

Background: The role of early secreted antigenic target of 6-kDa in tuberculosis is unclear.
Results: Early secreted antigenic target of 6 kDa induces interleukin-8 in lung epithelial cells via reactive oxygen species and protein kinases and promotes granuloma formation.
Conclusion: Early secreted antigenic target of 6 kDa induces interleukin-8 to promote granuloma formation.
Significance: Induction of interleukin-8 expression by early secreted antigenic target of 6 kDa is a potential mechanism for tuberculosis pathogenesis.
Keywords: Chemokines, Gene Regulation, Lung Injury, Oxidative Stress, Transcription Regulation, Tuberculosis, Interleukin-8
Abstract
Early secreted antigenic target of 6 kDa (ESAT-6) of Mycobacterium tuberculosis is critical for the virulence and pathogenicity of M. tuberculosis. IL-8, a major chemotactic cytokine for neutrophils and T lymphocytes, plays important roles in the development of lung injury. To further understand the role of ESAT-6 in lung pathology associated with tuberculosis development, we studied the effects of ESAT-6 on the regulation of IL-8 expression in lung epithelial cells. ESAT-6 induced IL-8 expression by increasing IL-8 gene transcription and mRNA stability. ESAT-6 induction of IL-8 promoter activity was dependent on nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) binding and sensitive to pharmacological inhibition of PKC and ERK and p38 MAPK pathways. ESAT-6 activated ERK and p38 MAPK phosphorylation and rapidly induced reactive oxygen species (ROS) production. Dimethylthiourea but not mannitol inhibited IL-8 induction by ESAT-6, further supporting the involvement of ROS in the induction of IL-8 expression. Exposure of mice to ESAT-6 induced localized inflammatory cell aggregate formation with characteristics of early granuloma concomitant with increased keratinocyte chemoattractant CXCL1 staining in bronchiolar and alveolar type II epithelial cells and alveolar macrophages. Our studies have identified a signal transduction pathway involving ROS, PKC, ERK, and p38 MAPKs and NF-κB and AP-1 in the ESAT-6 induction of IL-8 expression in lung epithelial cells. This has important implications for the understanding of lung innate immune responses to tuberculosis and the pathogenesis of lung injury in tuberculosis.
Introduction
Mycobacterium tuberculosis is the causative agent for tuberculosis, and the lung is a primary target for the infection, resulting in respiratory symptoms and chronic lung inflammation. An estimated 2 billion people worldwide are infected with M. tuberculosis, and in 2009 alone, of the 8.8 million people that became ill with tuberculosis, 1.4 million perished. ESAT-6 (early secreted antigenic target of 6 kDa), a protein secreted by M. tuberculosis, is a T cell antigen and a promising vaccine candidate (1, 2). However, it has also been implicated in the virulence and pathogenicity of M. tuberculosis. Based on its ability to bind to laminin and cause lysis of lung epithelial cells, ESAT-6 has been suggested to play roles in the dissemination of M. tuberculosis (3). In addition, ESAT-6 contributes to granuloma formation (4), a hallmark of tuberculosis pathology, through induction of epithelial cell matrix MMP-9 (metalloproteinase-9) in a zebrafish model of tuberculosis. ESAT-6 also regulates cytokine production by immune cells, such as induction of IL-1β secretion by macrophages (5) and inhibition of IFN-γ production by T-cells (6), indicating that it may have important roles in the innate and adaptive immune responses to M. tuberculosis infection.
The respiratory epithelium comprises ∼70 m2 in area and is vital for the maintenance of alveolar integrity, innate immune defense, and inflammation control through the production of surfactant and cytokines (7–9). IL-8, a member of the CXC chemokine family, serves as a chemoattractant for neutrophils, T-cells, and monocytes and controls trafficking of these cells to the sites of infection (10). Lung epithelial cells produce elevated levels of IL-8 following infection by a variety of respiratory viruses and bacteria (10). High IL-8 levels in the bronchoalveolar lavage fluid were associated with pulmonary tuberculosis (11) and high IL-8 levels in plasma correlated with increased mortality in M. tuberculosis infection (12). Pulmonary epithelial cells were identified as a major source of IL-8 production in response to M. tuberculosis infection (13). These data suggest that elevated IL-8 levels may be responsible for injury to lung architecture commonly seen in pulmonary tuberculosis patients. Infection of A549 lung epithelial cells by M. tuberculosis induces IL-8 production (13) that is dependent on reactive oxygen species and mitogen-activated protein kinase activation (14).
Enhanced neutrophil trafficking to sites of infection triggered by elevated IL-8 levels may be involved in the clearance of M. tuberculosis, but excess neutrophils may cause tissue damage by release of oxidants and proteases (15) and be detrimental to host protective immunity (16). If mycobacteria are not cleared, neutrophils may actually traffic the bacteria to other sites in the body, aiding in their dissemination (15). Oxidants generated by NADPH oxidase-dependent mechanisms are implicated in the clearance of mycobacteria (17). Considering the importance of IL-8 in innate immune responses to M. tuberculosis infection and its role in the development of lung injury, it is important to understand the mechanisms regulating IL-8 expression by M. tuberculosis. Although M. tuberculosis stimulates lung epithelial cells to produce IL-8 (13, 14), bacterial components responsible for the induction and the underlying mechanisms for IL-8 stimulation are not known. We hypothesized that ESAT-6 is an important modulator of IL-8 expression in lung epithelial cells. In this study, we found that ESAT-6 induced IL-8 levels in lung epithelial cells by increasing gene transcription and IL-8 mRNA stability. ESAT-6 induction of IL-8 expression was sensitive to pharmacological inhibition of protein kinase C and ERK and p38 mitogen-activated protein kinase (MAPK) signaling pathways. ESAT-6 induction of IL-8 expression was associated with the production of reactive oxygen species and inhibited by the hydroxyl radical scavenger dimethylthiourea. Administration of ESAT-6 into lungs of mice produced localized inflammatory cell aggregates concomitant with increased KC3 staining by lung epithelial cells and macrophages.
EXPERIMENTAL PROCEDURES
Cell Culture
NCI-H441 cells (HTB-174, ATCC), a human lung adenocarcinoma cell line with characteristics of bronchiolar (Clara) epithelial cells, and A549 cells (CCL-185, ATCC), a human lung adenocarcinoma cell line with certain characteristics of alveolar type II cells, were grown on plastic tissue culture dishes in RPMI 1640 and F12K medium, respectively, supplemented with 10% fetal bovine serum, penicillin (100 units/ml), streptomycin (100 μg/ml), and amphotericin B (0.25 μg/ml) in a humidified atmosphere of 95% room air and 5% CO2. Semiconfluent cells were placed in serum-free medium overnight (16–17 h) prior to treatment with ESAT-6.
Cell Viability
Cell viability was determined using the CellTiter96AQueous non-radioactive cell proliferation assay (Promega, Madison, WI). The colorimetric assay measures the reduction of the tetrazolium compound (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt), which is an indicator of the number of viable cells in culture. Cell death was determined by annexin V staining for apoptotic cells and propidium iodide staining for end stage apoptotic or necrotic cells. Cells were stained with FITC-labeled annexin V and propidium iodide using a kit (BD Biosciences) following the manufacturer's instructions. The apoptosis and viability of the cells were examined by flow cytometry analysis with FACSCalibur flow cytometer (BD Biosciences), using FlowJo software.
Materials
Recombinant ESAT-6 and CFP10 expressed in Escherichia coli were purified as described previously (18) and found to contain low LPS (39 pg/mg protein) by a limulus amebocyte assay and to be free of protein aggregates by fast liquid chromatography gel filtration (19). ESAT-6 preparations were essentially free of peptidoglycan by GC-MS/MS analysis. Purified ESAT-6 was prepared in Hanks' balanced salt solution (HBSS) at 2 mg/ml and stored at −76 °C. Lipofectamine 2000 was from Invitrogen. Protein kinase C inhibitors bisindolylmaleimide, Go6976, and Go6883 and mitogen-activated protein kinase inhibitors PD98059, SB203580, and SP600125 were from Calbiochem or LC Laboratories (Woburn, MA). Luciferase reporter plasmids containing −546/+44 and −133/+44 bp of the IL-8 gene were kindly provided by Dr. Naofumi Mukaida (Cancer Research Institute, Kanazawa University, Kanazawa, Japan). The IL-8 promoter fragments were subcloned into the promoterless pGL3luc(basic) vector (Promega). Antibodies against phosphorylated and total ERK, p38, and JNK mitogen-activated protein kinases and goat anti-rabbit alkaline phosphatase-conjugated secondary antibody were from Cell Signaling (Beverly, MA). Actin antibodies were from Santa Cruz Biotechnology, Inc.
Plasmids and Transient Transfection
Plasmids were amplified in E. coli Top10 strain (Invitrogen) and purified by anion exchange chromatography (Qiagen, Valencia, CA). Plasmids were transfected into cells along with pcDNA3.1 (Invitrogen), a β-galactosidase expression plasmid, by liposome-mediated DNA transfer using Lipofectamine 2000 according to the manufacturer's instructions. Luciferase and β-galactosidase activities of cell lysates were determined by chemiluminiscent assays (Promega (Madison, WI) and Tropix (Bedford, MA)). Luciferase activities of cell lysates were normalized to cotransfected β-galactosidase activity or protein content to correct for variations in transfection efficiency.
Site-directed Mutagenesis
Transcription factor binding sites in the IL-8 promoter were altered by site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). Mutated promoter fragments were sequenced to confirm the presence of mutations.
RNA Isolation and Northern Blotting
RNA was isolated using TRI-Reagent (Molecular Research Center Inc., Cincinnati, OH) and subjected to Northern blotting analysis according to a method described previously (20). IL-8 mRNA bands were detected with a PhosphorImager, and IL-8 and 18 S rRNA RNA bands were quantified using Quantity One Image Acquisition and Analysis Software (Bio-Rad). IL-8 mRNA levels were normalized to 18 S rRNA levels to correct for variations in the loading and transfer of RNA.
Enzyme-linked Immunosorbent Assay (ELISA)
The levels of IL-8 in cell medium were determined by ELISA, using a matched antibody pair according to the manufacturer's protocol (R&D Systems, Minneapolis, MN).
Fluorescence Microscopy
Cells were grown on Permanox coverslips and then maintained in serum-free medium overnight (16–17 h). Medium was replaced with fresh serum-free medium, and cells were incubated in the presence of 5 or 10 μm dihydroethidium for 1 h in the dark. Afterward, cells were rinsed twice with serum-free medium and incubated with ESAT-6 (5 μg/ml) for various times. Cells were rinsed twice with cold phosphate-buffered saline, and coverslips were air-dried briefly and mounted with Vectashield. Images were captured with a Nikon Eclipse TE2000-5 inverted fluorescent microscope equipped with an Ultra-VIEW LCI scanning confocal system (PerkinElmer Life Sciences) using 488-nm excitation and 568-nm emission filters. Imaging SuiteTM version 5.0 acquisition and processing software was used to acquire the images.
Isolation of Nuclei and Nuclear Run-on Assay
The methods for the isolation of nuclei and run-on transcription assay were as described previously (20, 21). Equal amounts of 32P-labeled total RNA were hybridized to nitrocellulose membranes containing immobilized linearized plasmids. After washing, radioactivity bound to membranes was quantified by PhosphorImager scanning.
Preparation of Nuclear Extracts and Electrophoretic Mobility Shift Assay (EMSA)
The methods for nuclear extract isolation (22) and electrophoretic mobility shift assay (23) were as described previously. The protein concentration of nuclear extracts was determined by the Bradford assay. Double-stranded oligonucleotides were end-labeled using [γ-32P]ATP and T4 polynucleotide kinase. The sense strand sequences of IL-8 promoter oligonucleotides (binding sites underlined) were as follows: AP-1, 5′-AGTGTGATGACTCAGGTTTG-3′ (−133/−114 bp); NF-κB, 5′-AATCGTGGAATTTCCTCTGA-3′ (−84/−65 bp). Protein-DNA complexes were formed by incubating 0.5–1 ng (100,000 cpm) of the labeled oligonucleotide with 5 μg of nuclear protein in 20 μl of binding buffer (13 mm Hepes, pH 7.9, containing 13% glycerol, 80 mm KCl, 5 mm MgCl2, 1 mm DTT, 1 mm EDTA) and 1 μg of poly(dI-dC) as nonspecific competitor DNA at 30 °C for 20 min. The protein-DNA complexes were resolved by non-denaturing polyacrylamide gel electrophoresis and visualized by autoradiography or PhosphorImager scanning.
Western Immunoblotting
Proteins (10–15 μg) were separated by SDS-PAGE on 10% BisTris gels using 3-morpholinopropanesulfonic acid as running buffer and electroblotted to PVDF membranes using an XCell II Mini-Cell apparatus (Novex, San Diego, CA). After blocking with 5% nonfat dry milk in TBS-T buffer, the membranes were incubated with primary antibodies or antisera at 1:1000 dilution at 4 °C overnight, followed by goat anti-rabbit alkaline phosphatase-conjugated secondary antibody at 1:2000 dilution for 1 h at room temperature. Protein bands were visualized by the enhanced chemifluorescence detection method (GE Healthcare) and quantified using Quantity One Image acquisition and analysis software (Bio-Rad).
Animals
Animal studies were approved by our institutional animal care and use committee. Female C57BL6 mice (6–8 weeks) (Taconic Farms) (n = 5) were anesthetized by intraperitoneal injection of ketamine (100 mg/kg) and xylazine (8.5 mg/kg) and administered 40 μl of HBSS, recombinant ESAT-6, or CFP10 in HBSS via intranasal inhalation. Mice were sacrificed 3 days after administration of ESAT-6 or CFP-10, and lungs were instilled with 5% formaldehyde in phosphate-buffered saline under a constant pressure of 20 cm H2O and stored in the fixative for 24 h. Formaldehyde-fixed lungs were embedded in paraffin, and 5-μm sections were cut for immunohistochemical analysis.
Immunohistochemistry
Immunohistochemical detection was performed with a kit (UltraVision Plus Detection System, Thermo Scientific) that uses horseradish peroxidase (HRP) and 3-amino-9-ethylcarbazole as a chromogenic substrate. Lung sections were serially incubated with KC polyclonal antibody (0.5 μg/ml) (1:100) (Biovision) or non-immune rabbit IgG, biotin-conjugated goat secondary antibody, and HRP-labeled streptavidin. Afterward, sections were stained with 3-amino-9-ethylcarbazole and counterstained with hematoxylin. Lung sections were also subjected to hematoxylin and eosin staining according to standard procedures.
Statistical Analyses
Data are shown as means ± S.D. or S.E. In experiments in which IL-8 promoter activity/mRNA/protein levels in untreated cells were arbitrarily considered as 100, statistical significance was analyzed by the one-sample t test. One-tailed p values of <0.05 were considered significant. Effects of ESAT-6 and CFP-10 on mouse weight were analyzed by the paired t test, and two-tailed p values of <0.05 were considered significant.
RESULTS
ESAT-6 Induces IL-8 mRNA and Protein Levels in Lung Epithelial Cells
ESAT-6 and its molecular partner, CFP10 (culture filtrate protein of 10 kDa), are encoded by genes located in region of difference 1 (RD1), which is deleted in the attenuated vaccine strain of Mycobacterium bovis bacillus Calmette-Guerin and secreted by the ESX-1 protein secretion system of M. tuberculosis (24) (25). Secretion of ESAT-6 together with CFP10 is associated with the virulence and pathogenicity of M. tuberculosis, and production of IL-8 by lung epithelial cells can contribute to the inflammation that is characteristic of tuberculosis and other lung diseases. Therefore, we studied the effects of ESAT-6 on IL-8 production by H441 lung epithelial cells. H441 lung epithelial cells possess certain morphologic and ultrastructural characteristics of bronchiolar (Clara) epithelial cells and express surfactant proteins A, B, and D, markers of type II and bronchiolar epithelial cells, making them suitable as a model for cells lining the distal lung epithelium.
ESAT-6 but not CFP10 at concentrations ranging from 1 to 5 μg/ml induced significant amounts of IL-8 mRNA compared with untreated cells (Fig. 1). Because ESAT-6 and CFP10 were purified from recombinant E. coli lysates and could contain LPS, we repeated experiments in the presence of polymyxin B, an inhibitor of LPS. Polymyxin B did not alter ESAT-6 stimulation of IL-8 mRNA expression, indicating that this effect is not due to LPS contamination. In separate experiments, polymyxin B (10 μg/ml) blocked LPS (1 μg/ml) induction of TNF-α production by human monocytes (data not shown). Because peptidoglycan is known to induce IL-8 expression, we wanted to know if recombinant ESAT-6 preparations contain peptidoglycan. GC-MS/MS analysis of muramic acid (a marker of peptidoglycan) levels (HBSS = 1.7 ng/0.5 ml; ESAT-6 = 1.96 ± 0.9 ng/5 μg of ESAT-6 in 0.5 ml of HBSS, n = 3) indicated that recombinant ESAT-6 is essentially free of peptidoglycan. We also found that ESAT-6 induced IL-8 mRNA and IL-8 protein levels in a dose- and time-dependent manner (Fig. 1, C–H). The inductive effect of ESAT-6 was apparent at 1 h of treatment and increased over time, as reflected in the increased IL-8 levels in the cell culture medium. Quantitative RT-PCR analysis in agreement with Northern blotting analysis indicated that ESAT-6 increased IL-8 mRNA levels in a time-dependent manner with significant inductive effects at 1 h of treatment (2.4–6-fold compared with control). Similarly ESAT-6 induced IL-8 expression in A549 lung epithelial cells that have certain characteristics of alveolar type II epithelial cells (data not shown). Collectively, these data indicated that ESAT-6 is a strong inducer of IL-8 expression in lung epithelial cells.
FIGURE 1.

Effects of ESAT-6 on IL-8 mRNA and protein levels. H441 cells were treated with medium or ESAT-6 (5 and 10 μg/ml), CFP-10 (5 μg/ml), polymyxin B (Pmx B) (10 μg/ml), or polymyxin B + ESAT-6 (5 μg/ml) for 6 h, and IL-8 mRNA levels were analyzed by Northern blotting. A representative Northern blot (A) and data (means ± S.D. (error bars), n = 2) (B) are shown. H441 cells were treated with or without ESAT-6 at the indicated concentrations for 6 h (C–E) or treated with ESAT-6 (5 μg/ml) for the indicated times (F–H). IL-8 mRNA and IL-8 protein levels in the culture medium were determined by Northern blotting and ELISA, respectively. Data shown are means ± S.D. (n = 2 for Northern blotting experiments, and n = 6 for ELISA measurements).
Because ESAT-6 is implicated in the lysis of lung epithelial cells (3, 26) and apoptosis of macrophages (27), we examined the effect of recombinant ESAT-6 on apoptosis and cell death by annexin V and propidium iodide staining. ESAT-6 at a concentration of 5 μg/ml and after 6 h of exposure did not induce cell apoptosis (control = 2.5% ± 0.28, ESAT-6 = 2.75% ± 1.1, n = 3–4) but increased necrotic cell death by ∼7% (control = 5.75 ± 0.62%, ESAT-6 = 13.0% ± 1.78, n = 3–4, p < 0.01), suggesting that the observed inductive effects of ESAT-6 on IL-8 expression are specific and not due to toxicity.
ESAT-6 Induces IL-8 Expression by Increasing Gene Transcription and mRNA Stability
We next determined whether ESAT-6 increases IL-8 mRNA levels through increased IL-8 gene transcription and/or mRNA stability. Using a transcription run-on assay in isolated nuclei, we found that ESAT-6 increased IL-8 gene transcription in a time-dependent manner with significant effect after 6 h (control = 1; ESAT-6 at 3 h = 2.5 ± 0.7; ESAT-6 at 6 h = 6.0 ± 4.0, n = 2) (Fig. 2A). In agreement with these data, ESAT-6 increased IL-8 promoter activity in transient transfection experiments (Fig. 2B). Because mRNA stability plays a significant role in the control of IL-8 mRNA induction by cytokines and other agents (28), we determined if ESAT-6 also increased IL-8 mRNA half-life in H441 cells after blockade of new transcription with actinomycin D. Our experiments to determine IL-8 mRNA half-life by [3H]uridine pulse-chase were not successful. IL-8 mRNA half-life increased from <1 h in control cells to >3 h in ESAT-6-treated cells (Fig. 2, C and D), indicating that enhanced mRNA stability contributes to ESAT-6-mediated increase of IL-8 mRNA levels. GAPDH mRNA stability (half-life in control and ESAT-6-treated cells = 2.5–3 h) was not affected by ESAT-6 treatment.
FIGURE 2.
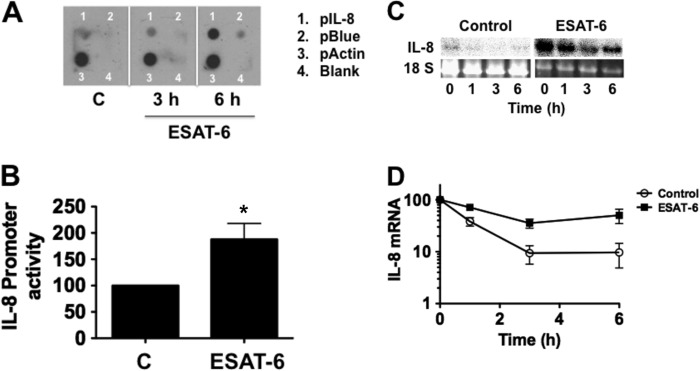
ESAT-6 increases IL-8 gene transcription and IL-8 mRNA stability. A, H441 cells were treated with or without ESAT-6 (5 μg/ml) for the indicated times, and the IL-8 gene transcription rate was determined by a nuclear run-on assay using isolated nuclei. Actin gene transcription rate was measured as a sample loading control. Data shown are representative of two independent experiments. pBlue, empty plasmid Bluescript. B, H441 cells transiently transfected with an IL-8-luciferase promoter plasmid (−546/+44 bp) were treated with or without ESAT-6 (5 μg/ml) for 6 h, and luciferase activity was measured and normalized to total cell protein. Data shown are means ± S.E. (error bars) (n = 4). *, p < 0.05 compared with control cells. C and D, H441 cells were treated with or without ESAT-6 (5 μg/ml) for 6 h to induce IL-8 mRNA, and then incubation continued in the presence of actinomycin D (5 μm) to block new transcription. Cells were collected at different times for isolation of RNA, and IL-8 and GAPDH mRNA levels were determined by Northern blotting. Data for GAPDH mRNA levels are not shown. Data shown are means ± S.E. (n = 3).
ESAT-6 Induction of IL-8 Promoter Activity Is Dependent on the Binding of AP-1 and NF-κB
A minimal IL-8 promoter region consisting of −133/+ 44 bp is necessary and sufficient for the basal and TNF-α induction of IL-8 promoter activity (29). The minimal promoter region contains binding sites for NFIL-6, NF-κB, and AP-1 transcription factors that act independently and synergistically to activate IL-8 promoter in a cell-specific manner in response to stimulatory agents (29). Of these transcription factors, NF-κB and AP-1 play critical roles in the induction of IL-8 promoter activity. We therefore analyzed the effects of ESAT-6 on the binding of NF-κB and AP-1 to IL-8 promoter by an electrophoretic mobility shift assay (Fig. 3A). H441 cells expressed constitutive levels of AP-1 and NF-κB DNA binding activities, and ESAT-6 treatment markedly increased AP-1 DNA binding activity but had only modest effects on NF-κB binding. To assess the roles of AP-1 and NF-κB, we mutated AP-1 and NF-κB binding sites in IL-8 promoter (−546/+44 bp). ESAT-6 increased activities of −546/+44 and −133/+44 bp IL-8 promoter constructs by a similar degree (∼2-fold compared with control). Mutations of AP-1 or NF-κB binding sites, individually or in combination, significantly decreased basal and ESAT-6-induced IL-8 promoter activity (Fig. 3B). Mutation of the NF-κB site alone and in combination with AP-1 drastically reduced basal promoter activity and rendered the promoter insensitive to ESAT-6 stimulation. Although mutation of the AP-1 site alone decreased basal promoter activity by greater than 75%, the promoter was sensitive to ESAT-6 induction, albeit to a significantly lesser degree than the wild type promoter. Together, these data indicated that both AP-1 and NF-κB are essential for ESAT-6 induction of the IL-8 promoter.
FIGURE 3.
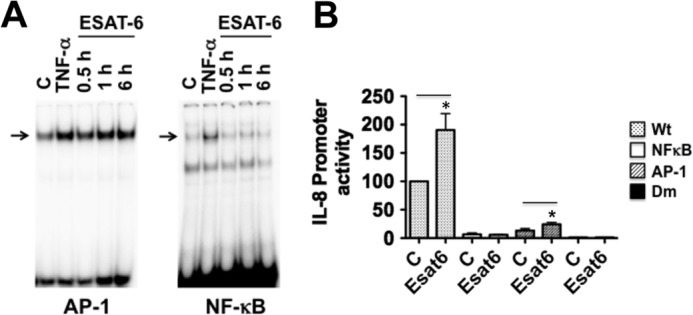
A, effect of ESAT-6 on IL-8 AP-1 and NF-κB DNA binding activities. H441 cells were treated with or without ESAT-6 (5 μg/ml) for the indicated times, and AP-1 and NF-κB DNA binding activities in the nuclear extracts were determined by an electrophoretic mobility shift assay. B, effect of ESAT-6 on the wild type and mutant IL-8 promoter activities. H441 cells were transiently transfected with wild type, AP-1, NF-κB, or AP-1 and NF-κB double mutant (Dm) IL-8-luciferase promoter plasmids (−546/+44 bp) and then treated with or without ESAT-6 (5 μg/ml) for 6 h. Luciferase activities were measured and normalized to total cell protein. Data shown are means ± S.E. (error bars) (n = 3–4). *, p < 0.05 compared with untreated cells.
Protein Kinase C and Mitogen-activated Protein Kinase Signaling Pathways Are Important for ESAT-6 Induction of IL-8 Expression
Protein kinase C (PKC) enzymes are intracellular serine-threonine protein kinases activated by multiple proinflammatory stimuli, such as TNF-α, lipopolysaccharide, and oxidants (30). Activation of PKC appears to be one of the key contributing factors for the development of lung injury (31). We hypothesized that ESAT-6 activation of PKC could be involved in the induction of IL-8 expression and tested this with pharmacological inhibitors of PKC. Bisindolylmaleimide and Go6983 inhibited IL-8 mRNA and protein levels by greater than 80% (Fig. 4). The effect of Go6976 on ESAT-6-induced changes was less than those of the other two PKC inhibitors, with more pronounced inhibition of levels of IL-8 mRNA than IL-8 protein (Fig. 4). These data indicated that PKC activation is an important step for the ESAT-6 induction of IL-8 expression.
FIGURE 4.
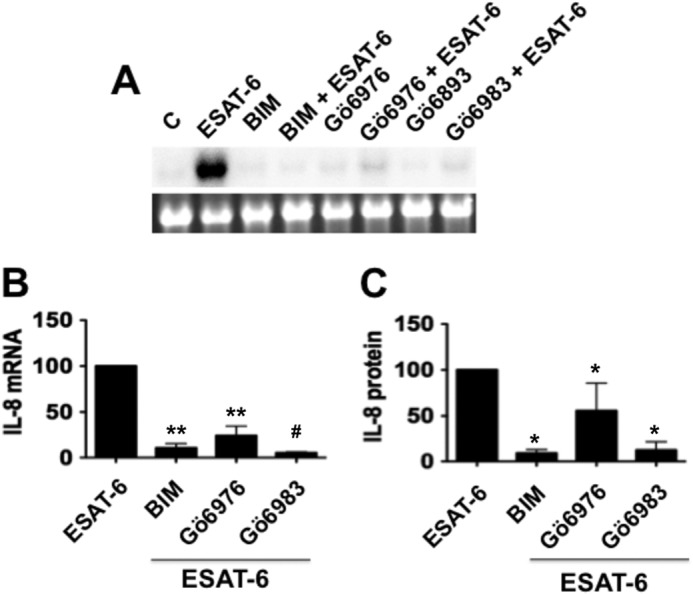
Effects of protein kinase C inhibitors on ESAT-6 induction of IL-8 mRNA and IL-8 protein expression. H441 cells were first treated with or without bisindolylmaleimide (5 μm), Go6976 (1 μm), or Go6983 for 1 h and then exposed to ESAT-6 (5 μg/ml) or medium alone for 6 h. IL-8 mRNA and IL-8 protein levels in the culture medium were determined by Northern blotting and ELISA, respectively. Data are means ± S.E. (error bars) (n = 3–4). A and B, effect on IL-8 mRNA levels. A representative Northern blot is shown. **, p < 0.01; #, p < 0.0001 compared with cells treated with ESAT-6 alone. C, effect on IL-8 levels in the medium. *, p < 0.05 compared with cells treated with ESAT-6 alone.
MAPK signaling cascades couple various cell surface stimuli to cellular responses and are regulated by protein kinase C (32), and ESAT-6 activates p38 MAPK in human lymphocytes (6). We determined if ESAT-6 induction of IL-8 expression was associated with activation of MAPKs and if pharmacological inhibitors of MAPKs affected it. In separate experiments, we evaluated the specificity of MAPK inhibitors at concentrations used in this study by determining their effects on phorbol myristate acetate (10 nm) activation of MAPKs and found that they displayed specificity of inhibition, except that ERK inhibitor PD98059 also inhibited JNK activation (data not shown). PD98059 is known to inhibit JNK MAPK (33). Western blotting analysis showed that ESAT-6 increased ERK and p38 phosphorylation, but the effect on JNK phosphorylation was modest (Fig. 5A). Similarly, inhibitors of ERK and p38 but not JNK MAPK significantly inhibited ESAT-6 induction of IL-8 mRNA and protein levels (Figs. 5, B–D). At a concentration as high as 50 μm, SP600125, a JNK MAPK inhibitor, failed to suppress ESAT-6 induction of IL-8 mRNA levels (data not shown). On the contrary, inhibition of JNK MAPK enhanced IL-8 protein but not mRNA levels (Fig. 5D). These findings indicated that ERK and p38 but not JNK MAPK signal transduction pathways are important for ESAT-6-mediated induction of IL-8 mRNA levels; however, the JNK pathway could contribute to the enhancement of IL-8 protein levels.
FIGURE 5.

Role of MAPKs in the ESAT-6 induction of IL-8 expression. A, effect of ESAT-6 on the phosphorylation of ERK and p38 MAPKs. H441 cells were treated with or without ESAT-6 (5 μg/ml) for different times, and the levels of phosphorylated ERK, p38, and JNK MAPKs were determined by Western immunoblotting. The blots were stripped and probed for tubulin, which served as a loading control. Representative blots from three independent experiments are shown. B–D, effects of MAPK inhibitors on ESAT-6 induction of IL-8 mRNA and IL-8 protein levels. H441 cells were treated with or without PD98059 (30 μm), SB203580 (10 μm), or SP600125 (10 μm) for 1 h and then exposed to medium or ESAT-6 (5 μg/ml) for 6 h. IL-8 mRNA and IL-8 protein levels in the medium were determined by Northern blotting and ELISA, respectively. Data of Northern blotting results (C) are means ± S.E. (error bars) (n = 3–4), and IL-8 ELISA data (D) are means ± S.D. (n = 2–4). *, p < 0.05; **, p < 0.01 compared with cells treated with ESAT-6 alone.
ERK and p38 MAPK Inhibitors Inhibit AP-1 DNA Binding Activity
We found that pharmacological inhibitors of ERK and p38 but not JNK MAPK suppressed ESAT-6 induction of IL-8 expression. We also found that ESAT-6 increased AP-1 DNA binding activity, and the AP-1 site is important for basal and ESAT-6 induction of IL-8 promoter activity. To determine if ESAT-6 activation of ERK and p38 MAPK pathways control induction of AP-1 activity, we tested the effects of pharmacological inhibitors on ESAT-6 induction of AP-1 DNA binding activity. We found that ERK inhibitor PD98059 and p38 inhibitor SB203580 reduced basal and ESAT-6-induced AP-1 activity, whereas JNK inhibitor SP600125 modestly inhibited induction of AP-1 activity (Fig. 6). These data indicated that ERK and p38 but not JNK MAPK pathways control the ESAT-6-dependent increase of AP-1 activity to induce IL-8 expression.
FIGURE 6.

Effects of MAPK inhibitors on AP-1 DNA binding to IL-8 promoter. H441 cells were first incubated with PD98059 (PD) (30 μm), SB203580 (SB) (10 μm), or SP600125 (SP) (10 μm) for 1 h and then treated with ESAT-6 (5 μg/ml) for 3 h (A) or 6 h (B). Nuclear proteins were isolated, and AP-1 binding to IL-8 promoter was determined by an electrophoretic mobility shift assay.
Intracellular Oxidants Mediate ESAT-6 Induction of IL-8 Expression
The intracellular redox state controls the activation of AP-1 and NF-κB to regulate expression of a wide variety of genes involved in inflammatory responses (34). Because ESAT-6 induction of IL-8 was associated with increases in AP-1 and NF-κB binding activities, and AP-1 and NF-κB are necessary for the induction of IL-8 promoter activity, we hypothesized that intracellular oxidants generated in response to ESAT-6 exposure may be involved in the induction of IL-8 expression. We tested the effects of hydroxyl radical scavengers on ESAT-6 induction of IL-8 expression and found that dimethylthiourea inhibited IL-8 expression, but mannitol did not (Fig. 7). Due to cell impermeability, mannitol can only scavenge extracellular hydroxyl radicals, whereas dimethylthiourea can also neutralize intracellular hydroxyl radicals. To obtain further evidence for the generation of oxidants in cells exposed to ESAT-6, we performed direct imaging of reactive oxygen species (ROS) production by confocal microscopy, using dihydroethidium (35), which specifically reacts with superoxide anion to form the red fluorescent product, 2-hydroethidium, that intercalates with DNA. Exposure of cells to ESAT-6 rapidly induced superoxide generation as visualized by red fluorescence, whereas control cells showed very low or undetectable fluorescence (Fig. 8).
FIGURE 7.
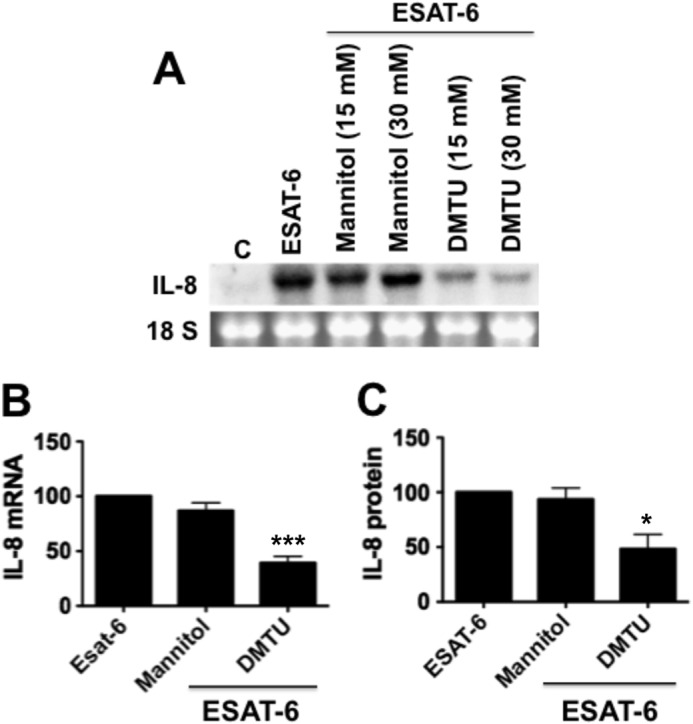
Effects of antioxidants on ESAT-6 induction of IL-8 mRNA and IL-8 protein levels. H441 cells were treated with or without mannitol (15 and 30 μm) or dimethylthiourea (DMTU) (15 and 30 μm) for 1 h and then exposed to ESAT-6 (5 μg/ml) for 6 h, and IL-8 mRNA and IL-8 protein levels in the medium were determined by Northern blotting and ELISA, respectively. Data shown are means ± S.E. (error bars) (n = 3–5). A and B, effect on IL-8 mRNA levels. ***, p < 0.001 for cells treated with DMTU (30 mm) + ESAT-6 compared with cells treated with ESAT-6 alone. C, effect on IL-8 levels in the medium. *, p < 0.05 for cells treated with DMTU (30 mm) + ESAT-6, compared with cells treated with ESAT-6 alone.
FIGURE 8.
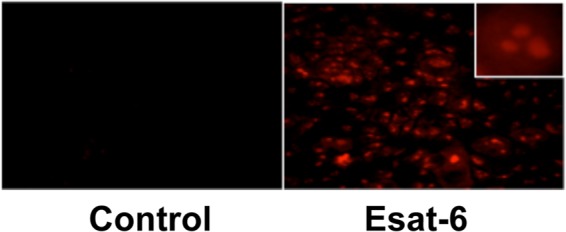
Effect of ESAT-6 on reactive oxygen species production in H441 cells. Fluorescence microscopic detection by dihydroethidium staining. H441 cells were first incubated with dihydroethidium and then exposed to medium alone or ESAT-6 (5 μg/ml). Images were captured with a fluorescence microscope. Images shown were obtained after a 2-min exposure of cells to ESAT-6. The inset shows an enlarged section of stained cells. Similar results were obtained in two other independent experiments.
ESAT-6 Induces Acute Lung Inflammation and Early Granuloma Formation in Mice
Our in vitro cell culture experiments demonstrated that ESAT-6 is a potent inducer of IL-8 expression. Because IL-8 is a chemoattractant for neutrophils, T-cells, and monocytes, we determined the effects of ESAT-6 on lung inflammatory responses in mice. There appears to be no information on the physiologic levels of ESAT-6. We used ESAT-6 in the range of 2.5–40 μg/mouse based on our in vitro cell culture data. Recombinant ESAT-6 or CFP-10 in HBSS or HBSS alone was administered into mouse lungs via the intranasal route, and lung histology and KC expression were determined by hematoxylin and eosin staining and immunostaining, respectively, after 3 days. Results indicated that ESAT-6, but not CFP-10 at a dose of 40 μg/mouse caused acute lung inflammation, as indicated by the presence of localized inflammatory cell aggregates (Fig. 9A). ESAT-6 at lower doses (2.5 and 10 μg) caused considerably less inflammation, as indicated by the presence of fewer inflammatory cell aggregates in the lung (data not shown). Inflammatory cell aggregates in lungs were found in all mice (n = 5) exposed to ESAT-6 but not CFP-10. Immunostaining demonstrated that ESAT-6, but not CFP-10, significantly increased KC staining in alveolar type II and bronchiolar epithelial cells and macrophages (Fig. 9B), indicating increased KC expression in vivo by these cells. Inflammatory cell aggregates appeared to contain macrophages and lymphocytes with the appearance of epithelioid cells, suggesting the identity of cell aggregates as early stage granuloma. We found that mice that received 40 μg of ESAT-6, but not CFP-10, suffered weight loss by ∼11% after 3 days (Table 1).
FIGURE 9.

ESAT-6 induces formation of inflammatory cell aggregates or early granuloma formation in mouse lungs. Light micrograph sections of lungs of mice 3 days after administration of 40 μg of ESAT-6 or CFP10. Control mice received HBSS only. A, hematoxylin and eosin staining. a and d, control lung. b and e, lung from CFP-10-treated mouse. c and f, lung from ESAT-6-treated mouse. Magnification was ×40 (a–c) and ×400 (d–f). B, KC immunostaining. a, control; b, lung from CFP-10-treated mouse; c, lung from ESAT-6-treated mouse; d, lung from ESAT-6-treated mouse showing cell aggregate. Staining in alveolar type II (arrow) and bronchiolar (arrowhead) epithelial cells and macrophages (open arrow) are shown. Enlarged sections of original photomicrographs (×200) are shown.
TABLE 1.
Effects of administration of ESAT-6 or CFP-10 (40 μg/mouse) on mouse weight
ESAT-6, CFP-10, or HBSS was intranasally administered into mice, and mice were weighed daily.
| Weight (mean ± S.E.) (n = 5) |
p value | ||
|---|---|---|---|
| Day 0 | Day 3 | ||
| g | g | ||
| Control | 17.6 ± 0.42 | 17.88 ± 0.33 | 0.0721 |
| CFP-10 | 18.36 ± 0.52 | 18.3 ± 0.47 | 0.642 |
| ESAT-6 | 17.7 ± 0.14 | 15.72 ± 0.31 | 0.0023 |
DISCUSSION
ESAT-6 is associated with the virulence and pathogenicity of M. tuberculosis (36–38). Recently, ESAT-6 was demonstrated to activate the inflammasome (5) and promote apoptosis and lysis (3) of lung epithelial cells. The lung epithelium contributes to the initiation and amplification of innate and inflammatory responses to various insults (8, 39, 40). IL-8 plays a key role in mediating inflammatory responses and is implicated in the pathogenesis of acute and chronic lung diseases (10). In this study, we found that ESAT-6 induced IL-8 mRNA and protein levels in A549 and H441 lung epithelial cells in a time- and concentration-dependent manner. Interestingly, CFP-10 that is secreted with ESAT-6 and functions as its molecular partner (41) had no effect on IL-8 expression. The inductive effect of ESAT-6 was not sensitive to polymyxin B, indicating that the effects are indeed due to ESAT-6 per se and not due to trace amounts of lipopolysaccharide that may be present in recombinant ESAT-6 preparations. ESAT-6 induction of IL-8 expression cannot be due to peptidoglycan contamination because ESAT-6 preparations were essentially free of peptidoglycan.
ESAT-6 increased the IL-8 gene transcription rate and IL-8 mRNA stability, indicating that both transcriptional and mRNA stability mechanisms control IL-8 induction. The importance of transcriptional mechanisms is further supported by the stimulatory effect of ESAT-6 on IL-8 promoter activity. Electrophoretic mobility shift experiments demonstrated that H441 cells express constitutive binding activities for AP-1 and NF-κB with the NF-κB binding activity present at a lower level than that of AP-1. ESAT-6 treatment increased AP-1 DNA binding activity, but its stimulatory effect on NF-κB binding activity was modest. Consistent with the functional roles for AP-1 and NF-κB in IL-8 induction, mutations of AP-1 and NF-κB reduced basal and ESAT-6-induced IL-8 promoter activity. These data also indicated that AP-1 and NF-κB function in a combinatorial/cooperative manner to induce IL-8 promoter activity in response to ESAT-6 stimulation. A relatively short sequence of −133/+41 bp of the 5′-flanking region is necessary and sufficient for the basal activity and stimulus-specific induction of IL-8 promoter activity (29). Transcription factors AP-1, NF-κB, and NFIL-6 function independently and synergistically to activate IL-8 promoter activity in a stimulus- and cell type-specific manner (29). Although NF-κB has been suggested to be essential for IL-8 induction, there are exceptions, as in the case of H2O2 induction of IL-8 in A549 cells (42). Our data demonstrating a modest increase in NF-κB binding activity in ESAT-6-treated cells suggested that IL-8 induction could occur independently of NF-κB. However, NF-κB is essential for basal promoter activity. AP-1 and NFIL-6 are known to synergize with NF-κB for maximal induction of IL-8 (29).
We also found that ESAT-6 treatment increased the half-life of IL-8 mRNA, indicating that enhanced mRNA stability contributes to the induction of IL-8 levels. Control of IL-8 mRNA degradation is an important mechanism for the regulation of IL-8 levels, and the p38 MAPK pathway and AU-rich elements located in the 3′-untranslated region have been found to be involved in the control of IL-8 mRNA stability (43). Activation of p38 MAPK by ESAT-6 could suggest a role for the p38 MAPK signaling pathway in the stabilization of IL-8 mRNA to increase IL-8 expression. A variety of agents, such as nitric oxide (44, 45), adenovirus (46), and Shiga toxin (47), increase IL-8 mRNA stability to increase IL-8 mRNA levels in epithelial cells, fibroblasts, and monocytic cells.
ESAT-6 associates with lung epithelial cells by binding to laminin (3) and inhibits IL-12 production in macrophages via TLR signaling by binding to TLR2 (48). The identities of ESAT-6-interacting protein(s) on lung epithelial cells and the ensuing signaling events are not known. We investigated the potential roles of signal transduction pathways involved in the ESAT-6 induction of IL-8 expression. We found that PKC inhibitors reduced IL-8 induction, indicating that ESAT-6 activation of PKC is a key step in the induction of IL-8 expression. Sensitivity to inhibition by bisindolylmaleimide, Go6976, and Go6983 indicated that calcium-dependent conventional protein kinase C enzymes are involved. Sensitivity to Go6976 indicated that the novel and calcium-independent PKCμ might also be involved. Activation of the PKC signaling pathway has been linked to the development of acute and chronic lung diseases, including those that are caused by a variety of environmental and infectious agents (30). Inhalation of occupational dusts, such as asbestos, silica, and allergens present in home and occupational environments, activate PKC signaling that probably contributes to the development of lung diseases (30). Activation or down-regulation of PKC has been implicated in the host inflammatory responses to M. tuberculosis infection (49) or survival of mycobacteria (50), respectively. Mycobacterial cord factor trehalose 6-monomycolate activated protein kinase C, resulting in the production of TNF-α in mouse lung (51). The M. tuberculosis 19-kDa lipoprotein induced ROS production via association with TLR2 and the atypical PKCζ (52). Our data have indicated that ESAT-6 is yet another mycobacterial component that activates PKC to elicit host inflammatory responses.
Our data demonstrated that ESAT-6 activated ERK and p38 and, to a lesser degree, JNK phosphorylation, and pharmacological inhibitors of ERK and p38 but not JNK MAPKs inhibited IL-8 induction, indicating that ERK and p38 MAPK activations are necessary for IL-8 induction. Although additional studies are required to map the exact sequence of signaling pathways, it is likely that PKC activation precedes MAPK activation. Previous studies have shown that activation of PKC by agonists and other agents mediates activation of MAPK. Induction of MUC5Ac by phorbol myristate acetate, a PKC agonist, was mediated via activation of MEK and ERK MAPK signaling pathways (53). Activation of p38 MAPK in epithelial cells is mediated via Src and PKC signaling pathways (54). Lipopolysaccharide induction of c-Jun and c-Fos in tracheal smooth muscle cells was mediated by PKC via activation of ERK, p38, and JNK MAPKs (55). The identities of PKC enzymes involved in the ESAT-6 induction of IL-8 expression remains to be investigated.
We found that ESAT-6-induced IL-8 expression requires ERK and p38 activation and is linked to an increase of AP-1 DNA binding activity. Although the JNK MAPK pathway is a known activator of AP-1, ERK and p38 MAPK pathways also regulate AP-1 activation via control of expression and activations of c-Jun and c-Fos (56–58). The involvement of a particular MAPK signaling pathway(s) in IL-8 induction appears to be dependent on the cell type and the stimulus. Apart from JNK, ERK and p38 MAPK pathways also play major roles in the regulation of IL-8 expression. Cadmium induction of IL-8 expression in airway epithelial cells is mediated by an NF-κB-independent but ERK MAPK-dependent pathway (59). JNK and ERK MAPK were found to control IL-8 induction by YKL-40, a chitinase-like glycoprotein, in bronchial epithelial cells (60). In U937 monocytic cells, Helicobacter pylori induces IL-8 expression primarily via the p38 MAPK pathway and NF-κB activation (61). Our studies indicated that ERK and p38 but not JNK MAPK pathways are required for an increase in AP-1 DNA binding to the IL-8 promoter. Mechanisms underlying ERK and p38 MAPK-mediated increase of AP-1 DNA binding activity remain to be investigated. H441 cells express c-Jun, c-Fos, Jun B, Jun D, Fra 1, and Fra 2 (23, 62), indicating that the AP-1 complex on the IL-8 promoter could be composed of combinations of these proteins. Inhibition of ESAT-6 increase of AP-1 DNA binding activity by PD 98059 and SB 203580 could be due to inhibition of expression and/or activities of one or more components of the AP-1 complex. The identities of AP-1 components targeted by PD 98059 and SB 203580 are not known. In mouse macrophages, H. pylori induces phosphorylation of c-Fos and formation of the AP-1 complex via ERK MAPK activation to induce expression of c-Myc (63). We found that inhibition of the JNK MAPK pathway did not suppress ESAT-6 induction of IL-8 levels. On the contrary, JNK MAPK inhibition enhanced ESAT-6 induction of IL-8 protein but not mRNA levels, suggesting that the effects are exerted at the posttranslational level. Inhibition of the JNK MAPK pathway induces IL-8 levels via enhanced stabilization of IL-8 mRNA in cystic fibrosis lung epithelial cells expressing high levels of tristetrapolin (64).
We found that ESAT-6 rapidly induced superoxide anion (O2⨪) production in cells, as demonstrated by dihydroethidium fluorescence. The associations between PKC and MAPK activation and ROS production indicate an intracellular signaling pathway that mediates ESAT-6 induction of IL-8 expression, although the exact sequence of the signaling events remains to be determined. ROS are composed of partially reduced metabolites of oxygen, such as superoxide anion, hydrogen peroxide, and hydroxyl radical, and are produced intracellularly as byproducts of normal aerobic metabolism by the mitochondrial electron transport chain (65). Elevated levels of ROS can also be produced as a consequence of exposure to environmental agents, or under pathological conditions, by the actions of enzymes, such as NADPH oxidase, 5-lipoxygenase, xanthine oxidase, and nitric-oxide synthases (66). ROS serve as important second messengers to control a wide range of physiological and pathological processes (66). Elevated ROS activate PKC and MAPK signaling pathways to target NF-κB (67) and AP-1 (68) transcription factor pathways to ultimately alter target gene expression. The major source for ROS and its role in the initiation of signal transduction to ultimately induce IL-8 expression in lung epithelial cells exposed to ESAT-6 remains to be determined.
The roles of intracellular ROS in mediating inflammatory responses to mycobacterial infection are not well understood. Recent studies have demonstrated that M. tuberculosis elicits inflammatory responses in monocytes/macrophages (69) and lung epithelial cells (70) via Toll-like receptor 2 and ROS-mediated activation of MAPK signaling pathways. Specifically, tuberculin purified protein derivative was found to elicit inflammatory responses in monocytes/macrophages via the TLR2-ROS-MAPK signaling pathway (69). Our studies have demonstrated that ESAT-6 is another mycobacterial protein that causes elevated intracellular ROS to induce IL-8 levels. The ability of ESAT-6 to induce IL-8 levels suggests that it could promote initiation of granuloma formation via IL-8-mediated recruitment of T cells, neutrophils, and monocytes. Furthermore, elevated IL-8 can induce release of matrix metalloproteinase-9 from neutrophils (71) for the recruitment of macrophages to contribute to nascent granuloma maturation. ESAT-6 induces epithelial cell matrix MMP-9 to recruit macrophages to promote granuloma formation in a zebrafish model of Mycobacterium marinum infection (4). Administration of neutralizing anti-IL-8 antibody suppressed the development of tuberculin skin reaction by reducing the number of neutrophils and lymphocytes at the injection site in rabbits, indicating that IL-8 is important for granuloma formation (72). ESAT-6 could also synergize cytokine-induced IL-8 production at later stages of infection to contribute to lung injury commonly seen in pulmonary tuberculosis.
ESAT-6 promoted the formation of inflammatory cell aggregates in lungs of mice concomitant with increased KC staining in macrophages and bronchiolar and alveolar type II epithelial cells. Cells in the aggregates had the appearance of epithelioid cells, indicating the identity of cell aggregates as early stage granuloma. Results of animal experiments, namely induction of KC in bronchiolar and alveolar type II epithelial cells and infiltration of inflammatory cells into lungs of mice exposed to ESAT-6, are consistent with the inductive effects of ESAT-6 on IL-8 levels in lung cells in vitro. Consistent with the lack of effects of CFP-10 on IL-8 levels in lung cells in vitro, CFP-10 did not significantly induce KC immunostaining or promote inflammatory cell infiltration into lungs of mice. Together, our in vitro cell culture and in vivo mouse studies indicated that ESAT-6 interacts with lung alveolar type II and bronchiolar epithelial cells to induce IL-8 production, which in turn promotes trafficking of macrophages and other cells to initiate granuloma formation in the lung. Results also demonstrated that ESAT-6 increased KC staining in macrophages, suggesting that increased KC expression by alveolar macrophages could also contribute to trafficking of macrophages and lymphocytes to initiate granuloma formation in lung. Our studies have identified ESAT-6-mediated IL-8 increase in alveolar type II and bronchiolar epithelial cells as a potential pathway for granuloma formation in M. tuberculosis infection, adding to the importance of epithelial cells in the pathogenesis of tuberculosis. Previously, ESAT-6 induction of MMP-9 in epithelial cells was shown to promote granuloma formation in zebrafish (4).
In conclusion, our studies have demonstrated that ESAT-6 induces IL-8 expression in H441 lung epithelial cells by increasing gene transcription and mRNA stability. IL-8 induction was dependent on NF-κB and AP-1 binding and sensitive to PKC and ERK and p38 MAPK inhibitors. ESAT-6 rapidly induced ROS production in cells, and IL-8 induction was inhibited by the hydroxyl radical scavenger dimethylthiourea. Our results have identified a signaling mechanism involving intracellular ROS, PKC, ERK and p38 MAPK, and AP-1/NF-κB pathways for ESAT-6 induction of IL-8; however, the exact sequence of signaling events remains to be elucidated. In agreement with results of our in vitro cell culture studies, ESAT-6 increased KC, a mouse homolog of IL-8, in lung epithelial cells and macrophages concomitant with inflammatory cell aggregate or granuloma formation in mice. ESAT-6 induction of IL-8 could be one of the mechanisms for granuloma formation and lung innate immune responses to tuberculosis and a contributing factor to the pathogenesis of pulmonary tuberculosis.
Acknowledgments
We thank Dr. Peter Barnes for helpful comments and review of the manuscript. We are grateful to Drs. Greg Dooley and Stephen J. Reynolds (Colorado State University, Fort Collins, CO) for determining muramic acid levels in recombinant ESAT-6 preparations by GC-MS/MS.
This work was supported, in whole or in part, by National Institutes of Health Grant AI099345 (to B. S.). This work was also supported by institutional funds (to V. B.).
- KC
- keratinocyte chemoattractant CXCL1
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- HBSS
- Hanks' balanced salt solution.
REFERENCES
- 1. Andersen P., Andersen A. B., Sørensen A. L., Nagai S. (1995) Recall of long-lived immunity to Mycobacterium tuberculosis infection in mice. J. Immunol. 154, 3359–3372 [PubMed] [Google Scholar]
- 2. Sørensen A. L., Nagai S., Houen G., Andersen P., Andersen A. B. (1995) Purification and characterization of a low molecular mass T-cell antigen secreted by Mycobacterium tuberculosis. Infect. Immun. 63, 1710–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kinhikar A. G., Verma I., Chandra D., Singh K. K., Weldingh K., Andersen P., Hsu T., Jacobs W. R., Jr., Laal S. (2010) Potential role for ESAT6 in dissemination of M. tuberculosis via human lung epithelial cells. Mol. Microbiol. 75, 92–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Volkman H. E., Pozos T. C., Zheng J., Davis J. M., Rawls J. F., Ramakrishnan L. (2010) Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science 327, 466–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mishra B. B., Moura-Alves P., Sonawane A., Hacohen N., Griffiths G., Moita L. F., Anes E. (2010) Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol. 12, 1046–1063 [DOI] [PubMed] [Google Scholar]
- 6. Peng H., Wang X., Barnes P. F., Tang H., Townsend J. C., Samten B. (2011) The Mycobacterium tuberculosis early secreted antigenic target of 6 kDa inhibits T cell interferon-γ production through the p38 mitogen-activated protein kinase pathway. J. Biol. Chem. 286, 24508–24518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boggaram V. (2003) Regulation of lung surfactant protein gene expression. Front. Biosci. 8, d751–d764 [DOI] [PubMed] [Google Scholar]
- 8. Martin L. D., Rochelle L. G., Fischer B. M., Krunkosky T. M., Adler K. B. (1997) Airway epithelium as an effector of inflammation. Molecular regulation of secondary mediators. Eur. Respir. J. 10, 2139–2146 [DOI] [PubMed] [Google Scholar]
- 9. Chroneos Z. C., Midde K., Sever-Chroneos Z., Jagannath C. (2009) Pulmonary surfactant and tuberculosis. Tuberculosis 89, S10–S14 [DOI] [PubMed] [Google Scholar]
- 10. Mukaida N. (2003) Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. Am. J. Physiol. Lung Cell Mol. Physiol. 284, L566–L577 [DOI] [PubMed] [Google Scholar]
- 11. Kurashima K., Mukaida N., Fujimura M., Yasui M., Nakazumi Y., Matsuda T., Matsushima K. (1997) Elevated chemokine levels in bronchoalveolar lavage fluid of tuberculosis patients. Am. J. Respir. Crit. Care Med. 155, 1474–1477 [DOI] [PubMed] [Google Scholar]
- 12. Friedland J. S., Hartley J. C., Hartley C. G., Shattock R. J., Griffin G. E. (1995) Inhibition of ex vivo proinflammatory cytokine secretion in fatal Mycobacterium tuberculosis infection. Clin. Exp. Immunol. 100, 233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wickremasinghe M. I., Thomas L. H., Friedland J. S. (1999) Pulmonary epithelial cells are a source of IL-8 in the response to Mycobacterium tuberculosis. Essential role of IL-1 from infected monocytes in a NF-κB-dependent network. J. Immunol. 163, 3936–3947 [PubMed] [Google Scholar]
- 14. Lee H. M., Shin D. M, Jo E. K. (2009) Mycobacterium tuberculosis induces the production of tumor necrosis factor-α, interleukin-6, and CXCL8 in pulmonary epithelial cells through reactive oxygen species-dependent mitogen-activated protein kinase activation. J. Bacteriol. Virol. 39, 1–10 [Google Scholar]
- 15. Lowe D. M., Redford P. S., Wilkinson R. J., O'Garra A., Martineau A. R. (2012) Neutrophils in tuberculosis. Friend or foe? Trends Immunol. 33, 14–25 [DOI] [PubMed] [Google Scholar]
- 16. Nandi B., Behar S. M. (2011) Regulation of neutrophils by interferon-γ limits lung inflammation during tuberculosis infection. J. Exp. Med. 208, 2251–2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang C. T., Cambier C. J., Davis J. M., Hall C. J., Crosier P. S., Ramakrishnan L. (2012) Neutrophils exert protection in the early tuberculous granuloma by oxidative killing of mycobacteria phagocytosed from infected macrophages. Cell Host Microbe 12, 301–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang X., Barnes P. F., Dobos-Elder K. M., Townsend J. C., Chung Y. T., Shams H., Weis S. E., Samten B. (2009) ESAT-6 inhibits production of IFN-γ by Mycobacterium tuberculosis-responsive human T cells. J. Immunol. 182, 3668–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang X., Barnes P. F., Huang F., Alvarez I. B., Neuenschwander P. F., Sherman D. R., Samten B. (2012) Early secreted antigenic target of 6 kDa protein of Mycobacterium tuberculosis primes dendritic cells to stimulate Th17 and inhibit Th1 immune responses. J. Immunol. 189, 3092–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boggaram V., Margana R. K. (1994) Developmental and hormonal regulation of surfactant protein C (SP-C) gene expression in fetal lung. Role of transcription and mRNA stability. J. Biol. Chem. 269, 27767–27772 [PubMed] [Google Scholar]
- 21. Greenberg M. E., Bender T. P. (1997) Identification of newly transcribed RNA. in Current Protocols in Molecular Biology (Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K., eds) pp. 4.10.11–14.10.11, John Wiley & Sons, Inc., New York: [DOI] [PubMed] [Google Scholar]
- 22. Schreiber E., Matthias P., Müller M. M., Schaffner W. (1989) Rapid detection of octamer binding proteins with “mini-extracts,” prepared from a small number of cells. Nucleic Acids Res. 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berhane K., Boggaram V. (2001) Identification of a novel DNA regulatory element in the rabbit surfactant protein B (SP-B) promoter that is a target for ATF/CREB and AP-1 transcription factors. Gene 268, 141–151 [DOI] [PubMed] [Google Scholar]
- 24. Harboe M., Oettinger T., Wiker H. G., Rosenkrands I., Andersen P. (1996) Evidence for occurrence of the ESAT-6 protein in Mycobacterium tuberculosis and virulent Mycobacterium bovis and for its absence in Mycobacterium bovis BCG. Infect. Immun. 64, 16–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Abdallah A. M., Gey van Pittius N. C., Champion P. A., Cox J., Luirink J., Vandenbroucke-Grauls C. M., Appelmelk B. J., Bitter W. (2007) Type VII secretion. Mycobacteria show the way. Nat. Rev. Microbiol. 5, 883–891 [DOI] [PubMed] [Google Scholar]
- 26. Hsu T., Hingley-Wilson S. M., Chen B., Chen M., Dai A. Z., Morin P. M., Marks C. B., Padiyar J., Goulding C., Gingery M., Eisenberg D., Russell R. G., Derrick S. C., Collins F. M., Morris S. L., King C. H., Jacobs W. R., Jr. (2003) The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc. Natl. Acad. Sci. U.S.A. 100, 12420–12425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Derrick S. C., Morris S. L. (2007) The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol. 9, 1547–1555 [DOI] [PubMed] [Google Scholar]
- 28. Hoffmann E., Dittrich-Breiholz O., Holtmann H., Kracht M. (2002) Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 72, 847–855 [PubMed] [Google Scholar]
- 29. Roebuck K. A. (1999) Regulation of interleukin-8 gene expression. J. Interferon Cytokine Res. 19, 429–438 [DOI] [PubMed] [Google Scholar]
- 30. Dempsey E. C., Cool C. D., Littler C. M. (2007) Lung disease and PKCs. Pharmacol. Res. 55, 545–559 [DOI] [PubMed] [Google Scholar]
- 31. Dempsey E. C., Newton A. C., Mochly-Rosen D., Fields A. P., Reyland M. E., Insel P. A., Messing R. O. (2000) Protein kinase C isozymes and the regulation of diverse cell responses. Am. J. Physiol. Lung Cell Mol. Physiol. 279, L429–L438 [DOI] [PubMed] [Google Scholar]
- 32. Cobb M. H., Goldsmith E. J. (1995) How MAP kinases are regulated. J. Biol. Chem. 270, 14843–14846 [DOI] [PubMed] [Google Scholar]
- 33. Salh B. S., Martens J., Hundal R. S., Yoganathan N., Charest D., Mui A., Gómez-Muñoz A. (2000) PD98059 attenuates hydrogen peroxide-induced cell death through inhibition of Jun N-terminal kinase in HT29 cells. Mol. Cell Biol. Res. Commun. 4, 158–165 [DOI] [PubMed] [Google Scholar]
- 34. Sen C. K., Packer L. (1996) Antioxidant and redox regulation of gene transcription. FASEB J. 10, 709–720 [DOI] [PubMed] [Google Scholar]
- 35. Peshavariya H. M., Dusting G. J., Selemidis S. (2007) Analysis of dihydroethidium fluorescence for the detection of intracellular and extracellular superoxide produced by NADPH oxidase. Free Radic. Res. 41, 699–712 [DOI] [PubMed] [Google Scholar]
- 36. Pym A. S., Brodin P., Brosch R., Huerre M., Cole S. T. (2002) Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol. Microbiol. 46, 709–717 [DOI] [PubMed] [Google Scholar]
- 37. Volkman H. E., Clay H., Beery D., Chang J. C., Sherman D. R., Ramakrishnan L. (2004) Tuberculous granuloma formation is enhanced by a mycobacterium virulence determinant. PLoS Biol. 2, e367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Davis J. M., Ramakrishnan L. (2009) The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 136, 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stadnyk A. W. (1994) Cytokine production by epithelial cells. FASEB J. 8, 1041–1047 [DOI] [PubMed] [Google Scholar]
- 40. Cheng D. S., Han W., Chen S. M., Sherrill T. P., Chont M., Park G. Y., Sheller J. R., Polosukhin V. V., Christman J. W., Yull F. E., Blackwell T. S. (2007) Airway epithelium controls lung inflammation and injury through the NF-κB pathway. J. Immunol. 178, 6504–6513 [DOI] [PubMed] [Google Scholar]
- 41. DiGiuseppe Champion P. A., Cox J. S. (2007) Protein secretion systems in Mycobacteria. Cell Microbiol. 9, 1376–1384 [DOI] [PubMed] [Google Scholar]
- 42. Lakshminarayanan V., Drab-Weiss E. A., Roebuck K. A. (1998) H2O2 and tumor necrosis factor-α induce differential binding of the redox-responsive transcription factors AP-1 and NF-κB to the interleukin-8 promoter in endothelial and epithelial cells. J. Biol. Chem. 273, 32670–32678 [DOI] [PubMed] [Google Scholar]
- 43. Winzen R., Kracht M., Ritter B., Wilhelm A., Chen C. Y., Shyu A. B., Müller M., Gaestel M., Resch K., Holtmann H. (1999) The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 18, 4969–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sparkman L., Boggaram V. (2004) Nitric oxide increases IL-8 gene transcription and mRNA stability to enhance IL-8 gene expression in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 287, L764–L773 [DOI] [PubMed] [Google Scholar]
- 45. Ma P., Cui X., Wang S., Zhang J., Nishanian E. V., Wang W., Wesley R. A., Danner R. L. (2004) Nitric oxide post-transcriptionally up-regulates LPS-induced IL-8 expression through p38 MAPK activation. J. Leukoc. Biol. 76, 278–287 [DOI] [PubMed] [Google Scholar]
- 46. Leland Booth J., Metcalf J. P. (1999) Type-specific induction of interleukin-8 by adenovirus. Am. J. Respir. Cell Mol. Biol. 21, 521–527 [DOI] [PubMed] [Google Scholar]
- 47. Thorpe C. M., Smith W. E., Hurley B. P., Acheson D. W. (2001) Shiga toxins induce, superinduce, and stabilize a variety of C-X-C chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect. Immun. 69, 6140–6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pathak S. K., Basu S., Basu K. K., Banerjee A., Pathak S., Bhattacharyya A., Kaisho T., Kundu M., Basu J. (2007) Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat. Immunol. 8, 610–618 [DOI] [PubMed] [Google Scholar]
- 49. Yadav M., Clark L., Schorey J. S. (2006) Macrophage's proinflammatory response to a mycobacterial infection is dependent on sphingosine kinase-mediated activation of phosphatidylinositol phospholipase C, protein kinase C, ERK1/2, and phosphatidylinositol 3-kinase. J. Immunol. 176, 5494–5503 [DOI] [PubMed] [Google Scholar]
- 50. Chaurasiya S. K., Srivastava K. K. (2009) Downregulation of protein kinase C-α enhances intracellular survival of Mycobacteria. Role of PknG. BMC Microbiol. 9, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sueoka E., Nishiwaki S., Okabe S., Iida N., Suganuma M., Yano I., Aoki K., Fujiki H. (1995) Activation of protein kinase C by mycobacterial cord factor, trehalose 6-monomycolate, resulting in tumor necrosis factor-α release in mouse lung tissues. Jpn. J. Cancer Res. 86, 749–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shin D. M., Yang C. S., Lee J. Y., Lee S. J., Choi H. H., Lee H. M., Yuk J. M., Harding C. V., Jo E. K. (2008) Mycobacterium tuberculosis lipoprotein-induced association of TLR2 with protein kinase Cζ in lipid rafts contributes to reactive oxygen species-dependent inflammatory signalling in macrophages. Cell Microbiol. 10, 1893–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hewson C. A., Edbrooke M. R., Johnston S. L. (2004) PMA induces the MUC5AC respiratory mucin in human bronchial epithelial cells, via PKC, EGF/TGF-α, Ras/Raf, MEK, ERK and Sp1-dependent mechanisms. J. Mol. Biol. 344, 683–695 [DOI] [PubMed] [Google Scholar]
- 54. Hofmann M., Zaper J., Bernd A., Bereiter-Hahn J., Kaufmann R., Kippenberger S. (2004) Mechanical pressure-induced phosphorylation of p38 mitogen-activated protein kinase in epithelial cells via Src and protein kinase C. Biochem. Biophys. Res. Commun. 316, 673–679 [DOI] [PubMed] [Google Scholar]
- 55. Lin W. N., Luo S. F., Lin C. C., Hsiao L. D., Yang C. M. (2009) Differential involvement of PKC-dependent MAPKs activation in lipopolysaccharide-induced AP-1 expression in human tracheal smooth muscle cells. Cell. Signal. 21, 1385–1395 [DOI] [PubMed] [Google Scholar]
- 56. Leppä S., Saffrich R., Ansorge W., Bohmann D. (1998) Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. EMBO J. 17, 4404–4413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Loesch M., Zhi H. Y., Hou S. W., Qi X. M., Li R. S., Basir Z., Iftner T., Cuenda A., Chen G. (2010) p38γ MAPK cooperates with c-Jun in trans-activating matrix metalloproteinase 9. J. Biol. Chem. 285, 15149–15158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen W., Bowden G. T. (1999) Activation of p38 MAP kinase and ERK are required for ultraviolet-B induced c-fos gene expression in human keratinocytes. Oncogene 18, 7469–7476 [DOI] [PubMed] [Google Scholar]
- 59. Cormet-Boyaka E., Jolivette K., Bonnegarde-Bernard A., Rennolds J., Hassan F., Mehta P., Tridandapani S., Webster-Marketon J., Boyaka P. N. (2012) An NF-κB-independent and Erk1/2-dependent mechanism controls CXCL8/IL-8 responses of airway epithelial cells to cadmium. Toxicol. Sci. 125, 418–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tang H., Sun Y., Shi Z., Huang H., Fang Z., Chen J., Xiu Q., Li B. (2013) YKL-40 induces IL-8 expression from bronchial epithelium via MAPK (JNK and ERK) and NF-κB pathways, causing bronchial smooth muscle proliferation and migration. J. Immunol. 190, 438–446 [DOI] [PubMed] [Google Scholar]
- 61. Hisatsune J., Nakayama M., Isomoto H., Kurazono H., Mukaida N., Mukhopadhyay A. K., Azuma T., Yamaoka Y., Sap J., Yamasaki E., Yahiro K., Moss J., Hirayama T. (2008) Molecular characterization of Helicobacter pylori VacA induction of IL-8 in U937 cells reveals a prominent role for p38MAPK in activating transcription factor-2, cAMP response element binding protein, and NF-κB activation. J. Immunol. 180, 5017–5027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vuong H., Patterson T., Adiseshaiah P., Shapiro P., Kalvakolanu D. V., Reddy S. P. (2002) JNK1 and AP-1 regulate PMA-inducible squamous differentiation marker expression in Clara-like H441 cells. Am. J. Physiol. Lung Cell Mol. Physiol. 282, L215–L225 [DOI] [PubMed] [Google Scholar]
- 63. Asim M., Chaturvedi R., Hoge S., Lewis N. D., Singh K., Barry D. P., Algood H. S., de Sablet T., Gobert A. P., Wilson K. T. (2010) Helicobacter pylori induces ERK-dependent formation of a phospho-c-Fos c-Jun activator protein-1 complex that causes apoptosis in macrophages. J. Biol. Chem. 285, 20343–20357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bhattacharyya S., Gutti U., Mercado J., Moore C., Pollard H. B., Biswas R. (2011) MAPK signaling pathways regulate IL-8 mRNA stability and IL-8 protein expression in cystic fibrosis lung epithelial cell lines. Am. J. Physiol. Lung Cell Mol. Physiol. 300, L81–L87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Turrens J. F. (2003) Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thannickal V. J., Fanburg B. L. (2000) Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 279, L1005–L1028 [DOI] [PubMed] [Google Scholar]
- 67. Martindale J. L., Holbrook N. J. (2002) Cellular response to oxidative stress. Signaling for suicide and survival. J. Cell Physiol. 192, 1–15 [DOI] [PubMed] [Google Scholar]
- 68. Mossman B. T., Lounsbury K. M., Reddy S. P. (2006) Oxidants and signaling by mitogen-activated protein kinases in lung epithelium. Am. J. Respir. Cell Mol. Biol. 34, 666–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang C. S., Shin D. M., Lee H. M., Son J. W., Lee S. J., Akira S., Gougerot-Pocidalo M. A., El-Benna J., Ichijo H., Jo E. K. (2008) ASK1-p38 MAPK-p47phox activation is essential for inflammatory responses during tuberculosis via TLR2-ROS signalling. Cell Microbiol. 10, 741–754 [DOI] [PubMed] [Google Scholar]
- 70. Lee H. M., Shin D. M., Kim K. K., Lee J. S., Paik T. H., Jo E. K. (2009) Roles of reactive oxygen species in CXCL8 and CCL2 expression in response to the 30-kDa antigen of Mycobacterium tuberculosis. J. Clin. Immunol. 29, 46–56 [DOI] [PubMed] [Google Scholar]
- 71. Chakrabarti S., Patel K. D. (2005) Regulation of matrix metalloproteinase-9 release from IL-8-stimulated human neutrophils. J. Leukoc. Biol. 78, 279–288 [DOI] [PubMed] [Google Scholar]
- 72. Larsen C. G., Thomsen M. K., Gesser B., Thomsen P. D., Deleuran B. W., Nowak J., Skødt V., Thomsen H. K., Deleuran M., Thestrup-Pedersen K. (1995) The delayed-type hypersensitivity reaction is dependent on IL-8. Inhibition of a tuberculin skin reaction by an anti-IL-8 monoclonal antibody. J. Immunol. 155, 2151–2157 [PubMed] [Google Scholar]