

Background: Significance of GPI-anchored protein enrichment in lipid rafts, which requires PGAP3-mediated structural remodeling of GPI, remains unclear.
Results: Reduced apoptotic cell clearance could be causal for autoimmunity occurring in PGAP3−/− mice.
Conclusion: GPI-anchored protein enrichment in lipid rafts has a significant role in immunity.
Significance: Analyzing PGAP3−/− mice will reveal biological significance of GPI-anchored protein enrichment in lipid rafts.
Keywords: Autoimmunity, Glycosylphosphatidylinositol Anchors, Lipid Raft, Lipid Structure, Phagocytosis, PGAP3
Abstract
Glycosylphosphatidylinositols (GPI) are complex glycolipids that are covalently linked to the C terminus of proteins as a post-translational modification and tether proteins to the plasma membrane. One of the most striking features of GPI-anchored proteins (APs) is their enrichment in lipid rafts. The biosynthesis of GPI and its attachment to proteins occur in the endoplasmic reticulum. In the Golgi, GPI-APs are subjected to fatty acid remodeling, which replaces an unsaturated fatty acid at the sn-2 position of the phosphatidylinositol moiety with a saturated fatty acid. We previously reported that fatty acid remodeling is critical for the enrichment of GPI-APs in lipid rafts. To investigate the biological significance of GPI-AP enrichment in lipid rafts, we generated a PGAP3 knock-out mouse (PGAP3−/−) in which fatty acid remodeling of GPI-APs does not occur. We report here that a significant number of aged PGAP3−/− mice developed autoimmune-like symptoms, such as increased anti-DNA antibodies, spontaneous germinal center formation, and enlarged renal glomeruli with deposition of immune complexes and matrix expansion. A possible cause for this was the impaired engulfment of apoptotic cells by resident peritoneal macrophages in PGAP3−/− mice. Mice with conditional targeting of PGAP3 in either B or T cells did not develop such autoimmune-like symptoms. In addition, PGAP3−/− mice exhibited the tendency of Th2 polarization. These data demonstrate that PGAP3-dependent fatty acid remodeling of GPI-APs has a significant role in the control of autoimmunity, possibly by the regulation of apoptotic cell clearance and Th1/Th2 balance.
Introduction
Hundreds of proteins are tethered to the cell surface membrane via a glycolipid structure called the GPI2 anchor in all eukaryotes, ranging from protozoa and fungi to mammals. All protein-linked GPI anchors share a common core structure, consisting of inositol phospholipid, glucosamine, mannose, and ethanolamine phosphate. GPI is synthesized and transferred to proteins in the endoplasmic reticulum. GPI-anchored proteins (GPI-APs) are then transported from the endoplasmic reticulum to the plasma membrane through the Golgi apparatus. The structure of the lipid part of the GPI anchor undergoes remodeling, which regulates the trafficking and localization of GPI-APs. Different remodeling processes occur in the endoplasmic reticulum and Golgi apparatus. Recently, the genes required for GPI remodeling were identified in mammalian cells (1). One type of remodeling is GPI fatty acid remodeling, in which an unsaturated fatty acid at the sn-2 position of the lipid is replaced with a saturated fatty acid, usually stearic acid (C18:0). This reaction occurs in the Golgi apparatus in mammalian cells. Post-GPI attachment to proteins 3 (PGAP3, a homologue of yeast Per1p) is involved in the removal of the unsaturated fatty acid (2). PGAP2 is involved in the addition of the saturated fatty acid at the sn-2 position (3).
At the plasma membrane, microdomains, termed lipid rafts, enrich cholesterol and sphingolipids that self-associate to form liquid-ordered structures with weak fluidity (4). The packing of lipids forms lipid rafts and allows them to be isolated as unique membrane fractions that are insoluble in nonionic detergents, termed detergent-resistant membrane (DRM) (5). GPI-APs are also enriched in lipid rafts and extracted in DRM. In the absence of PGAP3-dependent fatty acid remodeling, GPI-APs with an unsaturated chain are still expressed on the cell surface, but those proteins examined so far, such as CD55 (DAF), CD59, CD87 (urokinase-type plasminogen activator receptor), Thy-1, and HA-tagged placental alkaline phosphatase, are no longer enriched in DRM, indicating that fatty acid remodeling is critical for the enrichment of GPI-APs in lipid rafts (2, 6, 7). Conversely, the lack of GPI-APs in lipid rafts due to either defective GPI biosynthesis or PGAP3 function is not thought to affect the formation of lipid raft, based on the correct distribution of marker proteins such as caveolin, flotillin, and LAT into DRM (2, 6–8).
Lipid rafts are thought to function as platforms that sequestrate specific proteins and introduce and modulate signaling. This suggests that GPI-APs may also use raft-specific molecules to induce signaling, because GPI-APs lack intracellular domains (9). Indeed, several reports showed that cross-linking GPI-APs by antibodies could induce active or inhibitory signals intracellularly and that the GPI anchor was crucial for these signaling events. This indicated that enrichment of GPI-APs in the lipid rafts is crucial for their functions (10–12). To analyze the significance of GPI-AP enrichment in lipid rafts, cholesterol depletion is widely used to destroy lipid rafts, but this procedure simultaneously affects physiological membrane properties and is difficult to apply to in vivo studies. The replacement of the GPI anchor with a transmembrane peptide is another way to reduce the affinity of GPI-APs to lipid rafts, but only a few molecules can be examined in one study, and application for in vivo studies is also difficult. To overcome these problems, we generated PGAP3 knock-out mice. In these mice, GPI-APs do not undergo lipid remodeling and localize outside lipid rafts (6). PGAP3−/− mice exhibited minor morphological abnormalities such as short heads and kinked tails, abnormal reflexes such as limb grasping, and growth retardation (6). Homozygous males and females were fertile. Female PGAP3−/− mice were born normally according to Mendel's law, although fewer male PGAP3−/− mice were obtained for unknown reasons. Our previous report focused on T cell functions in PGAP3−/− mice and found that T cell development in the absence of PGAP3 was normal, but in vitro and in vivo T cell responses were enhanced, including alloreactive and antigen-specific immune responses (6). We followed PGAP3 knock-out mice over a long period and observed they tended to develop autoimmune symptoms. Here, we report that GPI-AP enrichment in lipid rafts induced by PGAP3-dependent fatty acid remodeling of the GPI anchor has a significant role in the control of autoimmunity possibly by regulating apoptotic cell clearance and the Th1/Th2 balance.
EXPERIMENTAL PROCEDURES
Sensitivity to Cold 1% Triton X-100
DRM were fractionated as described previously (12). Briefly, resident peritoneal macrophages (1 × 107) were lysed in cold buffer containing 1% Triton X-100. After centrifugation, supernatants were removed (S fractions, 1% Triton X-100-soluble fractions), and pellets were further solubilized in a buffer containing 60 mm n-octyl-β-d-glucoside (R fraction, 1% Triton X-100-resistant fractions). Thy-1 in the R and S fractions was analyzed by SDS-PAGE and immunoblotting. CD14 and transferrin receptor were detected using anti-mouse CD14 (BD Biosciences) and anti-transferrin receptor (Invitrogen), respectively.
Measurement of Autoantibodies in Sera
Mouse sera were diluted, and double-stranded DNA (dsDNA) or single-stranded DNA (ssDNA)-specific antibodies were quantified using a double-sandwich ELISA kit (Shibayagi, Shibukawa, Japan). The absorbance at 450 nm (620 nm as a reference wave length) was measured with an automated Micro-ELISA reader.
In Vitro Phagocytosis Assay
In vitro phagocytosis was performed as described previously (13). In brief, thymocytes (1 × 106 cells) from BALB/c mice younger than 12 weeks of age were incubated at 37 °C with 10 μm dexamethasone to induce apoptosis (14) and added to resident peritoneal macrophages (2.5 × 105 cells) cultured in 15 μ-slide 8 well chambers (ibidi, Verona, WI). After coculture for 1.5 h, the macrophages were thoroughly washed to remove surface-bound thymocytes, fixed, subjected to the TUNEL reaction, and observed by light microscopy. TUNEL staining was performed using an in situ cell death detection kit, fluorescein (Roche Applied Science). TUNEL-positive thymocytes were counted, and the phagocytosis index was determined as the number of TUNEL-positive apoptotic cells per macrophage. At least 150 macrophages per mouse were tested.
Immunohistochemical Analyses
For hematoxylin and eosin staining or periodic acid-Schiff staining, mouse tissues were fixed in 10% paraformaldehyde, 4% sucrose in 0.1 m phosphate buffer (pH 7.2), embedded in paraffin, and sectioned at 2 μm. For immunohistochemical analysis, frozen tissues were embedded in OCT compound (Sakura, Tokyo, Japan) and were cut on a cryostat to 8-μm-thick longitudinal sections and then fixed in 4% paraformaldehyde. Nonspecific binding was blocked with 3% fetal bovine serum (Thermo). To detect germinal centers (GC) in spleen, spleen sections were double-stained with anti-mouse B220 antibody conjugated with FITC (BD Biosciences) and biotinylated peanut agglutinin (PNA) (Vector Laboratories, Burlingame, CA), followed by Alexa594-conjugated streptavidin (Invitrogen). To detect the precipitation of immunocomplexes, frozen sections of kidney were stained with FITC-APure F(ab′) fragment of donkey anti-mouse IgG (H+L) and with FITC-conjugated donkey anti-rabbit IgG antibody (EMD Millipore, Billerica, MA) as control. To detect phagocytosis of apoptotic cells, macrophages were stained with rat anti-mouse CD68 (Serotec, Kidlington, UK), followed by Alexa594-conjugated streptavidin. TUNEL staining was performed using an In situ cell death detection kit, fluorescein (Roche Applied Science). Stained sections were mounted with Fluoromount (Diagnostic BioSystems, Pleasanton, CA) and observed by fluorescence microscopy (Olympus FLUOVIEW FV1000).
Intracellular Cytokine Staining
Splenocytes (5 × 106 cells in 2 ml) were cultured in 24-well plates (Iwaki) for 6 days with anti-CD3/anti-CD28. Splenocytes were harvested and stimulated with phorbol myristate acetate (50 ng/ml, Sigma) and ionomycin (2 μm, Sigma) in the presence of GolgiPlugTM (BD Biosciences) protein transport inhibitor containing brefeldin A for 5 h at 37 °C in a 5% CO2-humidified atmosphere. After stimulation, cells were harvested and stained with allophycocyanin (APC)-conjugated anti-mouse CD4 (BioLegend, San Diego). After washing with staining buffer (phosphate-buffered saline with 1% FBS and 0.09% NaN3), cells were fixed and permeabilized using a Cytofix/Cytoperm Plus Fixation/Permeabilization kit (BD Biosciences) and intracellularly stained with PE-conjugated anti-IL-4, PE-conjugated rat IgG1κ isotype, Alexa488-conjugated anti-IFN-γ, or Alexa488-conjugated rat IgG1κ Isotype (BioLegend). Cellular populations were analyzed on a flow cytometer (BD FACSCantoTM II; BD Biosciences) with FlowJo software (Treestar, Ashland, OR).
Staining of Regulatory T Cells
Splenocytes were stained with Mouse Treg FlowTM kit (FOXP3 Alexa Fluor® 488/CD4 APC/CD25 PE) (BioLegend).
Activation-induced Cell Death (AICD) Assay
AICD was determined by restimulation assay. Splenocytes were cultured in 24-well plates precoated with 10 μg/ml anti-CD3 and 2 μg/ml anti-CD28 for 2 days. The cultures were supplemented with 10 ng/ml IL-2 on day 3. Live cells were purified by density gradient centrifugation with Ficoll-PaqueTM PLUS (GE Healthcare) and then either left unstimulated or restimulated with anti-CD3 on day 6. After 16 h, cells were stained with APC-conjugated anti-TCRβ (eBioscience, San Diego), anti-CD4, or anti-CD8 mAbs (BioLegend). Apoptosis was determined by staining nuclear DNA using In situ cell death detecting kit, Fluorescein (Roche Applied Science). Cellular populations were analyzed on a flow cytometer with FlowJo software.
B Cell Proliferation Assay
B cells were freshly prepared from splenocytes using MACS B cell isolation kit (Miltenyi Biotec, Auburn, CA). B cells (0.5 × 105/well) were cultured in a 96-well plate for 48 h in the presence of LPS, anti-IgM, anti-CD40, or LPS + IL-4 for B cell proliferation assays. Cultures were pulsed with 2 μCi of methyl[3H]thymidine for the last 16 h of the culture period, and [3H]thymidine incorporation was measured. B cells were cultured in RPMI 1640 medium supplemented with 10% FCS, 50 μm 2-mercaptoethanol, 2 mm l-glutamine, and antibiotics.
Mice and Immunization
PGAP3 knock-out (PGAP3−/−) mice were generated as described previously (6) and were maintained on a C57BL/6 genetic background by backcrossing to C57BL/6 for more than six generations. To generate heterozygous Lck-Cre/PGAP3+/flox and cd19-Cre/PGAP3+/flox mice, PGAP3+/flox mice were crossed with transgenic mice expressing Cre under the control of the proximal lck promoter (active in T cells) (15) or the cd19 promoter (active in B cells) (16). We generated homozygous T or B cell-specific knock-out mice by mating heterozygous Lck-Cre/PGAP3+/flox or cd19-Cre/PGAP3+/flox mice, and we used these mice for our studies. All animal experiments were carried out according to the guidelines for the Care and Use of Laboratory Animals at Osaka University. For every immunization, PGAP3+/− littermate mice were used as controls. PGAP3−/− mice (16–22 weeks old) were immunized intraperitoneally with 100 μg of (4-hydroxy-3-nitrophenyl)acetyl (NP)-conjugated chicken γ-globulin (Biosearch Technologies, Novato, CA) emulsified with alum as a T cell-dependent antigen and were rechallenged with 5 μg of T cell-dependent antigen without adjuvant 2 weeks after primary immunization. For T cell-independent antigen immunization, mice were injected intraperitoneally with 100 μg of NP-conjugated aminoethylcarboxymethyl (AECM)-Ficoll (Biosearch Technologies). Sera were collected each week after the first immunization and were used for ELISA analysis.
Measurement of Ig Concentration
To measure NP-specific antibody in sera, total and high affinity NP-specific antibodies were determined by ELISA using either 16 NP-haptenated BSA (NP16-BSA)- or 4 NP-haptenated BSA (NP4-BSA)-coated 96-well plates. Plates were incubated with blocking buffer and then with diluted sera and purified Ig as standards. After washing, plates were incubated with alkaline phosphatase-conjugated, affinity-purified polyclonal goat anti-mouse IgM and IgG1.
Statistical Analysis
Unpaired Student's t test or nonparametric Wilcoxon rank-sum test were performed for statistical analysis, and the details are shown in the figure legend. All tests were analyzed using JMP 7 version 7.0.1 software (SAS, Cary, NC). Values were considered significant when p < 0.05.
RESULTS
GPI-APs Expressed in PGAP3−/− Mice Were Not Enriched in the DRM Fraction
Previously, we reported that in PGAP3−/− mice T cell development was normal, but in vitro and in vivo T cell responses were enhanced, including alloreactive and antigen-specific immune responses (6). We continued observation of PGAP3 knock-out mice over a long period and found that they tended to develop autoantibodies (see below). Although it was widely accepted that GPI-APs in PGAP3-deficient cells lose their characteristic nature and that GPI-AP Thy-1 is not enriched in lipid rafts in T cells from PGAP3−/− mice (2, 6, 7), we re-confirmed that this is the case with another GPI-AP, CD14, in PGAP3−/− mice before going into the detailed analysis of the mechanisms. Peritoneal macrophages were collected, and the DRM fraction was separated with cold 1% Triton X-100. CD14 was mainly detected in the DRM fraction in cells from PGAP3+/+ and PGAP3+/− mice, whereas it was mainly detected in the non-DRM fraction in cells from PGAP3−/− mice (Fig. 1), confirming that in the absence of fatty acid remodeling catalyzed by PGAP3 GPI-APs are localized outside lipid rafts in primary cells.
FIGURE 1.
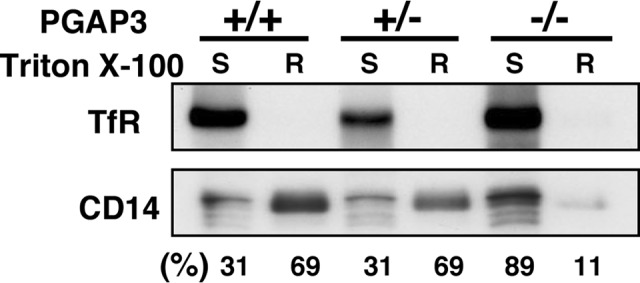
Fractionation of CD14 into Triton X-100-resistant (R) and -soluble (S) membranes. R and S fractions were analyzed by SDS-PAGE and immunoblotting. Transferrin receptor (TfR) was used as a marker for the detergent-soluble membrane fraction. Intensities of CD14 bands in R and S fractions were quantitatively measured by a CCD camera, and the each percentage of S to S plus R and R to S plus R was calculated. The result is representative of three independent experiments.
PGAP3−/− Mice Developed Anti-dsDNA and Anti-ssDNA Autoantibodies
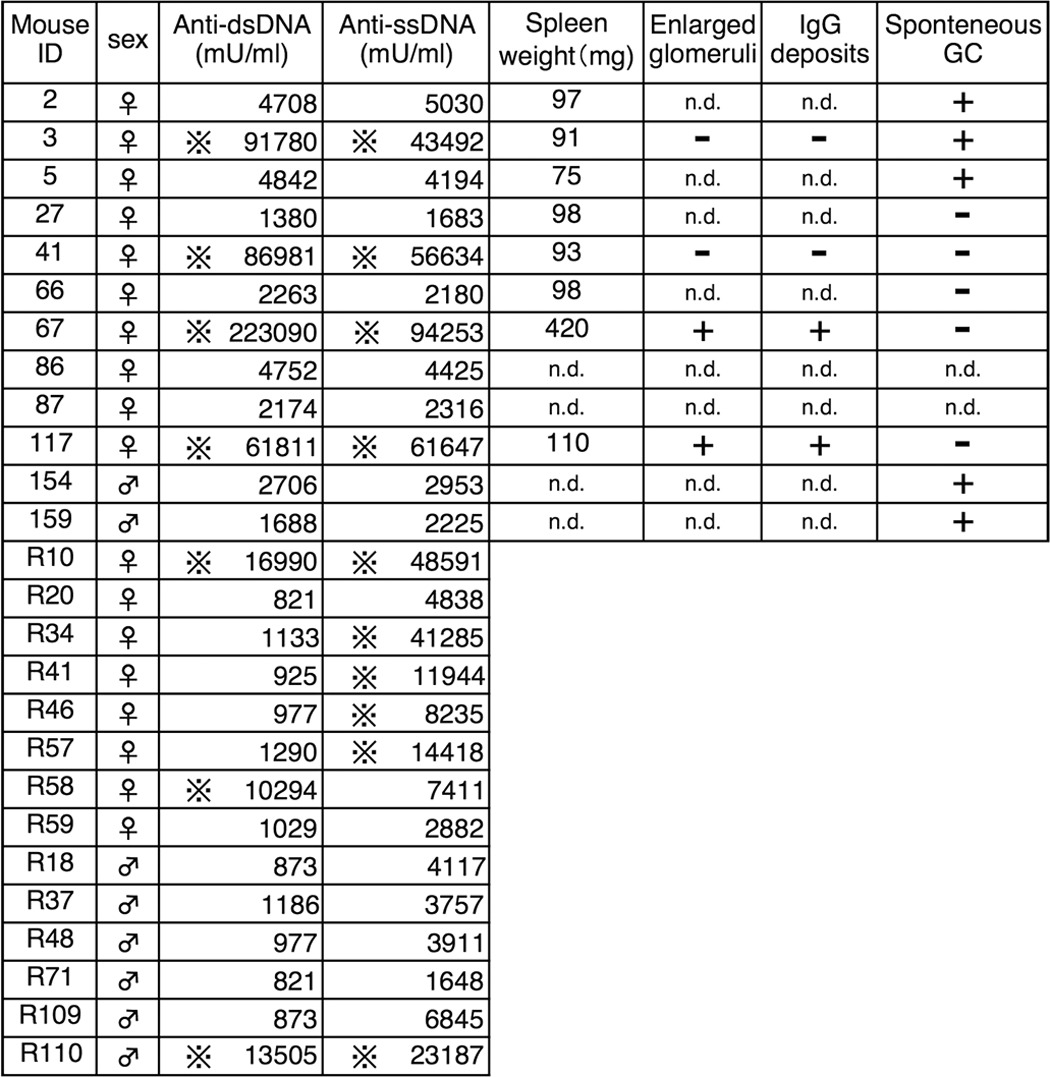
Because we wanted to understand what mechanisms caused enhanced T cells responses and how immune regulation was disturbed in PGAP3−/− mice, we measured the concentrations of autoantibodies to dsDNA or ssDNA in sera of mice 26–29 weeks of age as one of several screening experiments. Normal ranges were determined as the mean value ± 3 × S.D. calculated from values of PGAP3+/+ mice (dsDNA, 1.49 ± 3 × 1.23 units/ml; ssDNA, 3.33 ± 3 × 1.38 units/ml) (Fig. 2A and Table 1). Seven of 26 (7/26) PGAP3−/− mice had significantly high concentrations of anti-dsDNA, whereas 1/26 and 2/29 of age-matched PGAP3+/+ and PGAP3+/− mice had high values, respectively. Similarly, 10/26 PGAP3−/− mice had significantly high concentrations of anti-ssDNA, whereas 0/26 and 2/29 age-matched PGAP3+/+ and PGAP3+/− mice had high values, respectively. Six of 26 PGAP3−/− mice exhibited high levels of both anti-dsDNA and anti-ssDNA antibodies (0/26 in PGAP3+/+ and 1/29 in PGAP3+/− mice). In particular, higher onset rates of autoantibodies were observed in female mice (6/18 female mice versus 1/8 male mice for anti-dsDNA and 9/18 female mice versus 1/8 male mice for anti-ssDNA), which is often the case in autoimmune diseases (Table 1).
FIGURE 2.
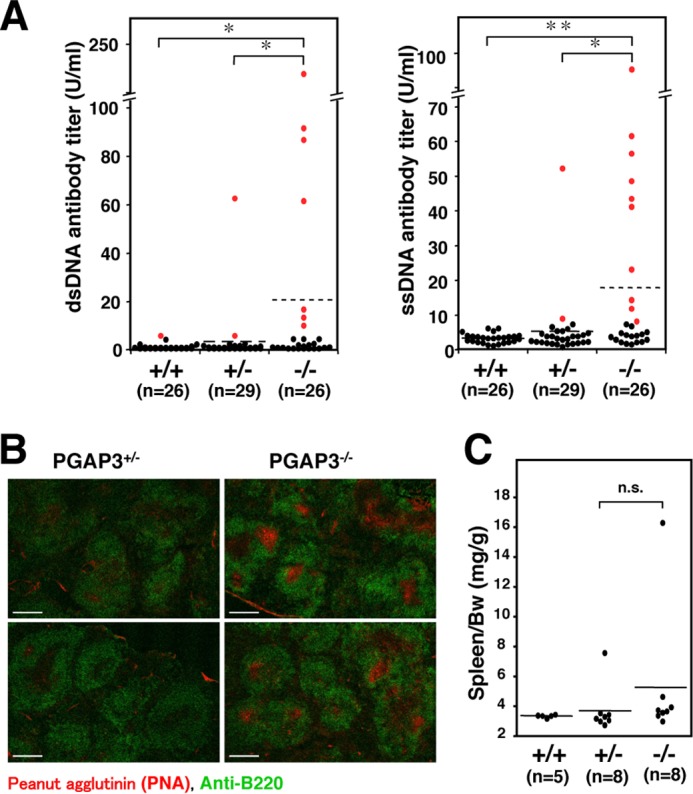
A significant number of aged PGAP3−/− mice developed autoantibodies and spontaneous GC formation. A, production of autoantibodies in aged PGAP3−/− mice. Titers of dsDNA-specific antibodies (left panel) and ssDNA-specific antibodies (right panel) in the serum of 26–29-week-old mice are shown as individual values (dot) or the mean (line). Normal ranges were determined as the mean value ± 3× S.D. The values above normal ranges are shown as red dots. *, p < 0.05; **, p < 0.005 (Wilcoxon rank sum test). B, immunohistochemical analysis of spleens. The spleen sections of 13-month-old PGAP3−/− and PGAP3+/− mice were stained with PNA (red) and antibody against B220 (green). Scale bar, 100 μm. The results are representative of two different PGAP3−/− mice and their controls. 5 of 10 PGAP3−/− mice examined had formed spontaneous GCs; in contrast, characteristic GCs were not observed in the 10 PGAP3+/− and 5 PGAP3+/+ mice examined. C, ratios of spleen to body weight are shown with the mean (line) and were analyzed using the Wilcoxon rank sum test. n.s., not significant.
TABLE 1.
Phenotypes of PGAP3−/− mice used in this study
The following symbols are used: × with four dots indicates over the normal range (mean ± 3× S.D.); plus indicates significant; minus indicates not significant; and n.d. means not determined.
Spontaneous GC Formation in PGAP3−/− Mice
The production of autoantibodies prompted us to examine whether spleen has any abnormality in PGAP3−/− mice. Sections from the spleens of mice at 13–15 months of age were prepared and double-stained with PNA and anti-B220 antibodies. The white pulps in spleens of PGAP3−/− mice were not significantly enlarged compared with PGAP3+/− mice, but 5 of 10 PGAP3−/− mice examined had formed spontaneous GCs that were stained with PNA (Fig. 2B and Table 1). In contrast, characteristic GCs were not observed in spleen sections from 10 PGAP3+/− and 5 PGAP3+/+ mice, although a small number of PNA+ cells were present in follicles (Fig. 2B). PGAP3−/− mice had not developed splenomegaly at the time of sacrifice (7 months or 13–15 months), and there was no significant statistical difference in spleen weight between PGAP3−/−, PGAP3+/−, and PGAP3+/+ mice (Fig. 2C). In addition, PGAP3−/− mice had no visible enlargement of inguinal, axillary, and mesenteric lymph nodes.
Enlarged Glomeruli with Deposition of Immunoglobulins in PGAP3−/− Mice
We next examined kidneys. The examination of kidney cryosections by periodic acid-Schiff staining revealed that two out of four 13–15-month-old PGAP3−/− mice that demonstrated high titers of anti-dsDNA and anti-ssDNA antibodies had statistically significant enlarged glomeruli (Fig. 3, A and B, and Table 1). In addition to the hypertrophy, pathological examination demonstrated that the space between the glomerular basement membrane and capillary endothelium was filled with diffused structures that indicated the matrix expansion in renal biopsies of PGAP3−/− mice, whereas the lumens remained clear in PGAP3+/− mice (Fig. 3A). Moreover, a significant deposition of immunoglobulins was observed in glomeruli of PGAP3−/− mice by immunofluorescence microscopy (Fig. 3C). Therefore, a significant number of aged PGAP3−/− mice developed symptoms similar to autoimmune disease.
FIGURE 3.

A significant number of aged PGAP3−/− mice developed enlarged glomeruli with deposition of immunoglobulins. A, kidney sections from 13-month-old PGAP3−/− and PGAP3+/− mice were stained with periodic acid-Schiff. Black lines delineate glomeruli. Scale bar, 100 μm. B, diameters of glomeruli were quantified. At least 30 glomeruli were measured for each mouse (upper panel). Nucleus numbers in each glomeruli were counted and quantified (lower panel). Each identification is indicated in PGAP3−/− mice (Table 1). +/+ and +/− were combined data of four PGAP3+/+ and PGAP3+/− mice, respectively. C, kidney sections from 13-month-old PGAP3+/− and PGAP3−/− mice were stained with FITC-conjugated antibodies anti-mouse IgG (H+L) or anti-rabbit IgG as a control (data not shown). Scale bar, 20 μm. The results of A–C are representative of four PGAP3−/− mice and their controls. **, p < 0.005 (Student's t test).
PGAP3−/− Mice Do Not Develop Hyper-reactive T Cell-dependent and T Cell-independent B Cell Responses
We then tried to reveal the mechanisms by which such autoimmune-like symptoms were caused. First, B cell development and responses were analyzed. In peripheral splenic B cells, percentages of T1, T2, and T3 B cell populations, mature and immature B cell populations, as well as marginal zone and follicular B cells were not different between PGAP3+/− and PGAP3−/− mice (data not shown). The ratios of pro-B to pre-B cells and pro-B plus pre-B to total B cells in the bone marrow were also normal in both groups of mice (data not shown). Thus, taken together with previous results that T cell development in the absence of PGAP3 was normal (6), PGAP3 deficiency did not appear to cause abnormal development of T and B cells. Next, we examined whether general T cell-dependent or -independent responses were impaired in PGAP3−/− mice. First, T cell-dependent B cell responses were analyzed. PGAP3+/− and PGAP3−/− mice were immunized with a hapten-protein carrier conjugate NP-conjugated chicken γ-globulin (17). Mice were bled, and anti-NP-specific serum antibody titers were assayed. Titers of total and high affinity anti-NP IgG1 were determined by their binding to NP16-BSA and NP4-BSA, respectively. Over the 6-week immunization period, there was no significant difference in levels of specific IgM and total or high affinity IgG1 anti-NP between PGAP3−/− and PGAP3+/− mice, although the values were slightly lower in PGAP3+/− mice. These results indicated that T cell-dependent B cell responses were not hyper-reactive (Fig. 4, A–C).
FIGURE 4.
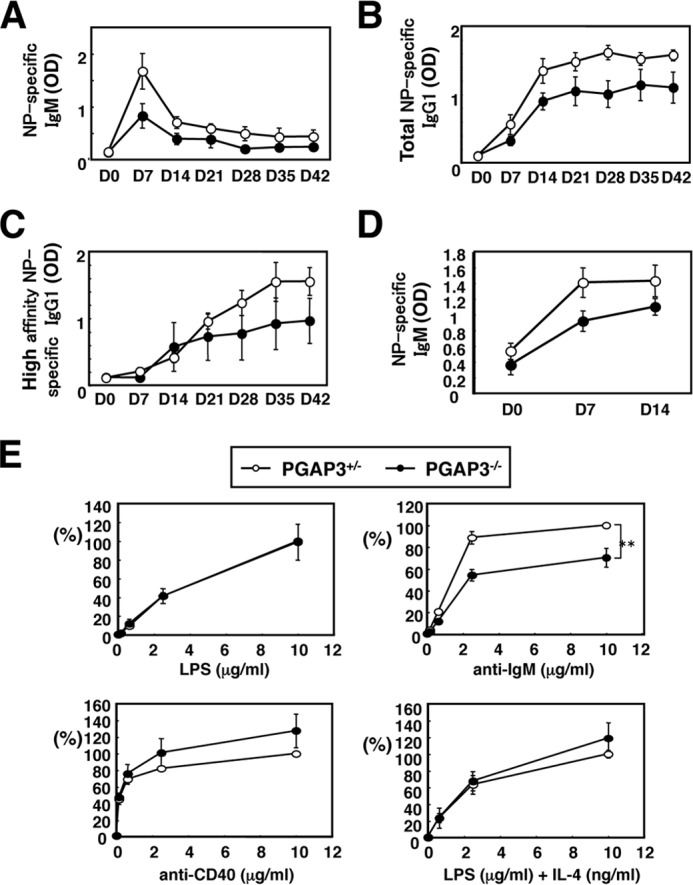
PGAP3−/− mice do not develop hyper-reactive T cell-dependent and T cell-independent B cell responses. A–C, T cell-dependent B cell responses are not hyper-reactive in PGAP3−/− mice. PGAP3+/− and PGAP3−/− mice were immunized with alum-precipitated NP-conjugated chicken γ-globulin. Serum levels of NP-specific IgM (A), total NP-specific IgG1 (B), or high affinity NP-specific IgG1 (C) were measured by ELISA. Each point represents the mean of four mice per genotype, and the mean values ± S.E. are shown. D, T cell-independent B cell responses were not hyper-reactive in PGAP3−/− mice. PGAP3+/− and PGAP3−/− mice were immunized with NP-AECM-Ficoll. Serum levels of NP-specific IgM were measured by ELISA. Each point represents the mean of four mice per genotype, and the mean values ± S.E. are shown. E, proliferation of B cells from PGAP3+/− and PGAP3−/− mice in response to various stimuli. Splenocytes from PGAP3−/− (filled circles) or PGAP3+/− (open circles) were freshly isolated and stimulated at 0.5 × 105/well for 48 h with LPS, anti-IgM, anti-CD40, or LPS + IL-4 at the indicated concentrations. Cell proliferation was analyzed by [3H]thymidine incorporation. Counts/min values of PGAP3+/− B cells under the highest stimuli dose were designated as 100%. The results represent the mean of four independent experiments. Error bars represent means ± S.E. (n = 4). **, p < 0.005 (Student's t test).
Next, T cell-independent B cell responses were analyzed by measuring NP-specific IgM production after immunization with NP-AECM-Ficoll. There was no difference in NP-specific IgM production between PGAP3+/− and PGAP3−/− mice (Fig. 4D). In addition, there was no significant difference in PGAP3+/− and PGAP3−/− mouse splenic B cell proliferation when stimulated in vitro by anti-CD40 antibodies or LPS in combination with or without IL-4. However, cross-linking of splenic B cells with anti-IgM antibodies induced a reduced proliferation of PGAP3−/− B cells, indicating that T cell-independent B cell responses were not hyper-reactive (Fig. 4E). Taken together, general B cell responses were not hyper-reactive, although the possibility that B cells were hyper-reactive to some specific antigens was not excluded.
PGAP3−/− Mice Exhibit a Tendency of Th2 Cell Differentiation
Next, we examined the populations and balance of Th1/Th2 cells, because overactivation of either Th1 or Th2 cells and/or skewed Th1/Th2 cytokine secretion can induce autoimmunity. Cytokine production patterns in CD4+ T cells demonstrated that IFNγ-producing Th1 but not IL-4-producing Th2 cells were reduced in PGAP3−/− mice, albeit we may need more mice to make the statistical significance more solid (Fig. 5, A and B). Thus, a tendency of Th2 cell differentiation was observed in PGAP3−/− mice (Fig. 5C).
FIGURE 5.
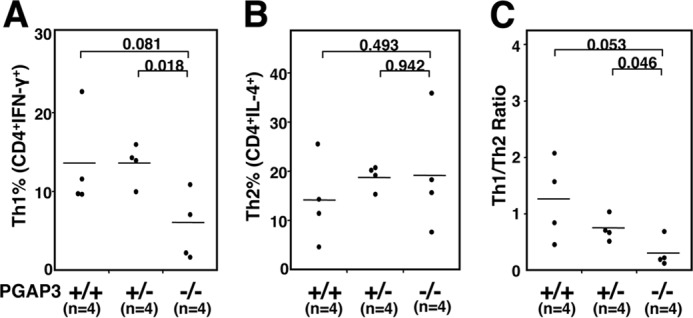
PGAP3−/− mice exhibit a bias toward Th2 cell differentiation. Naive T cells were primed for 6 days with anti-CD3/anti-CD28 and restimulated with 50 ng/ml phorbol myristate acetate and 2 μm ionomycin in the presence of brefeldin A for 5 h. Cells were fixed and stained with APC-conjugated anti-CD4, PE-conjugated anti-IL-4, and FITC-conjugated anti-IFN-γ antibodies after permeabilization. By gating on CD4-positive cells, percentages of lymphocytes stained with IFN-γ (A) and IL-4 (B) from PGAP3+/+, PGAP3+/−, and PGAP3−/− mice are shown. C, ratios of IFN-γ/IL-4 positive cells in CD4-positive cells from PGAP3−/− mice and their respective littermate controls are shown. The mean values are indicated by lines. Four PGAP3−/− mice that had high titers of both dsDNA- and ssDNA-specific antibodies (Mouse identification: 2, 3, 41, and 67 in Table 1), four PGAP3+/− mice and four PGAP3+/+ mice were examined. p values were determined by Student's t test.
AICD and CD4+CD25+ Regulatory T Cells Were Not Impaired in PGAP3−/− Mice
T cell apoptosis is believed to maintain homeostasis and self-tolerance in the immune system, and dysregulation of this system is also known to be causal for autoimmunity. Therefore, we investigated AICD in PGAP3−/− mice, but no difference in rates of apoptosis between PGAP3−/− and control lymphocytes was observed, suggesting that PGAP3−/− cells were not less susceptible to apoptosis than control cells (Fig. 6A).
FIGURE 6.
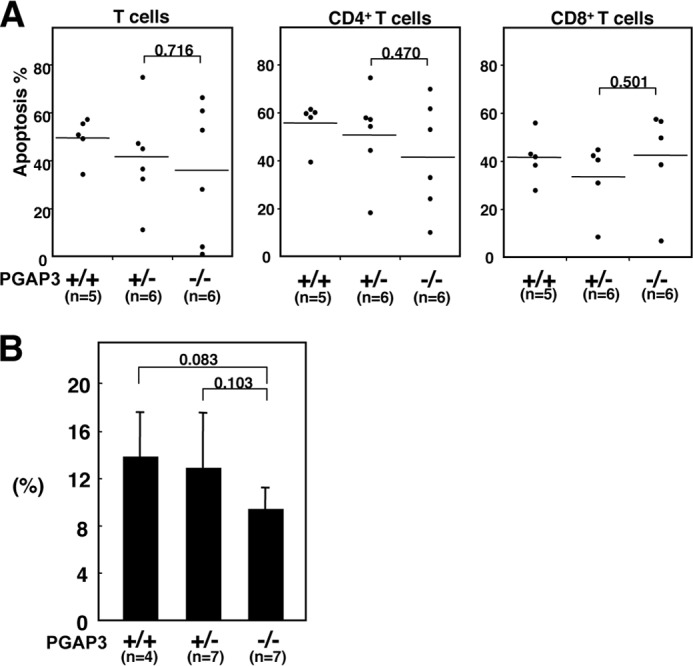
AICD and CD4+CD25+ regulatory T cells were not impaired in PGAP3−/− mice. A, AICD of cells from PGAP3−/− and control mice. Splenocytes were cultured in plates coated with anti-CD3 and anti-CD28 antibodies in the presence of recombinant IL-2 for 2 days. Apoptotic death was assessed by TUNEL staining 16 h after restimulation with anti-CD3 antibody on day 8. p values were determined by Student's t test. B, splenocyte cells were stained with Mouse Treg FlowTM kit. The ratio of CD4+CD25+FoxP3+ cells to CD4+T cells was quantified, and the mean value ± S.D. is shown. p values were determined by Student's t test.
We next investigated the possibility that a qualitative or quantitative imbalance of CD4+CD25+ regulatory T cells was causal for the production of autoimmune antibodies in aged PGAP3−/− mice. The absolute numbers of spleen cells and the percentage of CD4+ T cells to whole spleen lymphocytes and to CD3+ T cells in spleens of 12-month-old mice were not significantly different among PGAP3+/+, PGAP3+/−, and PGAP3−/− mice (Table 2). Similarly, the percentage of CD4+CD25+ T cells to total CD4+ cells in spleens was not significantly different among PGAP3+/+, PGAP3+/−, and PGAP3−/− mice, although the percentage in PGAP3−/− mice was somewhat lower than for PGAP3+/+ and PGAP3+/− mice (Fig. 6B).
TABLE 2.
Numbers of splenic lymphocytes and CD4+ T cells in 12-month-old PGAP3+/+, PGAP3+/−, and PGAP3−/− mice
There were no differences in splenic total lymphocytes and CD4+ T cell numbers between all mouse genotypes.
| Genotype | No. of spleen lymphocytes (×105) | CD4+ T cells/spleen lymphocytes | CD4+ T cells/CD3+ T cells |
|---|---|---|---|
| % | % | ||
| PGAP3+/+ | 1032 ± 140 (n = 5) | 19.95 ± 2.98 (n = 5) | 62.9 ± 6.91 (n = 5) |
| PGAP3+/− | 1050 ± 102 (n = 5) | 20.7 ± 4.47 (n = 8) | 66.8 ± 5.83 (n = 8) |
| PGAP3−/− | 918 ± 148 (n = 5) | 21.1 ± 2.33 (n = 8) | 65.3 ± 4.91 (n = 8) |
T and B Cell-specific PGAP3−/− Mice Do Not Develop Anti-DNA Antibodies
Because all hematopoietic lineages lack PGAP3 in PGAP3−/− mice, it was difficult to determine which lineages were responsible for the development of autoimmune symptoms. Therefore, we established T cell and B cell lineage-specific PGAP3 knock-out mice to investigate their contribution to autoimmune symptoms. Lck-Cre/PGAP3flox/flox mice lacking PGAP3 in T cells and cd19-Cre/PGAP3flox/flox mice lacking PGAP3 in B cells were used to measure titers of anti-dsDNA and anti-ssDNA antibodies in the sera of aged PGAP3−/− animals. Autoantibodies were not observed in Lck-Cre/PGAP3flox/flox, cd19-Cre/PGAP3flox/flox, or control mice. This indicated that the contribution of T and B cells alone might be minor and that a lack of PGAP3 in either T or B cell lineage was insufficient for the onset of autoimmune symptoms (Fig. 7).
FIGURE 7.
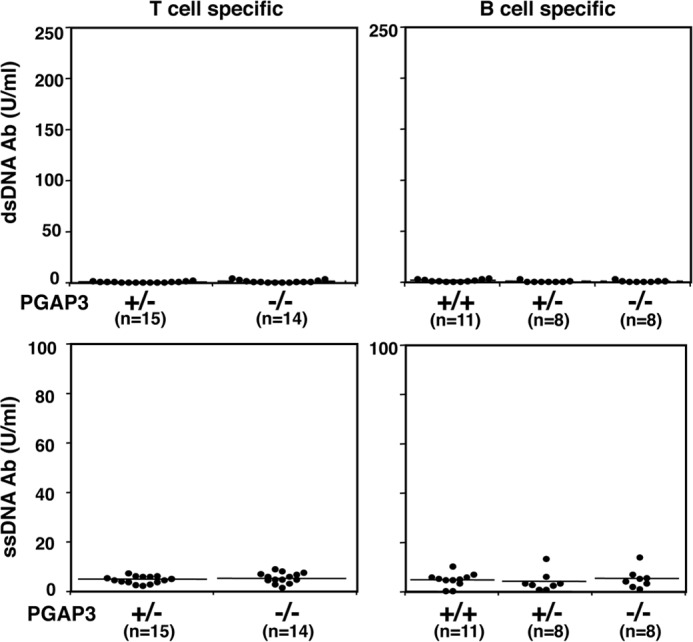
Production of autoantibodies in T- and B cell-specific PGAP3−/− mice. Titers of dsDNA-specific (upper panel) and ssDNA-specific antibodies (lower panel) in the sera of Lck-Cre/PGAP3flox/flox (left panel) and cd19-Cre/PGAP3flox/flox (right panel) mice and their respective littermate control mice at the age of 26–29 weeks are shown. The mean values are indicated by lines.
Reduced Efficiency of Apoptotic Cell Engulfment by Macrophages in PGAP3−/− Mice
Finally, we examined apoptotic cell clearance, because apoptotic and necrotic cells are strong candidates for sources of autoantigens that drive autoantibody responses in autoimmune disease. Inefficient clearance of apoptotic cells and the subsequent accumulation of apoptotic cell debris can provoke chronic inflammatory responses and may lead to a breakdown of self-tolerance (18). To examine the ability of resident peritoneal macrophages in PGAP3−/− mice to engulf apoptotic cells, we prepared prey TUNEL-positive apoptotic thymocytes from wild type mice. Incubation of apoptotic thymocytes with macrophages caused them to be engulfed by the macrophages (Fig. 8A). The relative phagocytosis index (EA/M, the mean number of engulfed apoptotic cells in total macrophages) of resident peritoneal macrophages from PGAP3−/− mice was lower (54.2%) than from PGAP3+/− mice (Fig. 8B). The relative percentage of macrophages harboring engulfed apoptotic thymocytes in total macrophages (MA/M) and the relative mean number of engulfed apoptotic cells per macrophage with engulfed apoptotic thymocytes (EA/MA) were also significantly decreased, compared with PGAP3+/− mice (Fig. 8B). These results indicated that clearance of apoptotic cells by peritoneal macrophages was defective in PGAP3−/− mice.
FIGURE 8.
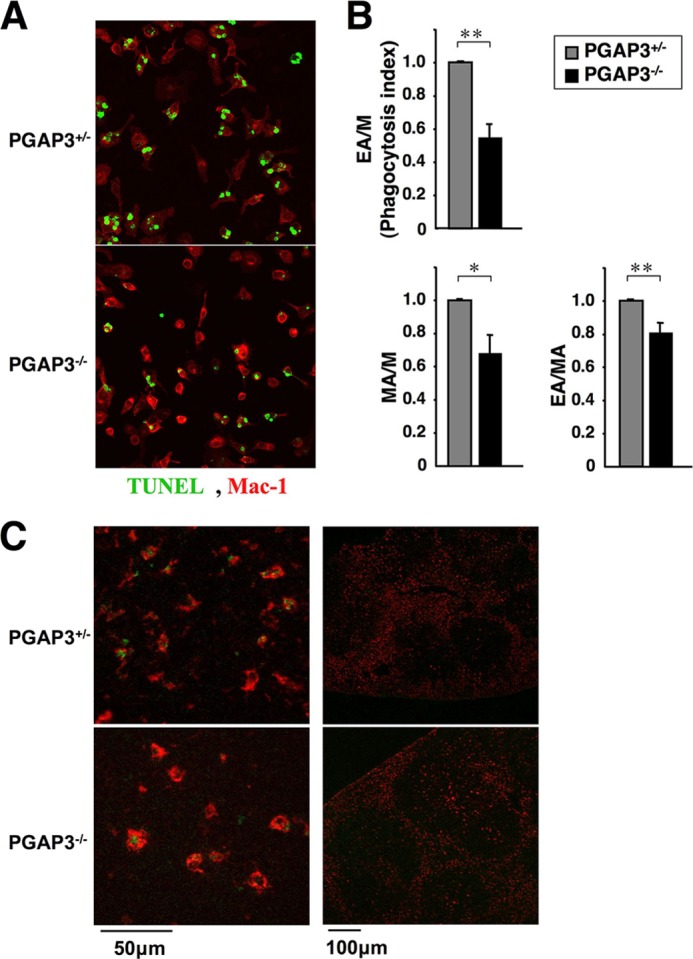
Engulfment of apoptotic cells by resident peritoneal macrophages. A, resident peritoneal macrophages from PGAP3−/− and control mice were cultured with dexamethasone-induced apoptotic thymocytes from wild type mice, stained by TUNEL method (green) and antibody against Mac-1 (red), and observed by microscope. Magnification, ×40. B, engulfment efficiency was quantified. M, total macrophages; MA, macrophages with engulfed apoptotic cells; EA, engulfed apoptotic cells. Phagocytosis index: the number of engulfed apoptotic cells per macrophage. The values of PGAP3+/− resident peritoneal macrophages cells were designated as 1, and the relative values are shown. The results are the average of eight independent experiments, and the mean values ± S.E. are shown. *, p < 0.05; **, p < 0.005 (Student's t test). C, engulfment of apoptotic cells by PGAP3-deficient tingible-body macrophages in GC. Spleen sections were prepared from 13- to 15-month-old PGAP3+/− or PGAP3−/− mice and stained with antibodies against CD68 (red) and TUNEL (green). Staining profiles were merged. Magnification, ×20. Left panels, cells in the GC. Scale bar, 50 μm. Right panels, total germinal centers. Scale bar, 100 μm. The results are representative of 10 PGAP3−/− mice and their controls.
GCs contain macrophages called “tingible-body” macrophages, which specifically express CD68. To assess the requirement for PGAP3 in tingible-body macrophage functions, we examined apoptotic cells and their association with tingible-body macrophages in spleen sections from 13- to 15-month-old mice. Immunofluorescence microscopy confirmed that CD68+ cells (red) in germinal centers engulfed TUNEL-positive cells (green), and no apparent difference of engulfment was observed between PGAP3+/+ and PGAP3−/− spleens (Fig. 8C). Most TUNEL-positive staining was localized within CD68+ macrophages, and almost no TUNEL-positive staining was present outside or on the surface of macrophages. CD68+ cells with engulfed materials were not enlarged, indicating no accumulation of apoptotic cells in the tingible-body macrophages in both PGAP3+/+ and PGAP3−/− spleens (Fig. 8C).
DISCUSSION
This study attempted to clarify the biological significance of GPI-AP enrichment in lipid rafts, especially in the immune system. GPI-APs require PGAP3 function to be enriched in lipid rafts that produce two saturated fatty acid chains of GPI by fatty acid remodeling. A defect in PGAP3 retains the surface expression of GPI-APs that harbor an unsaturated lipid chain instead of a saturated lipid chain and redistributes GPI-APs into non-DRM fractions (2). Therefore, knock-out of PGAP3 is a suitable way to analyze the biological significance of GPI-AP enrichment in lipid rafts.
In this study, we observed several new findings as follows: 1) a significant number of aged PGAP3−/− mice developed autoimmune-like symptoms, such as autoantibodies, spontaneous GC formation, and enlarged glomeruli deposited with immunoglobulin and accompanied by matrix expansion; 2) AICD and CD4+ regulatory T cells were not impaired, but the number of Th1 cells in aged PGAP3−/− mice was slightly but significantly decreased with possible Th2 polarization; 3) T cell-dependent and -independent B cell responses were not hyper-reactive in aged PGAP3−/− mice; 4) conditional targeting of PGAP3 in either B or T cells did not cause autoimmune-like symptoms, suggesting that cell lineages other than T and B cells might be responsible for the onset of autoimmune symptoms; 5) the efficiency of apoptotic cell engulfment by resident peritoneal macrophages was impaired in PGAP3−/− mice.
We previously reported that T cell development was normal in PGAP3−/− mice but that certain in vitro and in vivo T cell responses were enhanced, including alloreactive responses and antigen-specific immune responses. Nevertheless, we did not observe hyper-reactivity of B cells by T cell-dependent mechanisms. One possible explanation is that B cell responses were reduced in PGAP3−/− mice. This may be confirmed, as the proliferation of B cells stimulated by anti-IgM antibodies was significantly reduced (Fig. 4E) and T-cell-dependent and -independent B cell activations were apparently suppressed, although a significant difference was not observed (Fig. 4, A–D). Our previous study suggested expression levels of GPI-APs might be reduced in PGAP3−/− mice, depending on the cell lineage. The expression levels of CD48 and Thy-1 on different types of T cells and monocytes were decreased by 30–60%. The expression levels of CD48 on peripheral B cells were decreased similar with T cells (data not shown) and may be equivalent with that in heterozygous mice. Mice deficient for individual GPI-APs, such as CD48, Thy-1, CD59, Sca-1, and CD157, have been established and immune system functions analyzed (19–23). Because these heterozygous mice did not develop any phenotypes, decreased GPI-AP expression in PGAP3−/− mice would not cause obvious effects. Moreover, T and B cell lineage-specific conditional targeting of PGAP3 using Lck-Cre and cd19-Cre mice did not develop anti-DNA antibodies, suggesting that a lack of PGAP3-dependent T or B cell response might not be sufficient for autoimmunity, although the possibility that abnormal T or B cell signaling enhances autoimmunity or that a double deficiency of T and B cell lineages may be required for disease onset cannot be ruled out.
The engulfment efficiency of apoptotic cells by resident peritoneal macrophages was significantly reduced in PGAP3−/− mice. Apoptotic cells expose phosphatidylserine on their surface as an “eat me” signal to macrophages that respond by engulfing them. Using fibroblasts and epithelial cell lines as host cells to reconstitute engulfment, several molecules have been identified (MFG-E8/integrins, Tim4, stabilin 2, and BAI1) that bind to phosphatidylserine (13, 24–27). The clearance of apoptotic cells is critical as MFG-E8- and Tim4-deficient mice develop autoimmunity (28–30). Tingible-body macrophages in the spleen express Tim4 and MFG-E8, although MFG-E8 alone is required for their engulfment of apoptotic cells (30). In contrast, resident peritoneal macrophages express Tim4 but not MFG-E8 (24). Importantly, Tim4-deficient mice demonstrated impaired apoptotic cell clearance by peritoneal macrophages and B-1 cells but not by splenic Tim4-expressing APCs and macrophages (29). Moreover, the peritoneal Tim4-expressing cell-dependent clearance defect was sufficient to induce the production of autoantibodies. This may support our expectation that impaired clearance in peritoneal macrophages caused by a defect in PGAP3 might be sufficient for development of autoimmune-like conditions. We did not detect obvious defects of tingible-body macrophages in the spleen, indicating they are not significantly affected by the absence of PGAP3.
Engulfment of apoptotic cells is performed in sequential steps, including tethering and phagocytosis. In PGAP3−/− mice, numbers of engulfed apoptotic thymocytes per macrophage containing engulfed apoptotic cells (EA/MA) were significantly decreased, indicating that tethering might be impaired. Several GPI-APs, such as CD55, FcγRIIIb, ADP-ribosyltransferase RT6, and MAM domain-containing protein 1 (MAMDC1), have been linked with autoimmune disease (31–34). MAMDC1 is a poorly characterized gene that encodes a protein belonging to a novel subgroup of the Ig superfamily and contains six Ig-like domains followed by a single MAM domain (35). MAMDC1 exists ubiquitously in a variety of human tissues, including the immune system (34). Recently, MAMDC1 polymorphisms were found to associate with increased risk of systemic lupus erythematosus (34). CD55, widely expressed as a complement regulatory factor, functions as a suppressor for adaptive immune responses, and defects of CD55 significantly enhance T cell responses (31). We postulate that one of these GPI-APs or an unknown GPI-AP may be directly or indirectly involved in the tethering and/or engulfment processes of resident peritoneal macrophages, and deficiency of PGAP3 may generate higher numbers of nonengulfed apoptotic cells that may be immunogenic.
In conclusion, by analyzing PGAP3−/− mice, we demonstrated that GPI-AP enrichment in lipid rafts is not essential for differentiation of T and B cells but is important for maintenance of immune tolerance. Further analysis of PGAP3−/− mice might help identify GPI-APs and the precise mechanisms responsible for altered immune functions of PGAP3−/− mice.
Acknowledgments
We thank Drs. Masahito Ikawa, Hidetoshi Hasuwa, and Masaru Okabe (Genome Information Research Center, Osaka University) and Toshiki Moriyama (Health Care Center, Osaka University) for useful advice and discussions, and Akiko Kawai and Kiyo Kawata (Genome Information Research Center, Osaka University) for help in producing knock-out mice. We also thank all members of the Kinoshita laboratory for technical and spiritual support.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
- GPI
- glycosylphosphatidylinositol
- AICD
- activation-induced cell death
- APC
- allophycocyanin
- DRM
- detergent-resistant membrane
- GPI-AP
- GPI-anchored protein
- MAMDC1
- MAM domain containing protein 1
- NP
- (4-hydroxy-3-nitrophenyl)acetyl
- AECM
- aminoethylcarboxymethyl
- PGAP3
- post-GPI attachment to proteins 3
- PNA
- peanut agglutinin
- ssDNA
- single-stranded DNA
- GC
- germinal center
- PE
- phycoerythrin.
REFERENCES
- 1. Maeda Y., Kinoshita T. (2011) Structural remodeling, trafficking and functions of glycosylphosphatidylinositol-anchored proteins. Prog. Lipid Res. 50, 411–424 [DOI] [PubMed] [Google Scholar]
- 2. Maeda Y., Tashima Y., Houjou T., Fujita M., Yoko-o T., Jigami Y., Taguchi R., Kinoshita T. (2007) Fatty acid remodeling of GPI-anchored proteins is required for their raft association. Mol. Biol. Cell 18, 1497–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tashima Y., Taguchi R., Murata C., Ashida H., Kinoshita T., Maeda Y. (2006) PGAP2 is essential for correct processing and stable expression of GPI-anchored proteins. Mol. Biol. Cell 17, 1410–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Simons K., Ikonen E. (1997) Functional rafts in cell membranes. Nature 387, 569–572 [DOI] [PubMed] [Google Scholar]
- 5. Pike L. J. (2003) Lipid rafts: bringing order to chaos. J. Lipid Res. 44, 655–667 [DOI] [PubMed] [Google Scholar]
- 6. Murakami H., Wang Y., Hasuwa H., Maeda Y., Kinoshita T., Murakami Y. (2012) Enhanced response of T lymphocytes from Pgap3 knockout mouse: Insight into roles of fatty acid remodeling of GPI anchored proteins. Biochem. Biophys. Res. Commun. 417, 1235–1241 [DOI] [PubMed] [Google Scholar]
- 7. Seong J., Wang Y., Kinoshita T., Maeda Y. (2013) Implications of lipid moiety in oligomerization and immunoreactivities of GPI-anchored proteins. J. Lipid Res. 54, 1077–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abrami L., Fivaz M., Kobayashi T., Kinoshita T., Parton R. G., van der Goot F. G. (2001) Cross-talk between caveolae and glycosylphosphatidylinositol-rich domains. J. Biol. Chem. 276, 30729–30736 [DOI] [PubMed] [Google Scholar]
- 9. Simons K., Toomre D. (2000) Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39 [DOI] [PubMed] [Google Scholar]
- 10. Jones D. R., Varela-Nieto I. (1998) The role of glycosylphosphatidylinositol in signal transduction. Int. J. Biochem. Cell Biol. 30, 313–326 [DOI] [PubMed] [Google Scholar]
- 11. Robinson P. J. (1997) Signal transduction via GPI-anchored membrane proteins. Adv. Exp. Med. Biol. 419, 365–370 [DOI] [PubMed] [Google Scholar]
- 12. Hazenbos W. L., Murakami Y., Nishimura J., Takeda J., Kinoshita T. (2004) Enhanced responses of glycosylphosphatidylinositol anchor-deficient T lymphocytes. J. Immunol. 173, 3810–3815 [DOI] [PubMed] [Google Scholar]
- 13. Hanayama R., Tanaka M., Miwa K., Shinohara A., Iwamatsu A., Nagata S. (2002) Identification of a factor that links apoptotic cells to phagocytes. Nature 417, 182–187 [DOI] [PubMed] [Google Scholar]
- 14. Wyllie A. H. (1980) Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 284, 555–556 [DOI] [PubMed] [Google Scholar]
- 15. Hennet T., Hagen F. K., Tabak L. A., Marth J. D. (1995) T-cell-specific deletion of a polypeptide N-acetylgalactosaminyl-transferase gene by site-directed recombination. Proc. Natl. Acad. Sci. U.S.A. 92, 12070–12074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rickert R. C., Roes J., Rajewsky K. (1997) B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 25, 1317–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jacob J., Kassir R., Kelsoe G. (1991) In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J. Exp. Med. 173, 1165–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Muñoz L. E., Lauber K., Schiller M., Manfredi A. A., Herrmann M. (2010) The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat. Rev. Rheumatol. 6, 280–289 [DOI] [PubMed] [Google Scholar]
- 19. González-Cabrero J., Wise C. J., Latchman Y., Freeman G. J., Sharpe A. H., Reiser H. (1999) CD48-deficient mice have a pronounced defect in CD4+ T cell activation. Proc. Natl. Acad. Sci. U.S.A. 96, 1019–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beissert S., He H. T., Hueber A. O., Lellouch A. C., Metze D., Mehling A., Luger T. A., Schwarz T., Grabbe S. (1998) Impaired cutaneous immune responses in Thy-1-deficient mice. J. Immunol. 161, 5296–5302 [PubMed] [Google Scholar]
- 21. Holt D. S., Botto M., Bygrave A. E., Hanna S. M., Walport M. J., Morgan B. P. (2001) Targeted deletion of the CD59 gene causes spontaneous intravascular hemolysis and hemoglobinuria. Blood 98, 442–449 [DOI] [PubMed] [Google Scholar]
- 22. Stanford W. L., Haque S., Alexander R., Liu X., Latour A. M., Snodgrass H. R., Koller B. H., Flood P. M. (1997) Altered proliferative response by T lymphocytes of Ly-6A (Sca-1) null mice. J. Exp. Med. 186, 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Itoh M., Ishihara K., Hiroi T., Lee B. O., Maeda H., Iijima H., Yanagita M., Kiyono H., Hirano T. (1998) Deletion of bone marrow stromal cell antigen-1 (CD157) gene impaired systemic thymus independent-2 antigen-induced IgG3 and mucosal TD antigen-elicited IgA responses. J. Immunol. 161, 3974–3983 [PubMed] [Google Scholar]
- 24. Miyanishi M., Tada K., Koike M., Uchiyama Y., Kitamura T., Nagata S. (2007) Identification of Tim4 as a phosphatidylserine receptor. Nature 450, 435–439 [DOI] [PubMed] [Google Scholar]
- 25. Park S. Y., Jung M. Y., Kim H. J., Lee S. J., Kim S. Y., Lee B. H., Kwon T. H., Park R. W., Kim I. S. (2008) Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 15, 192–201 [DOI] [PubMed] [Google Scholar]
- 26. Park D., Tosello-Trampont A. C., Elliott M. R., Lu M., Haney L. B., Ma Z., Klibanov A. L., Mandell J. W., Ravichandran K. S. (2007) BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450, 430–434 [DOI] [PubMed] [Google Scholar]
- 27. Nakayama M., Akiba H., Takeda K., Kojima Y., Hashiguchi M., Azuma M., Yagita H., Okumura K. (2009) Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 113, 3821–3830 [DOI] [PubMed] [Google Scholar]
- 28. Hanayama R., Tanaka M., Miyasaka K., Aozasa K., Koike M., Uchiyama Y., Nagata S. (2004) Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 304, 1147–1150 [DOI] [PubMed] [Google Scholar]
- 29. Rodriguez-Manzanet R., Sanjuan M. A., Wu H. Y., Quintana F. J., Xiao S., Anderson A. C., Weiner H. L., Green D. R., Kuchroo V. K. (2010) T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 107, 8706–8711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miyanishi M., Segawa K., Nagata S. (2012) Synergistic effect of Tim4 and MFG-E8 null mutations on the development of autoimmunity. Int. Immunol. 24, 551–559 [DOI] [PubMed] [Google Scholar]
- 31. Liu J., Miwa T., Hilliard B., Chen Y., Lambris J. D., Wells A. D., Song W. C. (2005) The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J. Exp. Med. 201, 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Willcocks L. C., Lyons P. A., Clatworthy M. R., Robinson J. I., Yang W., Newland S. A., Plagnol V., McGovern N. N., Condliffe A. M., Chilvers E. R., Adu D., Jolly E. C., Watts R., Lau Y. L., Morgan A. W., Nash G., Smith K. G. (2008) Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J. Exp. Med. 205, 1573–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Matthes M., Hollmann C., Bertuleit H., Kühl M., Thiele H. G., Haag F., Koch-Nolte F. (1997) “Natural” RT6-1 and RT6-2 “knock-out” mice. Adv. Exp. Med. Biol. 419, 271–274 [PubMed] [Google Scholar]
- 34. Hellquist A., Zucchelli M., Lindgren C. M., Saarialho-Kere U., Järvinen T. M., Koskenmies S., Julkunen H., Onkamo P., Skoog T., Panelius J., Räisänen-Sokolowski A., Hasan T., Widen E., Gunnarson I., Svenungsson E., Padyukov L., Assadi G., Berglind L., Mäkelä V. V., Kivinen K., Wong A., Cunningham Graham D. S., Vyse T. J., D'Amato M., Kere J. (2009) Identification of MAMDC1 as a candidate susceptibility gene for systemic lupus erythematosus (SLE). PLoS One 4, e8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Litwack E. D., Babey R., Buser R., Gesemann M., O'Leary D. D. (2004) Identification and characterization of two novel brain-derived immunoglobulin superfamily members with a unique structural organization. Mol. Cell. Neurosci. 25, 263–274 [DOI] [PubMed] [Google Scholar]