

Summary
Corneal scarring due to injury is a leading cause of blindness worldwide and results from dysregulated inflammation and angiogenesis during wound healing. Here we demonstrate that the extracellular matrix metalloproteinase MMP12 (macrophage metalloelastase) is an important regulator of these repair processes. Chemical injury resulted in higher expression of the fibrotic markers α-smooth muscle actin and type I collagen, and increased levels of angiogenesis in corneas of Mmp12−/− mice compared with corneas of wild-type mice. In vivo, we observed altered immune cell dynamics in Mmp12−/− corneas by confocal imaging. We determined that the altered dynamics were the result of an altered inflammatory response, with delayed neutrophil infiltration during the first day and excessive macrophage infiltration 6 days later, mediated by altered expression levels of chemokines CXCL1 and CCL2, respectively. Corneal repair returned to normal upon inhibition of these chemokines. Taken together, these data show that MMP12 has a protective effect on corneal fibrosis during wound repair through regulation of immune cell infiltration and angiogenesis.
Key words: Corneal inflammation, Matrix metalloproteinases, Wound repair
Introduction
Corneal clarity and good visual quality are obtained through a highly ordered tissue architecture and lack of vascularization (Azar, 2006; Hassell and Birk, 2010). Millions of people worldwide have vision loss associated with corneal scarring and currently there are few non-surgical treatment options for those affected (Whitcher et al., 2001). Corneal injury occurs by a variety of mechanisms including infectious and noninfectious ulcers, incisional and laser surgery and trauma. Corneal epithelial and stromal cells are structurally supported by an extracellular matrix (ECM) that is composed of a mixture of molecules including collagens, laminin, fibronectin and heparan sulfate proteoglycans (Hassell and Birk, 2010). In addition to its structural role, the ECM serves as a reservoir for embedded cytokines and growth factors (Mott and Werb, 2004). Under normal conditions, proteinases mediate the maintenance of corneal tissue homeostasis through their proteolytic activities. Following injury, this proteolysis regulates aspects of the repair process including inflammation, neovascularization and remodeling (Sivak and Fini, 2002). Excessive proteolysis resulting in corneal scarring has been associated with loss of corneal clarity (Azar, 2006; Fini et al., 1998).
The matrix metalloproteinases (MMPs) represent the most prominent family of proteinases associated with corneal wound repair (Fini et al., 1998; Gordon et al., 2011; Mulholland et al., 2005). Human case studies have reported an increase in MMP expression and activity that correlates with severity of corneal disease (Brejchova et al., 2009; Gabison et al., 2005; Rohini et al., 2007). It is well established that MMPs can degrade all components of the extracellular matrix (Egeblad and Werb, 2002). More recently, the substrate repertoire of MMPs has expanded to include cell membrane-bound precursor forms of many growth factors, growth factor receptors, cell adhesion molecules and chemokines (Kessenbrock et al., 2010; Mott and Werb, 2004). As a result, MMPs are receiving more recognition for their important regulatory roles in various physiological and pathological processes including inflammation, neovascularization and cell proliferation.
MMP12 (macrophage metalloelastase) was first identified and characterized as a secreted protein from mouse peritoneal macrophages through its elastase activity (Werb and Gordon, 1975). It is a 54 kDa protease that shares many features typical of members of the MMP family including its chromosomal location, domain structure, and its ability to degrade ECM components. MMP12 differs from other MMPs with respect to its predominantly macrophage-specific pattern of expression and its ability to shed its C-terminal hemopexin domain upon processing (Shapiro, 1999; Shapiro et al., 1993).
MMP12 regulates fibrosis in several models of tissue injury (Dean et al., 2008; Hautamaki et al., 1997). In the Fas-induced model of acute lung injury, MMP12-deficient mice are protected from developing pulmonary fibrosis (Matute-Bello et al., 2007). After infection with the helminth parasite Schistosoma mansoni, MMP12-deficient mice exhibit reduced liver and lung fibrosis (Madala et al., 2010). In a murine model of cigarette smoke-induced emphysema, MMP12-deficient mice do not develop emphysema in response to long-term cigarette smoke exposure (Hautamaki et al., 1997). In humans, a minor allele of a single-nucleotide polymorphism (SNP) in MMP12 is positively associated with lung function in children with asthma and in adult smokers with chronic obstructive pulmonary disease (COPD) (Hunninghake et al., 2009).
Little is known about the role of MMP12 in the corneal repair process. MMP12 is upregulated in corneal injury: Scratch injury of rat corneal epithelium shows MMP12 expression in the periphery of healed epithelium (Lyu and Joo, 2005; Lyu and Joo, 2006), while alkali injury of rabbit corneas results in MMP12 expression by stromal myofibroblasts (Iwanami et al., 2009). Human corneal stromal fibroblasts treated with IL-1α to model early inflammation show a 10-fold increase in MMP12 expression compared with untreated fibroblasts (Mahajan et al., 2002). In the present study we tested the hypothesis that MMP12 significantly contributes to the recruitment of inflammatory cells and the negative angiogenic response in the process of corneal wound healing. We uncovered protective effects of MMP12 through its regulation of immune cell accumulation and activity, neovascularization, and fibrosis following injury.
Results
Mmp12 is expressed during corneal wound healing
We first examined the time course of Mmp12 expression in wild-type (WT) mouse corneas following chemical injury using a well-established alkali injury model for studying corneal inflammation, neovascularization and fibrosis (Carlson et al., 2003) (Fig. 1A). The injuries resulted in large corneal epithelial defects that healed completely by 8 days post-injury (Fig. 1A). At this time point, corneas had lost their normal transparency and avascularity, and were opaque and with peripheral neovascularization (Fig. 1A). Immunohistochemistry performed on unwounded corneas and corneas wounded 1 week prior demonstrated low MMP12 protein levels in native corneas and increased MMP12 protein levels following injury (Fig. 1B). MMP12-positive cells were observed in the corneal epithelium and stroma (Fig. 1B). We collected injured corneas at 2, 4, 6 and 8 days after injury and measured the time course of Mmp12 mRNA expression using quantitative real-time PCR (qPCR) analysis. We found a reproducible bimodal pattern of Mmp12 expression with highly elevated levels of expression at 2 and 6 days after injury (11-fold and 19-fold, respectively) and mildly elevated levels of expression at 4 and 8 days after injury (2-fold and 4-fold, respectively; Fig. 1C). Since macrophages are a major source of MMP12, it is interesting that the high expression levels of Mmp12 noted at 2 and 6 days after injury corresponded with time points of high macrophage recruitment (Lu et al., 2008; Nakao et al., 2005). Since resident macrophages are normally present in the cornea (Brissette-Storkus et al., 2002), we determined their ability to express Mmp12. Cells that were CD45+, CD11b+, F4/80+ were sorted by flow cytometry and analyzed for Mmp12 mRNA expression by qPCR (Fig. 1D). Resident macrophages of naïve WT corneas expressed Mmp12 while resident macrophages of Mmp12−/− mice lacked Mmp12 expression (Fig. 1D). These data support expression of Mmp12 both by native corneal macrophages as well as macrophages recruited from the peripheral circulation.
Fig. 1.
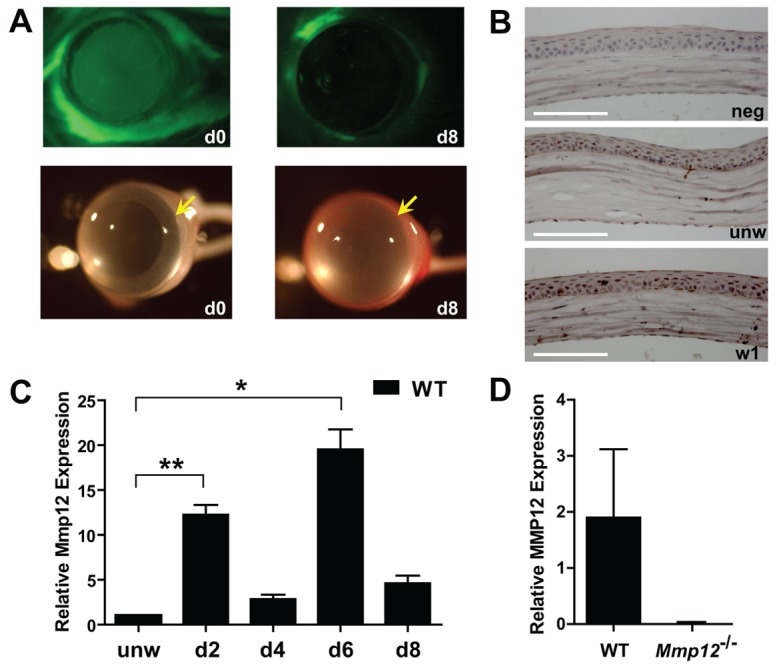
Mmp12 is expressed in a murine model of corneal epithelial and stromal injury. (A) The corneal injury model using filter paper soaked in sodium hydroxide applied to the central corneal results in a large, diffuse corneal epithelial defect immediately after injury (d0) seen with fluorescein staining (top panels), that is healed by 8 days after injury (d8). Photographs of the cornea (bottom panels) just prior to injury (d0) and 8 days after injury (d8) show diffuse cornea edema and peripheral neovascularization after injury. Yellow arrows indicate the boundary between the cornea and sclera. (B) Representative images of corneal tissue immunostained for MMP12 prior to injury (unw) and 1 week following injury (w1) and isotype control (neg). Scale bars: 100 µm. (C) Quantification of Mmp12 mRNA expression levels in wild-type mouse corneas after corneal injury. Relative levels (means ± s.e.m.); the means and ranges are: 12.3 (10.7–14.3), 2.8 (2.2–3.8), 19.5 (16.9–24.0) and 4.6 (3.1–6.0) for day 2 (d2), day 4 (d4), day 6 (d6) and day 8 (d8), respectively (n = 6), **P<0.005 and *P<0.05. (D) Mmp12 mRNA expression levels in sorted CD45+, CD11b+, F4/80+ cells isolated from unwounded corneas of WT and Mmp12−/− mice. Values are means ± s.e.m.; the mean and ranges are 1.89 (0.14–8) and 0 (0–0), respectively (n = 6).
MMP12 protects against corneal stromal myofibroblast transformation after injury
Because we found that MMP12 was expressed during corneal repair, we evaluated whether corneal fibrosis after chemical injury was altered in Mmp12−/− mice compared with WT mice. Wounds were generated and epithelial healing was monitored and compared by performing fluorescein staining and photographing the corneas immediately after injury, and 4 days, 2 weeks and 3 weeks after injury (Fig. 2A). The Mmp12−/− mice had delayed corneal epithelial closure at 4 days post-injury compared with WT mice, but demonstrated fully healed corneal epithelium by 2 and 3 weeks (Fig. 2A,B). Fibrotic markers, such as α-smooth muscle actin that marks myofibroblasts, begin to be expressed in the corneal stroma ∼7 days after injury (Stramer et al., 2005) and we found their expression to be maximal by 3 weeks. Therefore we collected corneas 3 weeks following chemical injury and stained using antibodies against fibrotic markers. We noted stronger expression of α-smooth muscle actin (SMA) in wounded Mmp12−/− corneas compared with WT corneas (Fig. 2C), while there was similar immunostaining for tenascin-C and fibronectin (Fig. 2C). Using qPCR analysis, we observed increased SMA expression in the wounded Mmp12−/− corneas as well as increased levels of collagen type 1 and collagen type 3, two other markers of corneal fibrosis (Fig. 2D). These results suggest that MMP12 is protective against corneal stromal myofibroblast transformation following injury.
Fig. 2.

Delayed epithelial healing and increased myofibroblast transformation in Mmp12−/− mice during corneal wound repair. (A) Photographs of scratch injured corneas just prior to injury (d0) and 4 days (d4), 2 weeks (w2) and 3 weeks (w3) after injury. (B) Hematoxylin and Eosin staining of WT and Mmp12−/− mouse corneas 3 weeks after injury. (C) Immunofluorescence of α-smooth muscle actin (SMA), tenasci and fibronectin in WT and Mmp12−/− mouse corneas 3 weeks after corneal injury. Nuclei were visualized by staining with DAPI (blue). Scale bars: 100 µm. (D) Quantification of expression levels of markers of fibrosis (SMA, COL1 and COL3) in wounded WT and Mmp12−/− mouse corneas 1 (w1; n = 6) and 3 (w3; n = 4) weeks after corneal injury. Results are means ± s.e.m.; *P<0.05.
MMP12 blunts the corneal angiogenic response to injury
To investigate how MMP12 protects against stromal fibrosis, we next examined the effect of MMP12 on corneal angiogenesis. Unlike other tissues, the cornea must mount an anti-angiogenic response during the repair of injury to prevent blood vessels from entering the cornea and decreasing its clarity. Indeed, MMP12 is the most potent MMP in degrading plasminogen to generate angiostatin, which inhibits microvascular endothelial cell proliferation (Cornelius et al., 1998). To determine if MMP12 contributes significantly to the negative angiogenic response we observed chemically injured corneas of WT and Mmp12−/− mice. Angiogenesis increased as early as 3 days after injury, with prominent angiogenesis 1 week after injury. Using an antibody against the endothelial marker CD31 on corneal flat mounts collected 1 week after injury, we observed that Mmp12−/− mice showed elevated levels of corneal neovascularization compared with WT mice (Fig. 3A,B), with significantly increased lengths of individual vessels and areas covered by neovascular vessels in injured Mmp12−/− corneas as compared with that in WT control mice.
Fig. 3.

MMP12 alters corneal angiogenesis following injury. (A) Micrographs of whole-mount corneal preparations stained for angiogenic endothelium (CD31) 6 days after injury. (B) Quantitative analysis of angiogenesis, showing lengths of individual vessels in injured Mmp12−/− corneas were 1.7 times longer compared with those in WT corneas (32±4.9 µm versus 18±1.2 µm; n = 11, P<0.05). Additional morphometric comparison of the area covered by CD31+ vessels in WT and Mmp12−/− corneas confirmed that the area was larger in the Mmp12−/− corneas as compared with that in WT control mice (10738±1245 pixels versus 6513±1017 pixels; n = 11, P<0.05). (C) Micrographs demonstrating corneal angiogenesis 6 days after injury in mice that underwent bone marrow transplantation. (D) Quantitative analysis of angiogenesis following bone marrow transplantation. After transplantation of WT and Mmp12−/− bone marrow cells into Mmp12−/− and WT recipient mice, respectively, neovascular lengths and areas were similar (41±8.9 µm versus 39±4.0 µm and 23273±7415 pixels versus 21850±4104 pixels, n = 7). Transplantation of Mmp12−/− bone marrow cells into Mmp12−/− recipient mice resulted in increased neovascular lengths compared with transplantation of WT bone marrow cells into WT donor mice (40±6.8 µm versus 23±6.8 µm and 12759±3455 pixels versus 10663±3698 pixels; n = 3, P<0.05). (E) Quantification of VEGFA and VEGFB mRNA expression levels in WT and Mmp12−/− mouse corneas 6 days after corneal injury. Results are means ± s.e.m., n = 8, **P<0.005. (F) Levels of VEGFA and plasminogen cleavage products in corneal lysates at 6 days post-injury. Western blots were probed with GAPDH as a loading control. (G) Corneal tissues at 1 and 4 days post-injury were homogenized and soluble lysates (10 µg per well) were subjected to gelatin zymography. Protein standards were used to identify inactive MMP on zymograms and are indicated with asterisks (**MMP9 and *MMP2). (H) Representative whole-mount micrographs stained for lymphangiogenic (LYVE-1) endothelium 6 days after injury. (I) Quantitative analysis of lymphangiogenesis, measured as vessel lengths pixel density. Values are means ± s.e.m. (n = 11). Scale bars: 10 µm.
Sources of MMP12 in the cornea following injury include resident corneal macrophages and macrophages recruited from the peripheral circulation (Brissette-Storkus et al., 2002; Chinnery et al., 2008; Hamrah et al., 2003). To determine the contribution of MMP12 produced by peripheral macrophages to corneal angiogenesis following injury, we reciprocally transplanted bone marrow cells from Mmp12−/− mice and WT mice into recipient mice that were lethally irradiated to generate bone marrow chimeric mice. To ensure engraftment, we waited 6 weeks before chemically injuring the corneas. Mmp12−/− recipient mice transplanted with Mmp12−/− bone marrow cells showed increased neovascular lengths compared with WT recipient mice transplanted with WT bone marrow cells (Fig. 3C,D). Interestingly, transplantation of Mmp12−/− bone marrow cells into WT recipient mice did not result in increased neovascular lengths compared with Mmp12−/− mice transplanted with WT bone marrow cells. Instead, neovascular lengths and areas similar to WT were observed. This partial reversal of neovascularization following bone marrow transplantation indicates that MMP12 produced from either bone marrow macrophages or corneal resident macrophages is sufficient to inhibit neovascularization.
We next determined the mechanism leading to the increased vascularization. Since VEGF is upregulated in inflamed and vascularized corneas in both human and animal models (Azar, 2006), we next analyzed levels of the vascular endothelial growth factors, VEGFA and VEGFB, in the wounded Mmp12−/− corneas. Six days after injury, levels of VEGFA mRNA in MMP12−/− corneas were twice that measured in WT corneas by qPCR and western blot (Fig. 3E,F). No difference was noted for VEGFB expression. Since MMP12 is proficient in degrading plasminogen to generate angiostatin in vitro (Cornelius et al., 1998), we evaluated plasminogen cleavage in wounded WT and MMP12−/− corneas by western blot. We observed that plasminogen was more effectively cleaved into plasmin heavy chain A by WT mice as compared with MMP12−/− mice (Fig. 3F).
MMP2 and MMP9 can increase the bioavailability of VEGF (Mott and Werb, 2004) and their enzymatic activities have been implicated in mediating corneal neovascularization (Kvanta et al., 2000; Zhang et al., 2002). Accordingly, we asked if MMP9 and MMP2 activities are elevated in wounded Mmp12−/− corneas. Both MMP2 and MMP9 activity levels were higher 1 day after injury compared with 4 days after injury by gelatin zymography. At both time points, although slightly more MMP2 activity was seen in the injured Mmp12−/− corneas, MMP9 activity levels were similar (Fig. 3G). Based on these results, we conclude that MMP12 does not have a dramatic effect on MMP2 and MMP9 activities in the cornea following chemical injury. Therefore, the increased angiogenesis noted in Mmp12−/− corneas compared with WT corneas is not likely due to an effect of MMP12 on MMP9 and MMP2 activities, but instead to increased VEGFA expression.
Lymphatic vessels are not usually present in the cornea, but corneal injury can result in lymphangiogenesis (Chen et al., 2011; Cursiefen et al., 2003). We stained flat mounts with the lymphatic vessel marker LYVE-1 to assess corneal lymphangiogenesis 7 days after injury (Fig. 3H). We observed that the average outgrowth lengths and morphometric comparison of the areas covered of LYVE-1 lymphatic vessels in injured Mmp12−/− corneas and WT corneas were similar (Fig. 3I). Therefore, unlike angiogenesis, MMP12 does not have a significant effect on corneal lymphangiogenesis following chemical injury.
Intravital imaging demonstrates altered neutrophil activity in MMP12-deficient mice
Our findings that MMP12 diminishes corneal fibrosis and angiogenesis following injury led us to ask what cell types are responsible for these protective effects of MMP12. Because macrophages are a major source of MMP12, we tested the role of MMP12 on inflammation and immune cell dynamics using an in vivo imaging platform [see Materials and Methods (Speier et al., 2008a; Speier et al., 2008b)]. We crossed c-fms-GFP mice (Sasmono et al., 2003), in which EGFP is driven under the CSF-1R promoter and highlights myeloid cells including neutrophils, monocytes and macrophages, with Mmp12−/− mice to generate WT and Mmp12−/− mice expressing EGFP-labeled myeloid cells. We visualized and monitored myeloid cell behavior in vivo in the corneal stroma and imaged individual cell activity (Fig. 4A; supplementary material Movies 1 and 2) in unwounded corneas and in wounded corneas at 1 day and 4 days after chemical injury. In unwounded WT corneas, myeloid cells were minimally motile and showed short track lengths, slow crawling velocity and little displacement (Fig. 4B,C). Myeloid cells of unwounded corneas deficient in MMP12 had a slight increase in all three of these dynamic parameters compared with myeloid cells of WT mice. One day after injury, we observed a large increase in track lengths, velocity, and displacement of WT myeloid cells (Fig. 4B,C). By contrast, MMP12−/− myeloid cells were much less dynamic after injury and exhibited decreased velocity and only a small increase in track lengths and displacement (Fig. 4B,C).
Fig. 4.

Myeloid cell dynamics are inhibited in injured Mmp12−/− mouse corneas. (A) Examples of migration of myeloid cells in the stroma of injured corneas – a high-migratory cell in a WT cornea (yellow arrow), and a low-migratory cell in a Mmp12−/− cornea (yellow arrow). See supplementary material Movies 1 and 2 for time-lapse recordings. Scale bars: 50 µm. (B) Representative tracts of migrating cells during 30 minutes of imaging. Each color represents one cell. (C) Motility analysis of myeloid cells showing average track lengths, average track lengths per minute, total displacement and velocity. (D) One day after injury, WT corneas have a greater proportion of c-fms-EGFP+ cells that are Gr-1+. Representative whole-mount micrographs show that most of the c-fms-EGFP+ cells are also positive for Gr-1 in WT corneas, whereas fewer are positive for Gr-1 in Mmp12−/− corneas. (E) Injured WT corneas have a higher percentage of Gr-1+ cells compared with Mmp12−/− corneas. The means ± s.e.m. percentage of Gr-1+ cells are shown and are 94±2.3% (WT; n = 9) and 64±13% (Mmp12−/−; n = 8), *P<0.05. Scale bars: 100 µm.
To determine the primary cell type of the highly dynamic cells noted 1 day after injury, we collected corneas and stained flat mount preparations of the mice expressing c-fms-EGFP with the marker Gr-1 to assess the relative populations of neutrophils and macrophages (Fig. 4D). Neutrophils represented a significant percentage of the myeloid cells present 1 day after injury and were present in higher abundance in the corneas of WT mice compared with Mmp12−/− mice (Fig. 4D,E). This result indicates that MMP12 has a critical role in promoting neutrophil activities acutely after injury.
MMP12 promotes the accumulation of neutrophils in wounded corneas
To visualize the effect of MMP12 on neutrophil infiltration before and after injury directly, we wounded Mmp12−/− mice and examined them by flat mount immunostaining for Gr-1. Rare neutrophils were present in the unwounded corneas of WT and MMP12−/− mice (Fig. 5A). One day after chemical injury, significantly fewer neutrophils (50%) accumulated in anterior stroma of MMP12−/− corneas compared with WT corneas (Fig. 5A,B). This result demonstrated the lack of neutrophils in WT and MMP12−/− unwounded corneas and confirmed their positive recruitment by MMP12 following injury.
Fig. 5.

MMP12 deficiency results in decreased neutrophil infiltration following injury. (A) Representative whole-mount micrographs of uninjured corneas and corneas injured 1 day prior from WT and Mmp12−/− mice. Neutrophils are absent in uninjured corneas and 1 day after injury fewer Gr-1+ cells accumulate in Mmp12−/− corneas. (B) Quantification of Gr-1 levels in wild-type and Mmp12−/− mouse corneas 1 day after injury. Mean pixel levels are shown and are 144804 (WT; n = 6) and 77522 (Mmp12−/−; n = 9), **P<0.005. (C) CXCL chemokine expression levels are altered in wounded Mmp12−/− corneas. Cytokine protein expression in WT and Mmp12−/− corneas 1 and 4 days after injury. Individual cytokines were spotted in duplicate and the identity of CXCL1 is indicated. Positive control spots are located at the corners. (D) Relative cytokine levels in corneas of WT and Mmp12−/− mice 1 and 4 days after injury. Duplicate spots were averaged. CXCL1 levels are indicated with an asterisk (*). (E) Effect of CXCL1 neutralization on neutrophil infiltration in the cornea. An antibody to CXCL1 or isotype control (rat IgG) was injected into the subconjunctival space of WT mice. After 2 hours, the corneas were wounded chemically. Representative whole-mount micrographs of the corneas demonstrate a decrease in Gr-1+ cells following CXCL1 antibody injection compared with isotype control. (F) Quantification of Gr-1 levels in corneas of rat IgG-treated and anti-CXCL1-treated mice. The numbers (means ± s.e.m.) of Gr-1+ cells per corneal section are shown, and are 44167±7387 (rat IgG2a; n = 4) and 13713±3034 pixels (anti-CXCL1; n = 4), *P<0.05. Scale bars: 10 µm.
MMP12 alters the expression of chemokine CXCL1
CXC chemokines are the major chemoattractants for neutrophils in inflammation (Allen et al., 2007). Therefore, we investigated whether altered expression of these chemokines might be a mechanism by which neutrophils are differentially recruited to corneas of MMP12−/− mice. We used a cytokine protein expression array with protein lysates collected from corneas of WT and Mmp12−/− mice 1 day and 4 days after chemical injury (Fig. 5C). Expression of CXCL1, but not of CXCL9, -10, -11, -12 and -13, was reduced in the wounded corneas of Mmp12−/− mice at both 1 day and 4 days. This result suggests that MMP12 regulates protein expression of CXCL1 the first few days after injury and may be a mechanism by which MMP12 increases neutrophil accumulation in wounded corneas.
To evaluate the importance of CXCL1 in mediating neutrophil recruitment to the cornea and migration through the corneal stroma, we injected a neutralizing antibody to CXCL1 or an isotype control into the subconjunctival space of WT mice 2 hours prior to chemical injury (Lin et al., 2007; Xue et al., 2007). Flat mount immunostaining showed that injured corneas of mice injected with the rat IgG2a isotype control antibody had abundant Gr-1-positive cells infiltrating into the central cornea (Fig. 5E). However, significantly fewer Gr1 cells (68±5.5%) infiltrated corneas of mice injected with anti-CXCL1 (Fig. 5F). This result demonstrates that CXCL1 has a significant role in mediating neutrophil recruitment to the corneal stroma following chemical injury.
MMP12 inhibits the accumulation of macrophages in wounded corneas
Macrophages are the major source of MMP12. We next investigated whether macrophage levels are also altered in the corneas of Mmp12−/− mice. When we stained flat mounts of healthy unwounded corneas with an antibody against the macrophage marker F4/80, we observed that resident macrophages were similar in number and distribution in unwounded corneas of WT and Mmp12−/− mice and were evenly distributed with regular spacing between cells (Fig. 6A,B). Following chemical injury, this orderly distribution pattern was disrupted and the macrophages migrated into and accumulated in the central cornea (Fig. 6A). Six days after injury, the number of macrophages in the central corneas of Mmp12−/− mice was twofold higher compared with corneas of injured WT mice (Fig. 6A,B). Flow cytometric analysis of immune cells from entire wounded corneas 6 days after injury confirmed an increase in macrophage levels in Mmp12−/− whole mouse corneas (Fig. 6C). These data suggest a functional role of MMP12 in negatively regulating the accumulation of macrophages into wounded corneas.
Fig. 6.
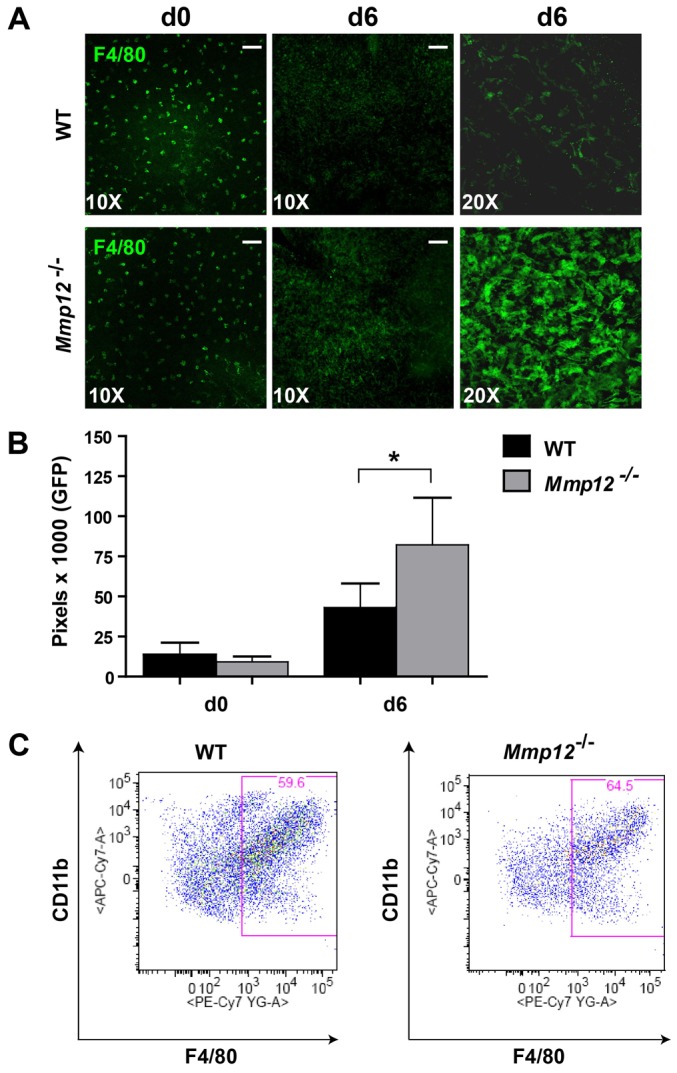
MMP12 deficiency results in increased macrophage infiltration. (A) Representative whole-mount micrographs of the central cornea demonstrating similar F4/80+ (green) cellular distribution and quantity in uninjured (day 0) corneas of WT and Mmp12−/− mice at 10× and 20× magnification. Six days after injury (d6), increased levels of F4/80+ cells are presents in corneas of Mmp12−/− mice. (B) Quantification of F4/80 levels in WT and Mmp12−/− mouse corneas before injury (d0) and 6 days after injury (d6). Mean pixel numbers are shown and are 13713 (WT) and 9246 (Mmp12−/−) for unwounded corneas (n = 7), and 43011 (WT) and 82241 (Mmp12−/−) 6 days after injury (n = 13 corneas), *P<0.05. (C) Corneal immune cells were isolated from corneas of WT (n = 3) and Mmp12−/− mice (n = 3) injured 6 days previously. FACS analysis of CD11b versus F4/80 after gating on live cells demonstrated CD11b+/F4/80+ = 59.5±0.088% and 64.6±0.24% for WT and Mmp12−/− mice, respectively, *P<0.05. Scale bars: 10 µm.
Bone marrow transplantation reverses the increased macrophage infiltration seen in Mmp12−/− mice
To determine the contribution of MMP12 produced by resident macrophages versus macrophages from the peripheral circulation to injured corneas, we performed bone marrow transplantation. Following lethal irradiation, bone marrow cells from Mmp12−/− mice and WT mice were reciprocally transplanted into recipient mice to generate bone marrow chimeric mice. As controls, bone marrow cells from Mmp12−/− donor mice were transplanted into Mmp12−/− recipient mice and bone marrow cells from WT donor mice were transplanted into WT recipient mice. Analysis of macrophage infiltration 6 days after injury revealed that transplantation with WT donor bone marrow cells reversed the increased macrophage infiltration seen in Mmp12−/− mice (Fig. 7A,B). Furthermore, WT mice transplanted with bone marrow cells from Mmp12−/− mice showed an approximate twofold increase in central corneal macrophage infiltration, similar to the results observed in non-transplanted Mmp12−/− mice (Fig. 7B). As expected, Mmp12−/− mice transplanted with bone marrow cells from donor Mmp12−/− mice had higher levels of macrophage infiltration compared with WT mice transplanted with bone marrow from donor WT mice (Fig. 7C,D). These results demonstrate that MMP12 produced by bone marrow-derived macrophages have an important function in inhibiting the recruitment of macrophages to injured corneas.
Fig. 7.

Bone marrow transplantation reverses the increased macrophage infiltration seen in Mmp12−/− mice. (A) Wild-type mice and Mmp12−/− mice were given transplants of bone marrow from Mmp12−/− mice or wild-type mice, respectively. Representative whole-mount micrographs of the central cornea of uninjured corneas (d0) and corneas injured 6 days previously (d6) demonstrate increased F4/80+ cell infiltration after Mmp12−/− bone marrow cells are transplanted into wild-type mice. (B) Quantification of corneal F4/80 levels following bone marrow transplantation before and after injury. Mean pixel levels are shown and are 5460 (WT to Mmp12−/−; n = 9) and 10634 (Mmp12−/− to WT; n = 11) for unwounded corneas, and 64028 (WT to Mmp12−/−; n = 8) and 199503 (Mmp12−/− to WT; n = 11) 6 days after injury (n = 13), *P<0.05. (C) Wild-type mice and Mmp12−/− mice were given transplants of bone marrow from wild-type or Mmp12−/− mice, respectively. Increased F4/80+ cell infiltration occurred in the injured corneas of Mmp12−/− mice. (D) Quantification of corneal F4/80 levels following bone marrow transplantation before and after injury show increased F4/80 levels in injured corneas of Mmp12−/− mice. Mean pixel levels are 15559 (WT to WT; n = 6) and 10274 (Mmp12−/− to Mmp12−/−; n = 7) for unwounded corneas, and 78531 (WT to WT; n = 7) and 99197 (Mmp12−/− to Mmp12−/−; n = 11) 6 days after injury (n = 6), *P<0.05. Scale bars: 10 µm.
MMP12 alters the expression of CCL2
Since CC chemokines are important inducers of the migration of monocytes and macrophages, we next asked if altered expression of these chemokines could account for the differential recruitment of macrophages to wounded corneas of Mmp12−/− mice. We collected RNA from unwounded corneas or from corneas wounded by chemical injury 6 days prior to collection. By qPCR analysis the mRNA expression levels of CCL7, -8, and -20 in unwounded healthy corneas were lower in Mmp12−/− mice compared with WT mice (Fig. 8A). However, wounded corneas of Mmp12−/− mice showed a significant 1.5-fold increase in CCL2 expression and 1.7-fold decrease in CCL8 expression compared with wounded WT corneas (Fig. 8A). There was no significant difference in CCL7 and CCL20 expression levels.
Fig. 8.

CCL chemokine expression levels are altered in wounded Mmp12−/− corneas. (A) Relative expression levels of CCL chemokine family members CCL2, CCL7, CCL8 and CCL20 in WT and Mmp12−/− corneas before injury (d0) and 6 days after injury (d6), as determined by qRT-PCR. Expression levels are relative to uninjured WT corneas. Results are means ± s.e.m. (n = 6), **P<0.005 and *P<0.05. (B) Cytokine expression in WT and Mmp12−/− corneas 1 day after injury. Individual cytokines are spotted in duplicate and the identity of CCL2 is indicated. Positive control spots are located at the corners. (C) Relative cytokine levels in corneas of WT and Mmp12−/− mice 4 days after injury. Duplicate spots were averaged. CCL2 levels are indicated with an asterisk (*). (D) Effect of CCL2 neutralization on macrophage infiltration in the cornea. An antibody to CCL2 or PBS control was injected in the subconjunctival space of WT mice. After 2 hours, the corneas were wounded chemically. The subconjunctiva were re-injected with an antibody to CCL2 or PBS control 3 days after injury and the corneas were collected 6 days after injury. Representative whole-mount micrographs of the corneas demonstrate decreased F4/80+ cells following CCL2 antibody injection compared with the PBS control. (E) Quantification of F4/80 levels in corneas of PBS-treated and anti-CCL2-treated mice. The mean number (± s.e.m.) of F4/80+ cells per corneal section are shown and are 128±38 pixels (PBS; n = 7) and 28±8 (anti-CCL2; n = 7), *P<0.05. Scale bars: 10 µm.
Similar to the change in CCL2 RNA expression levels, we observed a 1.6-fold increase in protein levels of CCL2 in protein lysates from corneas of Mmp12−/− mice wounded 1 day prior by chemical injury, compared to WT wounded corneas (Fig. 8B,C). CCL1, -3, -4, -5, -11 and -17 did not show increased expression in MMP12−/− mice (Fig. 8C).
To determine the importance of CCL2 in mediating macrophage recruitment to the cornea and migration through the corneal stroma, we injected a neutralizing antibody to CCL2 or PBS control into the subconjunctival space of WT mice 2 hours prior to chemical injury and again 3 days later. Injured corneas of mice injected with PBS had abundant infiltration of F4/80-positive cells into the central cornea (Fig. 8D). Infiltration of these cells showed a significant 68±8.9% reduction in F4/80-positive cells in corneas in mice injected with anti-CCL2 antibody compared with corneas of mice injected with PBS control (Fig. 8D,E). This result demonstrates that CCL2 has a significant role in mediating macrophage recruitment to the corneal stroma following chemical injury. MMP12 therefore decreases expression of CCL2, thus contributing to a mechanism by which it reduces macrophage accumulation in wounded corneas.
Discussion
The repair response to injury is a complex process that results in the activation of inflammatory and neovascular pathways. MMPs are expressed after injury and are important modulators of these repair pathways. Using a corneal chemical injury model, we observed significantly elevated levels of Mmp12 expression 2 and 6 days after injury corresponding to time points of high macrophage recruitment. We used MMP12 null mice to determine the role of MMP12 in repair mechanisms following injury. We observed that injured corneas of Mmp12−/− mice had elevated expression of a marker of myofibroblasts, α-smooth muscle actin, and increased neovascularization. To identify the cells responsible for the altered repair phenotypes, we imaged myeloid cell dynamics in WT and Mmp12−/− mouse corneas in vivo and observed blunted immune cell activities in Mmp12−/− corneas 1 day after injury. At this early time-point, the injured Mmp12−/− corneas had less neutrophil infiltration and we demonstrated that this owed to reduced expression of the chemokine CXCL1. Six days after injury, Mmp12−/− mouse corneas had more macrophage infiltration and this owed to elevated CCL2 chemokine levels. The mechanism is most likely the inactivation of these chemokines by MMP12 cleavage (Dean et al., 2008). Collectively, these results demonstrate a protective role of MMP12 in the inflammatory, neovascular, and fibrotic responses to corneal injury. Our findings suggest MMP12 as an important factor needed for the maintenance of corneal clarity following injury.
MMP12 inhibits angiogenesis
The cornea is normally devoid of blood vessels, which is important for the maintenance of corneal clarity. Angiogenesis depends on tightly regulated endothelial cell growth and can be switched on and off by angiogenic and anti-angiogenic factors. There is growing evidence that the proteolytic activities of several MMPs including MMP1, -2, -7, -9 and -14 promote angiogenesis (Kessenbrock et al., 2010). Here we show that MMP12 activity also regulates angiogenic processes following injury via altered VEGFA expression and plasminogen cleavage. VEGFA functions to recruit VEGFR1-expressing monocytes/macrophages causing the release of hemangiogenic growth factors and therefore has an crucial role in inducing inflammatory neovascularization (Cursiefen et al., 2004). MMP9, a major MMP involved in tumor angiogenesis, also regulates angiogenesis through its effects on VEGF. Conveyed by inflammatory cells, MMP9 functions to increase the bioavailability of VEGF during an angiogenic switch in a mouse model of multistage pancreatic carcinogenesis (Bergers et al., 2000). Although Mmp12−/− mice infected with S. mansoni have marked increases in MMP9 and MMP2 activity in their liver lysates (Madala et al., 2010), we did not find significantly increased MMP9 activity in wounded Mmp12−/− corneas. Increased MMP9 activity is therefore not responsible for the elevated VEGFA expression and suggests a more direct effect of MMP12 on VEGF. Plasminogen is cleaved into the functionally active molecules, plasmin and angiostatin, which are anti-angiogenic (Gabison et al., 2004). Altered plasminogen cleavage was observed in the Mmp12−/− mice and may be an important mechanism by which MMP12 affects corneal neovascularization. Since bone marrow transplantation partially reversed the increased angiogenesis noted in MMP12−/− mice, this provides evidence for an important function of MMP12 produced by bone marrow-derived macrophages in promoting corneal angiogenesis.
MMP12 regulation of inflammation is tissue-specific
The early recruitment of neutrophils observed in MMP12−/− mouse corneas in response to injury is beneficial because of its importance in the innate immune response to prevent infection (Li et al., 2006). This positive effect of MMP12 on neutrophil activities has also been observed in other tissue models of inflammation including the lung (Nénan et al., 2007; Warner et al., 2001), and the skin (Dean et al., 2008).
In contrast, MMP12 produced from bone marrow-derived macrophages has a negative effect on the recruitment of macrophages to injured corneas. Interestingly, the increased macrophage infiltration noted in the MMP12−/− mouse corneas contrasts with the reduction of macrophage infiltration noted in other tissue models of injury performed using MMP12−/− mice (Hautamaki et al., 1997; Longo et al., 2005). An aneurysm model in which calcium chloride (CaCl2) was applied to periaortic tissue in MMP12−/− mice and a pulmonary emphysema model in which MMP12−/− mice were exposed to cigarette smoke both resulted in a reduction of macrophage infiltration in the MMP12−/− mice. Furthermore, inhibition or loss of MMP12 activity resulting in reduced macrophage numbers has been observed in other injury models such as crescentic glomerulonephritis and chronic obstructive pulmonary disease (COPD) (Haq et al., 2011; Hautamaki et al., 1997; Kaneko et al., 2003). Whether elastin, a major connective tissue component of lung tissue, aortic tissue and kidney tissue, but not the cornea (Alexander and Garner, 1983), or other microenvironment differences play opposite effects on MMP12-mediated macrophage recruitment remain to be determined.
MMP12 confers protection following injury
Following injury, the activities of the MMPs have generally been regarded as tissue destructive. However, there is a growing body of evidence that MMPs have reparative properties following injury as well. The overexpression of the collagenase MMP14 in the corneal stroma also reduces corneal opacification and the expression of the fibrotic markers type III collagen and a-SMA (Galiacy et al., 2011). MMPs also facilitate axonal regeneration in vitro (Zuo et al., 1998). During skin repair, nitration of MMP13 promotes its release from endothelial cells and accelerates healing (Lizarbe et al., 2008). Significantly, although MMP12 appears to inhibit corneal fibrosis, its activity has been associated with the development of lung pathology. Future studies defining the molecular and cellular-specific differences should give insights into how dual destructive and reparative responses of MMP12 come to be tissue-specific during repair.
In conclusion, our data suggest that MMP12 may be a potential therapeutic target for the prevention of corneal fibrosis following injury through its effects on inflammation and angiogenesis. It is now important to determine if excess MMP12 delivered to the eye using intrastromal or subconjunctival injection of recombinant MMP12 can prevent or reduce corneal fibrosis following injury (Galiacy et al., 2011). Further studies using human corneal tissue will establish if gene transfer of MMP12 can used to reduce corneal fibrosis and be used as a potential medical therapeutic alternative to corneal transplantation for the treatment of corneal scarring.
Materials and Methods
Mice
Mice homozygous for the null allele of the MMP12 (Shipley et al., 1996) and c-fms-EGFP mice (Sasmono et al., 2003) in which EGFP is driven under the CSF-1R promoter were genotyped using published protocols and were backcrossed to FVB/n. All experiments were performed with 6- to 12-week old male and female mice and sibling wild-type littermates served as controls. Mice were maintained under pathogen-free conditions in the UCSF barrier facility. All animal experiments were conducted in accordance with procedures approved by the UCSF Institutional Animal Care and Use Committee.
Animal model of injury
Mice were anesthetized by isofluorane inhalation (Baxter Pharmaceutical, Deerfield, IL) and by topical application of 0.5% Proparacaine (Akorn Inc., Buffalo Grove, IL) placed on the cornea. An alkaline burn was created by applying filter paper 2.5 mm in diameter soaked in 0.1 N NaOH (Sigma, St. Louis, MO) for 30 seconds to the central cornea followed by rinsing with 250 µl of phosphate-buffered saline. After the chemical burn treatment, topical 0.5% Proparacaine was again placed on the cornea for anesthesia.
Immunohistochemistry
Paraffin-embedded tissue sections (5 µm) were deparaffinized, rehydrated, and incubated in 0.1 M citrate buffer (pH 6.0) for 20 minutes at 100°C for antigen retrieval. The sections were then incubated in 0.3% hydrogen peroxide for 30 minutes to block endogenous peroxidase activity. Slides were blocked in TNB blocking buffer for 30 minutes at room temperature and then incubated overnight with goat anti-mouse MMP-12 antibody (Santa Cruz Biotechnology, Dallas, TX) at 4°C. The following day, sections were incubated with biotinylated anti-goat IgG (Vector Labs, Burlingame, CA) for 1 hour. The sections were then incubated for 60 minutes at room temperature with Streptavidin-HRP diluted 1∶100 in TNB and for 10 minutes in 50 ml of Biotin Tyramide Working Solution from TSA Biotin System (PerkinElmer, Waltham, MA) to amplify signal. Staining was then developed using DAB Substrate (Invitrogen, Carlsbad, CA). Following development, slides were counterstained with Hematoxylin and Eosin for 5 minutes, dehydrated, and sealed.
RNA and real-time PCR
Corneal tissue was harvested and then freeze-fractured with a mortar and pestle. Total RNA was extracted using homogenization and Trizol reagent (Invitrogen, Carlsbad, CA) following the manufacturer's protocol and then subjected to DNase treatment (New England Biolabs, Ipswich, MA). For cDNA synthesis, total RNA was reverse transcribed using SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA). For real-time PCR analysis, the expression levels of MMP12, SMA, COL1, COL3, CCL2, CCL7, CCL8, CCL20, CXCL1, CXCL2, CXCL5, VEGFA, VEGFB and HPRT were determined using an Applied Biosystems 7500 Instrument (Carlsbad, CA) or Eppendorf Mastercycler realplex machine (Eppendorf, Hamburg, Germany). cDNA was measured from duplicate samples. Real-time PCR reactions included the following: 20 ng of diluted reverse transcription product, 2× SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA), and 250 nM of each forward and reverse PCR primer. For assays, reactions were incubated at 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles at 95°C for 15 seconds followed by 60°C for 1 minute, and then 95°C for 15 seconds. Relative quantification of expression was calculated with the 2−ΔΔCt method and the cycle threshold difference corrected for HPRT. Data are presented as fold change in gene expression normalized to HPRT. All experiments were performed in triplicate and used samples derived from 2 corneas. Primer sequences are listed in supplementary material Table S1 and were purchased from Integrated DNA Technologies (San Diego, CA).
MMP12 expression in resident corneal macrophages
Resident corneal macrophages were isolated from two corneas per sample. Following removal of other ocular tissue, corneas were minced with a scalpel and digested with collagenase type I at 37°C for 1 hour (82U per cornea; Sigma, St. Louis, MO), as previously described (Sun et al., 2012). Cells were washed with FACS staining buffer (Ca2+/Mg2+-free HBSS, 2% FBS) and incubated with Fc-blocking rat IgG (eBioscience, San Diego, CA). The cells were then stained with antibodies to CD45-FITC (eBioscience, San Diego, CA), F4/80-PeCy7 (Biolegend, San Diego, CA), and CD11b-APC-E780 (eBioscience, San Diego, CA). Macrophages (CD45+CD11b+F4/80+) were sorted into 100-cell aliquots using a FACS Aria II cell sorter (Becton Dickinson, Franklin Lakes, NJ) into a 96-well plate containing necessary reagents for lysis (SuperScript CellsDirect OneStep kit; Invitrogen, Grand Island, NY) and cDNA synthesis + PCR amplification (SuperScript III RT Platinum Taq kit; Invitrogen, Grand Island, NY), as described in the protocol for Biomark (Fluidigm, South San Francisco, CA). The resulting cDNA was then analyzed for MMP12 mRNA expression by SYBR Green real-time PCR. Reactions were run in a thermocycler at 95°C for 10 minutes, followed by 95°C for 15 seconds and 60°C for 60 seconds for 40 cycles. Quantification of relative expression was calculated using the 2-ΔΔCt method and normalized to GAPDH.
Whole-mount and confocal microscopy
Eyes were enucleated and the corneas were dissected to remove the lens, iris and retina. Four incisions were made equal distances apart to aid in flattening the corneas. Corneal tissue was then fixed with 4% paraformaldehyde in PBS at 4°C overnight. Immunostaining was performed using primary antibodies against F4/80 (Invitrogen, Carlsbad, CA), Gr-1 and CD31 (BD Pharmingen, San Jose, CA), LYVE-1 (R&D Systems, Minneapolis, MN), α-SMA and Tenascin (Sigma, St. Louis, MO), and Fibronectin (Millipore, Billerica, MA). Secondary antibodies of the appropriate class conjugated with Alexa Fluor 488 or Alexa Fluor 594 were obtained from Invitrogen or conjugated with Dylight 549 (Jackson ImmunoResearch Laboratories, West Grove, PA). Routine protocols included corneas stained with an isotype control or secondary antibodies alone. Corneas were then placed epithelial side-up, mounting medium was added, and coverslips were placed.
A confocal laser-scanning microscope (LSM 5 Pascal; Zeiss) was used to image the localization of Alexa Fluor 488, Alexa Fluor 594 and Dylight 549 in the central cornea. Optical sections (z = 48 µm) of confocal epifluorescence images were acquired sequentially with a 10× objective lens with image acquisition software (Zeiss). Thirteen optical sections were merged and viewed en face. 3D images were obtained and rotated using Bitplane Imaris software (version 7.3.1 for Windows X64, Bitplane Inc., South Windsor, CT). For F4/80, Gr-1, CD31 and LYVE-1 whole-mount stained images, the number of pixels per color was determined. In addition, for CD31 and LYVE-1 staining, the lengths of blood and lymphatic neovascularization were calculated using NIH ImageJ software and the innermost vessel of the limbal arcade was used as the border.
Isolation of corneal infiltrating cells and flow cytometry
Inflammatory cells were isolated from corneas 1 day and 6 days after injury. Two corneas from single mice were excised after injury, and residual iris material was removed. The corneas were combined and minced using a scalpel, and incubated 1 hour at 37°C in 100 ml collagenase type I (82 U per cornea; Sigma-Aldrich, St. Louis, MO), as described (Sun et al., 2012). The resulting cell suspensions were washed with FACS buffer (HBSS without Ca2+ or Mg2+, 2% FBS). Cells were incubated with Fc blocking antibody (eBioscience, San Diego, CA) and stained with antibodies to CD11 APC-eFluor 780, CD45 PerCP-Cy5.5, Ly-6G (Gr-1) PE, and F4/80 Antigen PE-Cy7 (eBioscience, San Diego, CA). Cell profiles were acquired on a LSRII flow cytometer (Becton Dickinson, Franklin Lakes, NJ), and were gated based on isotype marker. Cell numbers were analyzed using FlowJo analysis software (Tree Star, Inc., Ashland, OR).
Bone marrow transplantation
Chimeric mice in which host bone marrow was replaced by bone marrow from WT or Mmp12−/− mice were generated as previously described (Colnot et al., 2006). Briefly, bone marrow was harvested from 6- to 8-week-old WT or Mmp12−/− mice by flushing the femurs and tibias with 2% FBS in phosphate-buffered saline. Cells were centrifuged, incubated in red blood cell lysing buffer (R7757, Sigma, St. Louis, MO), washed in RPMI 1640 medium, and re-suspended in 2% FBS in phosphate-buffered saline. Bone marrow cells (2×106 cells) were injected in the tail vein of lethally irradiated (9 Gy) recipient mice. Corneal injury was performed 6 weeks later.
Protein extraction and proteome profiler array
Corneal tissue was freshly snap-frozen, sonicated in lysis buffer (50 mM Tris-HCl buffer pH 8.0, 1% NP-40, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS), and protease inhibitor cocktail (Complete, mini, EDTA-free, Roche, Mannheim, Germany), and gently agitated for 2 hours on a rocking platform shaker at 4°C. Supernatants were collected by centrifugation (12,000 rpm, 20 minutes, 4°C) and then stored at −80°C or quantified using the BCA protein assay (Pierce, Thermo Scientific, Rockford, IL). The Proteome Profiler Mouse Cytokine array kit (R&D Systems, Minneapolis, MN) was performed according to the manufacturer's instructions. Briefly, equal amounts of proteins (500 µg) from control and treated mice were diluted and incubated with the cytokine array overnight at 4°C. After washing of unbound material, cytokine antibody complexes bound by its cognate capture antibody on the membrane were detected by chemiluminescence. Arrays were analyzed using ImageJ software and the densities of duplicate spots were averaged and normalized to the values for the control spots.
In vivo inhibition of CXCL1 and CCL2
The neutralization dosages of the antibodies for our in vivo studies were empirically determined and based on prior studies (Lin et al., 2007; Xue et al., 2007). For inhibition of CXCL1, subconjunctival injections were performed on WT mice with 10 µg neutralizing antibody to CXCL1 (R&D Systems, Minneapolis, MN) or control rat IgG2a 2 hours prior to corneal chemical injury. The anti-CXCL1 antibody 50% neutralizing dose (ND50) is 0.05–0.25 µg/ml for 30 ng/ml recombinant mouse (rm) CXCL1. Twenty-four hours after chemical injury, the eyes were enucleated and the corneas were collected for whole-mount immunostaining with a primary antibody against Gr-1 as described above. For inhibition of CXCL1, the subconjunctival space of WT mice was injected with 2 µg neutralizing antibody to CXCL1 (R&D Systems, Minneapolis, MN) or PBS 2 hours prior to corneal chemical injury. The anti-CCL2 antibody 50% neutralizing dose (ND50) is 0.5–2.5 µg/ml for 0.04 µg/ml recombinant mouse CCL2. The subconjunctiva were re-injected with 2 µg neutralizing antibody to CXCL1 or PBS 3 days later and the corneas were collected 6 days after injury for whole mount immunostaining with a primary antibody against F4/80 as described above. Difference in the means was calculated, and percent inhibition was derived using Prism GraphPad software (San Diego, CA, USA).
Western blotting
Cells were lysed in RIPA buffer plus protease inhibitors (Roche, Germany) and protein concentration was measured using the BCA Protein Assay Kit (Thermo Scientific, Waltham, MA). Lysates were subjected to SDS–PAGE, transferred to PVDF membranes, blocked in 5% BSA or dry milk, incubated with primary antibody overnight and visualized using ECL Detection Reagents (Pierce, Rockford, IL). Exposures were acquired using a LAS-4000 Imager (Fuji). Antibodies used include: VEGF-A (Abcam, Cambridge, MA), Plasminogen (Abcam, Cambridge, MA), GAPDH (Cell Signaling Technology, Danvers, MA), and HRP anti-rabbit (GE Healthcare, San Francisco, CA).
Gelatin gel zymography
Gelatin gel zymography for detecting MMP2 and MMP9 was performed as previously described (Kheradmand et al., 2002). Whole corneal homogenates were lysed in 50 mM Tris-HCl buffer pH 8.0, 1% NP-40, 150 mM NaCl, 0.5% sodium deoxycholate and 0.1% SDS. Insoluble aggregates and nuclei were removed by centrifugation and protein in the supernatant was quantified by BCA protein assay (Pierce, Thermo Scientific, Rockford, IL). Samples (10 µg of protein) were added to non-denaturing loading buffer and separated in 10% Tris-glycine gels with 0.1% gelatin (Invitrogen, Grand Island, NY). Gels were developed by incubation for 30 minutes at room temperature in Novex Zymogram Renaturing Buffer (Invitrogen, Grand Island, NY) and followed by overnight incubation at 37°C in Novex Developing Buffer (Invitrogen). Gels were then fixed and stained with 30% ethanol and 10% acetic acid containing 0.5% w/v Coomassie Blue G-250 for 30 minutes at room temperature. Gels were then destained in 30% ethanol and 10% acetic acid. MMP2 and MMP9 activity appeared as clear bands. Digital images of the gels were captured using a scanner.
Confocal imaging of myeloid cells in the corneal stroma of live mice
The procedure for in vivo confocal imaging of the cornea was based on a previously published protocol (Speier et al., 2008a; Speier et al., 2008b). A Nikon C1si spectral confocal laser-scanning microscope equipped with four lasers was used and images were captured using an Argon laser, a 40× objective (NIR/0.8 NA W lens) and camera (Nikon D-eclipse C1Si). Mice were anesthetized with 2.5% isoflurane (Summit Anesthesia Solutions, Bend, OR) and 1.2% avertin (Sigma-Aldrich, St Louis, MO). The mouse head was restrained using a stereotaxic headholder (SG-4N, Narishige, Tritech Research, Los Angeles, CA) and ophthalmic gel (Genteal; Novartis, New York, NY) was applied to the cornea as an aqueous contact medium between the cornea and microscope objective. Body temperature was controlled using a heating pad. The eye was additionally stabilized by retracting the eyelids with forceps covered with a loop of polyethylene tubing. Images were captured continuously every 3.22 seconds for 20 to 90 minutes in 320×320 µm fields and the acquisition software Ez-C1 Gold Ver. 3.8 (Nikon) was used to collect images. We performed confocal imaging employing the minimum required laser-power and scan-time necessary. Respiratory motion artifacts were corrected for using Bitplane Imaris (version 7.3.1 for Windows X64, Bitplane Inc., South Windsor, CT) drift correction and myeloid cell migration was analyzed automatically with spot detection at a minimum diameter of 8 µm and Brownian movement algorithm with a maximum distance of 20 µm. We also used Imaris to track the movement of the myeloid cells expressing EGFP (green). To construct tracking plots, we selected 15–20 representative cells on Imaris that were present for most of the imaging period and the X and Y position of these cells at each time point was normalized to the origin (0,0). The normalized X and Y positions were then plotted using Prism software (GraphPad Software, Inc., La Jolla, CA). The average track length was defined as the cumulative distance traveled by a cell over a given time. Displacement was defined as the straight-line distance of a cell from its starting point after any given time. Velocity was calculated as the displacement of a cell over several time steps. For each genotype, one field in at least seven different mice was analyzed.
Statistical analysis
Statistical analysis was performed with two-tailed t-tests to compare mean values (Prism, GraphPad Software, La Jolla, CA). A P-value less than 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Bennie Jeng, Lalitha Muthusubramaniam, Anna Ward and Neeraj Ramakrishnan for technical assistance and support and Ying Yu for assistance in maintaining the mouse colony. We thank Sebastian Peck and Dr. Gaetano Faleo for their advice and assistance with in vivo confocal imaging and Dr. Marja Lohela for advice with histology and intravital imaging analysis.
Footnotes
Author contributions
M.C. and Z.W. designed the study. M.C., J.L., A.B., A.C., I.M., and J.L. performed the experiments. M.C., J.L., and Z.W. contributed to manuscript preparation.
Funding
This work was supported by grants from the National Institutes of Health [grant numbers K08 EY018858 to M.F.C., EY002162 to M.F.C., and P01 AI053194 to Z.W.]; That Man May See Foundation [to M.F.C.]; and The Alta California Foundation [to M.F.C.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.128033/-/DC1
References
- Alexander R. A., Garner A. (1983). Elastic and precursor fibres in the normal human eye. Exp. Eye Res. 36, 305–315 10.1016/0014-4835(83)90015-5 [DOI] [PubMed] [Google Scholar]
- Allen S. J., Crown S. E., Handel T. M. (2007). Chemokine: receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 25, 787–820 10.1146/annurev.immunol.24.021605.090529 [DOI] [PubMed] [Google Scholar]
- Azar D. T. (2006). Corneal angiogenic privilege: angiogenic and antiangiogenic factors in corneal avascularity, vasculogenesis, and wound healing (an american ophthalmological society thesis). Trans. Am. Ophthalmol. Soc. 104, 264–302 [PMC free article] [PubMed] [Google Scholar]
- Bergers G., Brekken R., McMahon G., Vu T. H., Itoh T., Tamaki K., Tanzawa K., Thorpe P., Itohara S., Werb Z. et al. (2000). Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2, 737–744 10.1038/35036374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejchova K., Liskova P., Hrdlickova E., Filipec M., Jirsova K. (2009). Matrix metalloproteinases in recurrent corneal melting associated with primary Sjörgen's syndrome. Mol. Vis. 15, 2364–2372 [PMC free article] [PubMed] [Google Scholar]
- Brissette-Storkus C. S., Reynolds S. M., Lepisto A. J., Hendricks R. L. (2002). Identification of a novel macrophage population in the normal mouse corneal stroma. Invest. Ophthalmol. Vis. Sci. 43, 2264–2271 [PMC free article] [PubMed] [Google Scholar]
- Carlson E. C., Wang I. J., Liu C. Y., Brannan P., Kao C. W., Kao W. W. (2003). Altered KSPG expression by keratocytes following corneal injury. Mol. Vis. 9, 615–623 [PubMed] [Google Scholar]
- Chen L., Hann B., Wu L. (2011). Experimental models to study lymphatic and blood vascular metastasis. J. Surg. Oncol. 103, 475–483 10.1002/jso.21794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery H. R., Humphries T., Clare A., Dixon A. E., Howes K., Moran C. B., Scott D., Zakrzewski M., Pearlman E., McMenamin P. G. (2008). Turnover of bone marrow-derived cells in the irradiated mouse cornea. Immunology 125, 541–548 10.1111/j.1365-2567.2008.02868.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colnot C., Huang S., Helms J. (2006). Analyzing the cellular contribution of bone marrow to fracture healing using bone marrow transplantation in mice. Biochem. Biophys. Res. Commun. 350, 557–561 10.1016/j.bbrc.2006.09.079 [DOI] [PubMed] [Google Scholar]
- Cornelius L. A., Nehring L. C., Harding E., Bolanowski M., Welgus H. G., Kobayashi D. K., Pierce R. A., Shapiro S. D. (1998). Matrix metalloproteinases generate angiostatin: effects on neovascularization. J. Immunol. 161, 6845–6852 [PubMed] [Google Scholar]
- Cursiefen C., Chen L., Dana M. R., Streilein J. W. (2003). Corneal lymphangiogenesis: evidence, mechanisms, and implications for corneal transplant immunology. Cornea 22, 273–281 10.1097/00003226-200304000-00021 [DOI] [PubMed] [Google Scholar]
- Cursiefen C., Chen L., Borges L. P., Jackson D., Cao J., Radziejewski C., D'Amore P. A., Dana M. R., Wiegand S. J., Streilein J. W. (2004). VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J. Clin. Invest. 113, 1040–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean R. A., Cox J. H., Bellac C. L., Doucet A., Starr A. E., Overall C. M. (2008). Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 112, 3455–3464 10.1182/blood-2007-12-129080 [DOI] [PubMed] [Google Scholar]
- Egeblad M., Werb Z. (2002). New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174 [DOI] [PubMed] [Google Scholar]
- Fini M. E., Cook J. R., Mohan R. (1998). Proteolytic mechanisms in corneal ulceration and repair. Arch. Dermatol. Res. 290 Suppl, S12–S23 10.1007/PL00007449 [DOI] [PubMed] [Google Scholar]
- Gabison E., Chang J. H., Hernández-Quintela E., Javier J., Lu P. C., Ye H., Kure T., Kato T., Azar D. T. (2004). Anti-angiogenic role of angiostatin during corneal wound healing. Exp. Eye Res. 78, 579–589 10.1016/j.exer.2003.09.005 [DOI] [PubMed] [Google Scholar]
- Gabison E. E., Mourah S., Steinfels E., Yan L., Hoang-Xuan T., Watsky M. A., De Wever B., Calvo F., Mauviel A., Menashi S. (2005). Differential expression of extracellular matrix metalloproteinase inducer (CD147) in normal and ulcerated corneas: role in epithelio-stromal interactions and matrix metalloproteinase induction. Am. J. Pathol. 166, 209–219 10.1016/S0002-9440(10)62245-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiacy S. D., Fournié P., Massoudi D., Ancèle E., Quintyn J. C., Erraud A., Raymond-Letron I., Rolling F., Malecaze F. (2011). Matrix metalloproteinase 14 overexpression reduces corneal scarring. Gene Ther. 18, 462–468 10.1038/gt.2010.159 [DOI] [PubMed] [Google Scholar]
- Gordon G. M., Austin J. S., Sklar A. L., Feuer W. J., LaGier A. J., Fini M. E. (2011). Comprehensive gene expression profiling and functional analysis of matrix metalloproteinases and TIMPs, and identification of ADAM-10 gene expression, in a corneal model of epithelial resurfacing. J. Cell. Physiol. 226, 1461–1470 10.1002/jcp.22306 [DOI] [PubMed] [Google Scholar]
- Hamrah P., Huq S. O., Liu Y., Zhang Q., Dana M. R. (2003). Corneal immunity is mediated by heterogeneous population of antigen-presenting cells. J. Leukoc. Biol. 74, 172–178 10.1189/jlb.1102544 [DOI] [PubMed] [Google Scholar]
- Haq I., Lowrey G. E., Kalsheker N., Johnson S. R. (2011). Matrix metalloproteinase-12 (MMP-12) SNP affects MMP activity, lung macrophage infiltration and protects against emphysema in COPD. Thorax 66, 970–976 10.1136/thx.2011.159087 [DOI] [PubMed] [Google Scholar]
- Hassell J. R., Birk D. E. (2010). The molecular basis of corneal transparency. Exp. Eye Res. 91, 326–335 10.1016/j.exer.2010.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hautamaki R. D., Kobayashi D. K., Senior R. M., Shapiro S. D. (1997). Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 277, 2002–2004 10.1126/science.277.5334.2002 [DOI] [PubMed] [Google Scholar]
- Hunninghake G. M., Cho M. H., Tesfaigzi Y., Soto-Quiros M. E., Avila L., Lasky-Su J., Stidley C., Melén E., Söderhäll C., Hallberg J. et al. (2009). MMP12, lung function, and COPD in high-risk populations. N. Engl. J. Med. 361, 2599–2608 10.1056/NEJMoa0904006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanami H., Ishizaki M., Fukuda Y., Takahashi H. (2009). Expression of matrix metalloproteinases (MMP)-12 by myofibroblasts during alkali-burned corneal wound healing. Curr. Eye Res. 34, 207–214 10.1080/02713680802687809 [DOI] [PubMed] [Google Scholar]
- Kaneko Y., Sakatsume M., Xie Y., Kuroda T., Igashima M., Narita I., Gejyo F. (2003). Macrophage metalloelastase as a major factor for glomerular injury in anti-glomerular basement membrane nephritis. J. Immunol. 170, 3377–3385 [DOI] [PubMed] [Google Scholar]
- Kawashima M., Kawakita T., Higa K., Satake Y., Omoto M., Tsubota K., Shimmura S., Shimazaki J. (2010). Subepithelial corneal fibrosis partially due to epithelial-mesenchymal transition of ocular surface epithelium. Mol. Vis. 16, 2727–2732 [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K., Plaks V., Werb Z. (2010). Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141, 52–67 10.1016/j.cell.2010.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheradmand F., Rishi K., Werb Z. (2002). Signaling through the EGF receptor controls lung morphogenesis in part by regulating MT1-MMP-mediated activation of gelatinase A/MMP2. J. Cell Sci. 115, 839–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvanta A., Sarman S., Fagerholm P., Seregard S., Steen B. (2000). Expression of matrix metalloproteinase-2 (MMP-2) and vascular endothelial growth factor (VEGF) in inflammation-associated corneal neovascularization. Exp. Eye Res. 70, 419–428 10.1006/exer.1999.0790 [DOI] [PubMed] [Google Scholar]
- Li Z., Burns A. R., Smith C. W. (2006). Two waves of neutrophil emigration in response to corneal epithelial abrasion: distinct adhesion molecule requirements. Invest. Ophthalmol. Vis. Sci. 47, 1947–1955 10.1167/iovs.05-1193 [DOI] [PubMed] [Google Scholar]
- Lin M., Carlson E., Diaconu E., Pearlman E. (2007). CXCL1/KC and CXCL5/LIX are selectively produced by corneal fibroblasts and mediate neutrophil infiltration to the corneal stroma in LPS keratitis. J. Leukoc. Biol. 81, 786–792 10.1189/jlb.0806502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizarbe T. R., García-Rama C., Tarín C., Saura M., Calvo E., López J. A., López-Otín C., Folgueras A. R., Lamas S., Zaragoza C. (2008). Nitric oxide elicits functional MMP-13 protein-tyrosine nitration during wound repair. FASEB J. 22, 3207–3215 10.1096/fj.07-103804 [DOI] [PubMed] [Google Scholar]
- Longo G. M., Buda S. J., Fiotta N., Xiong W., Griener T., Shapiro S., Baxter B. T. (2005). MMP-12 has a role in abdominal aortic aneurysms in mice. Surgery 137, 457–462 10.1016/j.surg.2004.12.004 [DOI] [PubMed] [Google Scholar]
- Lu P., Li L., Kuno K., Wu Y., Baba T., Li Y. Y., Zhang X., Mukaida N. (2008). Protective roles of the fractalkine/CX3CL1-CX3CR1 interactions in alkali-induced corneal neovascularization through enhanced antiangiogenic factor expression. J. Immunol. 180, 4283–4291 [DOI] [PubMed] [Google Scholar]
- Lyu J., Joo C-K. (2005). Wnt-7a up-regulates matrix metalloproteinase-12 expression and promotes cell proliferation in corneal epithelial cells during wound healing. J. Biol. Chem. 280, 21653–21660 10.1074/jbc.M500374200 [DOI] [PubMed] [Google Scholar]
- Lyu J., Joo C. K. (2006). Expression of Wnt and MMP in epithelial cells during corneal wound healing. Cornea 25, Suppl 1S24–S28 10.1097/01.ico.0000247209.01262.4e [DOI] [PubMed] [Google Scholar]
- Madala S. K., Pesce J. T., Ramalingam T. R., Wilson M. S., Minnicozzi S., Cheever A. W., Thompson R. W., Mentink-Kane M. M., Wynn T. A. (2010). Matrix metalloproteinase 12-deficiency augments extracellular matrix degrading metalloproteinases and attenuates IL-13-dependent fibrosis. J. Immunol. 184, 3955–3963 10.4049/jimmunol.0903008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan V. B., Wei C., McDonnell P. J., 3rd (2002). Microarray analysis of corneal fibroblast gene expression after interleukin-1 treatment. Invest. Ophthalmol. Vis. Sci. 43, 2143–2151 [PubMed] [Google Scholar]
- Matute-Bello G., Wurfel M. M., Lee J. S., Park D. R., Frevert C. W., Madtes D. K., Shapiro S. D., Martin T. R. (2007). Essential role of MMP-12 in Fas-induced lung fibrosis. Am. J. Respir. Cell Mol. Biol. 37, 210–221 10.1165/rcmb.2006-0471OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishige N., Nomi N., Morita Y., Nishida T. (2011). Immunohistofluorescence analysis of myofibroblast transdifferentiation in human corneas with bullous keratopathy. Cornea 30, 1129–1134 10.1097/ICO.0b013e318213798d [DOI] [PubMed] [Google Scholar]
- Mott J. D., Werb Z. (2004). Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 16, 558–564 10.1016/j.ceb.2004.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland B., Tuft S. J., Khaw P. T. (2005). Matrix metalloproteinase distribution during early corneal wound healing. Eye (Lond.) 19, 584–588 10.1038/sj.eye.6701557 [DOI] [PubMed] [Google Scholar]
- Nakao S., Kuwano T., Tsutsumi-Miyahara C., Ueda S., Kimura Y. N., Hamano S., Sonoda K. H., Saijo Y., Nukiwa T., Strieter R. M. et al. (2005). Infiltrationof COX-2-expressing macrophages is a prerequisite for IL-1 beta-induced neovascularization and tumor growth. J. Clin. Invest. 115, 2979–2991 10.1172/JCI23298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nénan S., Lagente V., Planquois J. M., Hitier S., Berna P., Bertrand C. P., Boichot E. (2007). Metalloelastase (MMP-12) induced inflammatory response in mice airways: effects of dexamethasone, rolipram and marimastat. Eur. J. Pharmacol. 559, 75–81 10.1016/j.ejphar.2006.11.070 [DOI] [PubMed] [Google Scholar]
- Rohini G., Murugeswari P., Prajna N. V., Lalitha P., Muthukkaruppan V. (2007). Matrix metalloproteinases (MMP-8, MMP-9) and the tissue inhibitors of metalloproteinases (TIMP-1, TIMP-2) in patients with fungal keratitis. Cornea 26, 207–211 10.1097/01.ico.0000248384.16896.7d [DOI] [PubMed] [Google Scholar]
- Sasmono R. T., Oceandy D., Pollard J. W., Tong W., Pavli P., Wainwright B. J., Ostrowski M. C., Himes S. R., Hume D. A. (2003). A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood 101, 1155–1163 10.1182/blood-2002-02-0569 [DOI] [PubMed] [Google Scholar]
- Shapiro S. D. (1999). Diverse roles of macrophage matrix metalloproteinases in tissue destruction and tumor growth. Thromb. Haemost. 82, 846–849 [PubMed] [Google Scholar]
- Shapiro S. D., Kobayashi D. K., Ley T. J. (1993). Cloning and characterization of a unique elastolytic metalloproteinase produced by human alveolar macrophages. J. Biol. Chem. 268, 23824–23829 [PubMed] [Google Scholar]
- Shipley J. M., Wesselschmidt R. L., Kobayashi D. K., Ley T. J., Shapiro S. D. (1996). Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc. Natl. Acad. Sci. USA 93, 3942–3946 10.1073/pnas.93.9.3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivak J. M., Fini M. E. (2002). MMPs in the eye: emerging roles for matrix metalloproteinases in ocular physiology. Prog. Retin. Eye Res. 21, 1–14 10.1016/S1350-9462(01)00015-5 [DOI] [PubMed] [Google Scholar]
- Speier S., Nyqvist D., Cabrera O., Yu J., Molano R. D., Pileggi A., Moede T., Köhler M., Wilbertz J., Leibiger B. et al. (2008a). Noninvasive in vivo imaging of pancreatic islet cell biology. Nat. Med. 14, 574–578 10.1038/nm1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speier S., Nyqvist D., Köhler M., Caicedo A., Leibiger I. B., Berggren P. O. (2008b). Noninvasive high-resolution in vivo imaging of cell biology in the anterior chamber of the mouse eye. Nat. Protoc. 3, 1278–1286 10.1038/nprot.2008.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stramer B. M., Austin J. S., Roberts A. B., Fini M. E. (2005). Selective reduction of fibrotic markers in repairing corneas of mice deficient in Smad3. J. Cell. Physiol. 203, 226–232 10.1002/jcp.20215 [DOI] [PubMed] [Google Scholar]
- Sun Y., Karmakar M., Taylor P. R., Rietsch A., Pearlman E. (2012). ExoS and ExoT ADP ribosyltransferase activities mediate Pseudomonas aeruginosa keratitis by promoting neutrophil apoptosis and bacterial survival. J. Immunol. 188, 1884–1895 10.4049/jimmunol.1102148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner R. L., Lewis C. S., Beltran L., Younkin E. M., Varani J., Johnson K. J. (2001). The role of metalloelastase in immune complex-induced acute lung injury. Am. J. Pathol. 158, 2139–2144 10.1016/S0002-9440(10)64685-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werb Z., Gordon S. (1975). Elastase secretion by stimulated macrophages. Characterization and regulation. J. Exp. Med. 142, 361–377 10.1084/jem.142.2.361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcher J. P., Srinivasan M., Upadhyay M. P. (2001). Corneal blindness: a global perspective. Bull. World Health Organ. 79, 214–221 [PMC free article] [PubMed] [Google Scholar]
- Xue M. L., Thakur A., Cole N., Lloyd A., Stapleton F., Wakefield D., Willcox M. D. (2007). A critical role for CCL2 and CCL3 chemokines in the regulation of polymorphonuclear neutrophils recruitment during corneal infection in mice. Immunol. Cell Biol. 85, 525–531 10.1038/sj.icb.7100082 [DOI] [PubMed] [Google Scholar]
- Zhang H., Li C., Baciu P. C. (2002). Expression of integrins and MMPs during alkaline-burn-induced corneal angiogenesis. Invest. Ophthalmol. Vis. Sci. 43, 955–962 [PubMed] [Google Scholar]
- Zuo J., Ferguson T. A., Hernandez Y. J., Stetler-Stevenson W. G., Muir D. (1998). Neuronal matrix metalloproteinase-2 degrades and inactivates a neurite-inhibiting chondroitin sulfate proteoglycan. J. Neurosci. 18, 5203–5211 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.