously identified as a potent antagonist of both α6/α3β2β3 and α3β2 nAChRs, with IC50 values of 506 and 792 pm, respectively (24). We introduced additional substitutions identified in the present study to generate a series of PeIA analogs to gain further mechanistic insight into the binding of PeIA to α6/α3β2β3 and α3β2 nAChRs. Incorporation of substitutions that disfavor binding to the α3 subunit produced ligands that selectively targeted the α6/α3β2β3 subtype. These substitutions included A7V, N11R, and E14A. Successive substitutions were made in the sequence of PeIA(S9H,V10A). First, we incorporated an N11R substitution with the expectation that this triply substituted peptide would be substantially less potent on α3β2 nAChRs. However, contrary to these expectations, the addition of the N11R mutation to PeIA(S9H,V10A) resulted in only a ∼34-fold loss of potency for α3β2 receptors (Fig. 4A and Table 3). This value was substantially less than the ∼1,600-fold loss produced by the single N11R substitution in native PeIA. The N11R substitution in PeIA(S9H,V10A,N11R) also induced a 3-fold loss of potency for α6/α3β2β3 nAChRs (Fig. 4B and Table 3). We therefore synthesized two additional analogs incorporating A7V and E14A substitutions. PeIA(A7V,S9H,V10A,N11R) showed a ∼380-fold loss of potency for α3β2 receptors, relative to PeIA(S9H,V10A,N11R), whereas PeIA(S9H,V10A,N11R,E14A) showed only a ∼4-fold loss of potency (Fig. 4A and Table 3). Minor losses in potency of ≥3-fold were observed for inhibition of α6/α3β2β3 receptors for both analogs, relative to PeIA(S9H,V10A,N11R) (Fig. 4B and Table 3). Finally, all five substitutions were combined to produce PeIA(A7V,S9H,V10A,N11R,E14A). This analog showed a >15,000-fold difference in the IC50 values for α6/α3β2β3 versus α3β2 nAChRs (Fig. 5A and Table 3) and was essentially inactive on all other non-α6-containing nAChR subtypes, including α3β4, α4β2, α4β4, and α7 (Fig. 5B and Table 4). Interestingly, increased potency was observed for α6β4 nAChRs (Fig. 5B and Table 4), relative to native PeIA (24), suggesting that one or more of the five substitutions produced an interaction that enhanced binding to α6β4 nAChRs. The kinetics of block and unblock of α6/α3β2β3 and α3β2 nAChRs by PeIA(A7V,S9H,V10A,N11R,E14A) are illustrated in Fig. 6.
FIGURE 4.

Effect of combined substitutions of non-cysteine amino acids on the potency of α-Ctx PeIA for rat α6/α3β2β3 and α3β2 nAChRs. Xenopus oocytes expressing α6/α3β2β3 or α3β2 nAChRs were subjected to TEVC as described under “Experimental Procedures,” and the IC50 values for inhibition of the IACh by each PeIA analog were determined. A and B, concentration-response analysis for inhibition of α3β2 and α6/α3β2β3 nAChRs by PeIA(S9H,V10A,N11R), PeIA(A7V,S9H,V10A,N11R), and PeIA(S9H,V10A,N11R,E14A). The IC50 values are summarized in Table 3. Error bars, S.E. from 4–5 oocytes for each experimental determination.
TABLE 3.
Potencies of PeIA peptides with multiple substitutions on rat nAChRs expressed in Xenopus oocytes
| Peptide | α3β2 |
α6/α3β2β3 |
||||
|---|---|---|---|---|---|---|
| IC50 | 95% C.I. | IC50 ratio relative to PeIAa | IC50 | 95% C.I. | IC50 ratio relative to PeIAa | |
| PeIA | 19.2 nm | 17.2–21.5 | 0 | 17.2 nm | 15.1–19.5 | 0 |
| PeIA(S9H,V10A,N11R) | 34.0 nm | 31.3–37.4 | 0.2 | 1.48 nm | 1.18–1.16 | −1.1 |
| PeIA(A7V,S9H,V10A,N11R) | 10.1 μm | 6.90–17.1 | 2.7 | 2.53 nm | 2.14–2.98 | −0.8 |
| PeIA(S9H,V10A,N11R,E14A) | 106 nm | 79.2–142 | 0.7 | 4.91 nm | 4.28–5.61 | −0.5 |
| PeIA(A7V,S9H,V10A,N11R,E14A) | 30.9 μm | 22.8–42.1 | 3.2 | 2.16 nm | 1.98–2.35 | −0.9 |
a Indicates the log change in the ratio of IC50 values relative to PeIA. Negative values indicate an increase in potency, whereas positive values indicate a decrease in potency.
FIGURE 5.

Potency and selectivity profile of PeIA(A7V,S9H,V10A,N11R,E14A) for several rat nAChR subtypes. Xenopus oocytes expressing different nAChRs were subjected to TEVC as described under “Experimental Procedures,” and the IC50 values for inhibition of the IACh by PeIA(A7V,S9H,V10A,N11R,E14A) were determined. A, concentration-response analysis for inhibition of α6/α3β2β3 and α3β2 nAChRs. B, concentration-response analysis for inhibition of α3β4, α4β2, α4β4, α6β4, and α7 nAChRs. In B, the 10 μm data points for α3β4, α4β2, α4β4, and α7 nAChRs are shown staggered to avoid overlap. The IC50 values are summarized in Table 4. Error bars, S.E. from 4–5 oocytes for each experimental determination.
TABLE 4.
Potencies of PeIA[A7V,S9H,V10A,N11R,E14A] on rat nAChRs expressed in Xenopus oocytes
| nAChR subtype | IC50 | 95% C.I. | IC50 ratio (compared with α6/α3β2β3) |
|---|---|---|---|
| rα3β2 | 30.9 μm | 22.8–42.1 | >15,000 |
| rα3β4 | >10 μma | NDb | ND |
| rα4β2 | >10 μma | ND | ND |
| rα4β4 | >10 μma | ND | ND |
| rα6/α3β2β3 | 2.16 nm | 1.98–2.35 | 1 |
| rα6β4 | 43.8 nm | 37.4–51.2 | 20 |
| rα7 | >10 μma | ND | ND |
a Less than 50% block at 10 μm.
b ND, not determined.
FIGURE 6.

The ligand binding kinetics of block and unblock of α6/α3β2β3 and α3β2 nAChRs by PeIA(A7V,S9H,V10A,N11R,E14A) at three progressively higher toxin concentrations. A, in the presence of 100 nm PeIA(A7V,S9H,V10A,N11R,E14A), the average response to 100 μm ACh was 2.7 ± 0.3% (n = 3) of control responses for α6/α3β2β3 receptors. B, for α3β2 receptors, the average response to 100 μm ACh in the presence of 100 nm and 1 μm PeIA(A7V,S9H,V10A,N11R,E14A) was 98.3 ± 2.2% (n = 4) and 87.0 ± 3.5% (n = 4), respectively. Values are ± S.E. C, control.
Specific Amino Acids of PeIA Interact with the α6 Subunit
α-Conotoxins are globular ligands that interact with both the principal (α subunit) and the complementary (usually a β subunit) components of the acetylcholine binding pocket. Rat α6 and α3 subunits are highly homologous, and the N-terminal ligand binding domains contain 137 amino acid identities (67%) and 32 similarities (15%) (Fig. 7A). Three non-homologous residues in the ligand binding domain, positions 152, 184, and 195, have previously been identified as being important determinants of high affinity binding of α-Ctx MII(S4A,E11A,L15A) to rat α6/α3β2β3 nAChRs (31). The sequences of β2 and β3 are also shown to illustrate the complementary component of the ligand binding site (Fig. 7B). We sought to determine whether one or more of these three amino acids was responsible for the large differences in potencies of some of the PeIA analogs observed here. In particular, we sought to determine whether Arg in the 11th position of PeIA interacted with Glu152 of the α6 subunit and Lys152 of the α3 subunit. We tested PeIA(N11R) and PeIA(A7V,S9H,V10A,N11R,E14A) on mutants of chimeric α6/α3 and α3 subunits where particular non-conserved residues from α3 were replaced with those from α6 and vice versa. PeIA(A7V,S9H,V10A,N11R,E14A) was chosen over other analogs because of its large separation in IC50 values between α6/α3β2β3 and α3β2 nAChRs. The mutant subunits that were examined were α6E152K/α3, α6E152K,D184E,T195Q/α3, and α3K152E,E184D,Q195T, each expressed in combination with β2 and/or β3 subunits. When tested on α3K152E,E184D,Q195Tβ2 receptors, the concentration-inhibition curve for PeIA(N11R) shifted to the left by ∼8-fold, toward that of α6/α3β2β3 (Fig. 8A and Table 5). Conversely, the inhibition curve of PeIA(N11R) for α6E152K,D184E,T195Q/α3β2β3 receptors shifted to the right by ∼130-fold, toward that of α3β2 receptors (Fig. 8A and Table 5). Of the three residues of α6 that were replaced with those of α3, the E152K switch alone accounted for ∼25% of the loss of potency of PeIA(N11R) for α6E152K/α3β2β3 receptors, as indicated by the rightward shift of ∼30-fold in the inhibition curve (Fig. 8A and Table 5). The results of these experiments further suggested that the 11th position of PeIA interacts with residue 152 of the α6 and α3 subunits. For further confirmation that specific residues of PeIA were critical for high affinity binding to α6/α3β2β2 receptors, we performed the same experiments using PeIA(A7V,S9H,V10A,N11R,E14A) and obtained similar results except that the magnitude of the changes in the IC50 values was greater for this analog than for PeIA(N11R). The IC50 value for α3K152E,E184D,Q195T/β2 shifted to the left by ∼2,000-fold and approximated the value obtained for α6/α3β2β3 receptors (Fig. 8A and Table 5). In the case of the α6 mutants, the IC50 values for inhibition of α6E152K,D184E,T195Q/α3β2β3 and α6E152K/α3β2β3 by PeIA(A7V,S9H,V10A,N11R,E14A) shifted to the right by ∼2,500- and ∼140-fold, respectively (Fig. 8B and Table 5). Residue 152 is located in the B-loop of the α subunit, and residues 184 and 195 are located in the C-loop in β strands 9 and 10, respectively. In order to better appreciate the proximity of the α-Ctx to these residues, we constructed a homology model using the crystal structure coordinates of the A. californica AChBP complexed with an analog of PnIA (33). Residues of the AChBP were mutated to the homologous residues of rat α3 and α6 subunits, and residues of PnIA(A10L,D14K) were mutated to those of PeIA(A7V,S9H,V10A,N11R,E14A). The Lys side chain of residue 152 of the α3 subunit (Fig. 9A) and the side chain of Glu152 of the α6 subunit (Fig. 9B) were found in close proximity to the Arg11 side chain of the α-Ctx.
FIGURE 8.
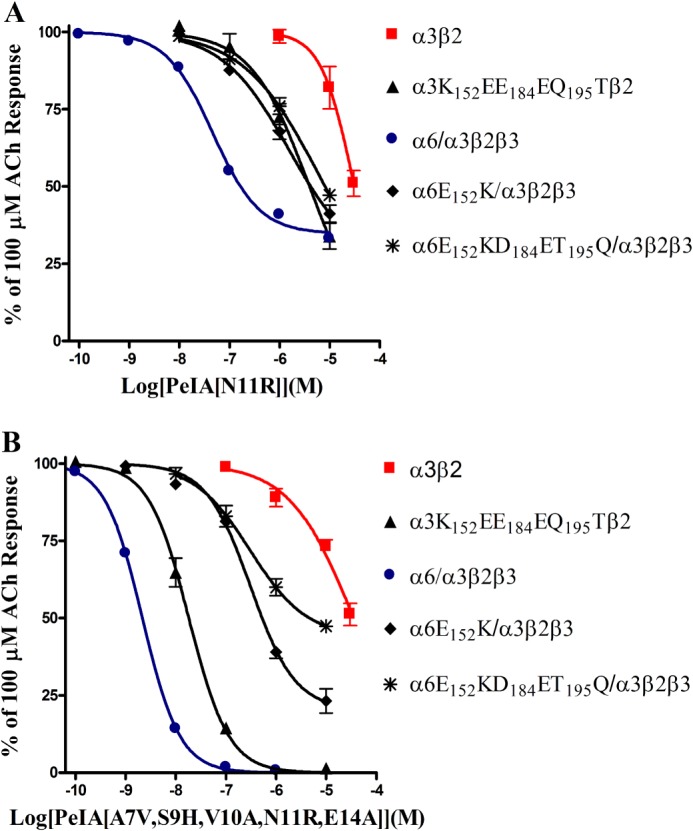
The sensitivity of α6/α3β2β3 and α3β2 nAChRs to inhibition by PeIA(N11R) and PeIA(A7V,S9H,V10A,N11R,E14A) is determined by three residues in the α6/α3 and α3 subunits. Xenopus oocytes expressing α6/α3β2β3, α3β2, and mutants of these receptors were subjected to TEVC as described under “Experimental Procedures,” and the inhibition of the IACh by PeIA(N11R) and PeIA(A7V,S9H,V10A,N11R,E14A) was determined. The IC50 values are summarized in Table 5. Error bars, S.E. from 4–5 oocytes for each experimental determination.
TABLE 5.
Potencies of PeIA analogs on rat α3β2 and α6β2 nAChR mutants expressed in Xenopus oocytes
| nAChR | IC50 values for PeIA(N11R) | 95% C.I. | IC50 values for PeIA(A7V,S9H,V10A,N11R,E14A) | 95% C.I. |
|---|---|---|---|---|
| rα3β2 | 30.9 μm | 22.8–42.1 | 35.6 μm | 25.0–50.1 |
| rα3K152E,E184D,Q195Tβ2 | 4.07 μm | 3.05–5.44 | 18.2 nm | 15.3–21.7 |
| rα6/α3β2β3 | 46.5 nm | 35.0–61.2 | 2.16 nm | 1.98–2.35 |
| rα6E152K/α3β2β3 | 1.53 μm | 0.360–6.49 | 311 nm | 215–450 |
| rα6E152K,D184E,T195Q/α3β2β3 | 6.20 μm | 0.234–164 | 5.47 μm | 3.20–9.36 |
FIGURE 9.
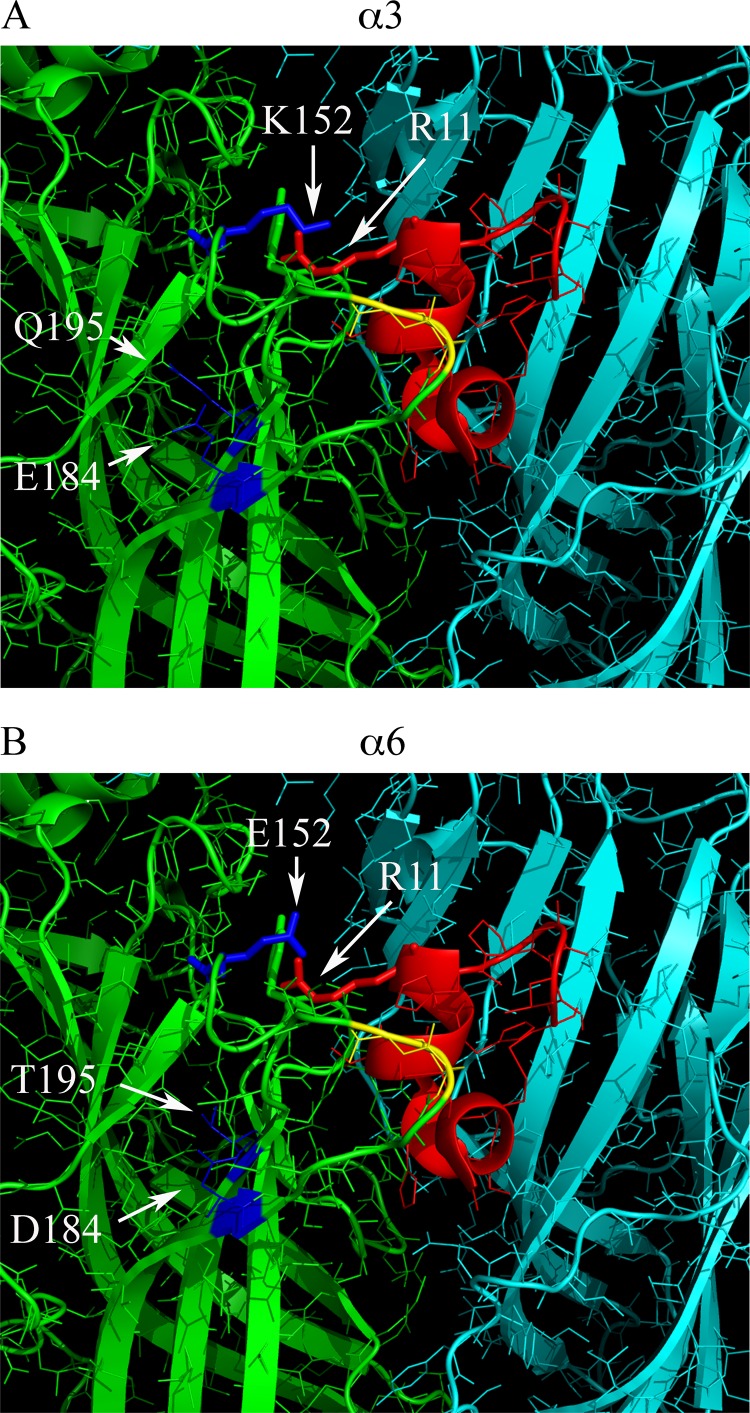
Homology binding model of PeIA(A7V,S9H,V10A,N11R,E14A) to rat α3β2 and α6β2 nAChRs. A, a 20 Å radius view of the ACh binding pocket of the A. californica AChBP complexed with α-Ctx PnIA(A10L,D14K) The principal subunit is shown in green, and the complementary subunit is shown in cyan. Residues 147–153 of the B-loop and 185–195 of the C-loop of the principal binding subunit were mutated to the homologous residues found in the rat α3 subunit. Residues of the C-loop shown in yellow are Cys189 and Cys190, and the residues shown in blue are Glu184 and Gln195 of the C-loop, and Lys152 of the B-loop, shown as a stick model. Residues of PnIA(A10L,D14K) were mutated to those of PeIA(A7V,S9H,V10A,N11R,E14A) and shown in red with Arg11 shown as a stick model. Note the close proximity of Lys152 of the α3 subunit and Arg11 of the α-Ctx, suggesting a possible interaction between the two. B, residues 147–153 of the B-loop and 185–195 of the C-loop of the AChBP principal binding subunit were mutated to the homologous residues found in the rat α6 subunit. Residues in blue are Asp184 and Thr195 of the C-loop and Glu152 of the B-loop. Mutagenesis of the AChBP and α-Ctx PnIA(A10L,D14K) as well as image rendering were performed as described under “Experimental Procedures.”
DISCUSSION
In this study, we used positional scanning mutagenesis of α-Ctx PeIA in conjunction with mutagenesis of α6 and α3 subunits to examine the interaction between PeIA and the nAChR subtypes α6/α3β2β3 and α3β2. Native PeIA has equal affinity for rat α6/α3β2β3 and α3β2 nAChRs; therefore, changes in potency for these receptor subtypes, resulting from mutations in the sequence of PeIA, can be used to identify functionally important residues. This strategy generated several PeIA analogs that were used to determine whether the differences in non-conserved amino acids of α6 and α3 subunits could be exploited to generate ligands that were selective for α6/α3β2β3 over the α3β2 subtype. We started with alanine scanning mutagenesis of PeIA. None of the three residues in the first Cys loop enhanced the peptide's ability to discriminate between the two nAChR subtypes, and substitution of His5 and Pro6 with Ala or Hyp, respectively, significantly reduced the potency of PeIA on both subtypes (Fig. 2, A and B, and Table 1). The amino acids Pro and His are thought to confer structural rigidity to the α-helical portion of α-Ctxs, and Pro in the 6th position is highly conserved among 4/7 α-Ctxs across species (Fig. 1). The fact that PeIA(H5N) retains activity whereas PeIA(P6A) and PeIA(P6O) do not suggests that Pro6, and not His5 (Figs. 2 (A and B) and 3 (A and B) and Tables 1 and 2), is critical for the α-helical structure of PeIA. Of the substitutions made in the first Cys loop, only H5N and A7V showed any discrimination between α6/α3β2β3 and α3β2 nAChRs (Fig. 3 (A and B) and Table 2).
Substitutions of amino acids in the second Cys loop showed some similarities with those of the first Cys loop. Replacing His12 with Ala essentially abolished activity for α6/α3β2β3 and α3β2 nAChRs (Fig. 2 (B and C) and Table 1), whereas replacing Pro13 with Ala, Hyp, or Ser had no effect (Figs. 2 (B and C) and 3 (A and B) and Tables 1 and 2), suggesting that, in contrast to the His and Pro in the first Cys loop, His12 is more important than Pro13 for structural rigidity. Alanine substitution of Asn11 and Glu14 had no effect on α6/α3β2β3 potency, whereas the S9A substitution increased potency (Fig. 3B and Table 2). For α3β2 nAChRs, S9A increased potency, whereas N11A had little effect, and E14A decreased potency (Fig. 2A and Table 1). Both α6/α3β2β3 and α3β2 nAChRs were inhibited less potently by PeIA(L15A) (Fig. 2, C and D, and Table 1). Non-Ala substitutions in the second Cys loop produced more significant changes in potency and selectivity. Substitution of Ser9 with Arg increased the potency for both receptor subtypes, whereas V10R nearly abolished activity (Fig. 3 (C and D) and Table 2). Finally, substitution of Asn11 with a positively charged amino acid, either Arg or Lys, abolished activity on α3β2 nAChRs but produced little change in potency for α6/α3β2β3 nAChRs (Fig. 3 (C and D) and Table 2). We note that the amino acid sequence of TxIB shows considerable homology with that of PeIA, differing by only six amino acids. TxIB is selective for α6/α3β2β3 nAChRs (35), whereas PeIA shows no discrimination between α6/α3β2β3 and α3β2 nAChRs (IC50 values of 11.1 and 9.70 nm, respectively) (24). Thus, some combination of these six differing residues must confer the selectivity of TxIB for the α6 subunit. TxIB has a Lys in the 11th position (Fig. 1), and this, taken together with the effects of the N11R substitution in PeIA, suggests that a positively charged amino acid in this position is important for conferring selectivity for the α6 subunit over the α3 subunit.
Further support for the importance of the 11th position was obtained through mutagenesis experiments of the receptor where N-terminal extracellular residues of the α6 subunit were replaced with those of the α3 subunit and vice versa. Replacing negatively charged Glu152 in the α6 subunit with positively charged Lys152 from the α3 subunit substantially reduced the potency of PeIA(N11R) for the α6E152K/α3β2β3 and α6E152K,D184E,T195Q/α3β2β3 mutants by 30- and 130-fold, respectively (Fig. 8A and Table 5). Conversely, the potency of PeIA(N11R) increased by ∼8-fold for the α3K152E,E184D,Q195Tβ2 mutant (Fig. 8A and Table 5). Although residue 152 appears to be a critical determinant of the ability of PeIA(N11R) to discriminate between α6/α3β2β3 and α3β2 nAChRs, other residues of the receptor also appear to be involved in the interaction of PeIA and its analogs with the nicotinic receptors as evidenced by the larger shifts in potencies of PeIA(A7V,S9H,V10A,N11R,E14A) for the different receptor subtypes and their mutants. The IC50 value for this analog for inhibition of α3K152E,E184D,Q195T/β2 decreased by >2,000-fold and approximated the value obtained for α6/α3β2β3 receptors (Fig. 8B and Table 5). This suggests that these three residues account for a large portion of the high affinity binding of PeIA(A7V,S9H,V10A,N11R,E14A) to the α6-β2 binding site.
A homology binding model was generated to gain a better understanding of the interaction between PeIA and its analogs with the three residues of the α6 and α3 subunits that affected ligand binding (Fig. 9). Although it is known that residue 152 of the α subunit can affect α-Ctx binding (31), this residue is located in the B-loop rather than the canonical ligand binding site composed of residues in the C-loop. However, as shown in Fig. 9, it appears that an interaction between residues of the B-loop, and in particular residue 152, may directly interact with α-Ctxs occupying the ACh binding pocket. Thus, when α-Ctxs have positively charged amino acids in the 11th position, such as TxIB or PeIA(N11R), binding is disfavored to α3-containing nAChRs potentially due to a repulsive charge-charge interaction with Lys152. The other two residues examined in this study, residues 184 and 195, are located in the C-loop but do not appear to interact directly with the α-Ctx because their side chains are oriented away from the α-Ctx. These residues are located in an area of the C-loop that has been proposed to act like a hinge allowing it to move toward the complementary subunit upon binding of agonists. Crystal structures of AChBPs complexed with different α-Ctxs show that the C-loop is even more open than its configuration in the absence of a bound α-Ctx (33, 36). Subtle changes in the position of the C-loop may be required to accommodate α-Ctxs of different sizes and amino acid compositions. The smaller Asp184 and Thr195, relative to Glu and Gln of the α3 subunit, respectively, may allow the C-loop of the α6 subunit to be more flexible and permit some α-Ctxs to bind more easily. Conversely, the C-loop of the α3 subunit may be more rigid, hindering α-Ctx binding. Other residues of the C-loop have been shown to play a more direct role in the binding of α-Ctxs. Beissner et al. (37) found that an Arg in position 185 of the α4 subunit prevents high affinity binding of several 4/7 α-Ctxs to the α4β2 subtype, and mutation to Ile, the residue found in the homologous position of α6 and α3 subunits, increased binding affinity. Last, the differing residues of the various α subunits may produce unique conformations of the C-loop itself, allowing α-Ctxs to bind to some receptor subtypes but not others, as suggested in a recent study examining the binding of α-Ctx BuIA to α6/α3β2β3 nAChRs (38).
We also note that TxIB has an Arg in the 9th position, and PnIA, which is selective for α6/α3β2β3 and α3β2 nAChRs over their β4-containing counterparts (20, 39), has Ala in this position (Fig. 1). Substitution of Ser9 with either Ala or Arg increased the potency for α6/α3β2β3 and α3β2 nAChRs (Figs. 2 (C and D) and 3 (C and D) and Tables 1 and 2) but decreased the potency for α6β4 nAChRs by ∼5- and 17-fold, respectively (Fig. 10). Thus, the 9th position appears to be important for discrimination between β2- and β4-containing nAChRs.
FIGURE 10.

The sensitivity of rat α6β4 to inhibition by PeIA, PeIA(S9A), and PeIA(S9R). Xenopus oocytes expressing α6β4 nAChRs were subjected to TEVC as described under “Experimental Procedures,” and the IC50 values for inhibition by PeIA and two analogs were determined. PeIA, PeIA(S9A), and PeIA(S9R) inhibited α6β4 with IC50 values of 262 (201–340) nm, 1.18 (0.947–1.147) μm, and 4.46 (3.30–6.02) μm, respectively. Values in parenthesis indicate the 95% confidence interval; error bars, S.E. from 4–5 oocytes for each experimental determination.
Combining several substitutions that preferentially favor binding to α6/α3β2β3 nAChRs or that preferentially eliminate activity on α3β2 nAChRs generated a ligand that was >15,000-fold more potent on α6/α3β2β3 nAChRs. This analog, PeIA(A7V,S9H,V10A,N11R,E14A), should prove to be a valuable pharmacological tool for distinguishing α6β2* from α3β2* nAChRs in tissues where these two subtypes are co-expressed. PeIA(A7V,S9H,V10A,N11R,E14A) is also essentially inactive on the other major CNS subtypes, including α3β4, α4β2, α4β4, and α7 nAChRs (Fig. 5B and Table 4). Unfortunately, the potencies for α6/α3β2β3 and α6β4 are separated by only 20-fold, and therefore, PeIA(A7V,S9H,V10A,N11R,E14A) cannot be used to distinguish between these two subtypes. However, very few α6β4 receptors are in the CNS and are found primarily in the visual system of rats (40, 41), the medial habenula of primates (42), and the hippocampus of adolescent mice (43) and can be pharmacologically distinguished from the α6β2* subtype using the α-Ctx analog BuIA(T5A,P6O) (43).
Current evidence suggests that, along with the α4β2* subtype, α6β2* receptors play an important role in modulating the release of dopamine in the nigrostriatal system, and loss of dopaminergic tone leads to disordered control of movement. In Parkinson disease and in animal models of Parkinson disease, the extent and severity of the disease correlates with a reduction of 125I-MII binding, suggesting a loss of α6β2* receptors (10, 11, 44). However, MII also potently binds to α3β2* nAChRs, which have also been suggested to contribute, although to a much lesser degree, to nigrostriatal dopamine release. The differential contribution of α6β2* and α3β2* nAChRs to this release has yet to be unequivocally determined for lack of highly selective ligands that distinguish between these two subtypes. This situation has now been remedied with the availability of PeIA(A7V,S9H,V10A,N11R,E14A), and this new analog may be particularly useful for differentiating the role of α6β2* from that of α3β2* nAChRs in the nigrostriatal system. Additionally, converging evidence from multiple studies suggests that drugs that target α6β2* may be useful in the pharmacotherapy of Parkinson disease and nicotine dependence (45, 46). However, compounds that target α6β2* would need to be devoid of activity on the α3β2* subtype to avoid unwanted cardiovascular and enteric side effects. The results of the present study should be useful in guiding the further development of compounds that selectively target α6β2* nAChRs.
This work was supported, in whole or in part, by National Institutes of Health Grants GM103801 and GM48677.

This article contains supplemental Table 1.
- nAChR
- nicotinic acetylcholine receptor
- α-Ctx
- α-conotoxin
- ACh
- acetylcholine
- AChBP
- acetylcholine-binding protein
- O and Hyp
- 4-trans-hydroxyproline
- Fmoc
- N-(9-fluorenyl)methoxycarbonyl
- C.I.
- confidence interval.
REFERENCES
- 1. Albuquerque E. X., Pereira E. F., Alkondon M., Rogers S. W. (2009) Mammalian nicotinic acetylcholine receptors. From structure to function. Physiol. Rev. 89, 73–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Champtiaux N., Han Z. Y., Bessis A., Rossi F. M., Zoli M., Marubio L., McIntosh J. M., Changeux J. P. (2002) Distribution and pharmacology of α6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J. Neurosci. 22, 1208–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Han Z. Y., Le Novère N., Zoli M., Hill J. A., Jr., Champtiaux N., Changeux J. P. (2000) Localization of nAChR subunit mRNAs in the brain of Macaca mulatta. Eur. J. Neurosci. 12, 3664–3674 [DOI] [PubMed] [Google Scholar]
- 4. Endo T., Yanagawa Y., Obata K., Isa T. (2005) Nicotinic acetylcholine receptor subtypes involved in facilitation of GABAergic inhibition in mouse superficial superior colliculus. J. Neurophysiol. 94, 3893–3902 [DOI] [PubMed] [Google Scholar]
- 5. Cox B. C., Marritt A. M., Perry D. C., Kellar K. J. (2008) Transport of multiple nicotinic acetylcholine receptors in the rat optic nerve. High densities of receptors containing α6 and β3 subunits. J. Neurochem. 105, 1924–1938 [DOI] [PubMed] [Google Scholar]
- 6. Cordero-Erausquin M., Pons S., Faure P., Changeux J. P. (2004) Nicotine differentially activates inhibitory and excitatory neurons in the dorsal spinal cord. Pain 109, 308–318 [DOI] [PubMed] [Google Scholar]
- 7. Azam L., McIntosh J. M. (2005) Effect of novel α-conotoxins on nicotine-stimulated [3H]dopamine release from rat striatal synaptosomes. J. Pharmacol. Exp. Ther. 312, 231–237 [DOI] [PubMed] [Google Scholar]
- 8. Perez X. A., Bordia T., McIntosh J. M., Quik M. (2010) α6β2* and α4β2* nicotinic receptors both regulate dopamine signaling with increased nigrostriatal damage. Relevance to Parkinson's disease. Mol. Pharmacol. 78, 971–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meyer E. L., Yoshikami D., McIntosh J. M. (2008) The neuronal nicotinic acetylcholine receptors α4* and α6* differentially modulate dopamine release in mouse striatal slices. J. Neurochem. 105, 1761–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kulak J. M., McIntosh J. M., Quik M. (2002) Loss of nicotinic receptors in monkey striatum after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine treatment is due to a decline in α-conotoxin MII sites. Mol. Pharmacol. 61, 230–238 [DOI] [PubMed] [Google Scholar]
- 11. Quik M., Polonskaya Y., Kulak J. M., McIntosh J. M. (2001) Vulnerability of 125I-α-conotoxin MII binding sites to nigrostriatal damage in monkey. J. Neurosci. 21, 5494–5500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gotti C., Guiducci S., Tedesco V., Corbioli S., Zanetti L., Moretti M., Zanardi A., Rimondini R., Mugnaini M., Clementi F., Chiamulera C., Zoli M. (2010) Nicotinic acetylcholine receptors in the mesolimbic pathway. Primary role of ventral tegmental area α6β2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J. Neurosci. 30, 5311–5325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Exley R., Clements M. A., Hartung H., McIntosh J. M., Cragg S. J. (2008) α6-containing nicotinic acetylcholine receptors dominate the nicotine control of dopamine neurotransmission in nucleus accumbens. Neuropsychopharmacology 33, 2158–2166 [DOI] [PubMed] [Google Scholar]
- 14. Liu L., Zhao-Shea R., McIntosh J. M., Gardner P. D., Tapper A. R. (2012) Nicotine persistently activates ventral tegmental area dopaminergic neurons via nicotinic acetylcholine receptors containing α4 and α6 subunits. Mol. Pharmacol. 81, 541–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jackson K. J., McIntosh J. M., Brunzell D. H., Sanjakdar S. S., Damaj M. I. (2009) The role of α6-containing nicotinic acetylcholine receptors in nicotine reward and withdrawal. J. Pharmacol. Exp. Ther. 331, 547–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Armishaw C. J. (2010) Synthetic α-conotoxin mutants as probes for studying nicotinic acetylcholine receptors and in the development of novel drug leads. Toxins 2, 1471–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Azam L., McIntosh J. M. (2009) α-Conotoxins as pharmacological probes of nicotinic acetylcholine receptors. Acta Pharmacol. Sin. 30, 771–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McIntosh J. M., Azam L., Staheli S., Dowell C., Lindstrom J. M., Kuryatov A., Garrett J. E., Marks M. J., Whiteaker P. (2004) Analogs of α-conotoxin MII are selective for α6-containing nicotinic acetylcholine receptors. Mol. Pharmacol. 65, 944–952 [DOI] [PubMed] [Google Scholar]
- 19. Luo S., Akondi K. B., Zhangsun D., Wu Y., Zhu X., Hu Y., Christensen S., Dowell C., Daly N. L., Craik D. J., Wang C. I., Lewis R. J., Alewood P. F., Michael McIntosh J. (2010) Atypical α-conotoxin LtIA from Conus litteratus targets a novel microsite of the α3β2 nicotinic receptor. J. Biol. Chem. 285, 12355–12366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo S., Nguyen T. A., Cartier G. E., Olivera B. M., Yoshikami D., McIntosh J. M. (1999) Single-residue alteration in α-conotoxin PnIA switches its nAChR subtype selectivity. Biochemistry 38, 14542–14548 [DOI] [PubMed] [Google Scholar]
- 21. Cartier G. E., Yoshikami D., Gray W. R., Luo S., Olivera B. M., McIntosh J. M. (1996) A new α-conotoxin which targets α3β2 nicotinic acetylcholine receptors. J. Biol. Chem. 271, 7522–7528 [DOI] [PubMed] [Google Scholar]
- 22. Talley T. T., Olivera B. M., Han K. H., Christensen S. B., Dowell C., Tsigelny I., Ho K. Y., Taylor P., McIntosh J. M. (2006) α-Conotoxin OmIA is a potent ligand for the acetylcholine-binding protein as well as α3β2 and α7 nicotinic acetylcholine receptors. J. Biol. Chem. 281, 24678–24686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McIntosh J. M., Plazas P. V., Watkins M., Gomez-Casati M. E., Olivera B. M., Elgoyhen A. B. (2005) A novel α-conotoxin, PeIA, cloned from Conus pergrandis, discriminates between rat α9α10 and α7 nicotinic cholinergic receptors. J. Biol. Chem. 280, 30107–30112 [DOI] [PubMed] [Google Scholar]
- 24. Hone A. J., Scadden M., Gajewiak J., Christensen S., Lindstrom J., McIntosh J. M. (2012) α-Conotoxin PeIA[S9H,V10A,E14N] potently and selectively blocks α6β2β3 versus α6β4 nicotinic acetylcholine receptors. Mol. Pharmacol. 82, 972–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuryatov A., Olale F., Cooper J., Choi C., Lindstrom J. (2000) Human α6 AChR subtypes. Subunit composition, assembly, and pharmacological responses. Neuropharmacology 39, 2570–2590 [DOI] [PubMed] [Google Scholar]
- 26. Kuryatov A., Lindstrom J. (2011) Expression of functional human α6β2β3* acetylcholine receptors in Xenopus laevis oocytes achieved through subunit chimeras and concatamers. Mol. Pharmacol. 79, 126–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dash B., Bhakta M., Chang Y., Lukas R. J. (2011) Identification of N-terminal extracellular domain determinants in nicotinic acetylcholine receptor (nAChR) α6 subunits that influence effects of wild-type or mutant β3 subunits on function of α6β2*- or α6β4*-nAChR. J. Biol. Chem. 286, 37976–37989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Deneris E. S., Boulter J., Swanson L. W., Patrick J., Heinemann S. (1989) Beta 3. A new member of nicotinic acetylcholine receptor gene family is expressed in brain. J. Biol. Chem. 264, 6268–6272 [PubMed] [Google Scholar]
- 29. Gotti C., Moretti M., Clementi F., Riganti L., McIntosh J. M., Collins A. C., Marks M. J., Whiteaker P. (2005) Expression of nigrostriatal α6-containing nicotinic acetylcholine receptors is selectively reduced, but not eliminated, by β3 subunit gene deletion. Mol. Pharmacol. 67, 2007–2015 [DOI] [PubMed] [Google Scholar]
- 30. Baddick C. G., Marks M. J. (2011) An autoradiographic survey of mouse brain nicotinic acetylcholine receptors defined by null mutants. Biochem. Pharmacol. 82, 828–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Azam L., Yoshikami D., McIntosh J. M. (2008) Amino acid residues that confer high selectivity of the α6 nicotinic acetylcholine receptor subunit to α-conotoxin MII[S4A,E11A,L15A]. J. Biol. Chem. 283, 11625–11632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hone A. J., Whiteaker P., Christensen S., Xiao Y., Meyer E. L., McIntosh J. M. (2009) A novel fluorescent α-conotoxin for the study of α7 nicotinic acetylcholine receptors. J. Neurochem. 111, 80–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Celie P. H., Kasheverov I. E., Mordvintsev D. Y., Hogg R. C., van Nierop P., van Elk R., van Rossum-Fikkert S. E., Zhmak M. N., Bertrand D., Tsetlin V., Sixma T. K., Smit A. B. (2005) Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an α-conotoxin PnIA variant. Nat. Struct. Mol. Biol. 12, 582–588 [DOI] [PubMed] [Google Scholar]
- 34. Pucci L., Grazioso G., Dallanoce C., Rizzi L., De Micheli C., Clementi F., Bertrand S., Bertrand D., Longhi R., De Amici M., Gotti C. (2011) Engineering of α-conotoxin MII-derived peptides with increased selectivity for native α6β2* nicotinic acetylcholine receptors. FASEB J. 25, 3775–3789 [DOI] [PubMed] [Google Scholar]
- 35. Luo S., Zhangsun D., Wu Y., Zhu X., Hu Y., McIntyre M., Christensen S., Akcan M., Craik D. J., McIntosh J. M. (2013) Characterization of a novel α-conotoxin from Conus textile that selectively targets α6/α3β2β3 nicotinic acetylcholine receptors. J. Biol. Chem. 288, 894–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hansen S. B., Sulzenbacher G., Huxford T., Marchot P., Taylor P., Bourne Y. (2005) Structures of aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 24, 3635–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beissner M., Dutertre S., Schemm R., Danker T., Sporning A., Grubmüller H., Nicke A. (2012) Efficient binding of 4/7 α-conotoxins to nicotinic α4β2 receptors is prevented by Arg185 and Pro195 in the α4 subunit. Mol. Pharmacol. 82, 711–718 [DOI] [PubMed] [Google Scholar]
- 38. Kim H. W., McIntosh J. M. (2012) α6 nAChR subunit residues that confer α-conotoxin BuIA selectivity. FASEB J. 26, 4102–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hone A. J., Meyer E. L., McIntyre M., McIntosh J. M. (2012) Nicotinic acetylcholine receptors in dorsal root ganglion neurons include the α6β4* subtype. FASEB J. 26, 917–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marritt A. M., Cox B. C., Yasuda R. P., McIntosh J. M., Xiao Y., Wolfe B. B., Kellar K. J. (2005) Nicotinic cholinergic receptors in the rat retina. Simple and mixed heteromeric subtypes. Mol. Pharmacol. 68, 1656–1668 [DOI] [PubMed] [Google Scholar]
- 41. Moretti M., Vailati S., Zoli M., Lippi G., Riganti L., Longhi R., Viegi A., Clementi F., Gotti C. (2004) Nicotinic acetylcholine receptor subtypes expression during rat retina development and their regulation by visual experience. Mol. Pharmacol. 66, 85–96 [DOI] [PubMed] [Google Scholar]
- 42. Quik M., Polonskaya Y., Gillespie A., Jakowec M., Lloyd G. K., Langston J. W. (2000) Localization of nicotinic receptor subunit mRNAs in monkey brain by in situ hybridization. J. Comp. Neurol. 425, 58–69 [DOI] [PubMed] [Google Scholar]
- 43. Azam L., Maskos U., Changeux J. P., Dowell C. D., Christensen S., De Biasi M., McIntosh J. M. (2010) α-Conotoxin BuIA[T5A;P6O]. A novel ligand that discriminates between α6β4 and α6β2 nicotinic acetylcholine receptors and blocks nicotine-stimulated norepinephrine release. FASEB J. 24, 5113–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bohr I. J., Ray M. A., McIntosh J. M., Chalon S., Guilloteau D., McKeith I. G., Perry R. H., Clementi F., Perry E. K., Court J. A., Piggott M. A. (2005) Cholinergic nicotinic receptor involvement in movement disorders associated with Lewy body diseases. An autoradiography study using [125I]α-conotoxin MII in the striatum and thalamus. Exp. Neurol. 191, 292–300 [DOI] [PubMed] [Google Scholar]
- 45. Wooters T. E., Smith A. M., Pivavarchyk M., Siripurapu K. B., McIntosh J. M., Zhang Z., Crooks P. A., Bardo M. T., Dwoskin L. P. (2011) bPiDI. A novel selective α6β2* nicotinic receptor antagonist and preclinical candidate treatment for nicotine abuse. Br. J. Pharmacol. 163, 346–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang L. Z., Campos C., Ly J., Ivy Carroll F., Quik M. (2011) Nicotinic receptor agonists decrease l-DOPA-induced dyskinesias most effectively in partially lesioned parkinsonian rats. Neuropharmacology 60, 861–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dutertre S., Nicke A., Lewis R. J. (2005) β2 subunit contribution to 4/7 α-conotoxin binding to the nicotinic acetylcholine receptor. J. Biol. Chem. 280, 30460–30468 [DOI] [PubMed] [Google Scholar]