

Background: PmrD binds to phospho-PmrA and sustains its phosphorylation state.
Results: Phospho-PmrA interacts with PmrD via several specific intermolecular interactions.
Conclusion: A steric inhibition mechanism was proposed for protecting phospho-PmrA against dephosphorylation.
Significance: This work provides novel data revealing how a connector protein protects an activated response regulator.
Keywords: Drug Resistance, Molecular Docking, NMR, Signal Transduction, Site-directed Mutagenesis, Protein-protein Interaction, Receiver Domain, Response Regulator, Site-directed Spin Labeling, Two-component System
Abstract
In bacteria, the two-component system is the most prevalent for sensing and transducing environmental signals into the cell. The PmrA-PmrB two-component system, responsible for sensing external stimuli of high Fe3+ and mild acidic conditions, can control the genes involved in lipopolysaccharide modification and polymyxin resistance in pathogens. In Klebsiella pneumoniae, the small basic connector protein PmrD protects phospho-PmrA and prolongs the expression of PmrA-activated genes. We previously determined the phospho-PmrA recognition mode of PmrD. However, how PmrA interacts with PmrD and prevents its dephosphorylation remains unknown. To address this question, we solved the x-ray crystal structure of the N-terminal receiver domain of BeF3−-activated PmrA (PmrAN) at 1.70 Å. With this structure, we applied the data-driven docking method based on NMR chemical shift perturbation to generate the complex model of PmrD-PmrAN, which was further validated by site-directed spin labeling experiments. In the complex model, PmrD may act as a blockade to prevent phosphatase from contacting with the phosphorylation site on PmrA.
Introduction
For all organisms, sensing and responding to environmental cues is essential to their survival. Compared with eukaryotic signaling cascades, which are mainly involved in mediating signals through specific Ser-, Thr-, and Tyr-phosphorylated residues among the related sub-tracts, in prokaryotic signaling systems, a distinct phosphorylation scheme predominates, the two-component system (TCS)2 (1–3). Hundreds of TCSs have been found in eubacteria, archaea, and a few eukaryotic organisms (4), and the TCS is the most prevalent system in bacteria for transducing external information into the cell and coping with environmental stresses (2, 5). A PmrA-PmrB TCS, dealing with cationic antimicrobial peptides and polymyxin resistance in pathogens (6), is responsible for sensing the external stimuli caused directly or indirectly by excess Fe3+, low Mg2+, and mild acidic environments (7–9). Under this TCS, pathogens can alter the composition of their cell walls to resist neutralization of polymyxin for drug resistance and allow bacterial survival within macrophages by reducing the affinity with cationic antimicrobial peptides (10, 11).
The increasing antibiotic resistance to Klebsiella pneumonia bacteria, a common cause of nosocomial bacterial infections causing pneumonia and urinary tract infections, especially in immunocompromised patients (12), led us to investigate how the virulence and drug resistance persist via the associated TCS. Similar to most response regulators, the PmrA of K. pneumoniae belonging to the OmpR-PhoB family is composed of an N-terminal receiver domain (Fig. 1) and a C-terminal effector/DNA-binding domain. Interestingly, at low Mg2+ concentration, the PhoP-PhoQ system promotes the expression of a small basic protein, PmrD (7), which can prevent the intrinsic dephosphorylation of phospho-PmrA and enhance the expression of PmrA-activated downstream genes (13). Therefore, PmrD is termed a “connector” because it connects the PmrA-PmrB and PhoP-PhoQ systems (14). Besides PmrD, other small-sized proteins that connect TCSs for Escherichia coli include SafA, which connects the PhoP-PhoQ and EvgS-EvgA systems (15); IraM, which connects PhoP-PhoQ and binds to response regulator RssB, thereby preventing the proteins from recruiting targets for degradation (16); and MzrA, which connects CpxR-Cpxa and OmpR-EnvZ systems (17). To date, the complicated mechanisms of cross-regulation among these TCSs are still not clear (14), and no detailed structural information is available about how the phosphorylation site of the response regulator is protected by the connector protein in the interconnecting signal transduction pathways such as PmrD and phospho-PmrA.
FIGURE 1.
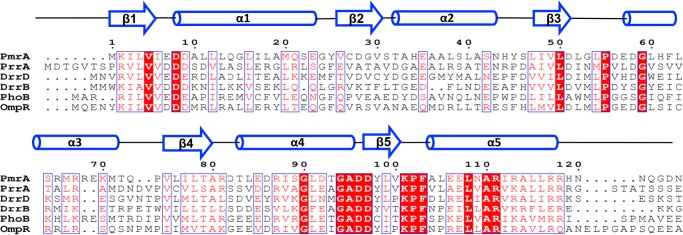
Primary sequence alignment of N-terminal receiver domains of PmrA and five other response regulators. The secondary structural elements of PmrAN are at the top of the figure. Identical residues are shown in white with a red background, and similar residues are shown in red. The alignment was generated by use of CLUSTAL-W (60) and ESPript (61).
Previously, we determined the solution structure for K. pneumoniae PmrD and mapped its binding sites when it bound to PmrAN (18). In this study, we present the structure of the BeF3−-activated N-terminal receiver domain of PmrA (PmrAN) solved by x-ray crystallography. The backbone assignment of PmrAN involved transverse relaxation-optimized spectroscopy-type three-dimensional NMR experiments with 2H,13C,15N-labeled PmrAN. With the chemical shift perturbations of PmrAN when binding with PmrD and structures of PmrD and PmrAN, we used HADDOCK (19) to generate a complex structure of the PmrD-PmrAN heterotetramer. Furthermore, using NMR hydrogen/deuterium exchange (HDX) exchange studies combined with a paramagnetic spin-labeling approach, we further confirmed the reliability of this complex model. We found that PmrAN interacts with PmrD via the residues near the active sites that are widely used by response regulators for intermolecular protein-protein interactions with histidine kinase. The results of this study can help reveal the molecular mechanisms of the interaction mode between connector proteins and phosphorylated response regulators in TCSs.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of the Recombinant Proteins
The coding regions of pmr and PmrAN (N-terminal 123 residues) were PCR-amplified from the genomic DNA of K. pneumoniae. The amplified gene products were cloned as an NdeI/XhoI fragment into the pET29b vector (Novagene). The resulting plasmids (pET-PmrD and pET-PmrAN) allowed for the in-frame fusion of each coding region containing an additional LEHHHHHH sequence at the C terminus to facilitate protein purification. The recombinant proteins were overexpressed in the host E. coli strain BL21(DE3) (Novagen) induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside at 37 °C (for PmrD) or 30 °C (PmrAN). After lysis with use of an M-110S microfluidizer (Microfluidics) and subsequent centrifugation, the overproduced proteins were purified from the soluble fraction by affinity chromatography on His-Bind resin (Novagene) and size-exclusion chromatography with a Superdex75 10/300 GL column. The eluted samples were dialyzed against 50 mm Tris (pH 8.0), 100 mm NaCl, and concentrated by use of Amicon (molecular weight of 5000; Millipore). For isotopically enriched samples, cells were grown in M9 minimal medium (20) containing 1 g/liter 15NH4Cl and 2 g/liter [13C]glucose. PmrD and PmrAN mutants were generated according to QuikChangeTM protocol (Stratagene). The authenticity of the recombinant proteins was verified by SDS-PAGE and mass spectrometry.
Crystallization of BeF3−-activated PmrAN
For crystallization trials, the BeF3−-activated PmrAN was first concentrated to 5 mg/ml in buffer containing 20 mm sodium phosphate, pH 6.0, and 30 mm NaCl with use of a Centricon concentrator (Millipore). The BeF3−-activated PmrAN was obtained by adding 5.3 mm BeCl2 (Fluka), 35 mm NaF, and 7 mm MgCl2. The hanging drop vapor diffusion method was then performed at 298 K by mixing 1 μl BeF3−-activated PmrAN with an equal volume of crystal screening solutions. Hexagonal crystals appeared after 3 days under the condition of 100 mm imidazole and 1 m sodium acetate at pH 6.5. The crystals belong to space group P41 and contain one PmrAN dimer per asymmetric unit. Collection of the cryogenic multiwavelength anomalous dispersion data involved use of an Area Detector Systems Quantum-315 charge-coupled device detector with a synchrotron radiation x-ray source at beamline BL13B1 at the National Synchrotron Radiation Research Center in Taiwan. X-ray diffraction data integration and scaling involved use of the HKL2000 package (21). The extension of initial phases to 1.99 Å and the preliminary auto-model building involved use of RESOLVE (22). XtalView (23) was used to examine electron density maps and for manual model building. Further refinement involved use of CNS (24) and PHENIX (25). After completion, the Rfactor of the final model for all reflections above 1σ between 25.38 and 1.70 Å resolution was refined to 16.5%, and an Rfree value of 17.5% was obtained with 5.2% randomly distributed reflections (26). The structures were visualized and plotted with use of PyMOL (27).
NMR Backbone Resonance Assignments of Free and Bound Forms of BeF3−-activated PmrAN
NMR experiments with 0.3 mm 2H,13C,15N-labeled PmrAN or in complex with PmrD (with a molar ratio of 1:2) in a Shigemi NMR tube (Allison Park, PA) were performed at 310 K with the use of Bruker AVANCE 600 and 800 NMR spectrometers (Bruker, Karlsruhe, Germany) equipped with a triple (1H, 13C, and 15N) resonance cryoprobe including a shielded z-gradient. All heteronuclear NMR experiments were performed as described (28). Sequence-specific assignment of the backbone atoms was achieved by independent connective analysis of transverse relaxation-optimized spectroscopy-type three-dimensional HNCA, HN(CO)CA, CBCA(CO)NH, HNCACB, HNCO, and HN(CA)CO experiments. The chemical shifts of individual spin systems (HN, N, Cα, Cβ, C′, and Hα) were collected manually, and the backbone resonances were assigned by visual inspection. Transverse relaxation-optimized spectroscopy-based NOESY and HNCO spectra were acquired on the triple-labeled PmrAN in complex with PmrD. From the resonance assignments for free PmrAN, most of the resonances in the bound PmrAN could be easily identified because their cross-peaks were well superimposed. For the residues with significant shift perturbations, their backbones were assigned by comparing NOE connectivities and carbonyl carbon chemical shifts between the free and bound forms. All NMR spectra were processed and analyzed by use of Topspin (Bruker Biospin), NMRPipe (29), and NMRView (30) software packages.
HDX Experiments
HDX experiments were initiated by adding D2O (99.9%) to lyophilized protein, which had been prepared at the required pH and buffer conditions. The concentration of 15N,13C,2H-labeled PmrAN was ∼100 μm, and the unlabeled PmrD was ∼200 μm. The 15N-1H selective optimized flip-angle short-transient heteronuclear multiple quantum coherence procedure (31) was used to obtain the 15N-1H correlation spectra with 1024 (t2) and 64 (t1) complex points and 4 scans at 310 K.
Chemical Shift Perturbation Experiments
To map the binding sites on PmrAN with PmrD, we collected 1H-15N HSQC spectra for 2H,15N-labeled PmrAN with unlabeled PmrD at a molar ratio of 1:2. We investigated and ruled out the possibility that the shift changes of PmrD were due to constituents of BeF3−. All spectra processing were analyzed by use of XWIN-NMR (Bruker Biospin) and SPARKY (32). Normalized chemical shift changes were calculated as follows.
The cut-off (0.135 ppm) was set as the S.D. for all chemical shift changes. All residues with values above the cut-off were considered affected by interaction with PmrD.
Complex Structure Determination using HADDOCK
The information-driven docking program HADDOCK (version 2.0) (19) was used to generate a PmrD-PmrAN complex model. The starting structures for docking were an x-ray crystal structure of PmrAN and the 10 lowest energy structures of PmrD, which were refined with the constraints measured from the chemical shift perturbations (18). From NMR titration data, different sets of ambiguous interaction restraints were used to generate the complex model. PmrD contained sets that included the active residues 4, 26, 48, 49, 50, 52, 67, 70, 71, 73, and 74 and passive residues 1, 24, 47, 64, and 66. The PmrAN model contained sets that included the active residues 8, 9, 53, 59, 99, 104, 105, 138, 139, 183, 189, 229, 234, and 236. The active residues were chosen on the basis of the chemical shift perturbation data and high solvent accessibility (>50%). The passive residues are the solvent-accessible surfaces neighboring active residues. During the rigid body energy minimization, 10,000 structures were calculated, and the 199 best solutions based on the intermolecular energy were used for the semi-flexible, simulated annealing, followed by explicit water refinement. The best 199 docked models were clustered by use of a cut-off r.m.s.d. of 3.5 Å. The clusters were ranked by the averaged HADDOCK score of their top 10 complex models (Table 1).
TABLE 1.
Docking and structural statics for the best 199 and best 10 PmrD-PmrAN model structures
| Docking statistics | Ensemble (199 structures) | Best 10 structures |
|---|---|---|
| Haddock score | −144.61 ± 19.86 | −181.352 ± 4.61 |
| Evdw (kcal/mol) | −82.18 ± 8.16 | −89.55 ± 8.22 |
| Eelec (kcal/mol) | −550.71 ± 79.78 | −694.77 ± 52.86 |
| Einter(kcal/mol) | −583.84 ± 80.92 | −738.89 ± 51.84 |
| EAIR (kcal/mol) | 48.24 ± 11.51 | 43.83 ± 3.37 |
| BSA (Å2) | 3143.53 ± 151.53 | 3378.55 ± 157.648 |
| r.m.s.d. from lowest energy structure (Å) | 0.984 | 0.847 ± 0.33 |
| No. of AIR violations > 0.3 Å | 1.23 ± 0.47 | 1.1 ± 0.3 |
| Deviations from idealized geometry | ||
| r.m.s.d. for bond angles | 0.5° | 0.5° |
| r.m.s.d. for bond lengths (Å) | 0.004 | 0.004 |
| Ramachandran analysis, residues in | ||
| Most favored regions (%) | 82.2 | 82.9 |
| Additionally allowed regions (%) | 15.8 | 14.9 |
| Generously allowed regions (%) | 1.4 | 1.4 |
| Disallowed regions (%) | 0.6 | 0.8 |
Site-directed Spin Labeling and Paramagnetic Relaxation Enhancement (PRE) Experiments
PmrDC54 constructs containing a single cysteine residue were generated by use of site-directed mutagenesis to convert existing extra cysteine residues to serine. Mutations were confirmed by DNA sequencing over the entire open reading frame and were found to be properly folded and capable of binding BeF3−-activated PmrAN as for wild-type PmrD. Purified mutants were modified with the thio-specific spin-label reagent MTSL ((1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-methyl) methanethiosulfonate)) purchased from Affymetrix-Anatrace. The protein solution was added to a 20-fold molar excess of MTSL solution dissolved in acetone. The spin-labeling reactions were performed in the dark at room temperature overnight. Excess MTSL was removed by extensive dialysis, and the sample was concentrated to 150 μm. For NMR HSQC experiments, the complex was prepared with a molar ratio of 1:2 of 15N,13C,2H triple-labeled PmrAN:PmrDC54 with MTSL or 15N,13C,2H triple-labeled PmrD: PmrAN D56C with MTSL. All experiments were repeated after the spin label was reduced with 5-fold excess concentrated ascorbic acid and DTT (2–5 μl from a concentrated stock; dilution < 1.25%) to NMR samples. Samples were placed at 25 °C for at least 1 h after the addition of reducing agent. The intensity ratios of paramagnetic versus diamagnetic spectra were obtained from the peak heights and further normalized before distance constraints were converted.
RESULTS
Structure of PmrAN and Dimer Interface
Beryllium fluoride, a phosphoryl analog (33), was used to produce an activated form of PmrAN for structural studies. The structure of PmrAN was solved with experimental phases from molecular replacement data at 1.70 Å. The initial phases of PmrAN were solved with use of the molecular replacement software MOLREP from the CCP4 suite (34). The overall structure of PmrAN adopts a typically (βα)5 topology and follows the signature CheY-like α/β-fold architecture of receiver domains shared by all response regulators (35). Alternating β-strands and α-helices in the primary structure fold into a central five-stranded parallel β-sheet surrounded by two α-helices on one side and three on the other side (Fig. 2A). The asymmetric unit has only one molecule that forms a dimer with another crystallographic symmetry-related molecule using the face formed by helix α4, strand β5, and helix α5 (Fig. 2A). The presence of a large number of intra- and intermolecular interactions involving hydrophobic and polar residues was revealed by analysis of the interface. The α4 helix of one monomer packs against the α5 helix of the other monomer through a hydrophobic patch formed between Leu-84 (α4), Ile-88 (α4), and Leu-91 (α5) and the aliphatic portions of Ala-104 (α4), Ala-110 (α5), and Ala-114 (α5) side chains (Fig. 2B). The interface is further stabilized by a number of side chains involved in attributive salt bridges, especially toward the center region, which creates an extensive network that surrounds the hydrophobic packing between the α4 and α5 helices. Intermolecular salt bridges are formed between Arg-87(α4)–Glu-107(α5), Asp-92(α4)–Arg-113(α5), and Asp-97(β5)–Arg-111(α5) at the outer sides (Fig. 2C). The x-ray structures of other members of the activated form of receiver domains in the OmpR-PhoB subfamily were reported previously, including PhoB and PhoP from E. coli and DrrB and DrrD from Thermotoga maritime (36–38). In all cases, the receiver domains form a dimer with an interface at α4-β5-α5, which reveals a common dimerization mechanism on activation with members of the OmpR-PhoB subfamily.
FIGURE 2.

X-ray crystal structure of PmrAN. A, ribbon structure of PmrAN shows the formation of a dimer with the interface at α4-β5-α5. The phosphate analog, BeF3−, is shown as a ball and stick figure in magenta, and Mg2+ is represented by a green sphere. B, hydrophobic interactions identified at the dimer interface. A hydrophobic patch (spheres) brings helices α4 (Leu-84, Leu-91, and Ile-88 colored in orange) and α5 (Ala-104, Ala-110, and Ala-114 colored in purple) together. C, the dimer interface is stabilized by an extensive network of salt bridges, Arg-87(α4)–Glu-107(α5), Asp-92(α4)–Arg-113(α5), and Asp-97(β5)–Arg-111(α5). D, an extended view of the active site. The Mg2+ is octahedrally coordinated to Asp-51, Gly-53, Asp-8, BeF3−, and two water molecules. The BeF3− moiety contacts with the side chain oxygen atom of Thr-79, the Nζ atom of Lys-101 and the backbone nitrogen atoms of Gly-53, Leu-52, and Ala-80. Carbon, oxygen, and nitrogen atoms are shown in green, red, and blue, respectively. E, active site electron density map (Fo − Fc) of the BeF3−-activated PmrAN calculated at 2σ is shown in gray within 1.7 Å of the residues depicted in sticks.
Coordination of the Active Site in PmrAN
PmrA is phosphorylated at Asp51, the residue that corresponds to the conserved phosphorylation site shared by the receiver domains of response regulators (39). In the crystal structure, Mg2+ binds to F1 of BeF3−, the side chain carboxyl oxygens of Asp-8 and Asp-51; the main chain carbonyl oxygen of Gly-53, a water molecule hydrogen-bonded to Asp-8 and another water molecule hydrogen-bonded to Asp-9, thus satisfying the octahedral coordination of Mg2+ (Fig. 2D). The BeF3− is non-covalently bound to Asp-51 centered within the active site. The conserved Lys-101 is involved in salt bridges with Asp-9 and F3 of BeF3−. The conserved Thr-79 forming a hydrogen bond with the F2 of BeF3−, involved in the activation mechanism in other response regulators, is in an inward orientation. The rotation of Thr-79 is accompanied by a rotation of the backbone N of the next residue, Ala-80, which forms a hydrogen bond with F3 of BeF3−. Ser/Thr and Phe/Tyr located in the β4-α4 loop and β5 (Thr-79 and Tyr-98 in PmrA, Fig. 2D) are the signature aromatic “switch” residues involved in rearrangements associated with phosphorylation (3, 40). In the PmrAN structure, Thr-79 is oriented toward the active site to coordinate with BeF3−, whereas Tyr-98 adopts an inward position, forming a hydrogen bond with the main chain carbonyl oxygen of Arg-81 situated at the middle of the β4-α4 loop. Crystallographic data are summarized in Table 2, and electron density for the active site of the refined model is shown in Fig. 2E.
TABLE 2.
Data collection and refinement statistics
| Data collection | |
|---|---|
| Space group | P41 |
| Cell dimensions | a = b 49.99 and c = 145.86 Å |
| Wavelength (Å) | 0.97315 |
| Resolution (Å) | 1.7 |
| Redundancy | 12.2 (11.9)a |
| I/σ(I) | 40.4 (6.7)a |
| Completeness (%) | 97.2 (94.5)a |
| Rmerge (%) | 6.6 (41.7) |
| Refinement | |
| Resolution (Å) | 25.38-1.70 |
| No. of reflections | 38,239 |
| Rwork/Rfree (%) | 16.5/17.5 |
| No. of atoms | |
| Protein | 2,232 |
| Ligands | 10 |
| Water | 424 |
| B factors | |
| Protein | 18.2 |
| Ligands | 14.6 |
| Water | 32.6 |
| r.m.s.d. | |
| Bond lengths (Å) | 0.006 |
| Bond angles | 1.09° |
| Ramachandran favored (%) | 99 |
| Ramachandran outliers (%) | 0 |
a Numbers in parentheses refer to the highest resolution shell.
1H, 13C, and 15N Assignments and Secondary Structure Comparisons between Solution and X-ray PmrAN Structures
We could not grow good crystals of PmrAN in complex with PmrD and therefore used NMR techniques to obtain detailed structural information and gain insight into the protein-protein interactions (41). Two-dimensional 15N-1H HSQC spectrum of PmrAN is well dispersed, which indicates a well folded structure. Following a standard sequential assignment procedure, all of the observed cross-peaks in the HSQC spectrum were completely assigned, which correspond to 97% of 1HN–15N pairs (115 of 119). The four unassigned residues were His-44, Leu-105, Gln-122, and Gly-123. Their cross-peaks disappeared, presumably because of solvent exposure. For the other backbone atoms, 98% of 1Hα resonances (116 of 119) and 98% of 13C′, 13Cα, and 13Cβ resonances (351 of 360) were assigned. Hα of Arg-87 was shifted up-field at 3.54 ppm, which is likely due to the shielding effect by Tyr-98 as seen in the crystal structure. The chemical shift index (29) based on Hα, Cα, Cβ, and C′ chemical shifts showed that PmrAN contains four β-strands (residues 2–7, 47–51, 74–79, and 95–100) and five α-helices (residues 10–22, 34–41, 59–70, 84–92, and 104–120), which are similar to those in the crystal structure.
Interaction between PmrD and PmrAN
Previously, we studied the binding stoichiometry of the PmrD-PmrAN complex in the presence of BeF3− (18). The binding ratio of 1:1 was determined by size-exclusion chromatography and 1H transverse relaxation time measurements. The equilibrium dissociation constant of ∼1 μm was measured by surface plasma resonance. To further identify the sites of interaction of PmrAN with PmrD, we performed chemical shift perturbation experiments, in which the chemical shift changes on the 15N,1HN correlation plot of the BeF3−-activated PmrAN were detected with the addition of 2-fold unlabeled PmrD (Fig. 3A). The addition of PmrD caused significant chemical shift changes, the intermediate exchange line broadening on several residues, and emerging cross-peaks for backbone 15N and 1HN resonances. At a molar ratio of 1 to 0.9, two separate NMR signals, one for the free and the other for the bound form, were clearly seen for some residues such as Asp-9, Val-49, Asp-56, and Gln-73. Therefore, we suggested that the interaction belongs to the slow exchange regime on the NMR timescale. The backbone resonances of Lys-2 and Leu-12 were missing because of peak broadening beyond detection. Leu-105 was not observed in the free form but appeared in the presence of PmrD, which suggests lower solvent accessibility of the amide proton of Leu-105 in the complex. We found no significant intermolecular NOEs in the three-dimensional NOESY-HSQC spectra, which may be due to weak binding of the protein complex. A total of 14 residues of PmrAN showed significant perturbations with the addition of a 2-fold molar ratio of PmrD (> 0.15 ppm) and were labeled on the chemical shift perturbation bar plot (Fig. 3B). Mapping these residues onto the structure showed that most of them are located near the phosphorylation site of PmrAN, which comprises the β1-α1 loop, N-terminal α1, β3-α3 loop, N-terminal α3, C-terminal β4, β4-α4 loop, C-terminal β5, β5-α5 loop and N-terminal α5 (Fig. 3C).
FIGURE 3.

The induced chemical shift perturbation of 2H,15N,13C-labeled PmrAN with the presence of PmrD. A, a section of overlaid 1H,15N TROSY-HSQC spectra of free PmrAN (cyan) binding to 2-fold molar ratio of unlabeled PmrD (red). The residues showing significant chemical shift perturbations are labeled with a one-letter code for amino acids with the residue number. B, graph representing the weighted average of the chemical shift differences between the free and PmrD-bound PmrAN. The secondary structural elements of PmrAN are illustrated on top. C, ribbon structure of PmrAN with the residues showing weighted chemical shift perturbations > 0.135 ppm are in red.
HDX Study and Solvent Accessibility between the Free and Bound Forms of PmrAN
HDX rates can provide information about the solvent accessibility for different backbone amide protons. Here we used HDX to map the binding interface in PmrD-PmrAN. First, we used the free form of 15N-labeled PmrAN for the HDX study and found that ∼41 cross-peaks were water-inaccessible (Fig. 4A). Mapping these residues onto the PmrAN structure showed that they were mainly located on the central five-stranded parallel β-sheet or the interface mediated by the α4-β5-α5 face but not on the surface near the phosphorylation site. Next, we added unlabeled PmrD into 15N-labeled PmrAN and repeated the same experiment under the same conditions and compared the results with the HDX data for free PmrAN. Most of the water-inaccessible residues were similar to those in the free PmrAN (Fig. 4B), so these residues are not involved in binding with PmrD. However, three residues, Leu-11, Phe-103, and Arg-81, showed a lower water-accessible rate in the presence of PmrD (Fig. 4B), presumably because of the steric occlusion effect of PmrD. Interestingly, Leu-11, Arg-81, and Phe-103 are all close to the phosphorylation site and are consistent with the binding interface determined by chemical shift perturbation (Fig. 4C).
FIGURE 4.

Shown is a hydrogen/deuterium exchange study on the free (A) and PmrD-bound (B) PmrAN. The overlay of 1H,15N TROSY-HSQC spectra of protein samples incubated in 90% H2O, 10% D2O (black) or in 100% D2O for 5 min (red). Three PmrAN residues, which are water-inaccessible in the bound form only, are labeled in blue. C, ribbon representation of PmrAN monomer. The residues that are water-inaccessible in the free PmrAN are colored in blue, and the three residues that were water-inaccessible only in the PmrD-bound PmrAN are colored and labeled in red.
Modeling of the PmrD-PmrAN Complex
With formation of the PmrD-PmrAN complex, we observed significant broadening of NMR signals for both PmrD and PmrAN, which may be due to the slower tumbling of the complex and/or a contribution from intermediate timescale chemical exchange broadening. These significantly broadened NMR resonances limited the acquisition of intermolecular NOEs critical for determining the structure of multiprotein complexes, although this effect aided in identifying interfacial residues. Because of a lack of intermolecular NOEs, we generated the complex model of a PmrD-PmrAN heterotetramer using the experimental data-driven docking method of HADDOCK (19). The generated complex model is dominated by intermolecular interactions between residues located on the active site pockets of PmrAN and the solvent-exposed face of the β-barrel of PmrD (Fig. 5A). Seven intermolecular hydrogen bonds were identified: Ser-23(HG)–Asp-9(OD1), Ser-23(HG)–Asp-9(OD2), Ala-49(O)–Leu-105(HN), Asn-67(OD1)–Thr-83(HN), His-70(HD1)–Gly-53(O), His-70(HE2)–Asp-9(OD1), and Arg-75(HH12)–Glu-85(OE2) (Fig. 5B). In addition, two intermolecular salt bridges were found between Lys-64–Glu-31 with 2-fold symmetry (Fig. 5B). Therefore, the hydrogen bonds and salt bridges for each partner interact with appropriately charged residues on the corresponding interface, providing a specificity of structural arrangement for the interaction between PmrD and PmrAN.
FIGURE 5.

A, HADDOCK-derived structural model of PmrD-PmrAN complex, with PmrD shown in beige and light blue, and the PmrAN dimer is shown in lime green. B, intermolecular interactions between the two PmrDs (top) and between PmrD and PmrAN (bottom) in the complex structure. C, summary of the intermolecular interactions between the two PmrDs (top) and between PmrD and PmrAN (bottom).
PRE Effect of Site-directed Spin Labeling on PmrDC54 and PmrAN D56C
To double-confirm the orientations of the PmrD-PmrAN complex model, we used site-directed spin labeling with a nitroxide spin-label compound, MTSL, covalently attached to a Cys residue (42) for PRE experiments, which can be used as an independent biophysical method to obtain intermolecular long distance (<30 Å) information. PmrD contains three Cys residues (Cys-17, Cys-35, and Cys-54), and PmrAN contains only one Cys residue (Cys-27). To determine which Cys residues could be labeled under the conditions described under “Experimental Procedures,” we initially covalently linked the stable mutant 15N-labeled PmrD C35S and wild-type 15N-labeled PmrAN using MTSL. The backbone HN of Cys-54 in PmrD C35S was significantly shifted as compared with PmrD, so Cys-54 was able to bind with MTSL. However, Cys-27 of PmrAN did not show any shift perturbation on adding MTSL, so Cys-27 is unlikely to bind to MTSL. We therefore produced two mutants, C17S/C35S on PmrD (PmrDC54) and D56C on PmrAN (PmrAN D56C), for PRE experiments. Spin labeling did not change the protein-protein interaction because the 1H-15N HSQC spectrum of PmrAN in complex with MTSL-labeled PmrDC54 with a reduced proxyl group was similar to that of PmrAN in complex with unlabeled PmrD (Fig. 6A) and vice versa. The unpaired electron of MTSL caused line-broadening effects on nearby residues in a distance-dependent manner. Line-broadening effects on the 15N-labeled PmrAN were observed in the complexes with MTSL-labeled PmrDC54 (Fig. 6A). The superimposed spectra of the reduced (black) and oxidized (red) states showed significant R2 relaxation enhancement effects. With the spin label on Cys-54 of PmrD, the signals on several residues of PmrAN, such as Asp-82, Leu-84, Val-100, Phe-103, and Leu-105, were significantly reduced in the oxidized state (Fig. 6, A and B). Residues Asp-82, Leu-84, Val-100, and Phe-103 were all close to the active site of PmrAN, as seen in the crystal structure. With MTSL attached to D56C of PmrAN, three peaks corresponding to PmrD residues Gly-24–Ala-25 and the side chain amide peak of Trp-3 were severely attenuated (data not shown). These data suggest that the interaction brings the binding surface of PmrD (the open β-barrel area) (18) and the active site of PmrAN close to each other. In conclusion, the observed intermolecular PRE data are generally consistent with the HADDOCK-derived complex structure (Fig. 6C) and hence can provide an independent biophysical method to confirm the long range distance information between corresponding regions of the complex model.
FIGURE 6.

PRE study on PmrD-PmrAN complex. A, an expanded region of superimposed 1H-15N HSQC spectra of 15N-labeled PmrAN in complex with PmrDC54 with (paramagnetic state, in red) or without (diamagnetic state, in black) MTSL spin labeling. The residues that disappeared in the paramagnetic state are labeled. B, a bar plot of intensity ratio of paramagnetic and diamagnetic cross-peaks (Ipara/Idia) versus residue number of PmrAN. Unassigned and overlapped residues are shown as short gray bars. C, structural model of PmrD-PmrAN complex obtained by HADDOCK docking. The MTSL spin label sites, Cys-54 of two PmrDs, are shown as magenta spheres, and the PmrAN residues with Ipara/Idia < 0.5 are shown as yellow spheres.
DISCUSSION
Several crystal structures of receiver domains of response regulators have been solved (43–46), but little structural information is available regarding how they interact with each other in multiprotein complexes. As well, knowledge of the structure of the receiver domain in complex with the connector protein is limited. Several groups have reported that the active site from the receiver domain of the response regulator is widely used for intermolecular protein-protein interactions with histidine kinase. Here, we present the crystal structure of PmrAN, in which a hydrogen bond couples with the carbonyl oxygen atom of Arg-81 situated on the β4-α4 loop and side chain of the aromatic residue Tyr-98, which is similar to the “T-loop-Y” model observed for the activation mechanism of a response regulator (47). In the model, the conformation of the β4-α4 loop was gated by phosphorylation and the position of the active site residue Thr-79, thus resulting in an inward orientation of the aromatic switch residue (47). Next, we demonstrated that PmrAN uses a similar surface to interact with the TCS connector protein PmrD (Fig. 7A). Therefore, PmrD is likely a competitive inhibitor of histidine kinase because it binds with the response regulator at a similar interface surrounding the phosphorylation site (48). In the PmrD-PmrAN complex model, the phosphoacceptor Asp-51 faces the open mouth area of the β-barrel of the PmrD structure, which reflects the protection mode of the connector protein on the phosphorylated response regulator. The orientation of PmrAN relative to PmrD can be attributed to three specific interactions. The first interaction is between the α1 and the β5-α5 loop region of PmrAN and loop 4 and N-terminal β1 in PmrD. The second interaction is between the β1-α1 loop of PmrAN and loop 2 of PmrD. The third interaction is between the β4-α4 loop and β3-α3 loop containing the Asp-51 phosphorylation site of PmrAN and C-terminal β6, loop 6, N-terminal β1, and β3 of PmrD. Interactions 2 and 3 create an environment that hides the phosphoryl analog of BeF3− between PmrD and PmrAN. The binding interface of PmrAN is also similar to that observed in the crystal structure of the DHp domain in the ThkA-TrrA complex and Spo0F in the Spo0B-Spo0F complex from Bacillus subtilis, in which TrrA and Spo0F are the response regulators. In the PmrD-PmrAN complex, the phosphoacceptor Asp-51 in PmrA faces the His-70 of PmrD, similar to the phosphodonor His in the HK DHp domain (His-547 in ThkA) (48, 49). Hydrogen bonds and electrostatic interactions are important for stability and specificity of protein-protein interactions (50, 51). Consistently, we identified seven intermolecular hydrogen bonds and five salt bridges in our HADDOCK complex model (Fig. 5C), which emphasizes the role these interactions play in the recognition and specificity of PmrD connector proteins.
FIGURE 7.

A proposed physical blockage mechanism of PmrA dephosphorylation inhibited by PmrD. A, the monomeric form of the PmrD-PmrAN complex is shown. His-70 of PmrD and Asp-51 from PmrAN are in red and blue, respectively. B, the crystal structure of the response regulator Spo0F in complex with the RapH phosphatase. The phosphorylation site Asp-52 is shown in blue, and the residue Gln-47 in RapH, which is responsible for dephosphorylating Spo0F, is in red. C, a stick diagram shows the intermolecular interactions near the active site residue Asp-51 of PmrAN. The nitrogen and oxygen atoms are in blue and red, respectively. The carbon atoms of His-70 from PmrD are in green and are colored gray for others. The Hδ1 and Hϵ2 of His-70 in PmrD form H-bonds (yellow dashed lines) with the carbonyl groups of two active site residues (Asp-9 and Gly-53) of PmrAN. The interactions demonstrate how PmrD can protect the phosphate group analog BeF3− (magenta) and the divalent cation Mg2+ (green sphere) on PmrAN and prevent it from dephosphorylation.
With response regulators, the phosphatase reaction is generally considered to proceed essentially as a reversal of the phospho-His phosphotransfer reaction, with water or hydroxide taking the place of the histidine imidazole side chain (52). Some of the histidine kinases have phosphatase activities toward their respective phosphorylated response regulators. The dephosphorylated kinases could promote a transition state with water in place of the phosphorylated histidine side chain, leading to hydrolysis instead of phosphorylation (53). For example, the structural basis of the response regulator Spo0F dephosphorylation by RapH phosphatases was revealed by the RapH-Spo0F crystal structure (Fig. 7B). In the structure, the side chain of RapH Gln-47 inserts into the Spo0F active site. RapH Gln-47 may orient water for direct in-line hydrophilic attack on the Spo0F phosphoaspartate 54 phosphorous atom and thus cause hydrolysis of the phosphoryl group (54). In these cases, the interactions are primarily responsible for the specific histidine kinase-response regulator or connector-response regulator binding.
The receiver domain of PmrA could form a cooperative, intermolecular dimer of a response regulatory component that interacts to convert an inherently weak set of protein-protein interactions into a stronger binding force that permits the formation of a functional viable complex. Activation of a response regulator by a “pre-existing population shift” may be a general mechanism for two-component signaling (55). Selective binding to a low populated conformation hidden with traditional structural methods may be a general model for ligand binding (56). Therefore, PmrD may selectively bind to the phosphorylated higher energy active conformation of PmrAN and prevents its dephosphorylation by phosphatase such as PmrB.
In our previous study, we proposed a model of the PmrD-PmrAN protein complex and examined how PmrD regulates phospho-PmrA. Here, we propose that PmrD is a phosphatase inhibitor that specifically binds the phosphorylated active site and acts as a molecular barricade that inhibits interaction between the phosphatase and phosphorylatable Asp residue of PmrA and thus prevents intermolecular dephosphorylation. PmrD might inhibit dephosphorylation by sterically hindering the transfer of phosphate from Asp-51 of PmrA to phosphatase PmrB. The large size of PmrD and its predicted binding interface with PmrA are consistent with the model that inhibition occurs by a steric hindrance. The model shows three H-bonds between PmrD and residues near phosphorylation sites of PmrAN. For example, the HD1 of His-70 in PmrD formed H-bonds with the carbonyl group of Gly-53 in PmrAN. In addition, the Hϵ2 of His-70 in PmrD formed an H-bond with OD1 of Asp-9 in PmrAN (Fig. 7C). A divalent cation close to the active site is required to add or remove phosphoryl groups in the receiver domain of a response regulator (57). Therefore, the steric hindrance effect provided by PmrD could prevent the dephosphorylation of PmrA mediated by the cation. As well, nucleophilic attack by a water molecule is the likely mechanism of autodephosphorylation in response regulators (58, 59). The side chain of His-70 in PmrD likely maintains the phosphoryl group stability by sterically inhibiting access of the water molecule, which executes a nucleophilic attack on the phosphorus and causes hydrolysis of the phosphoryl group. These interactions may explain how PmrD stabilizes the activated PmrAN protein and thus prevents dephosphorylation of phosphate on Asp-51 from a phosphatase such as PmrB. PmrD may inhibit dephosphorylation of the response regulator by acting as a simple physical blockade to sterically block access to the phosphoacceptor Asp residue on the receiver domain of the response regulator, thus precluding close contact of phosphatase with the phosphorylation site on PmrAN.
Acknowledgments
The NMR spectra were obtained at the High Field Nuclear Magnetic Resonance Center supported by the National Research Program for Genomic Medicine. We thank Laura Smales for copyediting the manuscript. We are grateful for access to synchrotron radiation beamlines BL13B1 at the National Synchrotron Radiation Research Center in Taiwan.
This work was supported by grants from Academia Sinica and the National Science Council (NSC 97-2311-B-001-013-MY3) in Taiwan and in part by the Ministry of Education (Taiwan) under the ATU plan.
The atomic coordinates and structure factors (code 3W9S) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Chemical shifts of PmrAN were deposited in the BioMagResBank under accession number BMRB 18922.
- TCS
- two-component system
- PmrA
- polymyxin B resistant protein A
- PRE
- paramagnetic relaxation enhancement
- PmrD
- polymixin B resistant protein D
- PmrAN
- the N-terminal receiver domain of PmrA
- HDX
- hydrogen/deuterium exchange
- r.m.s.d
- root mean square deviation
- MTSL
- (1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-methyl)methanethiosulfonate).
REFERENCES
- 1. Perraud A. L., Weiss V., Gross R. (1999) Signalling pathways in two-component phosphorelay systems. Trends Microbiol. 7, 115–120 [DOI] [PubMed] [Google Scholar]
- 2. Stock A. M., Robinson V. L., Goudreau P. N. (2000) Two-component signal transduction. Annu. Rev. Biochem. 69, 183–215 [DOI] [PubMed] [Google Scholar]
- 3. West A. H., Stock A. M. (2001) Histidine kinases and response regulator proteins in two-component signaling systems. Trends Biochem. Sci. 26, 369–376 [DOI] [PubMed] [Google Scholar]
- 4. Wurgler-Murphy S. M., Saito H. (1997) Two-component signal transducers and MAPK cascades. Trends Biochem. Sci. 22, 172–176 [DOI] [PubMed] [Google Scholar]
- 5. Hoch J., Silhavy T. (eds) (1995) Two-Component Signal Transduction, pp. 129–144, ASM Press, Washington, D.C [Google Scholar]
- 6. Roland K. L., Martin L. E., Esther C. R., Spitznagel J. K. (1993) Spontaneous pmrA mutants of Salmonella typhimurium LT2 define a new two-component regulatory system with a possible role in virulence. J. Bacteriol. 175, 4154–4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kox L. F., Wösten M. M., Groisman E. A. (2000) A small protein that mediates the activation of a two-component system by another two-component system. EMBO J. 19, 1861–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wösten M. M., Kox L. F., Chamnongpol S., Soncini F. C., Groisman E. A. (2000) A signal transduction system that responds to extracellular iron. Cell 103, 113–125 [DOI] [PubMed] [Google Scholar]
- 9. Perez J. C., Groisman E. A. (2007) Acid pH activation of the PmrA/PmrB two-component regulatory system of Salmonella enterica. Mol. Microbiol. 63, 283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Helander I. M., Kato Y., Kilpeläinen I., Kostiainen R., Lindner B., Nummila K., Sugiyama T., Yokochi T. (1996) Characterization of lipopolysaccharides of polymyxin-resistant and polymyxin-sensitive Klebsiella pneumoniae O3. Eur. J. Biochem. 237, 272–278 [DOI] [PubMed] [Google Scholar]
- 11. Zhou Z., Ribeiro A. A., Lin S., Cotter R. J., Miller S. I., Raetz C. R. (2001) Lipid A modifications in polymyxin-resistant Salmonella typhimurium: PMRA-dependent 4-amino-4-deoxy-L-arabinose, and phosphoethanolamine incorporation. J. Biol. Chem. 276, 43111–43121 [DOI] [PubMed] [Google Scholar]
- 12. Asensio A., Oliver A., González-Diego P., Baquero F., Pérez-Díaz J. C., Ros P., Cobo J., Palacios M., Lasheras D., Cantón R. (2000) Outbreak of a multiresistant Klebsiella pneumoniae strain in an intensive care unit: antibiotic use as risk factor for colonization and infection. Clin. Infect. Dis. 30, 55–60 [DOI] [PubMed] [Google Scholar]
- 13. Kato A., Groisman E. A. (2004) Connecting two-component regulatory systems by a protein that protects a response regulator from dephosphorylation by its cognate sensor. Genes Dev. 18, 2302–2313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mitrophanov A. Y., Groisman E. A. (2008) Signal integration in bacterial two-component regulatory systems. Genes Dev. 22, 2601–2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Itou J., Eguchi Y., Utsumi R. (2009) Molecular mechanism of transcriptional cascade initiated by the EvgS/EvgA system in Escherichia coli K-12. Biosci. Biotechnol. Biochem. 73, 870–878 [DOI] [PubMed] [Google Scholar]
- 16. Bougdour A., Cunning C., Baptiste P. J., Elliott T., Gottesman S. (2008) Multiple pathways for regulation of sigmaS (RpoS) stability in Escherichia coli via the action of multiple anti-adaptors. Mol. Microbiol. 68, 298–313 [DOI] [PubMed] [Google Scholar]
- 17. Gerken H., Charlson E. S., Cicirelli E. M., Kenney L. J., Misra R. (2009) MzrA: a novel modulator of the EnvZ/OmpR two-component regulon. Mol. Microbiol. 72, 1408–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luo S. C., Lou Y. C., Cheng H. Y., Pan Y. R., Peng H. L., Chen C. (2010) Solution structure and phospho-PmrA recognition mode of PmrD from Klebsiella pneumoniae. J. Struct. Biol. 172, 319–330 [DOI] [PubMed] [Google Scholar]
- 19. Dominguez C., Boelens R., Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 20. Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Vol. 3, p. A3, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 21. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode in Methods in Enzymology (Charles W., Carter, ed.), pp. 307–326, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 22. Terwilliger T. (2000) Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McRee D. E. (1999) XtalView Xfit - A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125, 156–165 [DOI] [PubMed] [Google Scholar]
- 24. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 25. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 28. Kay L. E. (1995) Pulsed field gradient multi-dimensional NMR methods for the study of protein structure and dynamics in solution. Prog. Biophys. Mol. Biol. 63, 277–299 [DOI] [PubMed] [Google Scholar]
- 29. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 30. Johnson B. A., Blevins R. A. (1994) NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 31. Schanda P., Kupce E., Brutscher B. (2005) SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J. Biomol. NMR 33, 199–211 [DOI] [PubMed] [Google Scholar]
- 32. Goddard T. D., Kneller D. G. (2008) SPARKY 3, University of California, San Francisco [Google Scholar]
- 33. Yan D., Cho H. S., Hastings C. A., Igo M. M., Lee S. Y., Pelton J. G., Stewart V., Wemmer D. E., Kustu S. (1999) Beryllofluoride mimics phosphorylation of NtrC and other bacterial response regulators. Proc. Natl. Acad. Sci. U.S.A. 96, 14789–14794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 35. Volz K. (1993) Structural conservation in the CheY superfamily. Biochemistry 32, 11741–11753 [DOI] [PubMed] [Google Scholar]
- 36. Bachhawat P., Stock A. M. (2007) Crystal structures of the receiver domain of the response regulator PhoP from Escherichia coli in the absence and presence of the phosphoryl analog beryllofluoride. J. Bacteriol. 189, 5987–5995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barbieri C. M., Mack T. R., Robinson V. L., Miller M. T., Stock A. M. (2010) Regulation of response regulator autophosphorylation through interdomain contacts. J. Biol. Chem. 285, 32325–32335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bachhawat P., Swapna G. V., Montelione G. T., Stock A. M. (2005) Mechanism of activation for transcription factor PhoB suggested by different modes of dimerization in the inactive and active states. Structure 13, 1353–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cho H. S., Pelton J. G., Yan D., Kustu S., Wemmer D. E. (2001) Phosphoaspartates in bacterial signal transduction. Curr. Opin. Struct. Biol. 11, 679–684 [DOI] [PubMed] [Google Scholar]
- 40. Robinson V. L., Buckler D. R., Stock A. M. (2000) A tale of two components: a novel kinase and a regulatory switch. Nat. Struct. Biol. 7, 626–633 [DOI] [PubMed] [Google Scholar]
- 41. O'Connell M. R., Gamsjaeger R., Mackay J. P. (2009) The structural analysis of protein-protein interactions by NMR spectroscopy. Proteomics 9, 5224–5232 [DOI] [PubMed] [Google Scholar]
- 42. Battiste J. L., Wagner G. (2000) Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry 39, 5355–5365 [DOI] [PubMed] [Google Scholar]
- 43. Buckler D. R., Zhou Y., Stock A. M. (2002) Evidence of intradomain and interdomain flexibility in an OmpR/PhoB homolog from Thermotoga maritima. Structure 10, 153–164 [DOI] [PubMed] [Google Scholar]
- 44. Toro-Roman A., Mack T. R., Stock A. M. (2005) Structural analysis and solution studies of the activated regulatory domain of the response regulator ArcA: a symmetric dimer mediated by the α4-β5-α5 face. J. Mol. Biol. 349, 11–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bent C. J., Isaacs N. W., Mitchell T. J., Riboldi-Tunnicliffe A. (2004) Crystal structure of the response regulator 02 receiver domain, the essential YycF two-component system of Streptococcus pneumoniae in both complexed and native states. J. Bacteriol. 186, 2872–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Birck C., Chen Y., Hulett F. M., Samama J. P. (2003) The crystal structure of the phosphorylation domain in PhoP reveals a functional tandem association mediated by an asymmetric interface. J. Bacteriol. 185, 254–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dyer C. M., Dahlquist F. W. (2006) Switched or not?: the structure of unphosphorylated CheY bound to the N terminus of FliM. J. Bacteriol. 188, 7354–7363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yamada S., Sugimoto H., Kobayashi M., Ohno A., Nakamura H., Shiro Y. (2009) Structure of PAS-linked histidine kinase and the response regulator complex. Structure 17, 1333–1344 [DOI] [PubMed] [Google Scholar]
- 49. Varughese K. I., Tsigelny I., Zhao H. (2006) The crystal structure of beryllofluoride Spo0F in complex with the phosphotransferase Spo0B represents a phosphotransfer pretransition state. J. Bacteriol. 188, 4970–4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Janin J., Chothia C. (1976) Stability and specificity of protein-protein interactions: the case of the trypsin-trypsin inhibitor complexes. J. Mol. Biol. 100, 197–211 [DOI] [PubMed] [Google Scholar]
- 51. Chothia C., Wodak S., Janin J. (1976) Role of subunit interfaces in the allosteric mechanism of hemoglobin. Proc. Natl. Acad. Sci. U.S.A. 73, 3793–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lukat G. S., Lee B. H., Mottonen J. M., Stock A. M., Stock J. B. (1991) Roles of the highly conserved aspartate and lysine residues in the response regulator of bacterial chemotaxis. J. Biol. Chem. 266, 8348–8354 [PubMed] [Google Scholar]
- 53. Stock J. B., Ninfa A. J., Stock A. M. (1989) Protein phosphorylation and regulation of adaptive responses in bacteria. Microbiol. Rev. 53, 450–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Parashar V., Mirouze N., Dubnau D. A., Neiditch M. B. (2011) Structural basis of response regulator dephosphorylation by Rap phosphatases. PLoS Biol. 9, e1000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stock A. M., Guhaniyogi J. (2006) A new perspective on response regulator activation. J. Bacteriol. 188, 7328–7330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li P., Martins I. R., Amarasinghe G. K., Rosen M. K. (2008) Internal dynamics control activation and activity of the autoinhibited Vav DH domain. Nat. Struct. Mol. Biol. 15, 613–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bourret R. B. (2010) Receiver domain structure and function in response regulator proteins. Curr. Opin. Microbiol. 13, 142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stock A. M., Martinez-Hackert E., Rasmussen B. F., West A. H., Stock J. B., Ringe D., Petsko G. A. (1993) Structure of the Mg2+-bound form of CheY and mechanism of phosphoryl transfer in bacterial chemotaxis. Biochemistry 32, 13375–13380 [DOI] [PubMed] [Google Scholar]
- 59. Wolanin P. M., Webre D. J., Stock J. B. (2003) Mechanism of phosphatase activity in the chemotaxis response regulator CheY. Biochemistry 42, 14075–14082 [DOI] [PubMed] [Google Scholar]
- 60. Thompson J. D., Higgins D. G., Gibson T. J. (1994) Improved sensitivity of profile searches through the use of sequence weights and gap excision. Comput. Appl. Biosci. 10, 19–29 [DOI] [PubMed] [Google Scholar]
- 61. Gouet P., Courcelle E., Stuart D. I., Métoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]