

Background: The molecular mechanisms for islet β-cell compensation and failure are not fully known.
Results: FoxO1/PPARγ signaling regulates key β-cell genes, with this network being up-regulated in nondiabetic insulin-resistant rats and impaired in rodents with diabetes.
Conclusion: We examine the potential for the FoxO1/PPARγ network as a feature of β-cell compensation and failure.
Significance: We identify targets for prevention of type 2 diabetes.
Keywords: Chromatin Immunoprecipitation (ChiP), Gene Expression, Pancreatic Islets, Pyruvate Carboxylase, Signal Transduction, PDX1, Immunofluorescence, Mouse Models
Abstract
The molecular mechanisms and signaling pathways that drive islet β-cell compensation and failure are not fully resolved. We have used in vitro and in vivo systems to show that FoxO1, an integrator of metabolic stimuli, inhibits PPARγ expression in β-cells, thus transcription of its target genes (Pdx1, glucose-dependent insulinotropic polypeptide (GIP) receptor, and pyruvate carboxylase) that are important regulators of β-cell function, survival, and compensation. FoxO1 inhibition of target gene transcription is normally relieved when upstream activation induces its translocation from the nucleus to the cytoplasm. Attesting to the central importance of this pathway, islet expression of PPARγ and its target genes was enhanced in nondiabetic insulin-resistant rats and markedly reduced with diabetes induction. Insight into the impaired PPARγ signaling with hyperglycemia was obtained with confocal microscopy of pancreas sections that showed an intense nuclear FoxO1 immunostaining pattern in the β-cells of diabetic rats in contrast to the nuclear and cytoplasmic FoxO1 in nondiabetic rats. These findings suggest a FoxO1/PPARγ-mediated network acting as a core component of β-cell adaptation to metabolic stress, with failure of this response from impaired FoxO1 activation causing or exacerbating diabetes.
Introduction
Peroxisome proliferator-activated receptor γ (PPARγ)2 is a member of the nuclear hormone receptor family of ligand-inducible transcription factors and plays a pivotal role in diverse biological processes such as glucose and lipid homeostasis, inflammation, and cellular proliferation and differentiation (1, 2). In addition to adipocytes and several other cell types, it is expressed in islet β-cells (3). We have shown that PPARγ in β-cells regulates transcription of the prodifferentiation transcription factor Pdx1 (4, 5) and the receptor for the incretin hormone glucose-dependent insulinotropic polypeptide (GIP) (6). Others have reported that PPARγ regulates expression of the anaplerotic enzyme pyruvate carboxylase in adipose tissue (7), but whether this occurs in β-cells is unknown. Collectively, these genes are key controllers of β-cell mass, mitochondrial fuel metabolism, and insulin secretion, suggesting the potential for a central role of PPARγ in β-cell compensation and/or failure. Consistent with this proposal, we have shown increased expression of PPARγ and its target genes in islets from rodents with successful β-cell adaption to an experimental reduction of β-cell mass (4, 6) and to obesity and insulin resistance (6, 8) and decreased expression in a diabetic rodent model (6, 8). Also, clinical trials with thiazolidinedione PPARγ agonists (9) have shown a high success rate at stabilizing β-cell function and preventing type 2 diabetes (10).
Understanding why β-cell PPARγ expression is defective under hyperglycemic conditions requires characterizing its upstream regulation. In adipocytes, PPARγ gene expression is under inhibitory control by FoxO1 (11, 12). FoxO1 is a member of the family of winged-helix/forkhead transcription factors that serve important roles in cellular differentiation, proliferation, apoptosis, and the response to cellular stress in many tissues. FoxO1 is highly expressed in β-cells and is a key regulator of β-cell development, mass, and function (13–16). The best described FoxO1 target gene in β-cells is Pdx1 (17). The mechanism is reported to be competition for the FoxA2-binding element on the Pdx1 promoter, with phosphorylation of FoxO1 resulting in nuclear export and release of its blockade of FoxA2-dependent Pdx1 expression. However, the evidence is mostly correlative: EMSA and competition assays for FoxO1 and FoxA2 on the Pdx1 promoter, and imaging studies showing cytoplasmic FoxO1 in Pdx1-positive β-cells and nuclear FoxO1 in Pdx1-negative β-cells (14, 17).
FoxO1 activity is modulated by post-translational modifications and protein-protein interactions that impact its intracellular trafficking and function (18). In the β-cell, insulin or growth factor PI3K/Akt-induced FoxO1 phosphorylation leads to its translocation from the nucleus to cytoplasm, relieving its inhibition of target gene transcription (14). The incretin hormones GIP and glucagon-like peptide 1 (GLP-1) also activate this sequence (19, 20). Another is the balance between oxidative stress-induced FoxO1 acetylation and deacetylation (21). Thus, FoxO1 orchestrates β-cell transcriptional regulatory pathways for adaptive responses to many hormones, growth factors, and metabolic stresses. Whereas much is known about FoxO1 activity in β-cells, much less is known regarding its downstream target genes.
In this study we provide evidence for a central role for PPARγ in FoxO1-dependent regulation of Pdx1 expression and other key β-cell genes. Also, we show strong nuclear localization of FoxO1 in β-cells along with reduced expression of PPARγ and its target genes in diabetic rats, suggesting a novel mechanism for β-cell failure.
EXPERIMENTAL PROCEDURES
Cell Culture and in Vitro Assays
INS-1 (832/13) cells and βTC6 cells were maintained as described previously (5). PPARγ and FoxO1 siRNA were carried out in INS-1 cells using pooled SMART selection-designed siRNA duplexes with UU 3-overhang and 5′-phosphate on the antisense strand that targeted rat FoxO1 (NM_012560) and rat PPARγ (NM_013124) (Dharmacon RNA Technology). Transfections were carried out with Dharmafect 4 transfection reagent for 24 h at final siRNA concentrations of 100 pm. The protocol was two siRNA transfections at 0 h and 48 h; control cells were transfected with scrambled siRNA.
EMSA used nuclear extracts from rat-derived INS-1 cells and mouse-derived βTC6 cells with PAGE-purified oligonucleotides for the mouse PPARγ promoter containing the putative FoxO1 binding site (forward, 5′-GTCAGATAGATAAACAAATTT-3′ and reverse, 5′-CTATCTATTTGTTTAAAATCG-3′) labeled by end filling with [α-32P]dCTP (PerkinElmer Life Sciences) as described previously (5). PPARγ specificity was determined with rabbit polyclonal FoxO1 antibody (Chemicon, AB4130) and competition studies that added increasing amounts of unlabeled dsDNA oligonucleotide sequences.
ChIP was performed with βTC6 cells using ChIP-IT kit (Active Motif) and a previously described method (5). Rabbit polyclonal FoxO1 antibody (Chemicon) was added to aliquots of precleared 300–500-bp chromatin fragments and incubated overnight, with parallel samples incubated with the negative-control IgG provided with the kit. Protein G-agarose beads were added to the mixture for 1.5 h at 4 °C. After reversing cross-links, DNA was isolated and PCR performed using primers for the mouse PPARγ promoter. Primer sequences were: forward, 5′-AACATCAACCATTGGAATACTACTGC-3′ and reverse, 5′-TGGTTGAAACTCACACATCTGAAG-3′ with an expected 281-bp PCR product. PCR conditions were 1 cycle at 94 °C for 3 min, 40 cycles at 94 °C for 30 s, 62 °C for 1 min, 72 °C for 1 min. A mouse-specific positive control was performed using a kit based on the binding of the transcription factor EFI-α by anti-RNA polymerase II (Active Motif).
Luciferase reporter gene assays were carried out in 60–70% confluent INS-1 cells in 6-well plates incubated overnight in antibiotic-free medium. Co-transfections were performed with the FoxO1 overexpression cassette (pcDNA3-FLAG-FKHR from Dr. Kun-Liang Guan, University of Michigan) and the Pdx1 promoter pTAL-PPRE-Pdx-1 luciferase plasmid (Dr. Roland Stein, Vanderbilt University) or 3× PPRE-luciferase reporter vector (Dr. Speigelman laboratory, Addgene) with Lipofectamine 2000 transfection reagent (Invitrogen). Renilla luciferase reporter plasmid (pRL-TK, Promega) was included (0.05 μg) in all transfections as internal control. Cells were lysed 48 h after transfection, and luciferase assay performed in a TD 20/20 luminometer (Turners Design) using a dual luciferase assay kit (Promega). Firefly luciferase activity was normalized to Renilla luciferase and expressed as relative luciferase activity of the reporter constructs with and without transfection of the FoxO1 overexpression cassette.
Animal Studies
All protocols were approved by the University of Vermont Institutional Animal Care and Use Committee. FoxO1-haplodeficient mice (17) and mice that overexpress a constitutively nuclear FoxO1 transgene (FoxO1S253A) in β-cells and liver (13, 22) were obtained from D. Accili (Columbia University) on a mixed background and were backcrossed for >10 generations to the C57BL/6 background. Mice with PPARγ deficiency restricted to pancreatic epithelium (PANC PPARγ−/−) were generated by crossing Pdx1-Cre mice (original source, D. Melton, Harvard University) and mice with two floxed PPARγ alleles as detailed previously (5). Some animals underwent glucose tolerance testing after an overnight fast that consisted of 2 g/kg intraperitoneal glucose with serum glucose measured at 0, 30, 60, 90, and 120 min (Freestyle glucose meter). Zucker lean (ZL, fa/+ or +/+) and Zucker fatty (ZF, fa/fa) male rats (Harlan) underwent 60% pancreatectomy (Px) or sham Px surgery at 6 weeks of age as described previously (8). Islets were isolated by pancreas duct perfusion with collagenase, Histopaque gradient separation, and hand picking.
Islet Expression Studies
Immunoblot and quantitative PCR analyses were performed as described previously (5). Immunoblot antibodies were rabbit polyclonal anti-Pdx1 (1:2000, Upstate/Millipore), mouse monoclonal anti-PPARγ (1:1000, Chemicon/Millipore), and rabbit polyclonal anti-FoxO1 (1:1000, Chemicon/Millipore), followed by goat anti-mouse/rabbit-HRP-conjugated antibody (Bio-Rad). Pyruvate carboxylase affinity blotting was performed using HRP-conjugated streptavidin. Detection was by chemiluminescence using HyperFilm-ECL (Amersham Biosciences). Membranes were stripped and reprobed to establish equivalent loading using anti-β-actin (Sigma). Islet quantitative PCR was carried out in a PTC-200 Peltier Thermal Cycler (MJ Research) using cDNAs, Taq polymerase (Promega), and primer combinations (sequences available on request). The thermal cycle program was denaturing step at 95 °C for 2 min followed by 35 cycles for PPARγ, 25 cycles for Pdx1, 30 cycles for FoxO1, at 94 °C for 15 s, 56 °C for 30 s, and 72 °C for 60 s, with an extension step of 5 min at 72 °C. Results are expressed relative to control gene expression (cyclophilin B).
GLP-1 Induction of Pdx1 Gene Expression
Isolated islets from PANC PPARγ−/− and WT mice were cultured overnight in RPMI 1640 medium, 10% FBS, 11.2 mm glucose, 100 units/ml penicillin, 100 μg/ml streptomycin, followed by overnight culture in serum-free medium with 3% BSA. Islets were incubated in serum-free medium with 3% BSA and 11.2 mm glucose plus freshly dissolved 100 nm mouse GLP-1 (Phoenix Pharmaceutical) or vehicle for 8 h followed by quantitative PCR analysis.
Pyruvate Carboxylase (PC) Activity
Freshly isolated islets from 3 week postsurgery sham and 60% Px ZF and ZL rats were frozen in liquid nitrogen and shipped to Dr. Ye Q. Liu at the University of Louisville. PC activity was measured as described previously (23).
Pancreas Immunofluorescence and Islet Morphometry
Excised pancreata were immersion-fixed overnight in 4.0% paraformaldehyde in 0.1 m phosphate buffer at 4 °C followed by embedding in paraffin. β-Cell mass was quantified using a previously detailed computer planimetric method (24). For multiple-labeling immunofluorescence studies of FoxO1 and Pdx1, sections were stained with guinea pig anti-insulin (Linco/Millipore) to mark β-cells, mouse anti-Pdx1 (Developmental Studies Hybridoma Bank, University of Iowa), and rabbit anti-FoxO1 (Cell Signaling, clone C29H4). Secondary antibodies were multiple-labeling grade anti-species-specific IgG conjugated to either CY2, CY3, or Alexa Fluor 647, respectively (Jackson ImmunoResearch or Molecular Probes/Invitrogen). Sections were imaged confocally using an LSM 510 META (University of Vermont Microscopy Imaging Center).
Statistical Analysis
Data are presented as mean ± S.E. or S.D., as indicated. Each data point from the animal studies represents an individual animal. Statistical significance was determined by the unpaired Student's t test or two-way ANOVA.
RESULTS
Identification and Characterization of FoxO1 Binding Site on Mouse PPARγ Promoter
The initial studies sought to identify a biological basis for FoxO1 regulation of PPARγ expression. The consensus binding sequence for FoxO1 is GTAAACA (25). We searched GenBank and identified a putative FoxO1 binding site within the mouse PPARγ promoter at positions −825 to −831 from the transcription start site (GenBank AY236530) with one nucleotide mismatch from the consensus sequence (Fig. 1A). MatInspector software identified homologous sequences in the rat and human PPARγ promoters. We confirmed that this sequence binds FoxO1 by performing EMSA and FoxO1 antibody inhibition studies using α-32P-labeled complementary strand oligomers of the identified sequence and nuclear extracts from mouse-derived βTC6 and rat-derived INS-1 cells (Fig. 1B). Also, the specificity of binding was established with a competition assay using excess unlabeled probe.
FIGURE 1.
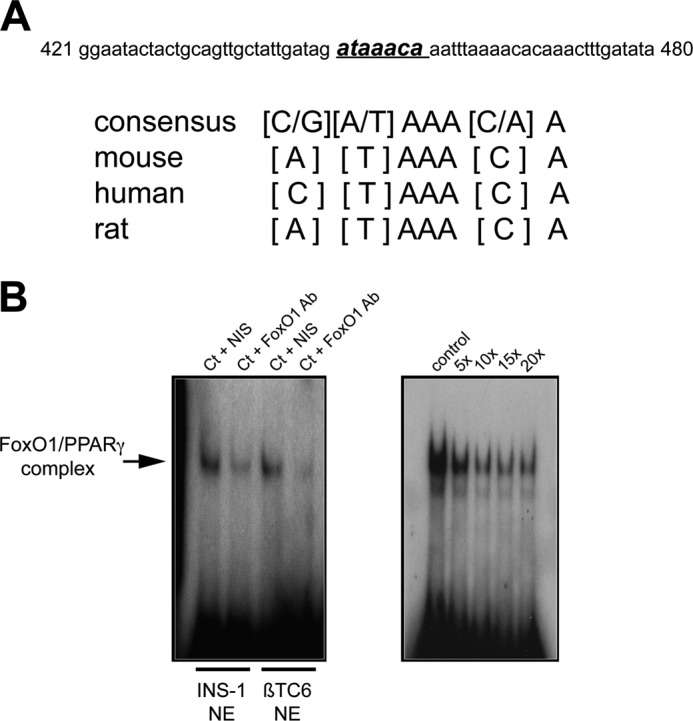
Identification of a FoxO1 binding site within the mouse PPARγ promoter. A, identified sequence in the mouse PPARγ promoter with one nucleotide mismatch from the FoxO1 consensus binding sequence. Homologous sequences within the rat and human PPARγ promoters also are shown. B, left, EMSA showing inhibition with FoxO1 antibody, using nuclear extracts (NE) from rat-derived INS-1 cells and mouse-derived βTC6 cells incubated with α-32P-labeled complimentary strand oligomers of the identified sequence. B, right, competition assay using increasing -fold amounts of unlabeled oligomers of the identified sequence. Ct, control; NIS, nonimmune serum.
ChIP assay was performed in βTC6 cells to confirm FoxO1 binds to this sequence in an intact insulin-containing cell line, using FoxO1 antibody-precipitated DNA fragments as the PCR template and a primer pair that spanned the region containing the FoxO1 binding site (Fig. 2A). Input DNA and the FoxO1 antibody-precipitated chromatin fragments both contained the correct 281-bp product in contrast to the nonimmune IgG negative control (Fig. 2B). Also shown is the positive control based on the binding of RNA polymerase II to the transcription factor EFI-α (Fig. 2C).
FIGURE 2.

ChIP assay confirming FoxO1 binds to the identified FoxO1 binding sequence in βTC6 cells. A, diagram of the ChIP assay showing the primers spanning a 281-bp sequence that contains the FoxO1 binding sequence in the mouse PPARγ promoter sequence. B, representative gel showing that input DNA and FoxO1 antibody-precipitated chromatin fragments contain the correct 281-bp product and the absence of a band with the control nonimmune IgG. C, positive control based on binding of RNA polymerase II to the transcription factor EFI-α (middle two lanes) and only a minimal product with the control IgG. Each lane represents an individual sample.
Expression studies confirmed FoxO1 inhibits PPARγ expression in an insulin-containing cell line. A FoxO1 siRNA protocol was established in INS-1 cells that resulted in a 60% knockdown of FoxO1 mRNA level and 47% reduction in FoxO1 protein by 96 h (Fig. 3A). PCR analysis showed a 2.0 ± 0.1-fold increase in PPARγ mRNA level compared with the scrambled siRNA control, and a similar increase in Pdx1 mRNA level: 2.2 ± 0.2-fold (Fig. 3B). Also, FoxO1 was overexpressed by transient transfection in INS-1 cells and assayed for PPARγ promoter activity using two PPRE luciferase reporters, Pdx1-PPRE and 3×PPRE. A 48-h overexpression of FoxO1 lowered Pdx1-PPRE driven luciferase activity by 41% compared with no FoxO1 overexpression (Fig. 3C). In addition, 3× PPARγ-PPRE reporter activity was decreased 44% with the FoxO1 overexpression (Fig. 3D). Together, these in vitro findings confirmed that FoxO1 binds to the identified binding site sequence and inhibits PPARγ promoter activity in insulin-containing cell lines.
FIGURE 3.
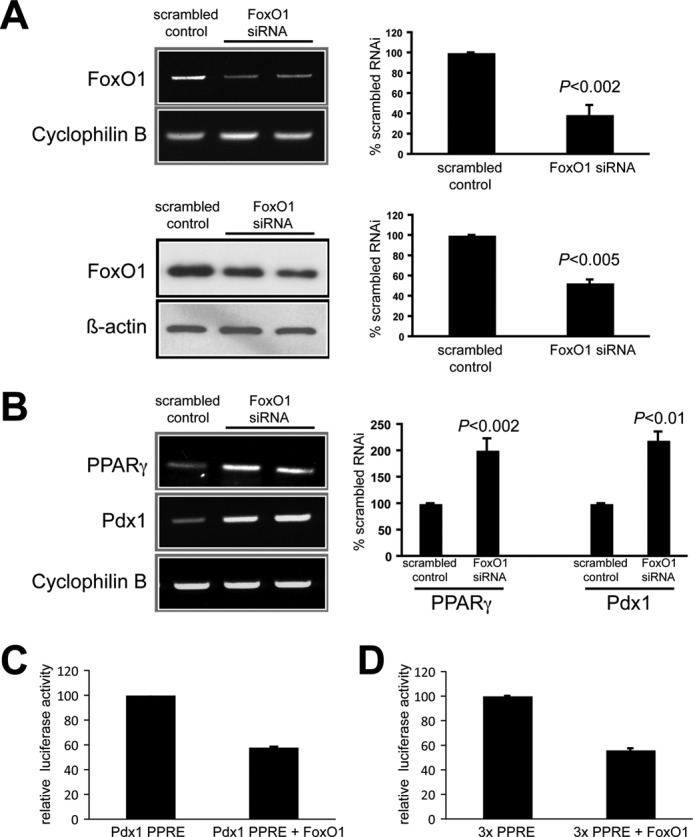
In vitro studies showing FoxO1 inhibits PPARγ expression in INS-1 cells. A, 96-h FoxO1 siRNA showing marked reductions in FoxO1 mRNA (top row, gel on left and mean ± S.D. (error bars) results of three experiments on right) and protein (bottom row, blot on left and mean ± S.D. results of three experiments on right) compared with control scrambled siRNA transfections. B, siRNA-induced reduction in FoxO1 expression resulting in a doubling of PPARγ and Pdx1 mRNA levels. Left, representative gel. Right, mean ± S.D. results of three experiments. Each lane on the gels represents individual samples. C, transfection of pTAL-PPRE-Pdx1 promoter luciferase reporter system in INS-1 cells, showing a large reduction in gene expression activity when cotransfected with pcDNA3-FLAG-FKHR FoxO1 overexpression cassette. n = 6, p < 0.001. D, very similar results obtained with the 3× PPRE-luciferase vector in INS-1 cells, again showing a large reduction in gene expression activity with cotransfection of the pcDNA3-FLAG-FKHR FoxO1 cassette. n = 6, p < 0.001.
In Vivo Studies
FoxO1 inhibition of PPARγ expression in native β-cells was investigated using isolated islets from genetically modified mice. We first tested global FoxO1-haploinsufficient mice. Null mice are embryonically lethal, but the haploinsufficient mice are normoglycemic with relatively normal pancreas morphology and β-cell mass, insulin content, and insulin secretion (17, 22). Islet lysates from 8-week-old FoxO1+/− mice had twice the PPARγ mRNA and protein levels of WT mice (Fig. 4). Pdx1 mRNA and protein levels also were increased 2-fold in the FoxO1+/− islets.
FIGURE 4.
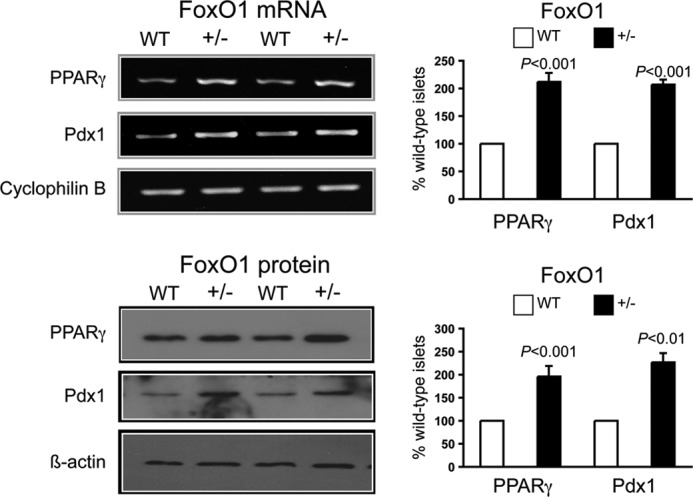
Increased islet PPARγ and Pdx1 expression in 8-week-old FoxO1-haploinsufficient mice. In agreement with the siRNA results in INS-1 cells, PPARγ and Pdx-1 mRNA (top panels) and protein (bottom panels) levels were doubled in isolated islets from global FoxO1-haploinsufficient mice. Left, representative gels showing results for two WT and two FoxO1-haploinsufficient mice. Right, mean ± S.D. results of three mice in each group.
We next studied mice that hyperexpress a constitutively nuclear FoxO1 transgene in β-cells and liver; the transgene encodes an amino acid substitution at the Ser253 phosphorylation site that prevents insulin-stimulated translocation of the mutant FoxO1 to the cytoplasm and is under control of the transthyretin promoter that directs expression to liver and β-cells (22). Others have shown that these mice on a mixed background have reductions in islet Pdx1 levels and early onset mild diabetes (13, 22). We backcrossed the transgene to the C57BL/6 background and confirmed glucose intolerance in 8–10-week-old mice, also noting a 25% reduction in β-cell mass compared with the littermate WT mice (Fig. 5A). As predicted, islet lysate immunoblots revealed a 45% reduction of PPARγ and a similar loss of Pdx1 versus WT islets (Fig. 5B).
FIGURE 5.

Reduced islet PPARγ and Pdx1 expression in mice that hyperexpress a constitutively nuclear FoxO1 transgene with amino acid substitution at the Ser253 phosphorylation site preventing insulin-stimulated translocation of the mutant FoxO1 to the cytoplasm, under control of the transthyretin promoter directing expression to the liver and β-cells. A, results in 8–10-week-old FoxO1S253A and WT mice showing glucose intolerance in the FoxO1S235A mice assessed by intraperitoneal glucose tolerance test (left) and a 25% reduction in the β-cell mass in FoxO1S253A mice (right). Values are mean ± S.E. (error bars). B, islet immunoblotting results for PPARγ and Pdx1 in WT and FoxO1S253A mice. Left, representative gels showing results for two WT and two FoxO1S253A mice. Right, mean ± S.D. results of three mice in each group.
PPARγ Is Required for GLP-1 Induction of Pdx1 Gene Expression in Vitro
We used GLP-1, a known mediator of FoxO1 nuclear translocation (20), to probe the FoxO1/PPARγ/Pdx1 system. Normal mouse islets were incubated for 8 h with or without 100 nm GLP-1(7–36 amide) at 11.1 mm glucose followed by real time PCR analysis. PPARγ and Pdx1 signals were increased 2–2.5-fold whereas MafA and FoxA2 were unchanged, showing the selectivity of the effect (Fig. 6A). We used this protocol with islets from PANC PPARγ−/− mice. In agreement with the prior results, GLP-1 stimulation nearly doubled PPARγ and Pdx1 mRNA expression in the floxed control mice. In contrast, in the PANC PPARγ−/− islets a GLP-1-induced increase in Pdx1 expression was absent (Fig. 6B), demonstrating that PPARγ is essential for in vitro GLP-1 activation of Pdx1 expression.
FIGURE 6.
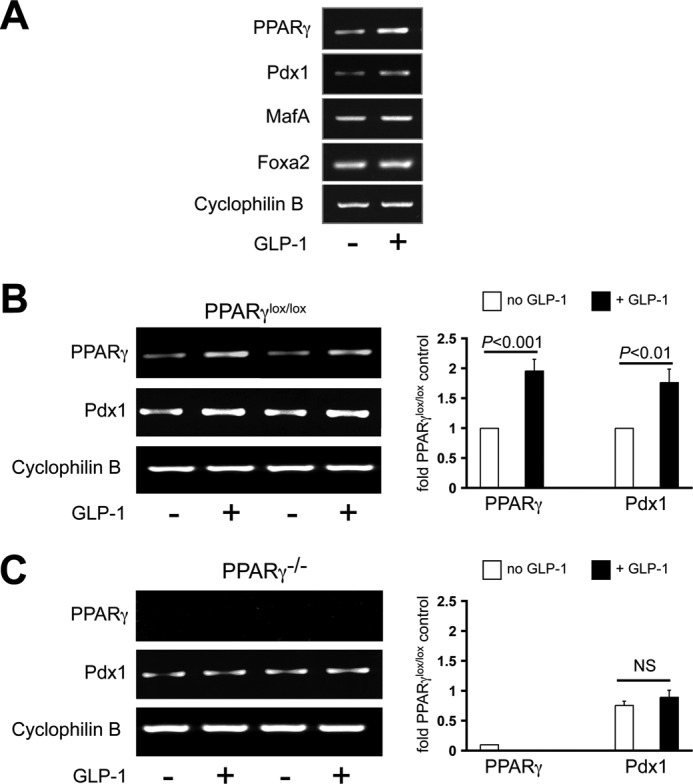
PPARγ is required for GLP-1 induction of Pdx1 expression in mouse islets. A, normal mouse islets were incubated 8 h with or without 100 nm GLP-1(7–36 amide) followed by real time PCR analysis. Expression of PPARγ and Pdx1 was doubled compared with no observable effect on MafA or Foxa2 expression. B and C, that protocol was used in isolated islets from 8-week-old floxed control mice (B) and PANC PPARγ−/− mice (C) with the normal GLP-1 induction of Pdx1 expression absent in the PANC PPARγ−/− islets. Left, representative gels with each lane representing islets from an individual mouse. Right, mean ± S.D. (error bars) results of three mice in each group.
PPARγ Regulates PC Expression in Islet β-Cells
We have shown that PPARγ transcriptionally regulates Pdx1 (4, 5) and GIP receptor genes (6) in β-cells. Others have shown PPARγ regulation of PC expression in white and brown adipose tissue (7). We previously studied ZF rats that are obese, hyperlipidemic, and insulin-resistant secondary to a mutated leptin receptor, but are virtually normoglycemic because of compensatory increases in β-cell mass and function (26, 27). Those studies showed that PC plays a critical role in their β-cell adaptive response by preventing dysmetabolism-induced defects in β-cell mitochondrial fuel metabolism (27). Also, reduced islet PC expression is found in rodents (28, 29) and humans with type 2 diabetes (30).
We investigated whether β-cell PC expression is PPARγ-regulated using an established siRNA for PPARγ in INS-1 cells that lowers PPARγ mRNA and protein 75% (4). Cells were treated 72 h with or without the PPARγ agonist troglitazone. PPARγ siRNA reduced PC mRNA band intensity >60% (Fig. 7A, first and fourth lanes) whereas troglitazone enhanced it 3-fold (second lane); that effect was lost when the PPARγ siRNA and troglitazone were used together (third lane).
FIGURE 7.
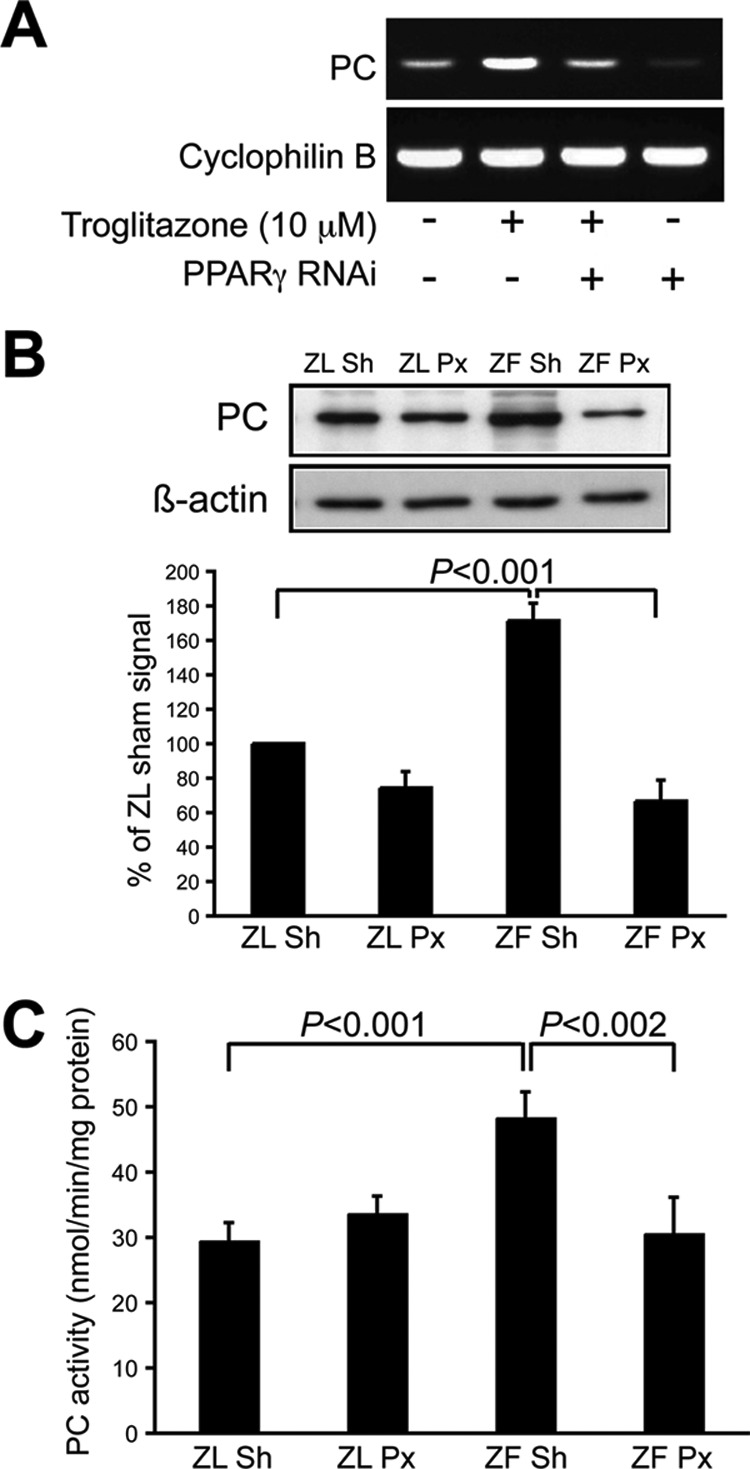
PPARγ regulates PC gene expression in β-cells. A, representative results of PPARγ siRNA in INS-1 cells with or without the PPARγ agonist troglitazone, showing a 60% reduction in PC mRNA with the PPARγ siRNA (first and fourth lanes) whereas troglitazone enhanced it 3-fold (second lane); the latter was lost when the PPARγ siRNA and troglitazone were used together (third lane). B, PC expression assessed with streptavidin A affinity labeling of PAGE-resolved islet extracts from 3 weeks after surgery 60% Px and sham ZL and ZF rats. Sham (Sh) ZF rats are obese and insulin-resistant, but normoglycemic because of compensatory increases in β-cell mass and function. In contrast, Px ZF rats become hyperglycemic (∼15 mm) by 3 weeks after surgery with the same obesity and free fatty acid levels as the normoglycemic ZF shams. As we have seen with other PPARγ-regulated genes in β-cells, the PC band intensity in the sham ZF islets was nearly twice that of the ZL rats and below normal in the diabetic Px ZF rats. Top, representative gel. Bottom, mean ± S.D. (error bars) results of four rats in each group. C, PC enzyme activity measured in islet extracts from 3 weeks after surgery 60% Px and sham ZL and ZF rats. Results are mean ± S.D. of three rats in each group.
We previously developed and characterized a diabetic rat model by performing a 60% Px in ZF rats (8). Px ZF rats became hyperglycemic (∼15 mm) by 3 weeks after surgery with the same free fatty acid levels and obesity as normoglycemic ZF shams. Additional controls were sham and 60% Px ZL rats that were euglycemic and metabolically indistinguishable. Islet immunoblots from 3 week postsurgery sham ZF rats had a doubling of PPARγ protein and its transcriptional target GIP receptor compared with both groups of ZL rats, whereas the diabetic Px ZF rats displayed 20–30% reductions versus the ZL rats (6), supporting our hypothesis of importance for PPARγ signaling in β-cell compensation and failure. We predicted a similar pattern for PC and have now assessed PC expression using streptavidin A affinity labeling of SDS-PAGE-resolved islet extracts from 3-week postsurgery Px and sham ZL and ZF rats. As expected, the band intensity in the sham ZF islets was nearly twice that of the ZL rats (171 ± 10%) whereas it was subnormal in the diabetic Px ZF rats (67 ± 12%, Fig. 7B). Comparable results were obtained for PC activity of islet extracts (Fig. 7C), with a 60% increase in sham ZF islets versus ZL rats (p < 0.001), but no increase in the Px ZF islets (p < 0.002 sham ZF versus Px ZF).
Nuclear Localization of FoxO1 in β-Cells of Hyperglycemic Rats
We sought an explanation for the markedly lowered expression of PPARγ and its target genes in diabetic ZF rats by examining the subcellular distribution of β-cell FoxO1 in 3-week postsurgery Px and sham ZL and ZF rats using confocal microscopy (Fig. 8). In both ZL groups, FoxO1 immunoreactivity was chiefly cytoplasmic (the hyperintense staining areas are erythrocytes in capillaries). In sham ZF rats, the FoxO1 staining was both nuclear and cytoplasmic. In contrast, in the Px ZF rats a striking increase in nuclear FoxO1 immunoreactivity was observed. In addition, in both ZL groups and sham ZF rats there was strong nuclear Pdx1 staining, whereas in the Px ZF rats the nuclear Pdx1 signal was markedly reduced. We conclude from these findings that in the diabetic ZF rats a defect in FoxO1 activation and/or translocation causes loss of the adaptive increase in the FoxO1/PPARγ pathway that is present in the nondiabetic insulin-resistant ZF rats.
FIGURE 8.
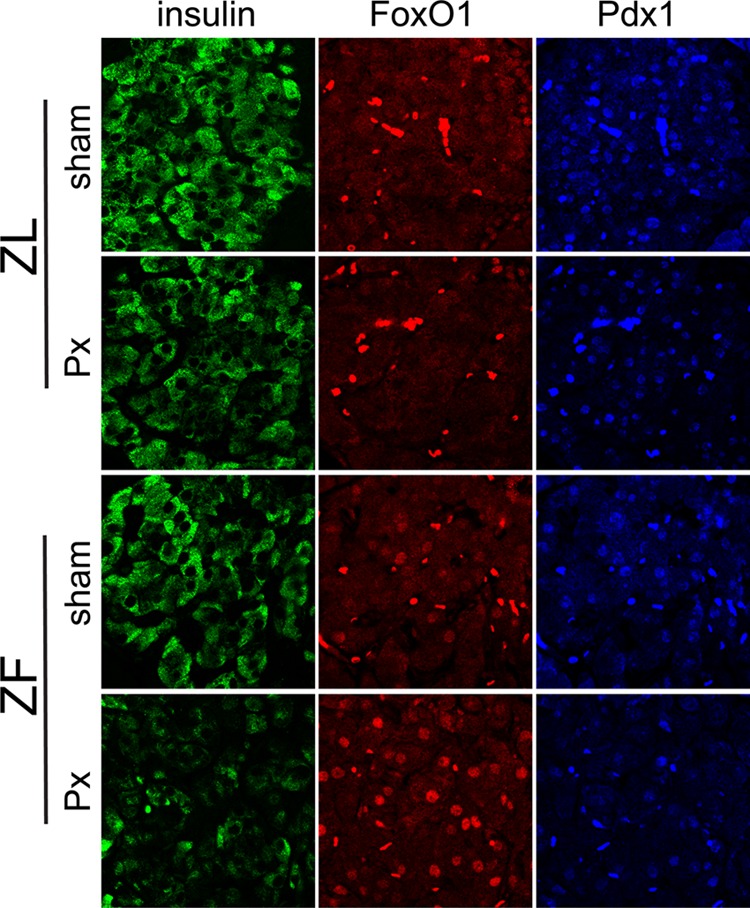
β-Cell FoxO1 and Pdx1 subcellular distribution in normoglycemic and diabetic rats. Representative islet confocal fields (n = 3/group) showing mainly cytoplasmic FoxO1 immunoreactivity and nuclear Pdx1 immunoreactivity in the β-cells of normoglycemic sham-operated and 60% Px ZL rats (top panels). In the β-cells of normoglycemic ZF-sham rats, there was both nuclear and cytoplasmic FoxO1 distribution along with strongly nuclear Pdx1. However, in the hyperglycemic ZF-Px rats (bottom panel), FoxO1 staining was principally nuclear along with decreased Pdx1 immmunoreactivity. For reference, β-cells are stained green. Bright, autofluorescing bodies are erythrocytes in islet capillaries. Field widths are 138 μm.
DISCUSSION
The current results have established PPARγ expression in β-cells is under transcriptional restraint by FoxO1. That conclusion is supported by our having identified a FoxO1 binding site in the mouse PPARγ promoter with homologous sequences in rats and humans, and in vitro studies demonstrating FoxO1 binding and inhibition of PPARγ expression. Also, transgenic mice with altered FoxO1 expression or defective islet FoxO1 nuclear translocation exhibited changes in islet PPARγ expression and parallel findings for its target gene Pdx1. Of particular importance was our demonstrating that PPARγ is necessary for GLP-1 mediated augmentation of Pdx-1 expression in vitro, as GLP-1 is an established FoxO1 activator (20). We have also shown that PPARγ regulates PC gene expression in β-cells, as has previously been characterized using molecular and in vivo techniques in white and brown adipose (7), making it is the third PPARγ target gene we have identified in β-cells along with the Pdx1 (4, 5) and GIP receptor (6) genes.
These results are especially notable because of the importance of the identified PPARγ target genes for regulating β-cell development and postnatal β-cell mass and function. Pdx1 is an essential transcription factor for pancreas morphogenesis and postnatally is a key regulator of β-cell differentiation, maturation, and survival (31). It is required for the normal adaptive increase in β-cell mass and function to insulin resistance (32, 33). Distal from Pdx1 is a network of genes that impact diverse aspects of β-cell signaling, mass, and function (34) including glucokinase with its well known regulatory effects on glycolysis, insulin secretion, and β-cell proliferation (35).
PC catalyzes mitochondrial pyruvate conversion to malate. In most cells, the normal pathway for pyruvate metabolism is through pyruvate dehydrogenase with entry into the citric acid cycle for ATP production. However in β-cells there is unusually high PC expression so that 50% of pyruvate undergoes carboxylation (36) with entry into the anaplerotic pathways of insulin secretion (37). Obese, insulin-resistant, hyperlipidemic ZF rats normally do not develop diabetes because of compensatory increases in β-cell mass and function (27). We previously looked for the Randle effect (inhibition of pyruvate dehydrogenase activation by elevated levels of free fatty acid) in islets from ZF rats and found 30% lowered pyruvate dehydrogenase activity, but, paradoxically, glucose oxidation and glucose-induced insulin secretion were increased. The explanation was increased PC expression with diversion of pyruvate metabolism to the anaplerotic pathways (this study and Ref. 27). We observed the same protective effect in high glucose-cultured rat islets (38). Also, Liu and co-workers showed a role for this enzyme in the β-cell mass and functional adaptation to a 60% Px in rats (23, 39). As such, PC is believed to play a key role in the β-cell adaptation to insulin resistance and dysmetabolism by preserving mitochondrial fuel metabolism (37, 40).
The β-cell GIP receptor mediates that incretin hormone regulation of insulin secretion and glucose tolerance. GIP receptor gene knock-out mice are glucose-intolerant (41). Also, GIP receptor allelic variations correlate with the heterogeneity in serum glucose and insulin levels after oral glucose challenge in nondiabetic humans (42).
The second aspect of our results relates to evidence that this signaling pathway is enhanced with successful β-cell compensation to insulin resistance, whereas defective nuclear translocation of FoxO1 resulting in persistent inhibition of PPARγ expression is a feature of failed β-cell compensation and hyperglycemia. We previously studied ZF rats made hyperglycemic by a 60% pancreatectomy and observed subnormal islet expression of PPARγ and its target gene GIP receptor versus the hyperexpression in nondiabetic ZF rats (6). The current study has shown the same pattern for PC along with reduced nuclear Pdx1 immunostaining in Px ZF rats, which are in accord with our previous report showing subnormal PC and Pdx1 mRNA expression in Px ZF islets (8).
Insight into the impaired PPARγ expression with hyperglycemia came with our finding a distinct change in the subcellular localization of FoxO1 in β-cells of ZF diabetic rats, specifically, instead of the nuclear and cytoplasmic pattern seen in the control rats, there was an intense nuclear distribution. This finding is not unique to the Px ZF model, as nuclear FoxO1 and reduced overall islet FoxO1 expression were found in insulin-resistant diabetic mice created by deletion of insulin receptors in GLUT4-expressing tissues (43). Also, below normal islet expression of Pdx1, GIP receptor, and PC is well known to occur in animals (6, 28, 29, 44) and humans (30, 45, 46) with type 2 diabetes. Based on this collective evidence, we propose that nuclear retention of FoxO1 impairing PPARγ-mediated defenses against metabolic stresses is an unrecognized feature of failed β-cell adaptation.
The mechanism of the defective nuclear to cytoplasmic translocation of FoxO1 with diabetes is not known, as the regulation of FoxO1 activity related to compartmentalization in β-cells is multifactorial related to several post-translational modifications and protein interactions (14, 18, 21). Best known for the β-cell is insulin or growth factor PI3K/Akt pathway-induced phosphorylation causing nuclear export that removes its inhibition of target gene transcription (14). Supporting a defect at this level of the signaling pathway is a study performed in New Zealand obese mice that became diabetic when fed a carbohydrate-rich diet; reductions in islet FoxO1 phosphorylation were noted (47). Furthermore, islets from these mice, and MIN 6 cells, incubated with high glucose and palmitate for 48 h underwent dephosphorylation of FoxO1 along with its upstream kinase Akt (47). Another study reported reduced insulin receptor and insulin receptor substrate 2 expression in islets from humans with type 2 diabetes, and it noted nuclear retention of FoxO1 in islets of mice with a β-cell-specific deletion of insulin receptors (48). Alternatively, FoxO1 activity and its subcellular compartmentation in β-cells is regulated by other serine/threonine kinases as well the balance between acetylation and deacetylation (21). Recently β-cell oxidative stress was linked to hyperexpression of a B55a-containing PP2A holoenzyme that dephosphorylates FoxO1 and promotes its nuclear location (49).
The importance of PPARγ in β-cells has been controversial. A β-cell-specific knock-out mouse for PPARγ based on a rat insulin promoter Cre/loxP recombinase system was reported to be normoglycemic with a standard chow diet and also after fat feeding (50). Another publication from these authors (51) using a tamoxifen-inducible Pdx1 Cre/loxP system that lowered islet PPARγ mRNA by 90% found no β-cell phenotype after 7 months of normal chow; glucose tolerance, insulin secretion, and β-cell mass were unchanged from WT mice as was expression of multiple islet genes including Pdx1, pyruvate carboxylase, and GIP receptor. In addition, 11 weeks of a high fat diet induced the same hyperglycemia, obesity, and insulin resistance in the PPARγ knock-out and WT mice. Also, there was no difference in β-cell mass or islet gene expression profiles between the fat-fed WT and PPARγ knock-out mice although the atypical finding of no effect of fat feeding on these parameters in WT mice makes these latter findings difficult to interpret. The authors concluded that PPARγ in β-cells is inconsequential. In contrast, we studied noninducible Pdx-1 Cre/loxP PPARγ-null mice (52) and found hyperglycemia at 8 weeks of age with standard chow along with near total absence of glucose-induced and GIP-induced insulin secretion in isolated islets, and a marked reduction in islet expression of Pdx-1 and GIP receptor (5, 6). There was no difference in β-cell mass or pancreas histology from control mice (5). As such, this mouse showed a phenotype of functional β-cell failure, with an expression profile that agreed exactly with the proposed signaling pathway in this study. Also, others have reported PPARγ regulation of gene expression for GLUT2 (53), glucokinase (54), and GPR40 (55) in β-cells, plus key modulatory effects of PPARγ in β-cell endoplasmic reticulum stress related to a cytokine-induced loss of SERCA2 expression have been shown (56, 57). In addition, numerous β-cell effects have been attributed to PPARγ from thiazolidinedione studies in islets, insulin-containing cell lines, and various animal models, although direct proof that the findings are PPARγ-mediated is mostly lacking. Thus, although the basis for the different β-cell phenotypes in the mouse models is unknown, we conclude that there is extensive support for an important regulatory role for PPARγ in β-cells, especially in the adaptation response to metabolic stresses.
In summary, our results are consistent with a novel mechanism for β-cell compensation and failure based on an integral relationship between FoxO1 and PPARγ, whereby FoxO1 exerts hierarchal control over several key β-cell genes, and PPARγ is a vital intermediate. FoxO1 is well poised to be a master transcriptional regulator over β-cell adaptive responses, as its expression and activity are influenced by numerous stimuli such as nutrient overload, growth factors, incretin hormones, and oxidative stress (13, 14). Downstream from PPARγ is a network of genes that exert important regulatory control over prodifferentiation processes, incretin effects, glucose and mitochondrial fuel metabolism, and β-cell compensation to obesity and insulin resistance (13, 27, 32, 33). We have shown that islet PPARγ and its target genes are hyperexpressed in nondiabetic insulin-resistant rats (present study and Refs. 6, 8) and rats during the β-cell adaptation after a partial pancreatectomy (4, 6), plus we and others have shown impaired expression of these same genes in diabetic animals (6, 29, 44) and humans with type 2 diabetes (30, 45, 46). Collectively, these findings support, but do not prove, a FoxO1/PPARγ-mediated transcriptional network functioning as a core element of how β-cells adapt to metabolic stress, with failure of this response causing or exacerbating diabetes.
Acknowledgments
We thank Dr. Domenico Accili (Columbia University) for the FoxO1 mouse models, Drs. Richard Mortenson and Sheng Duan (University of Michigan) for the PANC PPARγ−/− mouse model, Dr. Ye Q. Liu (Louisville University) for performing the pyruvate carboxylase activity measurements, Dr. Kun-Liang Guan (University of Michigan) for the FoxO1 expression plasmid, Dr. Roland Stein (Vanderbilt University) for the Pdx-1 promoter plasmid, and Dr. Chris Newgard (Duke University) for the INS-1 cells. The 3X PPRE-luciferase reporter vector developed by Dr. Bruce Spiegelman (Dana-Farber Cancer Institute) was obtained from Addgene. The Pdx1 monoclonal antibody developed by Dr. O. Madsen was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD, National Institutes of Health, and maintained by The University of Iowa, Department of Biology.
This work was supported, in whole or in part, by National Institutes of Health Grant DK56818 (to J. L. L.). This work was also supported by the American Diabetes Association (to M. P.).
- PPARγ
- peroxisome proliferator-activated receptor γ
- GIP
- glucose-dependent insulinotropic polypeptide
- GLP-1
- glucagon-like peptide 1
- PANC PPARγ−/−
- pancreas epithelium-specific PPARγ knockout mice
- PC
- pyruvate carboxylase
- PPRE
- peroxisome proliferator-activated receptor response element
- Px
- partial pancreatectomy
- ZF
- Zucker fatty
- ZL
- Zucker lean.
REFERENCES
- 1. Feige J. N., Gelman L., Michalik L., Desvergne B., Wahli W. (2006) From molecular action to physiological outputs: peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 45, 120–159 [DOI] [PubMed] [Google Scholar]
- 2. Tontonoz P., Spiegelman B. M. (2008) Fat and beyond: the diverse biology of PPARγ. Annu. Rev. Biochem. 77, 289–312 [DOI] [PubMed] [Google Scholar]
- 3. Dubois M., Pattou F., Kerr-Conte J., Gmyr V., Vandewalle B., Desreumaux P., Auwerx J., Schoonjans K., Lefebvre J. (2000) Expression of peroxisome proliferator-activated receptor γ (PPARγ) in normal human pancreatic islet cells. Diabetologia 43, 1165–1169 [DOI] [PubMed] [Google Scholar]
- 4. Moibi J. A., Gupta D., Jetton T. L., Peshavaria M., Desai R., Leahy J. L. (2007) Peroxisome proliferator-activated receptor-γ regulates expression of PDX-1 and NKX6.1 in INS-1 cells. Diabetes 56, 88–95 [DOI] [PubMed] [Google Scholar]
- 5. Gupta D., Jetton T. L., Mortensen R. M., Duan S. Z., Peshavaria M., Leahy J. L. (2008) In vivo and in vitro studies of a functional peroxisome proliferator-activated receptor γ response element in the mouse pdx-1 promoter. J. Biol. Chem. 283, 32462–32470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gupta D., Peshavaria M., Monga N., Jetton T. L., Leahy J. L. (2010) Physiologic and pharmacologic modulation of glucose-dependent insulinotropic polypeptide (GIP) receptor expression in β-cells by peroxisome proliferator-activated receptor (PPAR)-γ signaling: possible mechanism for the GIP resistance in type 2 diabetes. Diabetes 59, 1445–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jitrapakdee S., Slawik M., Medina-Gomez G., Campbell M., Wallace J. C., Sethi J. K., O'rahilly S., Vidal-Puig A. J. (2005) The peroxisome proliferator-activated receptor-γ regulates murine pyruvate carboxylase gene expression in vivo and in vitro. J. Biol. Chem. 280, 27466–27476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Delghingaro-Augusto V., Nolan C. J., Gupta D., Jetton T. L., Latour M. G., Peshavaria M., Madiraju S. R., Joly E., Peyot M. L., Prentki M., Leahy J. (2009) Islet β cell failure in the 60% pancreatectomised obese hyperlipidaemic Zucker fatty rat: severe dysfunction with altered glycerolipid metabolism without steatosis or a falling β cell mass. Diabetologia 52, 1122–1132 [DOI] [PubMed] [Google Scholar]
- 9. Yki-Järvinen H. (2004) Thiazolidinediones. N. Engl. J. Med. 351, 1106–1118 [DOI] [PubMed] [Google Scholar]
- 10. Leahy J. L. (2009) Thiazolidinediones in prediabetes and early type 2 diabetes: what can be learned about that disease's pathogenesis. Curr. Diab. Rep. 9, 215–220 [DOI] [PubMed] [Google Scholar]
- 11. Armoni M., Harel C., Karni S., Chen H., Bar-Yoseph F., Ver M. R., Quon M. J., Karnieli E. (2006) FOXO1 represses peroxisome proliferator-activated receptor-gamma1 and -gamma2 gene promoters in primary adipocytes. A novel paradigm to increase insulin sensitivity. J. Biol. Chem. 281, 19881–19891 [DOI] [PubMed] [Google Scholar]
- 12. Fan W., Imamura T., Sonoda N., Sears D. D., Patsouris D., Kim J. J., Olefsky J. M. (2009) FoxO1 transrepresses peroxisome proliferator-activated receptor γ transactivation, coordinating an insulin-induced feed-forward response in adipocytes. J. Biol. Chem. 284, 12188–12197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Okamoto H., Hribal M. L., Lin H. V., Bennett W. R., Ward A., Accili D. (2006) Role of the forkhead protein FoxO1 in β cell compensation to insulin resistance. J. Clin. Invest. 116, 775–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kitamura T., Ido Kitamura Y. (2007) Role of FoxO proteins in pancreatic β cells. Endocr. J. 54, 507–515 [DOI] [PubMed] [Google Scholar]
- 15. Kitamura T., Kitamura Y. I., Kobayashi M., Kikuchi O., Sasaki T., Depinho R. A., Accili D. (2009) Regulation of pancreatic juxtaductal endocrine cell formation by FoxO1. Mol. Cell. Biol. 29, 4417–4430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Al-Masri M., Krishnamurthy M., Li J., Fellows G. F., Dong H. H., Goodyer C. G., Wang R. (2010) Effect of forkhead box O1 (FoxO1) on β cell development in the human fetal pancreas. Diabetologia 53, 699–711 [DOI] [PubMed] [Google Scholar]
- 17. Kitamura T., Nakae J., Kitamura Y., Kido Y., Biggs W. H., 3rd, Wright C. V., White M. F., Arden K. C., Accili D. (2002) The forkhead transcription factor FoxO1 links insulin signaling to Pdx1 regulation of pancreatic β cell growth. J. Clin. Invest. 110, 1839–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao Y., Wang Y., Zhu W. G. (2011) Applications of post-translational modifications of FoxO family proteins in biological functions. J. Mol. Cell. Biol. 3, 276–282 [DOI] [PubMed] [Google Scholar]
- 19. Kim S. J., Winter K., Nian C., Tsuneoka M., Koda Y., McIntosh C. H. (2005) Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic β-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor FoxO1, and down-regulation of Bax expression. J. Biol. Chem. 280, 22297–22307 [DOI] [PubMed] [Google Scholar]
- 20. Buteau J., Spatz M. L., Accili D. (2006) Transcription factor FoxO1 mediates glucagon-like peptide-1 effects on pancreatic β-cell mass. Diabetes 55, 1190–1196 [DOI] [PubMed] [Google Scholar]
- 21. Qiang L., Banks A. S., Accili D. (2010) Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J. Biol. Chem. 285, 27396–27401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakae J., Biggs W. H., 3rd, Kitamura T., Cavenee W. K., Wright C. V., Arden K. C., Accili D. (2002) Regulation of insulin action and pancreatic β-cell function by mutated alleles of the gene encoding forkhead transcription factor FoxO1. Nat. Genet. 32, 245–253 [DOI] [PubMed] [Google Scholar]
- 23. Liu Y. Q., Han J., Epstein P. N., Long Y. S. (2005) Enhanced rat β-cell proliferation in 60% pancreatectomized rats by increased glucose metabolic flux through pyruvate carboxylase. Am. J. Physiol. Endocrinol. Metab. 288, E471–478 [DOI] [PubMed] [Google Scholar]
- 24. Jetton T. L., Everill B., Lausier J., Roskens V., Habibovic A., LaRock K., Gokin A., Peshavaria M., Leahy J. L. (2008) Enhanced β-cell mass without increased proliferation following chronic mild glucose infusion. Am. J. Physiol. Endocrinol. Metab. 294, E679–687 [DOI] [PubMed] [Google Scholar]
- 25. Barthel A., Schmoll D., Unterman T. G. (2005) FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 16, 183–189 [DOI] [PubMed] [Google Scholar]
- 26. Jetton T. L., Lausier J., LaRock K., Trotman W. E., Larmie B., Habibovic A., Peshavaria M., Leahy J. L. (2005) Mechanisms of compensatory β-cell growth in insulin-resistant rats: roles of Akt kinase. Diabetes 54, 2294–2304 [DOI] [PubMed] [Google Scholar]
- 27. Liu Y. Q., Jetton T. L., Leahy J. L. (2002) β-Cell adaptation to insulin resistance: increased pyruvate carboxylase and malate-pyruvate shuttle activity in islets of nondiabetic Zucker fatty rats. J. Biol. Chem. 277, 39163–39168 [DOI] [PubMed] [Google Scholar]
- 28. MacDonald M. J., Efendić S., Ostenson C. G. (1996) Normalization by insulin treatment of low mitochondrial glycerol phosphate dehydrogenase and pyruvate carboxylase in pancreatic islets of the GK rat. Diabetes 45, 886–890 [DOI] [PubMed] [Google Scholar]
- 29. MacDonald M. J., Tang J., Polonsky K. S. (1996) Low mitochondrial glycerol phosphate dehydrogenase and pyruvate carboxylase in pancreatic islets of Zucker diabetic fatty rats. Diabetes 45, 1626–1630 [DOI] [PubMed] [Google Scholar]
- 30. MacDonald M. J., Longacre M. J., Langberg E. C., Tibell A., Kendrick M. A., Fukao T., Ostenson C. G. (2009) Decreased levels of metabolic enzymes in pancreatic islets of patients with type 2 diabetes. Diabetologia 52, 1087–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson J. D., Ahmed N. T., Luciani D. S., Han Z., Tran H., Fujita J., Misler S., Edlund H., Polonsky K. S. (2003) Increased islet apoptosis in PDX1+/− mice. J. Clin. Invest. 111, 1147–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kulkarni R. N., Jhala U. S., Winnay J. N., Krajewski S., Montminy M., Kahn C. R. (2004) PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J. Clin. Invest. 114, 828–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brissova M., Blaha M., Spear C., Nicholson W., Radhika A., Shiota M., Charron M. J., Wright C. V., Powers A. C. (2005) Reduced PDX-1 expression impairs islet response to insulin resistance and worsens glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 288, E707–714 [DOI] [PubMed] [Google Scholar]
- 34. Khoo C., Yang J., Weinrott S. A., Kaestner K. H., Naji A., Schug J., Stoffers D. A. (2012) Research resource: the pdx1 cistrome of pancreatic islets. Mol. Endocrinol. 26, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Porat S., Weinberg-Corem N., Tornovsky-Babaey S., Schyr-Ben-Haroush R., Hija A., Stolovich-Rain M., Dadon D., Granot Z., Ben-Hur V., White P., Girard C. A., Karni R., Kaestner K. H., Ashcroft F. M., Magnuson M. A., Saada A., Grimsby J., Glaser B., Dor Y. (2011) Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab. 13, 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. MacDonald M. J. (1993) Glucose enters mitochondrial metabolism via both carboxylation and decarboxylation of pyruvate in pancreatic islets. Metabolism 42, 1229–1231 [DOI] [PubMed] [Google Scholar]
- 37. Jitrapakdee S., Wutthisathapornchai A., Wallace J. C., MacDonald M. J. (2010) Regulation of insulin secretion: role of mitochondrial signalling. Diabetologia 53, 1019–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu Y. Q., Moibi J. A., Leahy J. L. (2004) Chronic high glucose lowers pyruvate dehydrogenase activity in islets through enhanced production of long chain acyl-CoA: prevention of impaired glucose oxidation by enhanced pyruvate recycling through the malate-pyruvate shuttle. J. Biol. Chem. 279, 7470–7475 [DOI] [PubMed] [Google Scholar]
- 39. Xu J., Han J., Long Y.S., Epstein P. N., Liu Y. Q. (2008) The role of pyruvate carboxylase in insulin secretion and proliferation in rat pancreatic γ cells. Diabetologia 51, 2022–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cline G. W. (2011) Fuel-stimulated insulin secretion depends on mitochondrial activation and the integration of mitochondrial and cytosolic substrate cycles. Diabetes Metab. 35, 458–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miyawaki K., Yamada Y., Yano H., Niwa H., Ban N., Ihara Y., Kubota A., Fujimoto S., Kajikawa M., Kuroe A., Tsuda K., Hashimoto H., Yamashita T., Jomori T., Tashiro F., Miyazaki J., Seino Y. (1999) Glucose intolerance caused by a defect in the entero-insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proc. Natl. Acad. Sci. U.S.A. 96, 14843–14847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saxena R., Hivert M. F., Langenberg C., Tanaka T., Pankow J. S., Vollenweider P., Lyssenko V., Bouatia-Naji N., Dupuis J., Jackson A. U., Kao W. H., Li M., Glazer N. L., Manning A. K., Luan J., Stringham H. M., Prokopenko I., Johnson T., Grarup N., Boesgaard T. W., Lecoeur C., Shrader P., O'Connell J., Ingelsson E., Couper D. J., Rice K., Song K., Andreasen C. H., Dina C., Köttgen A., Le Bacquer O., Pattou F., Taneera J., Steinthorsdottir V., Rybin D., Ardlie K., Sampson M., Qi L., van Hoek M., Weedon M. N., Aulchenko Y. S., Voight B. F., Grallert H., Balkau B., Bergman R. N., Bielinski S. J., Bonnefond A., Bonnycastle L. L., Borch-Johnsen K., Böttcher Y., Brunner E., Buchanan T. A., Bumpstead S. J., Cavalcanti-Proença C., Charpentier G., Chen Y. D., Chines P. S., Collins F. S., Cornelis M. J., Crawford G., Delplanque J., Doney A., Egan J. M., Erdos M. R., Firmann M., Forouhi N. G., Fox C. S., Goodarzi M. O., Graessler J., Hingorani A., Isomaa B., Jørgensen T., Kivimaki M., Kovacs P., Krohn K., Kumari M., Lauritzen T., Lévy-Marchal C., Mayor V., McAteer J. B., Meyre D., Mitchell B. D., Mohlke K. L., Morken M. A., Narisu N., Palmer C. N., Pakyz R., Pascoe L., Payne F., Pearson D., Rathmann W., Sandbaek A., Sayer A. A., Scott L. J., Sharp S. J., Sijbrands E., Singleton A., Siscovick D. S., Smith N. L., Sparsø T., Swift A. J., Syddall H., Thorleifsson G., Tönjes A., Tuomi T., Tuomilehto J., Valle T. T., Waeber G., Walley A., Waterworth D. M., Zeggini E., Zhao J. H.; GIANT consortium; MAGIC investigators, Illig T., Wichmann H. E., Wilson J. F., van Duijn C., Hu F. B., Morris A. D., Frayling T. M., Hattersley A. T., Thorsteinsdottir U., Stefansson K., Nilsson P., Syvänen A. C., Shuldiner A. R., Walker M., Bornstein S. R., Schwarz P., Williams G. H., Nathan D. M., Kuusisto J., Laakso M., Cooper C., Marmot M., Ferrucci L., Mooser V., Stumvoll M., Loos R. J., Altshuler D., Psaty B. M., Rotter J. I., Boerwinkle E., Hansen T., Pedersen O., Florez J. C., McCarthy M. I., Boehnke M., Barroso I., Sladek R., Froguel P., Meigs J. B., Groop L., Wareham N. J., Watanabe R. M. (2010) Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat. Genet. 42, 142–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Talchai C., Xuan S., Lin H. V., Sussel L., Accili D. (2012) Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150, 1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zangen D. H., Bonner-Weir S., Lee C. H., Latimer J. B., Miller C. P., Habener J. F., Weir G. C. (1997) Reduced insulin, GLUT2, and IDX-1 in β-cells after partial pancreatectomy. Diabetes 46, 258–264 [DOI] [PubMed] [Google Scholar]
- 45. Yang B. T., Dayeh T. A., Volkov P. A., Kirkpatrick C. L., Malmgren S., Jing X., Renström E., Wollheim C. B., Nitert M. D., Ling C. (2012) Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol. Endocrinol. 26, 1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shu L., Matveyenko A. V., Kerr-Conte J., Cho J. H., McIntosh C. H., Maedler K. (2009) Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with down-regulation of GIP- and GLP-1 receptors and impaired β-cell function. Hum. Mol. Genet. 18, 2388–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kluth O., Mirhashemi F., Scherneck S., Kaiser D., Kluge R., Neschen S., Joost H. G., Schürmann A. (2011) Dissociation of lipotoxicity and glucotoxicity in a mouse model of obesity associated diabetes: role of forkhead box O1 (FoxO1) in glucose-induced β cell failure. Diabetologia 54, 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Folli F., Okada T., Perego C., Gunton J., Liew C. W., Akiyama M., D'Amico A., La Rosa S., Placidi C., Lupi R., Marchetti P., Sesti G., Hellerstein M., Perego L., Kulkarni R. N. (2011) Altered insulin receptor signalling and β-cell cycle dynamics in type 2 diabetes mellitus. PLoS One 6, e28050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yan L., Guo S., Brault M., Harmon J., Robertson R. P., Hamid R., Stein R., Yang E. (2012) The B55α-containing PP2A holoenzyme dephosphorylates FoxO1 in islet β-cells under oxidative stress. Biochem. J. 444, 239–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rosen E. D., Kulkarni R. N., Sarraf P., Ozcan U., Okada T., Hsu C. H., Eisenman D., Magnuson M. A., Gonzalez F. J., Kahn C. R., Spiegelman B. M. (2003) Targeted elimination of peroxisome proliferator-activated receptor γ in β cells leads to abnormalities in islet mass without compromising glucose homeostasis. Mol. Cell. Biol. 23, 7222–7229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Welters H. J., El Ouaamari A., Kawamori D., Meyer J., Hu J., Smith D. M., Kulkarni R. N. (2012) Rosiglitazone promotes PPARγ-dependent and -independent alterations in gene expression in mouse islets. Endocrinology 153, 4593–4599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ivashchenko C. Y., Duan S. Z., Usher M. G., Mortensen R. M. (2007) PPAR-γ knockout in pancreatic epithelial cells abolishes the inhibitory effect of rosiglitazone on caerulein-induced acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G319–326 [DOI] [PubMed] [Google Scholar]
- 53. Kim H. I., Kim J. W., Kim S. H., Cha J. Y., Kim K. S., Ahn Y. H. (2000) Identification and functional characterization of the peroxisomal proliferator response element in rat GLUT2 promoter. Diabetes 49, 1517–1524 [DOI] [PubMed] [Google Scholar]
- 54. Kim H. I., Cha J. Y., Kim S. Y., Kim J. W., Roh K. J., Seong J. K., Lee N. T., Choi K. Y., Kim K. S., Ahn Y. H. (2002) Peroxisomal proliferator-activated receptor-γ up-regulates glucokinase gene expression in β-cells. Diabetes 51, 676–685 [DOI] [PubMed] [Google Scholar]
- 55. Kim H. S., Hwang Y. C., Koo S. H., Park K. S., Lee M. S., Kim K. W., Lee M. K. (2013) PPAR-γ activation increases insulin secretion through the up-regulation of the free fatty acid receptor GPR40 in pancreatic β-cells. PLoS One 8, e50128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Evans-Molina C., Robbins R. D., Kono T., Tersey S. A., Vestermark G. L., Nunemaker C. S., Garmey J. C., Deering T. G., Keller S. R., Maier B., Mirmira R. G. (2009) Peroxisome proliferator-activated receptor γ activation restores islet function in diabetic mice through reduction of endoplasmic reticulum stress and maintenance of euchromatin structure. Mol. Cell. Biol. 29, 2053–2067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kono T., Ahn G., Moss D. R., Gann L., Zarain-Herzberg A., Nishiki Y., Fueger P. T., Ogihara T., Evans-Molina C. (2012) PPAR-γ activation restores pancreatic islet SERCA2 levels and prevents β-cell dysfunction under conditions of hyperglycemic and cytokine stress. Mol. Endocrinol. 26, 257–271 [DOI] [PMC free article] [PubMed] [Google Scholar]