

Abstract
Phosphodiesterases (PDEs) limit vasodilation in response to a variety of signaling cascades by metabolizing the cyclic nucleotides cAMP and cGMP. The objective of this study was to test the hypothesis that NO regulates expression of PDE3A, a cGMP-inhibited PDE. Incubation of rat pulmonary artery smooth muscle cells (rPaSMCs) with the NO-donor compound S-nitroso-glutathione (GSNO) increased PDE3A gene expression in a dose- and time-dependent manner. NO-donors increased PDE3A protein levels. Total and milrinone inhibitable cAMP PDE activity were increased 2.8±0.1- and 2.0±0.1-fold respectively in extracts of rPaSMCs exposed to GSNO. The effects of GSNO on PDE3A gene expression were mimicked by the soluble guanylate cyclase (sGC) activators YC-1 and BAY 41-2272 and blocked by the sGC inhibitor ODQ. Incubation of rPaSMC with interleukin-1β and tumor necrosis factor-α induced PDE3A gene expression, an effect which was inhibited by L-NIL, an antagonist of NO synthase 2, or ODQ. Actinomycin D, an inhibitor of RNA polymerase, blocked the GSNO-induced increase of PDE3A mRNA levels, whereas cycloheximide, an inhibitor of protein translation, did not. These observations suggest that NO modulates PDE3A gene expression via mechanisms dependent upon cGMP synthesis and gene transcription. Prolonged exposure to NO may alter the sensitivity of vascular smooth muscle to cGMP- or cAMP-dependent vasodilators, as well as PDE isoform-selective inhibitors.
Keywords: nitric oxide, cyclic guanosine monophosphate, GSNO, phosphodiesterase 3A, soluble guanylate cyclase, vasodilation
INTRODUCTION
Nitric oxide (NO) increases intracellular levels of cyclic guanosine monophosphate (cGMP) in vascular smooth muscle cells. NO acts, in part, by stimulating soluble guanylate cyclase (sGC) to synthesize cGMP leading to relaxation (1). Cyclic GMP is inactivated by cyclic nucleotide phosphodiesterases (PDEs), a complex family of enzymes which catalyze the hydrolysis of 3′,5′-cyclic nucleotides to their corresponding nucleoside 5′-monophosphates. PDEs are multi-domain proteins with distinct catalytic and regulatory sites (2). Eleven classes of PDEs are categorized on the basis of substrate specificity, kinetics, regulatory and immunological properties, and selective inhibition (3).
In vascular smooth muscle cells, PDEs limit vasodilation induced by a variety of signaling cascades by decreasing intracellular concentrations of cAMP and cGMP. Of the eleven classes of PDE isozymes described, at least eight have been identified in the pulmonary vasculature including PDEs 1, 2, 3, 4, 5, 7, 9, 11 (4–8). Expression of PDE6 is restricted to the eye and the pineal gland (9). Some classes of phosphodiesterases can metabolize both cAMP and cGMP, whereas PDEs 5, 6, and 9 selectively hydrolyze cGMP, and PDE4, PDE7 and PDE8 metabolize cAMP selectively (10).
PDE3 contributes most of total cAMP-specific PDE activity in rat pulmonary artery smooth muscle cells (11) and bovine pulmonary arteries (12). Since PDE3 has a higher affinity for cGMP than for cAMP but hydrolyzes cAMP at a 10-fold greater rate than cGMP, cGMP behaves as a competitive inhibitor of PDE3 (13, 14). As a result, increasing intracellular cGMP can subsequently alter the metabolism of intracellular cAMP by inhibiting the activity of PDE3.
There are two PDE3 genes (PDE3A and PDE3B) (15, 16). PDE3A and PDE3B are structurally similar, containing a catalytic domain at the carboxy terminus and an amino-terminal domain which is important for the intracellular localization of the enzyme. PDE3A is expressed in vascular smooth muscle cells (17), cardiomyocytes (15), lung parenchyma [18], and platelets (19), whereas PDE3B is expressed in adipocytes, hepatocytes, pancreatic cells, and vascular smooth muscle cells (17). There are 3 isoforms of PDE3A that are generated by transcription from alternative start sites in the PDE3A gene (14).
The aim of the current study was to characterize the influence of NO on the expression of PDE3A in rat pulmonary arterial smooth muscle cells (rPASMC). We hypothesized that NO induces PDE3A via the NO-cGMP pathway and may therefore be inhibitable by administration of ODQ. We observed that NO-donor compounds markedly induced PDE3A gene and protein expression, as well as milrinone-inhibitable cAMP hydrolysis via mechanisms involving cGMP and gene transcription.
MATERIALS AND METHODS
Reagents
GSNO, DETA-NO, YC-1 and BAY 41-2272 were obtained from Alexis Corp. (San Diego, CA). L-N6-(1-iminoethyl)lysine (L-NIL) and 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) were obtained from Calbiochem-Novabiochem Corp. (La Jolla, CA). The membrane-permeable analogues of cGMP and cAMP, 8-Br-cGMP and 8-Br-cAMP, were purchased from Biomol Research Laboratories Inc. (Plymouth Meeting, PA), and actinomycin D and cycloheximide were obtained from Sigma-Aldrich (St. Louis, MO). Interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) were obtained from R&D Systems Inc. (Minneapolis, MN).
Cell culture
Cultures of primary rPaSMC were prepared from explants of endothelium- and adventitia-stripped pulmonary arteries of adult Sprague-Dawley rats, as described previously (20). Cells were maintained in RPMI 1640 medium supplemented with 10% NuSerum (Collaborative Biomedical Products, Bedford, MA), 112 units/ml penicillin, and 112 units/ml streptomycin. Cells passaged between 6 and 12 times were used for experiments.
Quantitative RT-PCR
RNA was isolated from rPaSMC using Trizol reagent, and cDNA was generated with MMLV reverse transcriptase (Promega Corp., Madison, WI) and random primers (Promega). Quantitative PCR was performed with the ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA) using primers for PDE3A (Applied Biosystems) and SYBR® Green PCR Master Mix (Applied Biosystems). Post-amplification dissociation curves were performed to verify the presence of a single amplification product, cDNA corresponding to ribosomal RNA (18S) was detected with 18S VIC MGB primers (Applied Biosystems) and Taqman® Universal PCR Master Mix (Applied Biosystems). Changes in PDE3A mRNA levels normalized to 18S ribosomal RNA levels were determined using the relative Ct method.
Immunoblotting
RPaSMC were harvested in Tris-buffered saline (TBS) containing 10 μl/ml proteinase inhibitor cocktail (Sigma-Aldrich). Samples were centrifuged at 8,000 g for 15 min at 4°C. Supernatant proteins (120 μg) were fractionated using 8% SDS-PAGE and transferred to nitrocellulose filters (Osmonics Inc., Minnetonka, MN). Filters were blocked at room temperature for 1 h in TBS (150 mM NaCl, 10 mM Tris, 0.05% NP40, 0.01% NaN3, 50 mg/ml BSA, 10 mg/ml ovalbumin, pH 7.4) and then incubated at room temperature for 1 h with a rabbit polyclonal antibody directed against the PDE3A protein (1:500, mouse anti PDE3A-CT, AA1024-1041; generously provided by Dr. V. C. Manganiello, NHLBI, NIH, Bethesda). Bound antibody was detected by incubation of the filters with horseradish peroxidase-conjugated antibody against rabbit immunoglobin (1:5000 in TBS, Cell Signaling, Beverly, MA) and visualized using chemiluminescence (Western Lightning™ Chemiluminescence Kit; PerkinElmer Life Sciences, Boston, MA) and exposure to X-ray film. Films were scanned using a Color Image Scanner (Agfa, Mortsel, Belgium) and the National Institutes of Health Image 1.6 software.
Phosphodiesterase activity
Cyclic AMP PDE activity was measured as previously described (21). To assess the contribution of PDE3A to total cAMP-PDE activity, PDE activity was measured in the presence and absence of milrinone (10 μM), a specific inhibitor of PDE3.
Statistical analysis
Data are reported as means+S.D. After approving the assumption of normality and equal variance across groups, differences were assessed using ANOVA followed by a Dunnett test. Values were considered significant if p<0.05. Statistics were performed using the software package SigmaStat (Jandel Corporation, San Rafael, CA).
RESULTS
Incubation of rat pulmonary artery smooth muscle cells with GSNO increases PDE3A gene expression
To investigate the ability of NO to regulate PDE3A gene expression, RNA was extracted from rPaSMCs following incubation without and with 100 μM GSNO, a NO-donor compound, for 1 hour to 24 hours. Maximal changes in PDE3A gene expression were noted at 4 hour of incubation with GSNO (Fig. 1A). Incubation of rPaSMCs with GSNO for 4 hours increased PDE3A gene expression in a dose-dependent manner. Maximal changes in gene expression were observed at the dose of 10 and 100 μM (Fig. 1B).
Fig. 1.

A) Incubation with GSNO increases PDE3A gene expression in rPaSMC in a time-dependent manner. RNA was extracted from untreated rPaSMCs and rPaSMCs incubated with 100 μM GSNO for 1 to 24 hours. Complementary DNA was generated and analyzed for PDE3A gene expression using quantitative PCR; n=3, means±S.D., *p<0.05 vs. control. B) Incubation with GSNO increases PDE3A gene expression in rPaSMC in a dose-dependent manner. RNA was extracted from untreated rPaSMCs and rPaSMCs incubated with 1, 10, and 100 μM GSNO for 4 hours. Complementary DNA was generated and analyzed for PDE3A gene expression using quantitative PCR; n=3; means±S.D.; *p<0.05 vs. control. C) Incubation of rPaSMC with GSNO increases PDE3A protein levels. Immunoblots were prepared from extracts of untreated rPaSMCs and rPaSMCs incubated with 100 μM GSNO for 4, 8 and 16 hours. Blots were reacted with an antibody directed against PDE3A. Incubation with GSNO led to increased expression of an approximately 120-kDa PDE3A-immunoreactive protein (upper panel). Densitometric measurement of the PDE3A-immunoreactive protein levels is shown in the lower panel. D) GSNO induces cAMP dependent PDE activity in rPaSMC. Protein extracts were prepared from untreated rPaSMC and rPaSMC exposed to 100 μM GSNO for 8 hours; n=3, means±S.D., *p<0.05 vs. control. Protein extracts were prepared from untreated rPaSMC and rPaSMC exposed to 100 μM GSNO for 8 hours. Cyclic AMP-metabolizing PDE activity was measured in the presence and absence of the PDE3 inhibitor milrinone (10 μM), and milrinone-inhibitable PDE activity is shown as a percent of activity in untreated rPaSMC; n=3, means±S.D.; *p<0.05 vs. control.
Prolonged incubation of rat pulmonary artery smooth muscle cells with GSNO increases PDE3A protein levels
To determine whether or not NO-mediated changes in PDE gene expression were correlated with changes in protein levels, PDE3A protein levels were measured in rPaSMCs exposed to 100 μM GSNO for 4, 8, and 16 hours and compared to simultaneously-harvested untreated cells (Fig. 1C). Using immunoblot techniques, we observed that levels of a 110 kDa PDE3A-immunoreactive protein increased after 4 and 8 hours exposure to GSNO and decreased after 16 hours. Incubation of rPaSMC with DETA-NO (100 μM) for 16 hours increased PDE3A protein levels 2.4±0.1-fold (p<0.05, data not shown).
Incubation of rat pulmonary artery smooth muscle cells with GSNO for 8 hours increases cAMP PDE activity
To determine the effect of prolonged NO exposure of rPaSMC on PDE3 activity, cAMP PDE enzyme activity was measured in cell extracts taken from rPaSMCs following incubation without and with 100 μM GSNO for 8 hours. Total cytoplasmatic cAMP PDE activity increased 2.0±0.1-fold in extracts of rPaSMCs exposed to GSNO for 8 h (p<0.05, Fig. 1D). Milrinone-inhibitable cAMP PDE activity, representing PDE3 enzyme activity, was 2.8±0.1-fold greater in GSNO-treated cells than in controls (p<0.05, Fig. 1D).
NO regulation of gene expression of PDEs 3A is mediated by a sGC-dependent mechanism
To investigate the role of sGC in the regulation of PDE gene expression by NO, studies were performed using ODQ, a selective inhibitor of sGC. RPaSMCs were pretreated with or without 10 μM ODQ for 30 min followed by incubation with or without 100 μM GSNO for 4 hours. Pretreatment with ODQ blocked the effect of GSNO on PDE3A gene expression (Fig. 2). Incubation with ODQ alone did not alter PDE3A gene expression. Incubation with the sGC stimulator YC-1 (50 μM) induced PDE3A gene expression 2.8±0.4-fold after 4 hours (p<0.05, data not shown). Incubation with the sGC stimulator BAY 41-2272 (50 μM) for 4 hours induced PDE3A gene expression 13±1.6-fold (Fig. 3). These results suggest that the effect of NO on gene expression of PDE3A is mediated by a cGMP- and sGC-dependent mechanism. The PKA inhibitor H89 (50 μM) blocked the induction of PDE3A gene expression by GSNO (Fig. 4).
Fig 2.
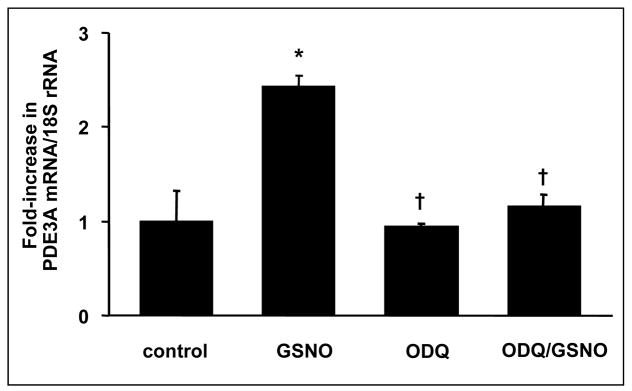
ODQ blocks GSNO-inducible PDE3A gene expression. After incubation with and without ODQ (10 μM) for 30 min, rPaSMC were stimulated with and without 100 μM GSNO for 4 hours. RNA was extracted, and complementary DNA was generated and analysed using quantitative PCR; n=3; means±S.D.; *p<0.05 vs. control, †p<0.05 vs. GSNO.
Fig. 3.

BAY-41 2272 induced PDE3A gene expression after 4 hours. RNA was extracted from rPaSMCs, and complementary DNA was generated and analysed using quantitative PCR; n=3, means±S.D., *p<0.05 vs. control.
Fig. 4.

H89 blocks GSNO-inducible PDE3A gene expression. After incubation with and without H89 (50 μM) for 30 min, rPaSMC were stimulated with and without 100 μM GSNO for 4 hours. RNA was extracted, and complementary DNA was generated and analysed using quantitative PCR; n=3; means±S.D.; *p<0.05 vs. control, †p<0.05 vs. GSNO.
To investigate the mechanisms involved in the regulation of PDE gene expression by NO, rPaSMCs were incubated without and with 8-Br-cGMP (1 mM) or 8-Br-cAMP (1 mM), membrane-permeable analogues of cGMP and cAMP respectively, for 2, 4 and 8 hours. We observed that incubation of rPaSMCs with 8-Br-cAMP increased PDE3A mRNA after 4 hours (Fig. 5), whereas 8-Br-cGMP elevated PDE3A expression beginning at 2 hours.
Fig. 5.

8-Br-cAMP (1 mM) increases PDE3A gene expression after 4 hours whereas 8-Br-cGMP (1 mM) induces after 2 and 4 hours. RNA was extracted from rPaSMCs, and complementary DNA was generated and analyzed using quantitative PCR; n=3, means±S.D., *p<0.05 vs. control.
Nitric oxide alters PDE gene expression via a transcription-dependent mechanism
To determine whether or not transcription or translation of mRNA contributed to the alterations in PDE gene expression observed following prolonged exposure to NO, studies were performed using actinomycin D (AD, 10 μM), an inhibitor of RNA polymerase, and cycloheximide (CHX, 1 μM), an inhibitor of protein translation. RNA was extracted from rPaSMCs pretreated with or without AD or CHX for 30 min prior to the 4 h incubation with GSNO. As shown in Fig. 6, pretreatment with AD blocked the effect GSNO on PDE3A gene expression whereas CHX had no effect. These results suggest that NO alters PDE gene expression in rPaSMC via a transcription-dependent mechanism.
Fig. 6.

Actinomycin D (AD) blocks and cycloheximide (CHX) does not alter GSNO-inducible PDE3A gene expression. After being incubated with and without AD (10 μM) or CHX (1 μM) for 30 min, rPaSMC were stimulated with and without 100 μM GSNO for 4 hours. RNA was extracted, and complementary DNA was generated and analyzed using quantitative PCR; n=4; means±S.D.; *p<0.05 vs. control, #p<0.05 vs. GSNO, †p<0.05 vs. CHX.
Prolonged incubation of rat pulmonary artery smooth muscle cells with interleukin-1β and tumor necrosis factor-α increases PDE3A gene expression
To investigate the ability of endogenously-produced NO by rPaSMC to alter PDE3A gene expression, RNA was extracted from rPaSMCs incubated with or without interleukin-1β (IL-1β, 20 ng/ml) and tumor necrosis factor-α (TNF-α, 100 ng/ml) for 8 hours. We observed that incubation with cytokines increased PDE3A gene expression (Fig. 7). Pretreatment of rPaSMCs with L-NIL (1 mM), an inhibitor of NOS2 markedly attenuated the ability of TNF-α and IL-1β to induce PDE3A gene expression. Similarly, ODQ (10 μM) inhibited the cytokine-mediated induction of PDE3A gene expression. These results suggest that inflammatory cytokines can increase PDE3A expression via NO- and cGMP-dependent mechanisms.
Fig. 7.

Cytokines induce PDE3A gene expression. RNA was extracted from rPaSMC after incubation with and without TNF-α (100 ng/ml) and IL-1β (20 ng/ml) for 8 hours. Cytokine-incubated rPaSMC were pretreated for 30 min with and without L-NIL (1 mM) or ODQ (10 μM). RNA was extracted and complementary DNA was generated and analyzed using quantitative PCR; n=3; means±S.D.; *p<0.05 vs. control, †p<0.05 vs. TNF/IL-1.
DISCUSSION
In the present study, prolonged exposure of rPaSMC to the NO-donor compound, GSNO, increased PDE3A gene expression in a dose- and time-dependent manner. Increased PDE3A gene expression was sGC-dependent. Exposure of rPaSMC to GSNO induced PDE3A protein expression. Increased PDE3A protein expression was accompanied by increased total cAMP PDE enzyme activity and increased milrinone-inhibitable cAMP PDE enzyme activity, representing increased PDE3 enzyme activity.
In a previous study using a transcriptional profiling approach, we found that incubation of rPaSMC with GSNO increased PDE3B and PDE4B mRNA levels while decreasing PDE7A gene expression. Some PDE genes were not included in the gene chip that we used, such as PDE1A and PDE8 through PDE11. Using the microarray approach we were not able to detect PDE5, which had previously been found in pulmonary vessels (22), or PDE4D gene expression, described previously in rat aortic vascular smooth muscle cells (23). Although PDE3A was represented on the microarray (Affymetrix rat U34A gene chips), and PDE3A is known as one of the main cAMP-hydrolyzing PDEs in the pulmonary artery (12), PDE3A gene expression was not detected by the array. However, using RT-PCR techniques we found that GSNO induces PDE3A gene expression. Since the effect of NO on PDE3A has not been examined in vascular smooth muscle cells, we further investigated the mechanisms by which NO regulates PDE3A expression in rPaSMC.
There are three isoforms of PDE3A (24). All of them have been identified in the heart (PDE3A1, PDE3A2 and PDE3A3) (15, 24). Only one of these has been detected in the cytosolic fraction of vascular smooth muscle cells and has been described as PDE3A2 with a molecular weight of 120 kDa (16, 17). PDE3A induced by GSNO in rPaSMC has a molecular weight of 118 kDa and is therefore most likely PDE3A2.
Incubation of rPaSMC with GSNO increased PDE3A gene expression consistently within 4 hours. The increase in PDE3A gene expression varied between 2- and 8-fold. This variation in the magnitude of the PDE3A gene response to GSNO exposure is most likely attributable to small variations in PDE3A mRNA levels in untreated cells of the control group.
Exposure of rPaSMC to TNF-α and IL-1β induces NOS2 gene expression and increases NO and cGMP levels (20). Induction of PDE3A gene expression in rPASMC after incubation with TNF-α and IL-1β was inhibited by L-NIL. Taken together, these observations suggest that concentrations of NO are sufficient to alter PDE3A mRNA levels in vivo.
Exposure of rPaSMCs to sGC activators YC-1 and BAY 41-2272 increased PDE3A gene expression, whereas a selective inhibitor of sGC enzyme activity, ODQ, blocked the effects of GSNO on gene expression of PDE3A. These results suggest that NO alters PDE3A gene expression, via a sGC-dependent mechanism. Since inhibition of sGC with ODQ did not decrease PDE3A gene expression after 8 hours, expression levels might be already on a baseline level.
We observed that the cAMP-dependent protein kinase (PKA) activator, 8-Br-cAMP, as well as 8-Br-cGMP, an activator of the cGMP-dependent protein kinase (PKG), increased PDE3A mRNA levels. Accumulation of cGMP induced by NO might increase PDE3A gene expression by direct stimulation of PKA, since inhibition of PKA with H89 blocked the effect of GSNO, or via inhibition of PDE3 activity leading to activation of PKA by rising cAMP levels.
Incubation with GSNO does not increase intracellular cAMP levels in whole cell extracts of rPaSMC (20). These findings support the hypothesis that cGMP directly activates PKA which induces PDE3A gene expression (25, 26), but it is also possible that inhibition of PDE3 by cGMP increases cAMP levels in specific compartments which may sufficient to activate PDE3A gene expression but are not detectable in whole cell extracts (27). In murine aorta, high concentrations of 8-Br-cGMP induced Ca2+-desensitization mainly via activation of PKA rather than PKG (28). In a previous study investigating the effect of NO on the cAMP-specific PDE4B (21), we found that PDE4B expression and activity were induced upon exposure to an NO donor compound via a mechanism dependent on sGC and PKA, but not on PKG.
Since the induction of PDE3A gene expression by GSNO was blocked by AD, PDE3A gene expression appears to be regulated via a transcription-dependent mechanism. In contrast, pre-incubation with CHX did not block increase in PDE3A gene expression suggesting that induction of PDE3A gene expression requires gene transcription but not new protein synthesis. One molecule regulating PDE3A gene expression in rat cardiomyocytes is ICER (inducible cAMP early repressor). Stability of ICER is regulated by PKA and is not translation-dependent (29). ICER protein elevation represses PDE3A gene expression (29). Cycloheximide might have blocked ICER protein expression interrupting the negative ICER-PDE3A feedback loop.
The ability of NO to alter PDE3A gene expression in PaSMCs may have important clinical implications. The phenomenon of rebound pulmonary hypertension following cessation of prolonged inhaled NO therapy is poorly understood (30). Several investigators have suggested that altered NOS enzyme activity may play a role (31, 32). Studies using PDE selective inhibitors suggest that PDEs may contribute to the rebound pulmonary hypertension (33, 34) and airway reactivity (35, 36). Schermuly et al. reported that combination of inhaled prostacyclin with simultaneous inhibition of PDE5 and PDE3 induced elevated cAMP levels and prolonged decrease of pulmonary artery pressure in rabbits with acute pulmonary hypertension (37). Their study showed that simultaneous inhibition of cAMP and cGMP hydrolyzing PDEs can result in an augmented and prolonged biological effect.
The increase in PDE3 enzyme activity associated with prolonged exposure to NO may represent a homeostatic mechanism to balance intracellular cyclic nucleotide levels. Chronic administration of NO-donor compounds, such as nitroglycerin, impairs the ability of vascular smooth muscle to respond to additional NO. The etiology of nitrate tolerance is believed to be multifactorial (30) with decreased sGC enzyme activity having an important role (38, 39, 40). Our results suggest that an increased activity of PDE3A leads to hydrolysation of cAMP and cGMP and decreases vascular response to NO-donors as well.
In summary, incubation of rPaSMCs with NO increased PDE3A gene expression, total cAMP PDE activity, and milrinone-inhibitable cAMP PDE activity. These results were mimicked by YC-1, as well as BAY 41-2272, and blocked by ODQ. These observations suggest that NO induces, via a sGC-and cGMP-dependent mechanism, PDE3A gene expression. NO-induced modulation of cGMP- and cAMP-metabolizing PDEs potentially alters the responsiveness to vasodilators and the sensitivity to PDE-selective inhibitors. We speculate that the increased cAMP PDE activity in rPaSMCs may have important implications for the prevention and treatment of rebound pulmonary hypertension after discontinuing NO inhalation.
Acknowledgments
This study was supported by a grant from the National Institutes of Health HL-74352 and a sponsored research agreement with INO Therapeutics, LLC (K. D. Bloch). We thank Dr. York Zausig for the helpful discussion of the manuscript.
Footnotes
Conflict of interests: None declared.
References
- 1.Ignarro LJ. Nitric oxide as a unique signaling molecule in the vascular system: a historical overview. J Physiol Pharmacol. 2002;53:503–514. [PubMed] [Google Scholar]
- 2.Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res. 2003;93:280–291. doi: 10.1161/01.RES.0000087541.15600.2B. [DOI] [PubMed] [Google Scholar]
- 3.Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100:309–327. doi: 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 4.Rabe KF, Tenor H, Dent G, Schudt C, Nakashima M, Magnussen H. Identification of PDE isozymes in human pulmonary artery and effect of selective PDE inhibitors. Am J Physiol. 1994;266:L536–L543. doi: 10.1152/ajplung.1994.266.5.L536. [DOI] [PubMed] [Google Scholar]
- 5.Smith SJ, Brookes-Fazakerley S, Donnelly LE, Barnes PJ, Barnette MS, Giembycz MA. Ubiquitous expression of phosphodiesterase 7A in human proinflammatory and immune cells. Am J Physiol Lung Cell Mol Physiol. 2003;284:L279–L289. doi: 10.1152/ajplung.00170.2002. [DOI] [PubMed] [Google Scholar]
- 6.D’Andrea MR, Qiu Y, Haynes-Johnson D, Bhattacharjee S, Kraft P, Lundeen S. Expression of PDE11A in normal and malignant human tissues. J Histochem Cytochem. 2005;53:895–903. doi: 10.1369/jhc.5A6625.2005. [DOI] [PubMed] [Google Scholar]
- 7.Phillips PG, Long L, Wilkins MR, Morrell NW. cAMP phosphodiesterase inhibitors potentiate effects of prostacyclin analogs in hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2005;288:L103–L115. doi: 10.1152/ajplung.00095.2004. [DOI] [PubMed] [Google Scholar]
- 8.Evgenov OV, Busch CJ, Evgenov NV, et al. Inhibition of phosphodiesterase 1 augments the pulmonary vasodilator response to inhaled nitric oxide in awake lambs with acute pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2006;290:L723–L729. doi: 10.1152/ajplung.00485.2004. [DOI] [PubMed] [Google Scholar]
- 9.Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Ther. 2006;109:366–398. doi: 10.1016/j.pharmthera.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Matsumoto T, Kobayashi T, Kamata K. Phosphodiesterases in the vascular system. J Smooth Muscle Res. 2003;39:67–86. doi: 10.1540/jsmr.39.67. [DOI] [PubMed] [Google Scholar]
- 11.Maclean MR, Johnston ED, Mcculloch KM, Pooley L, Houslay MD, Sweeney G. Phosphodiesterase isoforms in the pulmonary arterial circulation of the rat: changes in pulmonary hypertension. J Pharmacol Exp Ther. 1997;283:619–624. [PubMed] [Google Scholar]
- 12.Pauvert O, Salvail D, Rousseau E, Lugnier C, Marthan R, Savineau JP. Characterisation of cyclic nucleotide phosphodiesterase isoforms in the media layer of the main pulmonary artery. Biochem Pharmacol. 2002;63:1763–1772. doi: 10.1016/s0006-2952(02)00919-x. [DOI] [PubMed] [Google Scholar]
- 13.Beltman J, Becker DE, Butt E, et al. Characterization of cyclic nucleotide phosphodiesterases with cyclic GMP analogs: topology of the catalytic domains. Mol Pharmacol. 1995;47:330–339. [PubMed] [Google Scholar]
- 14.Movsesian MA. PDE3 cyclic nucleotide phosphodiesterases and the compartmentation of cyclic nucleotide-mediated signalling in cardiac myocytes. Basic Res Cardiol. 2002;97(S1):83–90. doi: 10.1007/s003950200035. [DOI] [PubMed] [Google Scholar]
- 15.Hambleton R, Krall J, Tikishvili E, et al. Isoforms of cyclic nucleotide phosphodiesterase PDE3 and their contribution to cAMP hydrolytic activity in subcellular fractions of human myocardium. J Biol Chem. 2005;280:39168–39174. doi: 10.1074/jbc.M506760200. [DOI] [PubMed] [Google Scholar]
- 16.Choi YH, Ekholm D, Krall J, et al. Identification of a novel isoform of the cyclic-nucleotide phosphodiesterase PDE3A expressed in vascular smooth-muscle myocytes. Biochem J. 2001;353:41–50. [PMC free article] [PubMed] [Google Scholar]
- 17.Liu H, Maurice DH. Expression of cyclic GMP-inhibited phosphodiesterases 3A and 3B (PDE3A and PDE3B) in rat tissues: differential subcellular localization and regulated expression by cyclic AMP. Br J Pharmacol. 1998;125:1501–1510. doi: 10.1038/sj.bjp.0702227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tarpey SB, Sawmiller DR, Kelly C, Thompson WJ, Townsley MI. Phosphodiesterase 3 activity is reduced in dog lung following pacing-induced heart failure. Am J Physiol Lung Cell Mol Physiol. 2003;284:L766–L773. doi: 10.1152/ajplung.00373.2002. [DOI] [PubMed] [Google Scholar]
- 19.Hung SH, Zhang W, Pixley RA, et al. New insights from the structure-function analysis of the catalytic region of human platelet phosphodiesterase 3A: a role for the unique 44-amino acid insert. J Biol Chem. 2006;281:29236–29244. doi: 10.1074/jbc.M606558200. [DOI] [PubMed] [Google Scholar]
- 20.Takata M, Filippov G, Liu H, et al. Cytokines decrease sGC in pulmonary artery smooth muscle cells via NO-dependent and NO-independent mechanisms. Am J Physiol Lung Cell Mol Physiol. 2001;280:L272–L278. doi: 10.1152/ajplung.2001.280.2.L272. [DOI] [PubMed] [Google Scholar]
- 21.Busch CJ, Liu H, Graveline AR, Bloch KD. Nitric oxide induces phosphodiesterase 4B expression in rat pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2006;290:L747–L753. doi: 10.1152/ajplung.00298.2005. [DOI] [PubMed] [Google Scholar]
- 22.Murray F, Maclean MR, Pyne NJ. Increased expression of the cGMP-inhibited cAMP-specific (PDE3) and cGMP binding cGMP-specific (PDE5) phosphodiesterases in models of pulmonary hypertension. Br J Pharmacol. 2002;137:1187–1194. doi: 10.1038/sj.bjp.0704984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu H, Palmer D, Jimmo SL, et al. Expression of phosphodiesterase 4D (PDE4D) is regulated by both the cyclic AMP-dependent protein kinase and mitogen-activated protein kinase signaling pathways. A potential mechanism allowing for the coordinated regulation of PDE4D activity and expression in cells. J Biol Chem. 2000;275:26615–26624. doi: 10.1074/jbc.M001634200. [DOI] [PubMed] [Google Scholar]
- 24.Wechsler J, Choi YH, Krall J, Ahmad F, Manganiello VC, Movsesian MA. Isoforms of cyclic nucleotide phosphodiesterase PDE3A in cardiac myocytes. J Biol Chem. 2002;277:38072–38078. doi: 10.1074/jbc.M203647200. [DOI] [PubMed] [Google Scholar]
- 25.Aizawa T, Wei H, Miano JM, Abe J, Berk BC, Yan C. Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ Res. 2003;93:406–413. doi: 10.1161/01.RES.0000091074.33584.F0. [DOI] [PubMed] [Google Scholar]
- 26.Sausbier M, Schubert R, Voigt V, et al. Mechanisms of NO/cGMP-dependent vasorelaxation. Circ Res. 2000;87:825–830. doi: 10.1161/01.res.87.9.825. [DOI] [PubMed] [Google Scholar]
- 27.Fischmeister R, Castro LR, Abi-Gerges A, et al. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res. 2006;99:816–828. doi: 10.1161/01.RES.0000246118.98832.04. [DOI] [PubMed] [Google Scholar]
- 28.Worner R, Lukowski R, Hofmann F, Wegener JW. cGMP signals mainly through cAMP kinase in permeabilized murine aorta. Am J Physiol Heart Circ Physiol. 2007;292:H237–H244. doi: 10.1152/ajpheart.00079.2006. [DOI] [PubMed] [Google Scholar]
- 29.Yan C, Miller CL, Abe J. Regulation of phosphodiesterase 3 and inducible cAMP early repressor in the heart. Circ Res. 2007;100:489–501. doi: 10.1161/01.RES.0000258451.44949.d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munzel T, Daiber A, Mulsch A. Explaining the phenomenon of nitrate tolerance. Circ Res. 2005;97:618–628. doi: 10.1161/01.RES.0000184694.03262.6d. [DOI] [PubMed] [Google Scholar]
- 31.Black SM, Heidersbach RS, McMullan DM, Bekker M, Johengen MJ, Fineman JR. Inhaled nitric oxide inhibits NOS activity in lambs: potential mechanism for rebound pulmonary hypertension. Am J Physiol. 1999;277:H1849–H1856. doi: 10.1152/ajpheart.1999.277.5.H1849. [DOI] [PubMed] [Google Scholar]
- 32.Ross GA, Oishi P, Azakie A, et al. Endothelial alterations during inhaled NO in lambs with pulmonary hypertension: implications for rebound hypertension. Am J Physiol Lung Cell Mol Physiol. 2005;288:L27–L35. doi: 10.1152/ajplung.00144.2004. [DOI] [PubMed] [Google Scholar]
- 33.Ivy DD, Kinsella JP, Ziegler JW, Abman SH. Dipyridamole attenuates rebound pulmonary hypertension after inhaled nitric oxide withdrawal in postoperative congenital heart disease. J Thorac Cardiovasc Surg. 1998;115:875–882. doi: 10.1016/S0022-5223(98)70369-1. [DOI] [PubMed] [Google Scholar]
- 34.Thelitz S, Oishi P, Sanchez LS. Phosphodiesterase-3 inhibition prevents the increase in pulmonary vascular resistance following inhaled nitric oxide withdrawal in lambs. Pediatr Crit Care Med. 2004;5:234–239. doi: 10.1097/01.pcc.0000124021.25393.2d. [DOI] [PubMed] [Google Scholar]
- 35.Mokry J, Mokra D, Nosalova G, Beharkova M, Feherova Z. Influence of selective inhibitors of phosphodiesterase 3 and 4 on cough and airway reactivity. J Physiol Pharmacol. 2008;59(Suppl 6):473–482. [PubMed] [Google Scholar]
- 36.Nejman-Gryz P, Grubek-Jaworska H, Glapinski J, Hoser G, Chazan R. Effects of the phosphodiestrase-4 inhibitor rolipram on lung resistance and inflammatory reaction in experimental asthma. J Physiol Pharmacol. 2006;57(Suppl 4):229–239. [PubMed] [Google Scholar]
- 37.Schermuly RT, Yilmaz H, Ghofrani HA, et al. Inhaled iloprost reverses vascular remodeling in chronic experimental pulmonary hypertension. Am J Respir Crit Care Med. 2005;172:358–363. doi: 10.1164/rccm.200502-296OC. [DOI] [PubMed] [Google Scholar]
- 38.Filippov G, Bloch DB, Bloch KD. Nitric oxide decreases stability of mRNAs encoding soluble guanylate cyclase subunits in rat pulmonary artery smooth muscle cells. J Clin Invest. 1997;100:942–948. doi: 10.1172/JCI119610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Axelsson KL, Andersson RG. Tolerance towards nitroglycerin, induced in vivo, is correlated to a reduced cGMP response and an alteration in cGMP turnover. Eur J Pharmacol. 1983;88:71–79. doi: 10.1016/0014-2999(83)90393-x. [DOI] [PubMed] [Google Scholar]
- 40.Schroder H, Leitman DC, Bennett BM, Waldman SA, Murad F. Glyceryl trinitrate-induced desensitization of guanylate cyclase in cultured rat lung fibroblasts. J Pharmacol Exp Ther. 1988;245:413–418. [PubMed] [Google Scholar]