

Background: Orally active proteasome inhibitors with novel mechanisms of action are in high clinical demand.
Results: K-7174, a homopiperazine derivative with a unique mode of proteasome inhibition, exhibits anti-myeloma effects via transcriptional repression of class I histone deacetylases.
Conclusion: K-7174 is an orally active proteasome inhibitor with a unique mechanism of action.
Significance: K-7174 may compensate for conventional proteasome inhibitors in cancer treatment.
Keywords: Cancer, Drug Resistance, Histone Deacetylase, Histone Deacetylase Inhibitors, Multiple Myeloma, Proteasome
Abstract
Bortezomib therapy is now indispensable for multiple myeloma, but is associated with patient inconvenience due to intravenous injection and emerging drug resistance. The development of orally active proteasome inhibitors with distinct mechanisms of action is therefore eagerly awaited. Previously, we identified homopiperazine derivatives as a novel class of proteasome inhibitors with a different mode of proteasome binding from bortezomib. In this study, we show that K-7174, one of proteasome inhibitory homopiperazine derivatives, exhibits a therapeutic effect, which is stronger when administered orally than intravenously, without obvious side effects in a murine myeloma model. Moreover, K-7174 kills bortezomib-resistant myeloma cells carrying a β5-subunit mutation in vivo and primary cells from a patient resistant to bortezomib. K-7174 induces transcriptional repression of class I histone deacetylases (HDAC1, -2, and -3) via caspase-8-dependent degradation of Sp1, the most potent transactivator of class I HDAC genes. HDAC1 overexpression ameliorates the cytotoxic effect of K-7174 and abrogates histone hyperacetylation without affecting the accumulation of ubiquitinated proteins in K-7174-treated myeloma cells. Conversely, HDAC inhibitors enhance the activity of K-7174 with an increase in histone acetylation. These results suggest that class I HDACs are critical targets of K-7174-induced cytotoxicity. It is highly anticipated that K-7174 increases the tolerability and convenience of patients by oral administration and has the clinical utility in overcoming bortezomib resistance as a single agent or in combination with HDAC inhibitors.
Introduction
Proteasome inhibition is now considered a unique and effective way to kill cancer cells that can tolerate conventional chemotherapy. Bortezomib is the first proteasome inhibitor (PI)2 approved for clinical application, and is now used for the treatment of multiple myeloma (MM) and mantle cell lymphoma (1–3). Although bortezomib therapy is a major advance in clinical oncology, there are several problems to be resolved as soon as possible (4, 5). Among them, off-target toxicities, patient inconvenience due to intravenous administration, and the development of both intrinsic and acquired resistance to bortezomib are emerging problems.
The introduction of novel orally active PIs with distinct mechanisms of action from bortezomib is an effective way to circumvent these issues. Recently, several novel PIs have been developed and are now undergoing clinical trials (6). Although some compounds are orally available, most of them are peptide derivatives and act via similar mechanisms of action as does bortezomib (7–12). In addition, point mutations in the proteasome β5 subunit gene underlying bortezomib resistance have been reported (13–15). Therefore, we still intend to develop novel orally bioavailable PIs with distinct mechanisms of action from bortezomib.
K-7174 (N,N′-bis-(E)-[5-(3,4,5-trimethoxy-phenyl)-4-pentenyl]homopiperazine) is a homopiperazine-derived small molecular compound developed as an orally active agent in vivo (see Fig. 1A for chemical structure) (16, 17). In a previous study, we have shown that K-7174 inhibits all three catalytic subunits of 20 S proteasome by direct binding, whereas bortezomib mainly acts on the β5 subunit (18). However, it is still unclear whether K-7174 has anti-myeloma activity and its underlying mechanism is different from that of bortezomib. The initial rationale for the use of bortezomib was the inhibition of NF-κB activity, since IκBα, which inactivates NF-κB, is a substrate of the proteasome complex (19). This scenario was challenged by the recent report of Hideshima et al. (20), in which bortezomib did not inactivate but rather activated the canonical NF-κB pathway in MM cells. Moreover, we have found that down-regulation of class I histone deacetylases (HDACs) is one of the major mechanisms of the cytotoxic action of bortezomib (21). It is likely that K-7174 exerts anti-myeloma action via similar mechanisms.
FIGURE 1.
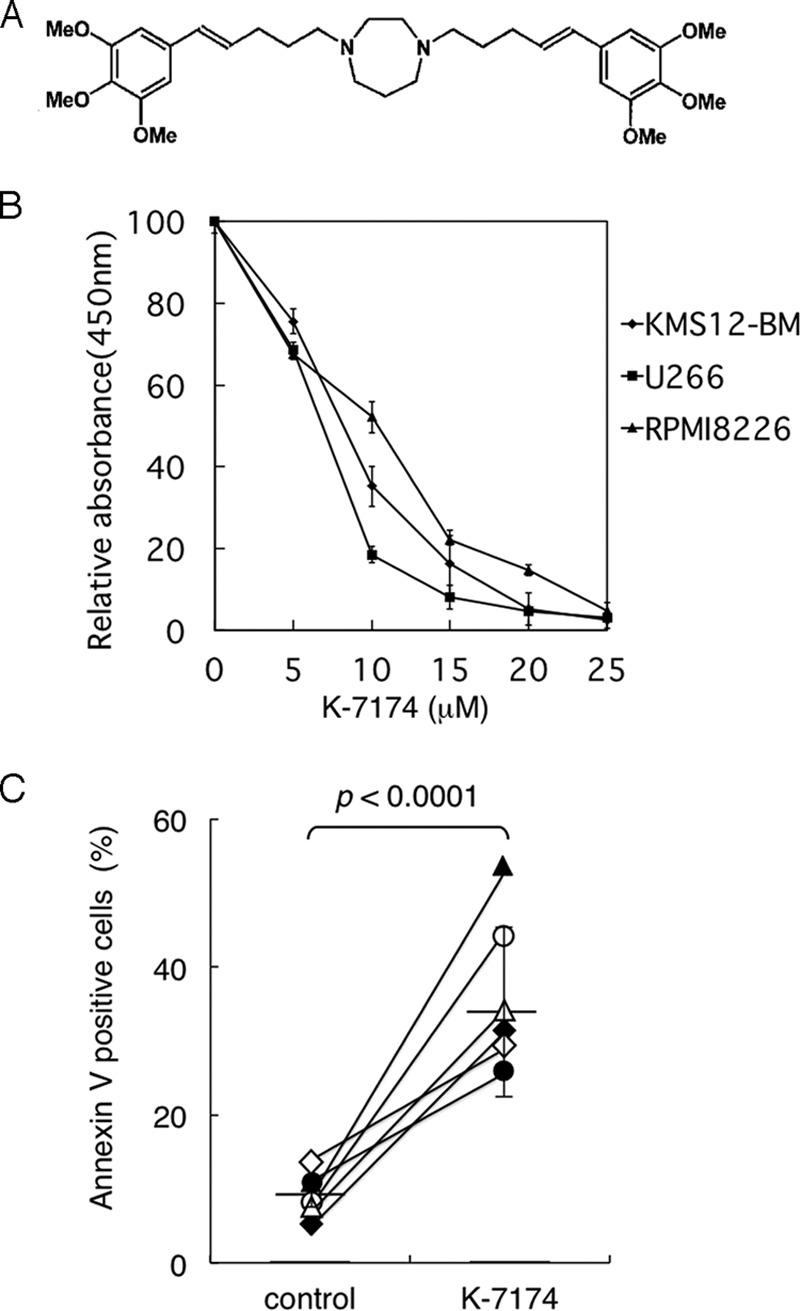
Anti-myeloma activity of K-7174 in vitro. A, chemical structure of K-7174. B, cell proliferation was measured by MTT assays after culturing MM cells with K-7174 for 72 h. Absorbance at 450 nm was analyzed with a microplate reader, and expressed as a percentage of the value of corresponding untreated cells. Panels show the dose-response curves of KMS12-BM (closed diamond), U266 (closed square), and RPMI8226 (closed triangle) cells. The mean ± S.D. (bars) of three independent experiments are shown. C, we cultured primary MM cells with either 10 μm K-7174 (K-7174) or the vehicle alone (control) for 48 h. Cell death/apoptosis was determined by reactivity with annexin-V on a flow cytometer. The y axis shows the proportion of annexin-V-positive cells (%). The values of individual samples are indicated as follows: patient 1, open circle; patient 3, open triangle; patient 4, open diamond; patient 5, closed circle; patient 7, closed triangle; and patient 8, closed diamond. Bars indicate the average values. p values were calculated by paired Student's t tests.
In this study, we demonstrated anti-myeloma activity of post-oral K-7174 and the utility in overcoming bortezomib resistance using a murine xenograft model. Moreover, we found that the cytotoxic activity of K-7174 largely depends on the expression levels of class I HDACs and the combination of K-7174 and HDAC inhibitors induces additive effects via the reduction of HDAC activity. These findings may provide a molecular basis and rationale for the use of K-7174 in myeloma treatment alone or in combination with HDAC inhibitors.
EXPERIMENTAL PROCEDURES
Cells and Cell Culture
We used 3 bona fide human MM cell lines, KMS12-BM, RPMI8226, and U266, in this study (22). These cell lines were purchased from the Health Science Research Resources Bank (Osaka, Japan). Primary CD138-positive MM cells were isolated from the bone marrow of patients at the time of the diagnostic procedure using the MACS system (Miltenyi Biotec, Gladbach, Germany). Informed consent was obtained in accordance with the Declaration of Helsinki, and the protocol was approved by the Institutional Review Board.
Drugs
The drugs used in this study and their sources are: K-7174 (Kowa, Tokyo, Japan), bortezomib (Millennium Pharmaceuticals, Cambridge, MA), 4-hydroperoxy cyclophosphamide (an active metabolite of cyclophosphamide) (Shionogi, Tokyo, Japan), melphalan (l-PAM) (Wako Biochemicals, Osaka, Japan), romidepsin (Gloucester Pharmaceuticals, Cambridge, MA), and vorinostat (Selleck Chemicals, Houston, TX).
Cell Proliferation Assay
Cell proliferation was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay using a Cell Counting Kit (Wako Biochemicals). Absorbance at 450 nm was analyzed with a microplate reader, and expressed as a percentage of the value of the corresponding untreated cells (23).
Assessment of Cell Death
Cells were washed with phosphate-buffered saline, and stained with allophycocyanin-conjugated annexin-V (Biovision, Mountain View, CA). Cell death/apoptosis was judged by annexin-V reactivity using a FACSaria flow cytometer (Becton Dickinson, Bedford, MA) as described previously (24).
Immunoblotting
Immunoblotting was carried out according to the standard method using the following antibodies: anti-acetyl histone H3 (Upstate Biotechnology/Millipore, Billerica, MA), anti-histone H3, anti-ubiquitin, anti-pro-caspase-8, -9, and -12, anti-HDAC2 (Cell Signaling Technology, Beverly, MA), anti-HDAC1 (Sigma), anti-HDAC3 (BD Transduction Laboratories, San Diego, CA), anti-CD138, anti-Sp1, and anti-GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA) (21).
Semi-quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total cellular RNA was isolated from 1 to 10 × 104 cells, reverse-transcribed into cDNA using SuperScript reverse transcriptase and oligo(dT) primers (Invitrogen), and subjected to subsequent semi-quantitative RT-PCR. Detailed information on the procedure was described previously (21).
Chromatin Immunoprecipitation Assays
We used the ChIP-IT Chromatin Immunoprecipitation Kit (Active Motif, Carlsbad, CA) to perform chromatin immunoprecipitation assays. In brief, cells were fixed with 1% formaldehyde at 37 °C for 5 min, and sonicated to obtain chromatin suspensions. After centrifugation, supernatants were incubated with antibodies of interest at 4 °C overnight. The mixture was then incubated with protein A-agarose beads at 4 °C for 1 h, and centrifuged to collect the beads. DNA fragments bound to the beads were purified with vigorous washing, and subjected to PCR as described previously (21).
Xenograft Murine Model
Cell suspensions in 100 μl of RPMI 1640 medium together with 100 μl of Matrigel basement membrane matrix (BD Biosciences) were inoculated subcutaneously into the right thigh of non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice (Charles River Laboratories, Wilmington, MA). Treatment was started after tumors were measurable (100–500 mm3) as described previously (21). K-7174 was administered intraperitoneally or orally in a volume of 400 μl of solution containing 3% DMSO and 97% sterile 0.9% NaCl once daily at various doses for 2 weeks. Bortezomib was administered intravenously twice a week via the tail vein at 0.5 mg/kg for 2 weeks. The control group received the vehicle (3% DMSO in 0.9% NaCl) alone on the same schedule. Caliper measurements of the longest perpendicular tumor diameters were performed every alternate day to estimate tumor volume using the following formula: 4/3π × (width/2)2 × (length/2), which represents the three-dimensional volume of an ellipse.
Establishment of Bortezomib-resistant MM Cell Lines
To confer bortezomib resistance to MM cells, we transduced with mutated PSMB5 cDNA as described previously (18, 25). A mutation was inserted at nucleotide position 322 (G/A) by PCR-based site-directed mutagenesis. Wild-type and mutated PSMB5 cDNAs were inserted into the lentiviral vector CSII-CMV-MCS-IRES-VENUS (kindly provided by Dr. Hiroyuki Miyoshi, RIKEN BioResource Center, Ibaraki, Japan) (26), and co-transfected into 293FT cells with packaging plasmids (Invitrogen) to produce infective lentiviruses in culture supernatants (27). RPMI8226 cells were infected by each viral supernatant for 24 h. We collected VENUS-positive cells by a FACSaria flow cytometer, seeded 1 cell/well in a 96-well plate to obtain single cell clones.
RESULTS
K-7174 Inhibits MM Cell Growth in Vitro
Following the demonstration of proteasome inhibitory action (18), we investigated whether K-7174 exerts anti-myeloma activities. First, we examined the cytotoxic effect of K-7174 on MM cell lines using MTT assays. K-7174 significantly inhibited the growth of three MM cell lines in a dose-dependent manner (Fig. 1B). Next, we confirmed the anti-myeloma activity of K-7174 using primary MM cells. CD138-positive cells isolated from bone marrow samples of 6 patients were cultured in the absence or presence of K-7174 for 2 days, followed by annexin-V staining to assess the induction of apoptosis. K-7174 significantly increased the percentage of annexin-V-positive cells in all cases examined (Fig. 1C).
K-7174 Inhibits Human MM Cell Growth in a Murine Xenograft Model
Having demonstrated the cytotoxicity of K-7174 against MM cells in vitro, we examined the in vivo efficacy using a xenograft mouse model. As described previously (21), we inoculated either RPMI8226 or U266 cells subcutaneously into NOD/SCID mice in the right thigh at 3 × 107 or 1 × 107 cells. When measurable tumors developed (usually after 4 days), either K-7174 (75 mg/kg) or vehicle (3% DMSO in 0.9% NaCl) was intraperitoneally administered once daily for 14 days (n = 3–4 in each group). We compared the tumor volume between vehicle-control and K-7174-treated groups on day 14. The tumor volume was significantly lower in the K-7174-treated group than in the vehicle-control group (Fig. 2, A and B). However, the K-7174-treated group had a significant body weight reduction after 10 days (data not shown). We therefore repeated the same set of experiments with reduced doses (30 and 50 mg/kg). Although no body weight reduction was observed, K-7174 failed to inhibit tumor growth at these doses (data not shown).
FIGURE 2.

Anti-myeloma activity of K-7174 in vivo. NOD/SCID mice were inoculated subcutaneously with 1 × 107 cells of U266 or 3 × 107 cells of RPMI8226 into the right thigh. Treatments were started on day 0, when tumors were measurable. Caliper measurements of the longest perpendicular tumor diameters were performed on alternate days to estimate the tumor volume (mm3) using the following formula: 4/3π × (width/2)2 × (length/2). A, mice were treated with 75 mg/kg of K-7174 intraperitoneally once daily for 14 days (n = 4) or the vehicle (3% DMSO in 0.9% NaCl) alone (Control) on the same schedule (n = 4). The y axis shows the average tumor volume in inoculated mice on the indicated days. The mean ± S.D. (bars) are shown. p values were calculated by one-way analysis of variance with the Student-Newman-Keuls multiple comparisons test. B, representative photographs of NOD/SCID mice inoculated with U266 cells on days 0 and 14 (original magnification: ×2). Inoculated tumors are indicated in circled regions. C, mice were treated with 50 mg/kg of K-7174 post-orally once daily for 14 days (U266 + K-7174, closed circle; n = 3, RPMI8226 + K-7174, closed triangle; n = 3), or the vehicle (3% DMSO in 0.9% NaCl) alone on the same schedule (U266 + control, open circle; n = 4, RPMI8226 + control, open triangle; n = 3). The y axis shows the average tumor volume in inoculated mice (bars indicate S.D.). p values were calculated by one-way analysis of variance with the Student-Newman-Keuls multiple comparisons test. Asterisks indicate p < 0.05 against the vehicle control. D, representative photographs of NOD/SCID mice inoculated with RPMI8226 cells on days 0 and 14 (original magnification: ×2). Inoculated tumors are indicated in circled regions. E, NOD/SCID mice were inoculated subcutaneously with 1 × 107 cells of U266 into the right thigh. When tumors were measurable, mice were treated with 50 mg/kg of K-7174 post-orally once daily (K-7174; n = 2) or the vehicle (3% DMSO in 0.9% NaCl) alone (Control; n = 2) for 3 days. We resected inoculated tumors and sorted human CD138-positive cells using a FACSaria flow cytometer for immunoblot analysis of cellular protein ubiquitination (Ubiq) and CD138 (hCD38) expression. We used lysates of U266 cells treated with 5 μm K-7174 (U266 + K-7174) or the vehicle control (U266) for 3 days in vitro as controls. The signal intensities were quantified by densitometry, normalized to those of the corresponding CD138, and shown as relative values setting the vehicle control (U266) to 1.0.
As K-7174 was originally developed for oral administration, we tested the therapeutic effects of post-oral K-7174 in tumor-inoculated mice. Either K-7174 (50 mg/kg) or vehicle (3% DMSO in 0.9% NaCl) was orally administered once daily for 14 days (n = 3–4 in each group). We determined the dose of K-7174 to be well tolerated by mice without obvious side effects including weight loss in pilot experiments (data not shown). The tumor volume was compared between vehicle-control and K-7174-treated groups for up to 28 days. As a result, tumor volume was significantly lower in the K-7174-treated group than in the vehicle-control group (Fig. 2, C and D). There were no obvious side effects including body weight reduction, leukocytopenia, thrombocytopenia, anemia and hepatic dysfunction in both vehicle-control and K-7174-treated groups (the data on day 21 are shown in Table 1). To examine the proteasome inhibitory effect of K-7174 in vivo, we determined the accumulation of ubiquitinated proteins in inoculated tumors after oral administration of K-7174. As expected, a 2–4-fold accumulation of ubiquitinated proteins was observed in tumor cells in the K-7174-treated group but not in the vehicle-control group (Fig. 2E). This suggests that K-7174 could effectively inhibit proteasome activity in vivo. Taken together, these data demonstrate the potent anti-myeloma activity of K-7174 in vivo and more importantly, that oral administration is more effective than intraperitoneal injection.
TABLE 1.
Biochemical parameters of mice treated with K-7174
We evaluated the indicated parameters in mice on day 21 of K-7174 treatment. Data are shown as the mean ± S.D. (n = 3 per group). p values were calculated by the Student's t test.
| Vehicle | K-7174 (50 mg/kg) | p value | |
|---|---|---|---|
| Body weight (g) | 23.0 ± 0.9 | 27.8 ± 0.7 | p > 0.05 |
| Peripheral blood count | |||
| WBC (×102/μl) | 28.8 ± 1.6 | 27.5 ± 1.3 | p > 0.05 |
| RBC (×104/μl) | 653.3 ± 90.2 | 694.8 ± 24.9 | p > 0.05 |
| HGB (g/dl) | 10.9 ± 1.5 | 11.6 ± 0.5 | p > 0.05 |
| HCT (%) | 32.6 ± 4.4 | 34.8 ± 1.7 | p > 0.05 |
| MCV (fl) | 49.9 ± 0.4 | 50.1 ± 0.9 | p > 0.05 |
| Mean corpuscular hemoglobin (pg) | 16.6 ± 0.1 | 16.6 ± 0.4 | p > 0.05 |
| Mean corpuscular hemoglobin concentration (g/dl) | 33.3 ± 0.1 | 33.2 ± 0.2 | p > 0.05 |
| Platelet (×104/μl) | 35.8 ± 9.7 | 26.5 ± 15.6 | p > 0.05 |
| Serum level | |||
| Aspartate aminotransferase (units/liter) | 113.8 ± 32.1 | 74.8 ± 10.0 | p > 0.05 |
| Alanine aminotransferase (units/liter) | 8.8 ± 2.8 | 10.3 ± 2.6 | p > 0.05 |
K-7174 Overcomes Bortezomib Resistance in Vitro and in Vivo
Because K-7174 appears to inhibit proteasome activity with a distinct mode from bortezomib (18), it is anticipated that K-7174 is effective for bortezomib-resistant MM cells. To substantiate this notion, we have established mutant and wild-type (WT) sublines from RPMI8226 cells lentivirally transduced with mutated and wild-type PSMB5. We selected representative sublines that express the highest levels of VENUS and PSMB5 as described previously (18). HDAC1 overexpression itself did not affect the proliferation potential and viability of myeloma cells (data not shown). Overexpression of wild-type PSMB5 conferred modest resistance to bortezomib but not K-7174 in RPMI8226 cells. Yet, sensitivity to bortezomib was significantly lower in the mutant subline than in the WT subline in MTT assays. In striking contrast, K-7174 induced cytotoxicity equally in WT and mutant sublines (Fig. 3A). Next, we confirmed the activity of K-7174 against bortezomib-resistant cells in vivo. We inoculated either WT or the mutant subline into NOD/SCID mice as described above. When measurable tumors developed, mice were randomly assigned to 3 groups: vehicle control, 0.5 mg/kg of bortezomib treated, and 50 mg/kg of K-7174 treated groups. Bortezomib strikingly inhibited tumor growth in mice inoculated with WT subline (Fig. 3B, left panel), but failed to do so when the mutant subline was inoculated (Fig. 3B, right panel). However, K-7174 significantly retarded tumor growth in mice inoculated with not only the WT subline (Fig. 3B, left panel) but also the mutant subline (Fig. 3B, right panel), indicating that K-7174 overcomes mutant PSMB5-confered bortezomib resistance in vivo.
FIGURE 3.

Cytotoxic effects of K-7174 on bortezomib-resistant MM cells. We established wild-type (WT) and mutant (mut) sublines from RPMI8226 by transducing with wild-type and mutated PSMB5 cDNA, respectively. A, cell proliferation was measured by MTT assays after culturing non-transduced cells and each subline with either K-7174 or bortezomib for 72 h. The y axis shows the relative absorbance setting wild-type values to 1.0. p values were calculated by one-way analysis of variance with the Student-Newman-Keuls multiple comparisons test. B, NOD/SCID mice were inoculated subcutaneously with 1 × 107 cells of wild-type (WT) or mutant (mutant) subline into the right thigh. Mice were treated with the vehicle (3% DMSO in 0.9% NaCl) alone (control, open triangle; n = 3), bortezomib, 0.5 mg/kg, intravenously twice a week (Bortezomib, closed square; n = 4), or K-7174, 50 mg/kg, post-orally once daily (K-7174, closed triangle; n = 4), respectively, for 2 weeks. The y axis shows the average tumor volume in inoculated mice (bars indicate S.D.). p values were calculated by one-way analysis of variance with the Student-Newman-Keuls multiple comparisons test. Asterisks indicate p < 0.05 against the vehicle control. C, primary MM cells were cultured with either K-7174 or bortezomib at the indicated doses for 48 h. Cell death/apoptosis was determined by reactivity with annexin-V on a flow cytometer. The y axis shows the proportion of annexin-V-positive cells (%).
Furthermore, we examined the effects of K-7174 on primary MM cells isolated from a patient who was heavily treated and became resistant to bortezomib. CD138-positive bone marrow mononuclear cells were cultured in the absence or presence of either K-7174 or bortezomib for 2 days, followed by annexin-V staining. Primary MM cells from 2 untreated patients were used as controls. Bortezomib induced apoptosis in control MM cells in a dose-dependent manner, but not in cells from a bortezomib-resistant patient (Fig. 3C, right panel). In contrast, K-7174 was able to induce apoptosis in primary MM cells from a bortezomib-resistant patient as well as untreated patients, although there was a variation in drug sensitivity (Fig. 3C, left panel). These results imply that K-7174 has a potential to overcome bortezomib resistance clinically.
Class I HDACs Are Critical Targets of K-7174-induced Cytotoxicity in MM Cells
We have previously shown that bortezomib treatment results in transcriptional repression of class I HDACs, which plays a critical role in bortezomib-mediated cytotoxicity in MM cells (21). We investigated whether this is the case with K-7174. To this end, we examined the expression of class I HDACs (HDAC1, -2, and -3) in MM cell lines during K-7174 treatment. Immunoblot analyses revealed that K-7174 down-regulated HDAC1 expression and reciprocally induced histone hyperacetylation in a dose- and time-dependent fashion (Fig. 4, A and B). To determine whether the down-regulation of HDAC expression occurred at transcriptional or post-transcriptional levels, we performed semi-quantitative RT-PCR analyses. As a result, K-7174 down-regulated class I HDAC genes (HDAC1, -2, and -3) at mRNA levels in all three MM cell lines in a time-dependent fashion (Fig. 4C). In addition, there were no significant changes in the expression of class II and III HDAC genes such as HDAC4, -5, -6, and SIRT1 during K-7174 treatment (Fig. 4D). K-7174 also decreased the expression levels of class I HDAC genes (HDAC1, -2, and -3) in CD138-positive cells isolated from the bone marrow of four MM patients (Fig. 4E). These results suggest that K-7174 specifically repressed the transcription of class I HDACs in MM cells.
FIGURE 4.

Cytotoxic effects of K-7174 depends on the expression levels of class I HDACs in MM cells. A, MM cell lines (KMS12-BM, U266, and RPMI8226) were cultured with K-7174 at the indicated doses for 48 h. Whole cell lysates were subjected to immunoblotting for the expression of HDAC1, acetylated histone H3, histone H3, and GAPDH (internal control). B, MM cell lines were cultured with K-7174 (5 μm for U266 and 10 μm for KMS12-BM and RPMI8226) for up to 3 days. Whole cell lysates were prepared at the given time points and subjected to immunoblotting as described above. C, total cellular RNA was isolated simultaneously in the experiments described in panel B and subjected to semi-quantitative RT-PCR analysis for the expression of HDAC1, HDAC2, HDAC3, and GAPDH (internal control). The amplified products were visualized by ethidium bromide staining after 2% agarose gel electrophoresis. The results of suboptimal amplification cycles, 35 cycles, are shown. The signal intensities of each band were quantified, normalized to those of the corresponding GAPDH, and shown as relative values setting Day 0 controls to 1.0. D, total cellular RNA was isolated from RPMI8226 cells in the experiments described in panel B and subjected to semi-quantitative RT-PCR analysis for the expression of HDAC4, HDAC5, HDAC6, SIRT1, and GAPDH (internal control). The amplified products were visualized by ethidium bromide staining after 2% agarose gel electrophoresis. The results of suboptimal amplification cycles (35 cycles) are shown. Detailed information of primers, including sequences, corresponding nucleotide positions and PCR product sizes, was described previously (21). E, we cultured primary MM cells with either 10 μm K-7174 (+) or the vehicle alone (−) for 48 h. The mRNA expression of class I HDACs was examined by RT-PCR as described above. F, RPMI8226 cells were transduced with either CSII-DsRed (mock) or CSII-DsRed-HDAC1 (HDAC1) vector as described previously (21). Transduced cells were cultured in the absence or presence of 5, 10, and 15 μm K-7174 for 48 h. Cells were harvested, stained with annexin-V, and subjected to flow cytometric analysis. The y axis shows the proportion of annexin-V positivity in the DsRed-positive fraction. The mean ± S.D. (bars) of three independent experiments are shown. p values were calculated by one-way analysis of variance with Student-Newman-Keuls multiple comparisons test. G, RPMI8226 cells were transduced with CSII-DsRed (mock), CSII-DsRed-HDAC1 (HDAC1), CSII-DsRed-HDAC2 (HDAC2), and CSII-DsRed-HDAC3 (HDAC3) vectors as described previously (21). Cell proliferation was measured by MTT assays after culture in the absence or presence of 5 and 10 μm K-7174 for 72 h. The y axis shows a percentage of the value of corresponding to untreated cells. The mean ± S.D. (bars) of three independent experiments are shown. p values were calculated by one-way analysis of variance with the Student-Newman-Keuls multiple comparisons test. Asterisks indicate p < 0.05 against the mock. H, whole cell lysates were prepared simultaneously in the experiments described in panel E and subjected to immunoblotting for the expression of ubiquitinated proteins (Ubiq), HDAC1, acetylated histone H3, and GAPDH (internal control).
To confirm the dependence of K-7174 action on class I HDAC expression, we performed gain-of-function of HDAC1, -2, and -3 using the lentiviral transduction system as described previously (21). Overexpression of each class I HDAC member significantly ameliorated K-7174-induced apoptosis in RPMI8226 cells (Fig. 4, F and G). In addition, we examined the status of histone acetylation in these cells. As anticipated, HDAC1 overexpression almost completely abrogated K-7174-induced histone hyperacetylation without affecting the accumulation of ubiquitinated proteins (Fig. 4H). Taken together, these results indicate that the anti-myeloma action of K-7174 is mediated via proteasome inhibition and subsequent transcriptional repression of class I HDACs.
K-7174 Induces Transcriptional Repression of Class I HDAC Genes via Caspase-8-mediated Degradation of Sp1
Next, we investigated the mechanisms of the transcriptional repression of class I HDAC genes by K-7174. We have previously demonstrated that Sp1 and GATA1 are major transcriptional activators of class I HDAC genes (28); however, MM cells barely expressed GATA transcription factors (data not shown). We therefore focused on the role of Sp1 in K-7174-induced down-regulation of HDAC gene expression (28, 29). To determine the binding of Sp1 to the promoter region of the HDAC1 gene, we performed chromatin immunoprecipitation assays. Sp1 was bound in the HDAC1 promoter regions of untreated MM cells, but dissociated from the promoter upon K-7174 treatment (Fig. 5A). These results suggest that K-7174 targets Sp1 but not GATA1 in MM cells, prompting us to examine the expression of Sp1 during K-7174 treatment. K-7174 markedly diminished the expression levels of Sp1 protein in all three MM cell lines in a time-dependent fashion (Fig. 5B), whereas Sp1 mRNA levels were unchanged (data not shown). To investigate the mechanisms of the decrease in Sp1 protein levels, we determined the activation of caspases in K-7174-treated MM cells, because the Sp1 protein is degraded by caspase-8 during bortezomib treatment (21). Immunoblot analyses revealed that K-7174 activated caspase-8 but not caspase-9 and -12 (Fig. 5C). The peptide inhibitor of caspase-8 (Z-IETD-fmk), but not caspase-9 (Z-LETD-fmk) and caspase-12 (Z-ATAD-fmk), was able to abrogate K-7174-induced down-regulation of Sp1, HDAC1, HDAC2, and HDAC3 (Fig. 5D) as well as cytotoxicity in RPMI8226 cells (Fig. 5E). These results suggest that K-7174 degraded Sp1 protein via the caspase-8-dependent pathway, leading in turn to transcriptional repression of class I HDAC genes in MM cells.
FIGURE 5.

K-7174 induces transcriptional repression of class I HDAC genes via caspase-8-mediated degradation of Sp1. A, MM cell lines were cultured with either K-7174 (+) or vehicle control (−) for 2 days, and subjected to chromatin immunoprecipitation assays. Chromatin suspensions were immunoprecipitated with anti-Sp1 and isotype-matched (IgG) antibodies. The resulting precipitants were subjected to PCR to amplify the promoter regions of the HDAC1 gene as described previously (21). The amplified products were visualized by ethidium bromide staining after 2% agarose gel electrophoresis. Representative data of 50 cycles are shown. Input indicates that PCR was performed with genomic DNA. B, MM cell lines were cultured with K-7174 (5 μm for U266 and 10 μm for KMS12-BM and RPMI8226) for up to 3 days. Whole cell lysates were prepared at the given time points and subjected to immunoblotting for Sp1 and GAPDH (internal control). C, RPMI8226 cells were cultured with 10 μm K-7174 for up to 3 days. Whole cell lysates were subjected to immunoblotting for pro-caspase-8, -9, and -12 and GAPDH (internal control). D, RPMI8226 cells were cultured with 100 μm Z-IETD-fmk (caspase-8 inhibitor), 100 μm Z-LEHD-fmk (caspase-9 inhibitor), 20 μm Z-ATAD-fmk (caspase-12 inhibitor), or vehicle alone (DMSO) for 48 h in the absence or presence of 10 μm K-7174. Whole cell lysates were subjected to immunoblotting for Sp1, HDAC1, HDAC2, HDAC3, and GAPDH (internal control). E, cell viability was determined after culturing RPMI8226 cells in the absence (Control) or presence of 10 μm K-7174 with 100 μm Z-IETD-fmk (IETD), 100 μm Z-LEHD-fmk (LEHD), 20 μm Z-ATAD-fmk (ATAD), or the vehicle control (DMSO) for 72 h. Absorbance at 450 nm was measured with a microplate reader, and expressed as a percentage of the value of the corresponding control cells. The mean ± S.D. (bars) of three independent experiments are shown. p values were calculated by one-way analysis of variance with the Student-Newman-Keuls multiple comparisons test.
Combination of K-7174 and HDAC Inhibitors Exerts Additive Cytotoxic Effects on MM Cells
Finally, we examined the combined effects of K-7174 and other therapeutic agents on MM. Isobologram analyses revealed that K-7174 showed additive cytotoxicity with HDAC inhibitors (romidepsin and vorinostat) in RPMI8226 cells, whereas no such effect was observed in combination with alkylating agents (cyclophosphamide and melphalan) (Fig. 6A). To understand the molecular mechanisms of the additive effects, we examined the expression of class I HDACs by immunoblotting and determined the cellular HDAC activity by monitoring the status of histone acetylation. Romidepsin markedly enhanced K-7174-induced hyperacetylation of histone H3 in RPMI8226 cells (Fig. 6B). These results suggest that class I HDACs are critical molecular targets of K-7174, and HDAC inhibitors may be helpful in enhancing therapeutic effects of K-7174.
FIGURE 6.
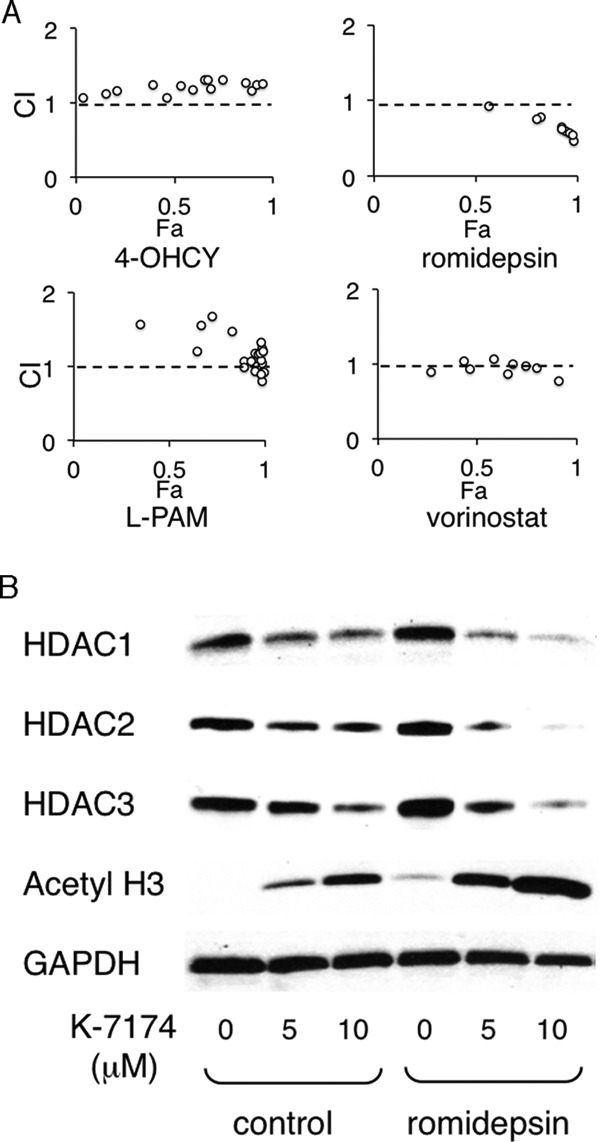
Additive cytotoxic effects of K-7174 and HDAC inhibitors. A, we cultured RPMI8226 cells with various concentrations of K-7174 in combination with 4-hydroperoxy cyclophosphamide (4-OHCY), melphalan (l-PAM), romidepsin, or vorinostat, and assessed for the cell viability after 72 h. Isobologram analysis was carried out using the CompuSyn software program to obtain the combination index (CI). The CI values of less than 1.0 and greater than 1.0 indicate synergism and antagonism, respectively, by definition (44). B, whole cell lysates were prepared simultaneously in the experiments described in panel A and subjected to immunoblotting for the expression of HDAC1, HDAC2, HDAC3, acetylated histone H3, and GAPDH (internal control).
DISCUSSION
In the present study, we show that (i) K-7174 is a novel orally active PI, which exhibits in vitro and in vivo cytotoxicity against MM cells, (ii) is more effective with oral administration than intraperitoneal injection in a murine xenograft model, (iii) is effective for bortezomib-resistant MM cell lines and primary MM cells, (iv) targets class I HDACs for cytotoxicity, and (v) induces additive effects with HDAC inhibitors. This is the first report demonstrating a model system to show that bortezomib resistance could be overcome by a PI and the critical role of class I HDACs in PI-induced cytotoxicity other than bortezomib. Taken together, K-7174 could be a novel therapeutic agent for MM, which allows for flexibility in the dosing schedule, increased patient convenience, and overcoming bortezomib resistance. In addition, K-7174 may be combined with other orally active anti-myeloma agents, such as lenalidomide and vorinostat, to establish the post-oral regimens suitable for treatment in the outpatient clinic.
K-7174 was originally reported as a specific inhibitor of GATA family transcription factors. Umetani et al. (16) reported that K-7174 inhibited cell adhesion via the down-regulation of vascular cell adhesion molecule-1 (VCAM-1) expression on endothelial cells. VCAM-1 is known to be a ligand for very late antigen-4, which was identified as a key molecule for cell adhesion-mediated drug resistance in MM cells (24); therefore, K-7174 may inhibit cell adhesion between stromal cells and MM cells and overcome drug resistance. In addition, Imagawa et al. (17) demonstrated that injection of K-7174 restored erythropoietin production and improved anemia of chronic disease in an in vivo mouse assay. As severe anemia is frequently observed in MM patients, K-7174 is also expected to improve anemia associated with MM.
Previous studies showed that the unfolded protein response is a major mechanism of anti-myeloma action of bortezomib (30); however, our studies indicate a critical role of class I HDACs in K-7174- and bortezomib-induced cytotoxicity (21). Their expressions were down-regulated at a transcriptional level via caspase-8-dependent degradation of the Sp1 protein. As a result, class I HDAC activity was reduced, leading to cytotoxicity in MM cells. We have also demonstrated that the caspase-12 inhibitor, which could inhibit ER stress-induced apoptosis, did not affect K-7174- and bortezomib-induced apoptosis but the caspase-8 inhibitor did. The caspase-8-mediated pathway is dominant over the unfolded protein response in our system. Supporting this idea, Mannava et al. (31) have recently reported that KLF9, which is indirectly transactivated by bortezomib, determines the response to bortezomib independently of the induction of ER stress. They have also shown that bortezomib-induced apoptosis largely depends on the class I HDAC activity. Moreover, Miller et al. (32) reported that caspase-8-dependent histone acetylation is essential for apoptosis induced by NPI-0052, a novel PI under preclinical investigation, in leukemia cells. Taken together, it appears that class I HDACs are critical targets of PI-induced cytotoxicity in hematological malignancies in general. However, the involvement of other mechanisms cannot be ruled out, because HDAC1 overexpression only partially ameliorated K-7174-induced apoptosis. Further investigation is required to fully elucidate the underlying mechanisms of K-7174 action in MM cells.
Although these findings indicate class I HDACs as valid therapeutic targets for MM, HDAC inhibitors, such as vorinostat and romidepsin, showed limited efficacy as single agents in clinical trials (33, 34). This may be attributable to the fact that the class I HDACs mediate various cellular events via not only deacetylation of core histones but also association with various non-histone proteins, causing a variety of side effects (35–37). The combination with PIs might be a strategy to overcome the limitation. On the other hand, HDAC inhibitors have attracted attention for their ability in restoring drug sensitivity in gefitinib-tolerant non-small cell lung cancer (38) and BCR-ABL-positive imatinib-resistant chronic myeloid leukemia stem cells (39). We have also shown that the HDAC inhibitor romidepsin could overcome bortezomib resistance conferred by HDAC1 overexpression in a murine xenograft model (21). Recently, Chesi et al. (40) demonstrated that the combination of HDAC inhibitors, vorinostat and panobinostat, with bortezomib would be effective in the treatment of relapsed MM using genetically engineered mouse models. These results suggest that HDAC inhibitors are effective in overcoming intrinsic drug resistance of malignant cells. Indeed, Harrison et al. (41) and Knop (42) have reported the successful results of clinical trials with romidepsin and bortezomib/dexamethasone for relapsed and/or refractory MM. These pre-clinical and clinical studies suggest that class I HDACs are effective therapeutic targets and the combination of PIs with HDAC inhibitors may represent the best treatment strategy for MM patients (21, 43).
Acknowledgments
We are grateful to Drs. Hiroaki Kimura and Naoya Shibayama (Jichi Medical School) for helpful discussions and technical advice and Akiko Yonekura for excellent assistance.
This work was supported in part by the High-Tech Research Center Project for Private Universities, Matching Fund Subsidy from MEXT, a Grant-in-Aid for Scientific Research from JSPS (to J. K. and Y. F.), the Adaptable and Seamless Technology transfer Program (A-STEP) from JST, and research grants from Japan Leukemia Research Fund, Takeda Science Foundation, Kano Foundation, Mitsui Life Social Welfare Foundation, and Osaka Cancer Foundation (to J. K.).
- PI
- proteasome inhibitor
- HDAC
- histone deacetylase
- MM
- multiple myeloma
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NOD/SCID
- non-obese diabetic/severe combined immunodeficiency
- PSMB5
- proteasome β5-subunit
- VCAM-1
- vascular cell adhesion molecule-1
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Moreau P., Richardson P. G., Cavo M., Orlowski R. Z., San Miguel J. F., Palumbo A., Harousseau J. L. (2012) Proteasome inhibitors in multiple myeloma. 10 years later. Blood 120, 947–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Friedberg J. W., Vose J. M., Kelly J. L., Young F., Bernstein S. H., Peterson D., Rich L., Blumel S., Proia N. K., Liesveld J., Fisher R. I., Armitage J. O., Grant S., Leonard J. P. (2011) The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood 117, 2807–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. San Miguel J. F., Schlag R., Khuageva N. K., Dimopoulos M. A., Shpilberg O., Kropff M., Spicka I., Petrucci M. T., Palumbo A., Samoilova O. S., Dmoszynska A., Abdulkadyrov K. M., Delforge M., Jiang B., Mateos M. V., Anderson K. C., Esseltine D. L., Liu K., Deraedt W., Cakana A., van de Velde H., Richardson P. G. (2013) Persistent overall survival benefit and no increased risk of second malignancies with bortezomib-melphalan-prednisone versus melphalan-prednisone in patients with previously untreated multiple myeloma. J. Clin. Oncol. 31, 448–455 [DOI] [PubMed] [Google Scholar]
- 4. Lonial S., Waller E. K., Richardson P. G., Jagannath S., Orlowski R. Z., Giver C. R., Jaye D. L., Francis D., Giusti S., Torre C., Barlogie B., Berenson J. R., Singhal S., Schenkein D. P., Esseltine D. L., Anderson J., Xiao H., Heffner L. T., Anderson K. C., SUMMIT/CREST Investigators (2005) Risk factors and kinetics of thrombocytopenia associated with bortezomib for relapsed, refractory multiple myeloma. Blood 106, 3777–3784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Richardson P. G., Briemberg H., Jagannath S., Wen P. Y., Barlogie B., Berenson J., Singhal S., Siegel D. S., Irwin D., Schuster M., Srkalovic G., Alexanian R., Rajkumar S. V., Limentani S., Alsina M., Orlowski R. Z., Najarian K., Esseltine D., Anderson K. C., Amato A. A. (2006) Frequency, characteristics, and reversibility of peripheral neuropathy during treatment of advanced multiple myeloma with bortezomib. J. Clin. Oncol. 24, 3113–3120 [DOI] [PubMed] [Google Scholar]
- 6. Hideshima T., Anderson K. C. (2011) Novel therapies in MM. From the aspect of preclinical studies. Int. J. Hematol. 94, 344–354 [DOI] [PubMed] [Google Scholar]
- 7. Kuhn D. J., Chen Q., Voorhees P. M., Strader J. S., Shenk K. D., Sun C. M., Demo S. D., Bennett M. K., van Leeuwen F. W., Chanan-Khan A. A., Orlowski R. Z. (2007) Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 110, 3281–3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khan M. L, Stewart A. K. (2011) Carfilzomib. A novel second-generation proteasome inhibitor. Future Oncol. 7, 607–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chauhan D., Catley L., Li G., Podar K., Hideshima T., Velankar M., Mitsiades C., Mitsiades N., Yasui H., Letai A., Ovaa H., Berkers C., Nicholson B., Chao T. H., Neuteboom S. T., Richardson P., Palladino M. A., Anderson K. C. (2005) A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 8, 407–419 [DOI] [PubMed] [Google Scholar]
- 10. Piva R., Ruggeri B., Williams M., Costa G., Tamagno I., Ferrero D., Giai V., Coscia M., Peola S., Massaia M., Pezzoni G., Allievi C., Pescalli N., Cassin M., di Giovine S., Nicoli P., de Feudis P., Strepponi I., Roato I., Ferracini R., Bussolati B., Camussi G., Jones-Bolin S., Hunter K., Zhao H., Neri A., Palumbo A., Berkers C., Ovaa H., Bernareggi A., Inghirami G. (2008) CEP-18770. A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib. Blood 111, 2765–2775 [DOI] [PubMed] [Google Scholar]
- 11. Kupperman E., Lee E. C., Cao Y., Bannerman B., Fitzgerald M., Berger A., Yu J., Yang Y., Hales P., Bruzzese F., Liu J., Blank J., Garcia K., Tsu C., Dick L., Fleming P., Yu L., Manfredi M., Rolfe M., Bolen J. (2010) Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 70, 1970–1980 [DOI] [PubMed] [Google Scholar]
- 12. Chauhan D., Singh A. V., Aujay M., Kirk C. J., Bandi M., Ciccarelli B., Raje N., Richardson P., Anderson K. C. (2010) A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 116, 4906–4915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lü S., Yang J., Song X., Gong S., Zhou H., Guo L., Song N., Bao X., Chen P., Wang J. (2008) Point mutation of the proteasome β5 subunit gene is an important mechanism of bortezomib resistance in bortezomib-selected variants of Jurkat T cell lymphoblastic lymphoma/leukemia line. J. Pharmacol. Exp. Ther. 326, 423–431 [DOI] [PubMed] [Google Scholar]
- 14. Oerlemans R., Franke N. E., Assaraf Y. G., Cloos J., van Zantwijk I., Berkers C. R., Scheffer G. L., Debipersad K., Vojtekova K., Lemos C., van der Heijden J. W., Ylstra B., Peters G. J., Kaspers G. L., Dijkmans B. A., Scheper R. J., Jansen G. (2008) Molecular basis of bortezomib resistance. Proteasome subunit β5 (PSMB5) gene mutation and over-expression of PSMB5 protein. Blood 112, 2489–2499 [DOI] [PubMed] [Google Scholar]
- 15. Franke N. E., Niewerth D., Assaraf Y. G., van Meerloo J., Vojtekova K., van Zantwijk C. H., Zweegman S., Chan E. T., Kirk C. J., Geerke D. P., Schimmer A. D., Kaspers G. J., Jansen G., Cloos J. (2012) Impaired bortezomib binding to mutant β5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia 26, 757–768 [DOI] [PubMed] [Google Scholar]
- 16. Umetani M., Nakao H., Doi T., Iwasaki A., Ohtaka M., Nagoya T., Mataki C., Hamakubo T., Kodama T. (2000) A novel cell adhesion inhibitor, K-7174, reduces the endothelial VCAM-1 induction by inflammatory cytokines, acting through the regulation of GATA. Biochem. Biophys. Res. Commun. 272, 370–374 [DOI] [PubMed] [Google Scholar]
- 17. Imagawa S., Nakano Y., Obara N., Suzuki N., Doi T., Kodama T., Nagasawa T., Yamamoto M. (2003) A GATA-specific inhibitor (K-7174) rescues anemia induced by IL-1β, TNF-α, or L-NMMA. FASEB J. 17, 1742–1744 [DOI] [PubMed] [Google Scholar]
- 18. Kikuchi J., Shibayama N., Yamada S., Wada T., Nobuyoshi M., Izumi T., Akutsu M., Kano Y., Sugiyama K., Ohki M., Park S-Y., Furukawa Y. (2013) Homopiperazine derivatives as a novel class of proteasome inhibitors with a unique mode of proteasome binding, PLoS One 8, e60649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hideshima T., Chauhan D., Richardson P., Mitsiades C., Mitsiades N., Hayashi T., Munshi N., Dang L., Castro A., Palombella V., Adams J., Anderson K. C. (2002) NF-κB as a therapeutic target in multiple myeloma. J. Biol. Chem. 277, 16639–16647 [DOI] [PubMed] [Google Scholar]
- 20. Hideshima T., Ikeda H., Chauhan D., Okawa Y., Raje N., Podar K., Mitsiades C., Munshi N. C., Richardson P. G., Carrasco R. D., Anderson K. C. (2009) Bortezomib induces canonical nuclear factor-κB activation in multiple myeloma cells. Blood 114, 1046–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kikuchi J., Wada T., Shimizu R., Izumi T., Akutsu M., Mitsunaga K., Noborio-Hatano K., Nobuyoshi M., Ozawa K., Kano Y., Furukawa Y. (2010) Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood 116, 406–417 [DOI] [PubMed] [Google Scholar]
- 22. Drexler H. G., Matsuo Y., MacLeod R. A. (2003) Persistent use of false myeloma cell lines. Hum. Cell 16, 101–105 [DOI] [PubMed] [Google Scholar]
- 23. Shimizu R., Kikuchi J., Wada T., Ozawa K., Kano Y., Furukawa Y. (2010) HDAC inhibitors augment cytotoxic activity of rituximab by up-regulating CD20 expression on lymphoma cells. Leukemia 24, 1760–1768 [DOI] [PubMed] [Google Scholar]
- 24. Noborio-Hatano K., Kikuchi J., Takatoku M., Shimizu R., Wada T., Ueda M., Nobuyoshi M., Oh I., Sato K., Suzuki T., Ozaki K., Mori M., Nagai T., Muroi K., Kano Y., Furukawa Y., Ozawa K. (2009) Bortezomib overcomes cell-adhesion-mediated drug resistance through down-regulation of VLA-4 expression in multiple myeloma. Oncogene. 28, 231–242 [DOI] [PubMed] [Google Scholar]
- 25. Ri M., Iida S., Nakashima T., Miyazaki H., Mori F., Ito A., Inagaki A., Kusumoto S., Ishida T., Komatsu H., Shiotsu Y., Ueda R. (2010) Bortezomib-resistant myeloma cell lines. A role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 24, 1506–1512 [DOI] [PubMed] [Google Scholar]
- 26. Kikuchi J., Shimizu R., Wada T., Ando H., Nakamura M., Ozawa K., Furukawa Y. (2007) E2F-6 suppresses growth-associated apoptosis of human hematopoietic progenitor cells by counteracting proapoptotic activity of E2F-1. Stem Cells 25, 2439–2447 [DOI] [PubMed] [Google Scholar]
- 27. Nonomura C., Kikuchi J., Kiyokawa N., Ozaki H., Mitsunaga K., Ando H., Kanamori A., Kannagi R., Fujimoto J., Muroi K., Furukawa Y., Nakamura M. (2008) CD43, but not P-selectin glycoprotein ligand-1, functions as an E-selectin counter-receptor in human B cell precursor leukemia NALL-1. Cancer Res. 68, 790–799 [DOI] [PubMed] [Google Scholar]
- 28. Wada T., Kikuchi J., Nishimura N., Shimizu R., Kitamura T., Furukawa Y. (2009) Expression levels of histone deacetylases determine the cell fate of hematopoietic progenitors. J. Biol. Chem. 284, 30673–30683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fulciniti M., Amin S., Nanjappa P., Rodig S., Prabhala R., Li C., Minvielle S., Tai Y. T., Tassone P., Avet-Loiseau H., Hideshima T., Anderson K. C., Munshi N. C. (2011) Significant biological role of Sp1 transactivation in multiple myeloma. Clin. Cancer Res. 17, 6500–6509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Obeng E. A., Carlson L. M., Gutman D. M., Harrington W. J., Jr., Lee K. P., Boise L. H. (2006) Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 107, 4907–4916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mannava S., Zhuang D., Nair J. R., Bansal R., Wawrzyniak J. A., Zucker S.N., Fink E.E., Moparthy K. C., Hu Q., Liu S., Boise L. H., Lee K. P., Nikiforov M. A. (2012) KLF9 is a novel transcriptional regulator of bortezomib- and LBH589-induced apoptosis in multiple myeloma cells. Blood 119, 1450–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller C. P., Rudra S., Keating M. J., Wierda W. G., Palladino M., Chandra J. (2009) Caspase-8 dependent histone acetylation by a novel proteasome inhibitor, NPI-0052. A mechanism for synergy in leukemia cells. Blood 113, 4289–4299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Richardson P., Mitsiades C., Colson K., Reilly E., McBride L., Chiao J., Sun L., Ricker J., Rizvi S., Oerth C., Atkins B., Fearen I., Anderson K., Siegel D. (2008) Phase 1 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk. Lymphoma. 49, 502–507 [DOI] [PubMed] [Google Scholar]
- 34. Niesvizky R., Ely S., Mark T., Aggarwal S., Gabrilove J. L., Wright J. J., Chen-Kiang S., Sparano J. A. (2011) Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer 117, 336–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wada T., Kikuchi J., Furukawa Y. (2012) Histone deacetylase 1 enhances microRNA processing via deactylation of DGCR8. EMBO Rep. 13, 142–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhaskara S., Knutson S. K., Jiang G., Chandrasekharan M. B., Wilson A. J., Zheng S., Yenamandra A., Locke K., Yuan J. L., Bonine-Summers A. R., Wells C. E., Kaiser J. F., Washington M. K., Zhao Z., Wagner F. F., Sun Z. W., Xia F., Holson E. B., Khabele D., Hiebert S. W. (2010) Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 18, 436–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delcuve G. P., Khan D. H., Davie J. R. (2013) Targeting class I histone deacetylases in cancer therapy. Expert. Opin. Ther. Targets 17, 29–41 [DOI] [PubMed] [Google Scholar]
- 38. Sharma S. V., Lee D. Y., Li B., Quinlan M. P., Takahashi F., Maheswaran S., McDermott U., Azizian N., Zou L., Fischbach M. A., Wong K. K., Brandstetter K., Wittner B., Ramaswamy S., Classon M., Settleman J. (2010) A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang B., Strauss A. C., Chu S., Li M., Ho Y., Shiang K. D., Snyder D. S., Huettner C. S., Shultz L., Holyoake T., Bhatia R. (2010) Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell 17, 427–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chesi M., Matthews G. M., Garbitt V. M., Palmer S. E., Shortt J., Lefebure M., Stewart A. K., Johnstone R. W., Bergsagel P. L. (2012) Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood 120, 376–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harrison S. J., Quach H., Link E., Seymour J. F., Ritchie D. S., Ruell S., Dean J., Januszewicz H., Johnstone R., Neeson P., Dickinson M., Nichols J., Prince H. M. (2011) A high rate of durable responses with romidepsin, bortezomib, and dexamethasone in relapsed or refractory multiple myeloma. Blood 118, 6274–6283 [DOI] [PubMed] [Google Scholar]
- 42. Knop S. (2011) From the observation DAC. Romidepsin revisited. Blood 118, 6231–6232 [DOI] [PubMed] [Google Scholar]
- 43. McConkey D. (2010) Proteasome and HDAC. Who's zooming who? Blood 116, 308–309 [DOI] [PubMed] [Google Scholar]
- 44. Chou T-C. (2010) Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446 [DOI] [PubMed] [Google Scholar]