

Abstract
We have previously discovered the naturally occurring antitussive alkaloid noscapine as a tubulin-binding agent that attenuates microtubule dynamics and arrests mammalian cells at mitosis via activation of the c-Jun NH2-terminal kinase pathway. It is well established that the p53 protein plays a crucial role in the control of tumor cell response to chemotherapeutic agents and DNA-damaging agents; however, the relationship between p53-driven genes and drug sensitivity remains controversial. In this study, we compared chemosensitivity, cell cycle distribution, and apoptosis on noscapine treatment in four cell lines derived from the colorectal carcinoma HCT116 cells: p53+/+ (p53-wt), p53−/− (p53-null), p21−/− (p21-null), and BAX−/− (BAX-null). Using these isogenic variants, we investigated the roles of p53, BAX, and p21 in the cellular response to treatment with noscapine. Our results show that noscapine treatment increases the expression of p53 over time in cells with wild-type p53 status. This increase in p53 is associated with an increased apoptotic BAX/Bcl-2 ratio consistent with increased sensitivity of these cells to apoptotic stimuli. Conversely, loss of p53 and p21 alleles had a counter effect on both BAX and Bcl-2 expression and the p53-null and p21-null cells were significantly resistant to the antiproliferative and apoptotic effects of noscapine. All but the p53-null cells displayed p53 protein accumulation in a time-dependent manner on noscapine treatment. Interestingly, despite increased levels of p53, p21-null cells were resistant to apoptosis, suggesting a proapoptotic role of p21 and implying that p53 is a necessary but not sufficient condition for noscapine-mediated apoptosis.
Introduction
Given the pivotal role of microtubules in biological processes such as intracellular transport, motility, morphogenesis, and mitotic spindle formation, treatment of cells with microtubule-targeting agents evokes activation of stress response pathways, cell cycle arrest, and induction of apoptosis (1). This accounts for the extensive use of microtubule-interfering agents in tumor chemotherapy. Typically, microtubule-interacting drugs (e.g., paclitaxel, Vinca alkaloids, etc.) alter microtubule dynamics and engage the cell cycle surveillance mechanisms to arrest cell division in mitosis. Many cancer cells possess genetic lesions in components of this pathway and thus fail to arrest in mitosis. Therefore, by targeting the spindle microtubules, chemotherapeutic agents can efficiently block cell cycle progression in normal cells with intact surveillance mechanisms while initiating programmed cell death in certain tumors to inhibit their aggressive growth (2, 3). In recent years, efforts have been devoted to elucidating signaling pathways that mediate the biological activities of microtubule-interfering agents (4).
It is becoming appreciated that the tumor suppressor protein p53 serves as a key player in the cellular response to a variety of extracellular and intracellular insults, such as DNA damage, oncogenic activation, and microtubule disruption (5, 6), exerting its function mainly through transcriptional activation of target genes, such as the cyclin-dependent kinase (CDK) inhibitor p21, for arresting the cell cycle and the proapoptotic protein BAX for inducing apoptosis (7, 8). Although drug-dependent microtubule disruption results in the up-regulation of p53 expression, the relationship between p53-driven genes and drug sensitivity remains controversial as the response is drug and cell type dependent. This is, in part, because not all drugs absolutely require p53 for their apoptotic function (9-11). In some settings, p53 loss can enhance drug-induced apoptotic cell death (12) and its loss correlates with multidrug resistance in many tumor types (13). The observation that this is a common defect in human tumors has spurred an active search for strategies aimed at directly activating cell death pathways downstream of p53. In this scenario, the role played by p21 is particularly intriguing because this protein can be activated by both p53-dependent and p53-independent mechanisms and can assume proapoptotic or antiapoptotic functions, depending on the cellular context (14).
Our laboratory has shown that noscapine, an opium alkaloid, alters microtubule assembly dynamics, arrests cells at mitosis, and causes apoptosis in many mammalian tumor cells (15). We have also reported that halogenated analogues of noscapine, in particular the brominated analogue, are ~10- to 40-fold more potent than noscapine (16). However, noscapine is further ahead in clinical trials (phase I/II) and the halogenated analogues are still in preclinical stages of development. Because the choice of most chemotherapeutic regimens is usually based on tumor origin without considering molecular determinants of drug sensitivity, we thought it worthwhile to investigate at this stage the relationships between drug sensitivity and various common cell cycle and apoptosis controlling genes. These results are immediately helpful in the clinic for accurately predicting the most effective drugs for an individual patient.
We have previously shown that the apoptotic cell death on noscapine treatment is accompanied by activation of c-Jun NH2-terminal kinases (JNK; ref. 17). There is an increasing body of evidence that points to an important role of JNK in coordinating the cellular response to stress by phosphorylating the transcription factors c-Jun and p53 (18, 19). We hypothesize that noscapine-mediated suppression of microtubule dynamics entails an irremediable damage, which results in p53 activation followed by induction of apoptotic cell death. Because a study of the role of genetic drug response determinants depends on the genetic context (20), we used cells with a well-defined isogenic background. Here we investigated the role of p53 and its two downstream effectors, p21 and BAX, in determining chemosensitivity of the apoptotic response on noscapine treatment in four isogenic variants of colon carcinoma HCT116 cells. Our results indicate that although p53 may be a necessary prerequisite for noscapine-induced apoptosis, it is not a sufficient condition for apoptosis. Our data point to an important role of p21 induction to affect p53-mediated apoptosis in colon adenocarcinoma cells on noscapine treatment.
Materials and Methods
Cells, viruses, plasmids, and reagents
The human colon cancer cell lines HCT116 p53+/+ (p53-wt), p53−/− (p53-null), p21−/− (p21-null), and BAX−/− (BAX-null) and the p21 plasmid construct pZL-WAF1 were gifts from Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD; refs. 21, 22). All cells were maintained in McCoy’s 5A medium supplemented with 10% fetal bovine serum and 2 mmol/L l-glutamine. Antibodies against α-tubulin and β-actin were from Sigma-Aldrich. All other primary antibodies against p53, p21, BAX, Bcl-2, and cyclin B1 were from Cell Signaling (Beverly, MA). Secondary antibodies were from Jackson ImmunoResearch, Inc. (West Grove, PA). Adenovirus-encoding wild-type p53 and empty vectors were amplified in human embryonic kidney 293 cells using the AdEasy system (Stratagene, La Jolla, CA).
Immunofluorescence microscopy
Immunofluorescence microscopy was done as previously described (16). After drug treatment, HCT116 p53-wt cells grown on poly-l-lysine–coated glass coverslips were fixed with cold (−20°C) methanol, washed with PBS, and blocked with 2% bovine serum albumin (BSA)/PBS. A mouse monoclonal antibody against α-tubulin (DM1A; Sigma, St. Louis, MO) was diluted 1:500 in 2% BSA/PBS and incubated for 2 h at 37°C, followed by incubation with a 1:200 dilution of a FITC-labeled goat anti-mouse immunoglobulin G antibody at 37°C for 1 h. Coverslips were then rinsed with 2% BSA/PBS and incubated with propidium iodide (0.5 μg/mL) for 15 min at room temperature before mounting with Aquamount (Lerner Laboratories, Pittsburgh, PA) containing 0.01% 1,4-diazobicyclo(2,2,2)octane (Sigma), and then examined using confocal microscopy.
In vitro cell proliferation and flow cytometric studies of cell cycle and apoptosis
The cell proliferation assay in a standard 96-well plate format using sulforhodamine B was done as previously described (16, 23, 24). The cell cycle status was also evaluated as previously described (23). Briefly, 2 × 106 cells were centrifuged, washed twice with ice-cold PBS, and fixed in 70% ethanol. Cells were then washed again with PBS twice and then incubated with propidium iodide (2 mg/mL) and RNase A (2 mg/mL) in PBS for 45 min before analyses on a FACSCalibur flow cytometer (Beckman Coulter, Inc., Fullerton, CA). The apoptotic cells were also quantified by flow cytometry using Alexa Fluor 488–conjugated Annexin V (Molecular Probes, Invitrogen, Eugene, OR).
Immunoblot analysis
Cells were then treated with 25 μmol/L noscapine for 0, 12, 24, and 48 h. At the appropriate time points, cells were lysed in 100 to 150 μL of lysis buffer containing freshly added protease inhibitors (10 μg/mL phenylmethylsulfonyl fluoride, 50 μg/mL aprotinin, and 1 mmol/L sodium orthovanadate). Protein concentration was determined using the Bradford reagent (Bio-Rad Laboratories, Hercules, CA). Samples containing 50 μg of total cellular protein were subjected to standard Western blotting analysis after SDS-PAGE. Positive antibody reactions were visualized with a horseradish peroxidase–conjugated secondary antibody and an enhanced chemiluminescence detection system according to the protocol of the manufacturer.
Results
Sensitivity of human colon cancer cells to noscapine depends on the p53/p21 status
We have previously shown that noscapine arrests mammalian cells in mitosis despite the presence of intact cellular microtubule arrays and targets them for apoptosis (15). Furthermore, in contrast to other microtubule-interacting agents such as paclitaxel, nocodazole, and vinblastine, noscapine attenuates microtubule dynamics without affecting the total tubulin polymer mass in cells (25). We first examined the effect of noscapine treatment over time on the spindle architecture and nuclear morphology of HCT116 colon cancer cells. Figure 1A shows representative confocal micrographs of HCT116 p53-wt cells treated with 25 μmol/L noscapine for the indicated hours. Normal radial arrays of interphase microtubules were seen at the start of treatment (time 0). In 12-h noscapine-treated cells, microtubule spindles, visualized by immunofluorescence staining of microtubules, looked abnormal and failed to assemble chromosomes at the metaphase plate (Fig. 1A). Mitotic figures started to appear, which showed both normal bipolar spindles and aberrant multipolar spindles, the latter we often observed in some cells. Some mitotic figures clearly showed the presence of lagging chromosomes despite a bipolar mitotic apparatus. Mitotic figures peaked at 24 h of treatment. After 48 h of drug treatment, fewer cells were found arrested in mitosis and we observed abnormally large multinucleated cells. In addition, many cells with fragmented nuclei and apoptotic morphologies were evident. In contrast, cells treated with the vehicle (DMSO) showed normal mitoses with a bipolar spindle apparatus and chromosomes congressing at the mid-plate followed by cytokinesis (data not shown).
Figure 1.
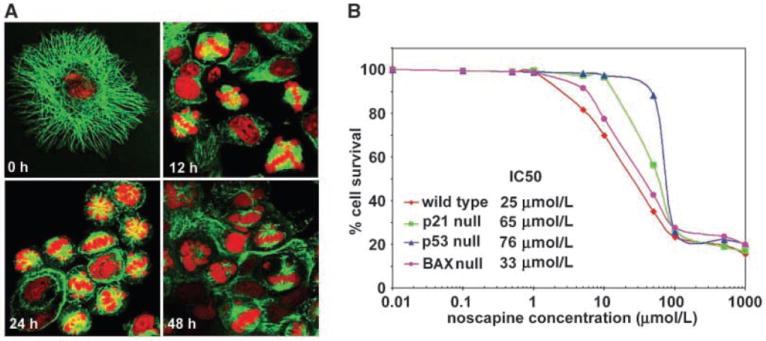
A, noscapine treatment of HCT116 colon cancer cells perturbed the spindle architecture and nuclear morphology. Confocal micrographs of HCT116 (p53 wt) cells treated with 25 μmol/L noscapine for 0, 12, 24, and 48 h. Intact normal radial arrays of microtubules and typical characteristic cellular morphologies of HCT116 cells are apparent at time 0 of treatment. As the time of noscapine treatment increases from 12 to 24 to 48 h, derangement of microtubule arrays (green) is evident. Mitotic figures begin to appear at 12 h and are abundant at 24 h. Forty-eight hours of treatment grossly perturbs the nuclear morphology as seen by the formation of large multilobed nuclei and the appearance of small spheroids of DNA (red) fragments, indicative of apoptosis. B, noscapine inhibits proliferation of HCT116 colon cancer cells. The percentage of cell proliferation at the indicated drug concentrations, compared with untreated cells, was measured by the standard sulforhodamine B assay. The four cell lines (p53-wt, p53-null, p21-null, and BAX-null) were grown in 96-well formats and treated with gradient concentrations of noscapine for 48 h. Plot of percent cell survival versus noscapine concentrations used for the determination of IC50 values. Points, average of three independent experiments.
Next, we investigated if these spindle defects caused an inhibition of the growth of HCT116 cells and its isogenic variants on noscapine treatment using sulforhodamine B assay. Figure 1B shows dose-response curves for the HCT116 cells (p53-wt, p53-null, p21-null, and BAX-null) following a 48-h exposure to gradient noscapine concentrations. The p53-wt cells were most sensitive (IC50 value of 25 μmol/L) to the antiproliferative effects of noscapine. In contrast, the p53-null cells were most resistant and displayed an IC50 of 76 μmol/L. The IC50 value of noscapine for p21-null cells was 63 μmol/L, which was also ~2.5-fold higher than that for the p53-wt cells, suggesting reduced chemosensitivity. The BAX-null cells displayed an IC50 value of 33 μmol/L, which was comparable to the wild-type cells. Thus, our results from the growth inhibition experiments concluded that the p53, BAX, and p21 status of colon cancer cells influences their sensitivity to noscapine cytotoxicity.
Noscapine induces G2-M arrest and apoptosis in a p53/p21–dependent manner
The induction of apoptosis is a major event in tumor cell response to cytotoxic agents, including the tubulin-binding drugs (26). We have shown that the tubulin-binding agent noscapine arrests cell cycle progression at the G2-M phase preceding apoptosis in mammalian cells (15, 17, 25). To investigate the roles of p53 and p21 in the negative regulation of cellular growth on noscapine treatment, isogenic variants of the HCT116 cells were treated with 25 μmol/L noscapine and their cell cycle profiles were analyzed at 0, 12, 24, and 48 h of treatment. Figure 2A shows the cell cycle profile of all the four cell types on noscapine treatment over time in a three-dimensional disposition. As can be seen in Fig. 2A, mitotic figures in p53-wt cells reached a peak at ~12 h, accompanied by an emergence of sub-G1 (<2N DNA) population. This sub-G1 population in p53-wt cells peaked to ~52% at 48 h. The p53-null cells, on the other hand, showed minimal sub-G1 population (~25%) even at 48 h of noscapine treatment despite accumulation of cells in the G2-M phase at 24 h. Our results depict that BAX-null cells showed a quite similar response as seen for p53-wt cells. The BAX-null cells accumulated in the G2-M phase, peaking at 12 h of noscapine treatment. At 24 h, we observed a significant reduction of the G2-M cell population with a concomitant increase of the sub-G1 population in BAX-null cells. This sub-G1 population reached a maximum of ~45% at 48 h of noscapine treatment, suggesting a reasonably good apoptotic response in these cells. The above findings are suggestive of an important role of p53 and of BAX-independent pathways in noscapine-induced apoptosis.
Figure 2.
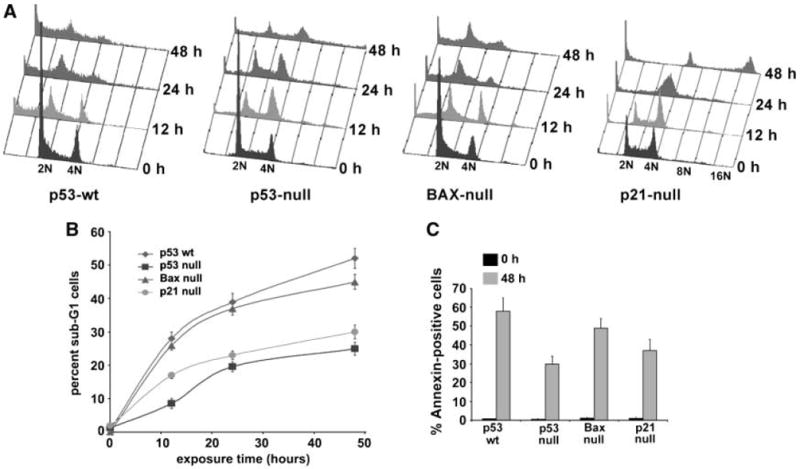
Noscapine alters the cell cycle profile of HCT116 colon cancer cells. A, effect of 25 μmol/L noscapine over time on the cell cycle profile of HCT116 cells (p53-wt, p53-null, p21-null, and BAX-null) shown in a three-dimensional disposition. Cells were harvested for analysis at the noted hours, fixed and stained with propidium iodide, and analyzed by flow cytometry [fluorescence-activated cell sorting (FACS)] using the Cell Quest Software. X-axis, intensity of propidium iodide fluorescence, which is indicative of the total DNA content of cells in different phases of the cell cycle. Y-axis, number of cells detected for a given DNA content. Z-axis, time points (i.e., 0, 12, 24, and 48 h). Cells residing in the G0-G1 phase of the cell cycle are designated with 2N and cells in the G2 phase or in mitosis are designated with 4N. Less than 2N DNA is indicative of hypodiploid or sub-G1 population, a diagnostic of apoptosis. As the arrested mitotic cells plummet from a peak at ~12 h in p53-wt cells, the number of apoptotic cells increases in concordance. The p53-wt cells show maximum sensitivity to noscapine treatment whereas the p53-null cells are significantly resistant to the apoptotic effects of noscapine. The BAX-null cells showed an appreciable sub-G1 population. The p21-null cells also escape the effect of the drug and do not succumb to apoptosis. Interestingly, they traverse continually through the cell cycle and result in an accumulation of a polyploid population. Representative results of three experiments done in triplicate. B, quantitative graphical representation of percentage of sub-G1 population that is reflective of dying apoptotic cells. Points, average of three independent experiments; bars, SD (P < 0.05). C, noscapine induces apoptosis as revealed by the externalization of phosphatidylserine measured by the Annexin V staining assay. A graphical representation of the extent of apoptosis by quantitating the Annexin-positive cells on 48-h noscapine treatment of the various isogenic variants of the HCT116 cells. Columns, average of three independent experiments; bars, SD (P < 0.05).
The p21-null cells showed a time-dependent increase of G2-M population up to 24 h of noscapine exposure. Interestingly, the p21-null cells continued to traverse through the cell cycle and did not exit mitoses but rather underwent multiple rounds of DNA synthesis, accumulating large amounts of DNA as evidenced by the presence of a 16N polyploid peak at 48 h (Fig. 2A). The p21-null cells showed ~30% sub-G1 population at 48 h, which conforms to the resistance of these cells to succumb to apoptosis. This reduction of apoptosis correlated with the reduced chemosensitivity to noscapine shown by the high IC50 value in p21-null cells. The progression of the entire cell cycle as a function of time in terms of the percentage of G0-G1, S, G2-M, and sub-G1 populations in all isogenic variants of HCT116 cells is shown in Table 1. Figure 2B is a graphical representation of percent sub-G1 cells that are indicative of apoptosis in all four cell types.
Table 1.
Effect of noscapine on the cell cycle progression of HCT116 cancer cells
| Cell cycle variables, % | p53 wt
|
p53 null
|
p21 null
|
Bax null
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 h | 12 h | 24 h | 48 h | 0 h | 12 h | 24 h | 48 h | 0 h | 12 h | 24 h | 48 h | 0 h | 12 h | 24 h | 48 h | |
| Sub-G1 | 0.9 ± 0.2 | 27.8 ± 2.8 | 39.2 ± 4.5 | 51.8 ± 5.3 | 1.3 ± 0.3 | 8.6 ± 2.5 | 19.6 ± 3.4 | 25.0 ± 2.6 | 2.0 ± 0.3 | 17.0 ± 2.1 | 22.9 ± 3.1 | 29.7 ± 4.2 | 0.2 ± 0.1 | 26.4 ± 3.2 | 37.7 ± 4.1 | 44.7 ± 4.9 |
| G1 | 57.7 ± 2.4 | 26.0 ± 2.0 | 27.5 ± 2.1 | 19.3 ± 2.3 | 49.5 ± 3.2 | 26.8 ± 4.4 | 24.5 ± 5.6 | 14.2 ± 4.8 | 24.7 ± 2.8 | 20.9 ± 4.6 | 4.3 ± 1.4 | 2.5 ± 0.9 | 52.7 ± 5.1 | 29.7 ± 3.6 | 27.3 ± 4.2 | 23.6 ± 3.8 |
| S | 15.2 ± 3.4 | 16.5 ± 2.8 | 11.9 ± 3.7 | 10.9 ± 2.4 | 16.9 ± 3.7 | 13.6 ± 4.1 | 25.0 ± 4.2 | 14.7 ± 3.7 | 25.7 ± 3.1 | 16.1 ± 4.4 | 19.2 ± 2.6 | 3.3 ± 1.1 | 18.8 ± 3.3 | 12.1 ± 2.4 | 14.4 ± 3.1 | 16.5 ± 2.8 |
| G2-M | 20.4 ± 1.8 | 20.0 ± 2.7 | 16.0 ± 3.2 | 9.9 ± 3.8 | 23.0 ± 3.6 | 37.3 ± 4.3 | 24.5 ± 2.8 | 20.5 ± 3.4 | 35.0 ± 3.3 | 36.0 ± 2.9 | 34.8 ± 3.6 | 15.0 ± 2.7 | 21.3 ± 4.5 | 20.8 ± 3.9 | 9.9 ± 4.8 | 8.9 ± 4.2 |
| Polyp | 6.0 ± 1.5 | 6.8 ± 3.2 | 7.5 ± 2.7 | 9.5 ± 3.4 | 5.4 ± 2.1 | 10.6 ± 3.9 | 7.5 ± 3.2 | 21.0 ± 6.8 | 11.1 ± 2.7 | 13.6 ± 1.8 | 21.0 ± 3.4 | 50.8 ± 4.5 | 7.4 ± 2.7 | 11.3 ± 2.4 | 11.1 ± 3.2 | 9.3 ± 1.7 |
NOTE: Cells were treated with 25 μmol/L noscapine for the indicated time (h) before being stained with propidium iodide for cell cycle analysis. Values shown are mean ± SE.
We next attempted to correlate the differences in sub-G1 population observed using cell cycle experiments with those obtained from Annexin V staining of apoptotic cells in all four isogenic cell types. Biochemically, the early apoptotic process is characterized by loss of lipid asymmetry between the two leaflets of plasma membrane, where phosphatidylserine translocates from the inner leaflet and gets displayed on the outer leaflet. Using flow cytometry, Alexa Fluor 488–labeled Annexin V was used to quantitate the apoptotic population at 48 h of noscapine treatment in all four cell types. Figure 2C shows a graphical representation of the percentage of Annexin V–positive cells obtained from the four variants of colon cancer cells treated with either the vehicle or noscapine for 48 h. The vehicle-treated cultures of all cell types contained very few apoptotic cells as evident from Fig. 2C. On the other hand, cells treated with noscapine for 48 h showed ~58% (p53-wt), ~30% (p53-null), ~49% (BAX-null), and ~37% (p21-null) Annexin-positive cells. The extent of apoptosis as seen by Annexin V staining in all four cell lines correlated with the extent of sub-G1 population obtained from cell cycle experiments.
Noscapine treatment alters levels of cell cycle and apoptosis regulatory proteins
We next examined the effect of noscapine on the expression of some cell cycle and apoptosis regulatory proteins in all four isogenic HCT116 cell lines. The time-dependent effects of exposure to 25 μmol/L noscapine on p53, BAX, Bcl-2, p21, and cyclin B1 levels are shown in Fig. 3. In p53-wt cells, immunoblot analysis showed distinctly elevated p53 levels as early as 12 h after initiation of noscapine treatment. We further evaluated whether the growth inhibitory effect of noscapine on colon cells with wild-type p53 status was associated with an up-regulation of p21, an inhibitor of cyclin-CDK complexes involved in G1-G2 phase progression (27). Following activation of p53, a significant increase in p21 expression over time was also detectable in p53-wt cells. The elevated level of p21 indicated that the stabilized p53 protein was transcriptionally active. The BAX gene, a proapoptotic member of the Bcl-2 family, is an important target for p53. Our results from the Western blot analysis (Fig. 3) showed a time-dependent elevation of BAX protein level and a pronounced down-regulation of Bcl-2 on noscapine treatment of p53-wt cells. The cyclin B1 levels increased up to 12 h followed by a decline to almost negligible levels at 48 h in p53-wt cells (Fig. 3). Because cyclin B1 plays a crucial role in progression through mitosis of mammalian cells, our results were in concordance with the mitotic activity of p53-wt cells over time on noscapine treatment.
Figure 3.
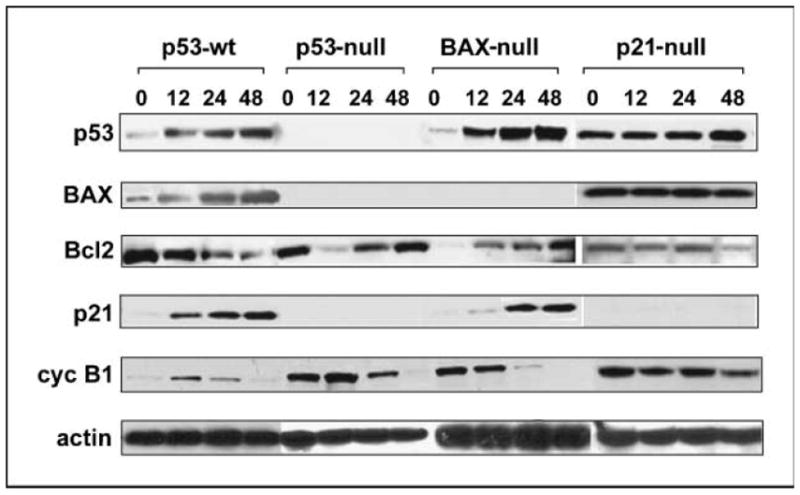
Immunoblot analysis of untreated and noscapine-treated HCT116 cells. All the four cell types were grown in the presence of 25 μmol/L noscapine for 0, 12, 24, and 48 h. After the indicated times, cells were lysed and total protein was extracted, separated by SDS-PAGE, electrotransferred onto polyvinylidene difluoride membrane, and subjected to immunoblotting with the indicated primary antibodies followed by incubation with horseradish peroxidase–conjugated secondary antibodies. β-Actin was used as a loading control.
Conversely, loss of p53 by homologous deletion in p53-null cells does not show any detectable levels of p21 and BAX proteins, which are the direct downstream effectors of p53. Because BAX is tightly regulated by p53, the absence of p53 results in lack of detectable BAX levels. Although p21 is normally regulated by p53, there is plenty of evidence of alternative p53-independent up-regulation of p21 that is mostly dependent on the treatment type (28). Noscapine treatment apparently does not induce the p53-independent pathway of inducing p21 expression, and thus the lack of detectable p21 levels. In p53-null cells, the levels of the Bcl-2 decreased drastically and transiently at 12 h of drug-treatment before restoring back to the basal levels at 48 h of treatment (Fig. 3). This restoration of Bcl-2 can partly be a reason for the reduction of apoptotic response in p53-null cells as evidenced by the minimal sub-G1 population and a severe reduction of Annexin-positive cells at 48 h. The cyclin B1 levels in p53-null cells were slightly up-regulated at 12 h followed by a strong decline up to 48 h of noscapine exposure (Fig. 3).
In BAX-null cells, noscapine induced an accumulation of p53 and p21 levels by 48 h (Fig. 3). These cells displayed drug sensitivity despite the absence of BAX. The Bcl-2 levels also increased on noscapine treatment in a time-dependent manner in BAX-null cells. Our immunoblot results show that the cyclin B1 levels inversely correlated with p21 levels in BAX-null cells (Fig. 3).
In p21-null cells, noscapine treatment did not have any change in the BAX levels similar to p53 levels, which also did not change over time except a slight increase at 48 h (Fig. 3). Although p21 is not known to play a role in the regulation of BAX, the p21-null cells are known to have high basal levels of p53 and BAX (29). They also lack the G1-S checkpoint (30). The high levels of BAX could be due to the high basal levels of p53 because BAX expression is tightly regulated by p53. The Bcl-2 levels showed a time-dependent decrease following noscapine treatment (Fig. 3). Noscapine treatment resulted in a cyclical behavior of cyclin B1 levels over time in p21-null cells. Most importantly, despite the presence of p53, the p21-null cells showed a significant reduction in percent sub-G1 population as depicted by cell cycle and Annexin staining experiments. Furthermore, p21-null cells had a higher IC50, showing reduced sensitivity to noscapine treatment.
Reintroduction of p53 restores noscapine-induced apoptosis in p53-null cells
Given the role of p53 in the induction of apoptosis in response to suppression of microtubule dynamics by noscapine, we investigated whether reintroduction of p53 in p53-null cells would restore noscapine-induced apoptosis. Toward this goal, we infected p53-null cells with an adenovirus encoding for p53 followed by a time-dependent exposure of noscapine to evaluate whether restoration of p53 could enhance noscapine-induced apoptosis in p53-null cells. As shown in Fig. 4A, there was a complete restoration of the apoptotic effects of noscapine on reintroduction of p53 gene. The percentage of sub-G1 cells at 48 h in noscapine treated-infected p53-null cells was ~48% as compared with ~27% in drug-treated, empty vector–infected p53-null cells (Fig. 4B). In addition, there occurred an increase in the number of Annexin-positive cells (~51%) in adenovirus-infected p53-null cells compared with ~25% Annexin-positive cells in empty vector–infected cells (Fig. 4C). It is interesting to note that the percent apoptosis in p53-infected p53-null cells was in close agreement with that observed in p53-wt cells. Immunoblot analysis of cell lysates of adenovirus-infected p53-null cells showed a restoration of near wild-type amounts of p53 at 0 h of treatment (Fig. 4D). Because adenoviral promoters vary in their potency from one cell type to another, we cannot really expect them to maintain wild-type steady-state levels of the proteins of interest studied. The significant p53 up-regulation at 12 h of noscapine treatment is clearly visible, which then levels off. This perhaps might be due to the saturation of regulated transcription levels of p53 on drug treatment among many other reasons. Therefore, it is obvious that downstream genes of p53, such as p21 and BAX, are also not sufficiently controlled by reintroduced p53. Although the BAX expression was almost unchanged over time of noscapine treatment, Bcl-2 was decreased at 48 h of drug treatment in infected cells (Fig. 4D). The cyclin B1 levels remained unchanged until 24 h, but a slight decrease was observed at 48 h (Fig. 4D). The empty vector–infected p53-null controls did not show any expression for p53, BAX, and p21. However, Bcl-2 levels showed a decrease at 12 h followed by an increase at 24 and 48 h of noscapine treatment (Fig. 4D). Cyclin B1 expression in the empty vector–transfected p53-null cells increased at 12 h followed by a decline up to 48 h of treatment (Fig. 4D). Surprisingly, despite a little deviation in the protein levels as compared with the wild-type scenario, a complete rescue of cell cycle kinetics and apoptosis to full wild-type extent in p53 null cells on reintroduction of p53 was observed (Fig. 4A–C).
Figure 4.
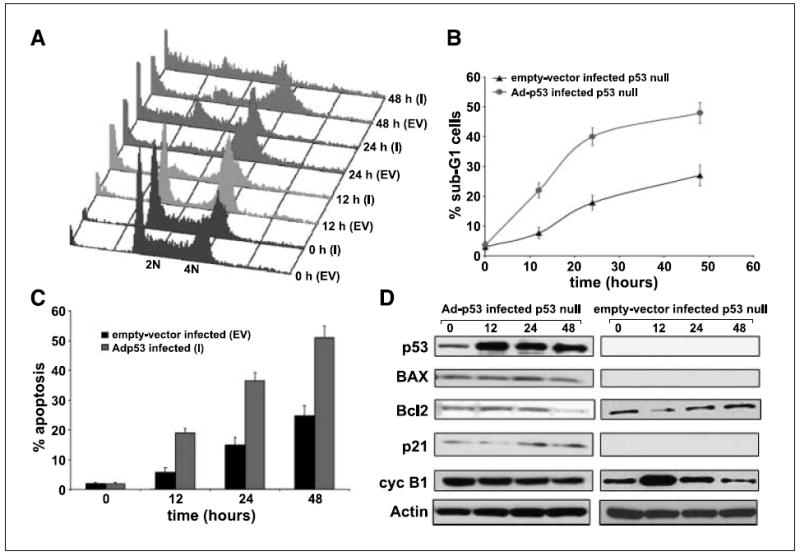
Reintroduction of the p53 wt gene in p53-null cells causes a complete restoration of the apoptotic response. p53-null cells were infected with an adenovirus (Ad) encoding the wild-type p53 gene and were subjected to noscapine exposure for 0, 12, 24, and 48 h. An empty vector control was used in the experiments to null the background effects of the adenovirus vector. A, three-dimensional FACS profile of Adp53-infected (I) and empty vector–infected (EV) cells for 0, 12, 24, and 48 h. B, quantitation of the percent sub-G1 population for Adp53-infected and empty vector–infected p53-null cells. C, graphical representation of the extent of apoptosis in Adp53-infected cells and empty vector–infected controls as evaluated by Annexin V staining followed by FACS analysis. Columns, average of three independent experiments; bars, SD (P < 0.05). D, immunoblot analysis of cell lysates from Adp53-infected and empty vector–infected p53-null cells.
p21 is necessary but not sufficient for p53-mediated noscapine-induced apoptosis
The results above suggest an important role of p21 in noscapine-mediated p53-dependent apoptosis. Our hypothesis was that p53 induction might be a necessary but not sufficient condition for causing apoptosis on noscapine treatment. To test this hypothesis, we transfected p21-null cells with a p21 plasmid construct and evaluated whether reintroduction of p21 could enhance apoptosis in p21-null cells. Our results showed that apoptotic response in these cells was restored by the transfection of p21 gene into p21-null cells thereby confirming the proapoptotic role of p21 on noscapine treatment. The percentages of sub-G1 cells (Fig. 5A and B) and Annexin-positive cells (Fig. 5C) were significantly higher in p21-transfected p21-null cells as compared with the empty vector–transfected p21-null cells (~51% versus ~28% sub-G1 cells, ~48% versus 32% Annexin-positive cells). The immunoblot analysis showed a progressive increase of p53 expression on noscapine treatment of p21-transfected p21-null cells as well as the empty vector–transfected p21-null cells (Fig. 5D). The Bcl-2 expression declined over time of treatment in both the p21-transfected and empty vector–transfected p21-null cells (Fig. 5D). The p21 levels in the p21-transfected cells were significantly higher at 24 h of noscapine treatment (Fig. 5D). Our results suggest that an increased p21 expression is necessary for p53-mediated apoptosis on noscapine treatment. These results thus identify molecular determinants of the in vitro response to noscapine showing a role for the p53/p21 system in the apoptotic response and provide evidence for a substantive proapoptotic role of p21 in noscapine-induced apoptosis.
Figure 5.
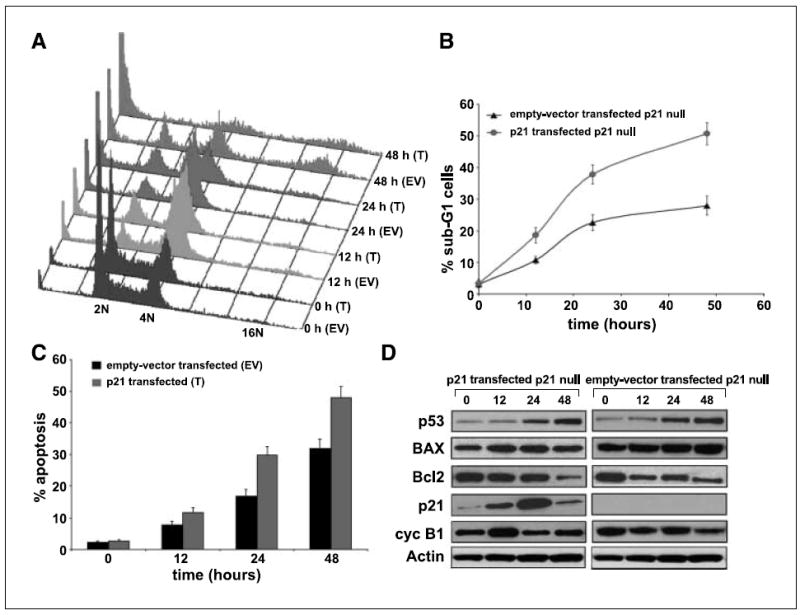
Up-regulation of p53 is a necessary but not sufficient condition for noscapine-mediated apoptosis. The p21 gene is a crucial player mediating the apoptotic effects downstream of the p53 gene. Reintroduction of p21 in p21-null cells significantly increased the sub-G1 population and the extent of apoptosis as revealed by cell cycle experiments and Annexin V staining. A, cell cycle profiles of p21-transfected p21-null cells (T) along with their empty vector–transfected controls. B, graphical representation of the quantitation of the sub-G1 population. C, graphical depiction of the percent Annexin-positive cells for p21-transfected cells and empty vector–transfected controls on noscapine treatment for 0, 12, 24, and 48 h. Columns, average of three independent experiments; bars, SD (P < 0.05). D, immunoblot analysis of cell lysates from p21-transfected p21-null cells and empty vector–transfected controls.
We next asked if p21 was sufficient to restore apoptosis in the absence of p53 in p53-null cells. To address this, we transfected p53-null cells with a p21 plasmid construct and measured the sub-G1 population by following the cell cycle of the transfected cells over time. Our results showed that noscapine-induced apoptosis was not restored by p21 alone in the absence of p53 (Fig. 6A). The sub-G1 populations of the p21-transfected p53-null cells and empty vector–transfected p53-null controls were comparable. Figure 6B depicts a graphical representation of the percentage of sub-G1 cells in the transfected and empty vector–transfected controls. This result further strengthened that p21 alone is not sufficient; rather, both p53 and p21 determine the sensitivity to noscapine-induced apoptosis.
Figure 6.
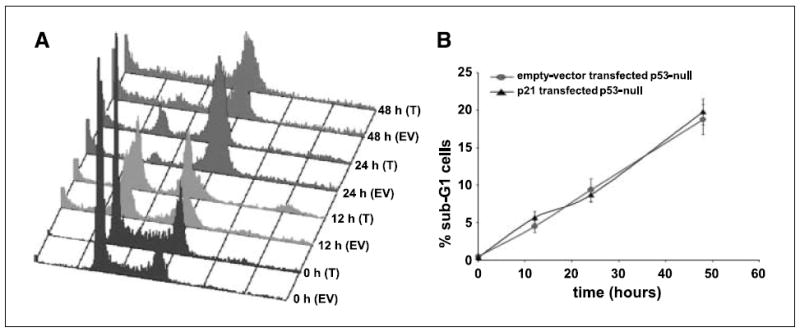
A, cell cycle profiles on noscapine treatment over time of p21-transfected p53-null cells along with the empty vector–transfected controls. B, quantitation of the percent sub-G1 population for the p21-transfected and empty vector–transfected p53-null cells. Introduction of p21 in p53-null cells is not sufficient to restore apoptosis as seen by the absence of an increased sub-G1 population on p21 transfection.
Discussion
Colorectal cancer is considered relatively resistant to chemotherapy, compared with other types of human malignancies such as ovarian cancer and esophageal cancer (31). Thus, attempts at improving the survival of patients affected by this disease depend largely on strategies targeting tumor cell resistance, which cannot be rationally planned without a detailed knowledge of the mechanisms underlying this phenomenon. A current paradigm about cancer chemotherapy indicates disabling of the intrinsic apoptotic pathways as a key factor in the response of tumor cells to anti-cancer drugs (26, 32). Therefore, strategies aiming to reestablish the capability of the cell to activate a cell death program are an active area of research.
Epidemiologic data show that p53 is mutated in over half of all human tumors (33). Among the remaining tumors, although wild-type p53 is expressed, the pathways of p53-mediated cell cycle arrest or apoptosis are defective. In the clinic, the functional status of p53 has been related to prognosis, progression, and therapeutic response of tumors (34). All these characteristics make p53 an important molecular target for tumor suppression and drug development. Although it is well established that p53 is an important mediator of apoptosis under some conditions, the downstream effectors of the p53 pathway in the cell death process are not completely known. Several p53-inducible genes have been identified, including mdm-2 (35), GADD45 (36), BAX (7), and p21 (37, 38). Recent evidence suggests that, apart from inducing arrest, p21 may participate in apoptosis in both p53-dependent and p53-independent pathways (39, 40). p53-induced apoptosis results from overlapping downstream pathways that both suppress mitogenic and survival signaling and promote proapoptotic signaling. In this context, p53 can up-regulate the proapoptotic Bcl-2 family member BAX (7) and possibly transcriptionally repress the antiapoptotic protein Bcl-2 (41). Therefore, p53-mediated up-regulation of BAX and perhaps concomitant down-regulation of Bcl-2 are pivotal in shifting the ratio of BAX/Bcl-2 in the cell and ultimately favoring apoptosis.
The present study was designed to delineate the role of p53 and its two downstream targets, p21 and BAX, in the response of colorectal carcinoma cells to noscapine treatment. To achieve this goal, we used isogenic variants of the HCT116 colon cancer cells (i.e., p53-wt, p53-null, p21-null, and BAX-null cells). Treatment of p53-wt cells with noscapine resulted in the maximum apoptotic response whereas p53-null cells were most resistant to apoptosis. This clearly agrees with the premise that because p53 is one of the major regulators of apoptosis, expression of this tumor suppressor sensitizes cells to apoptosis in response to stress, such as the pharmacologic suppression of microtubule dynamics on noscapine treatment. The accumulation of p53 is an early and critical step for p53 activation. In our study, noscapine treatment resulted in p53 accumulation in a time-dependent manner accompanied by an increase in the levels of its two well-studied transcriptional targets, p21 and BAX. On the other hand, the p53-null cells displayed a loss of the growth-inhibitory and apoptotic effects of noscapine, indicating that the p53 gene plays a crucial role in mediating the effects of noscapine. We also found an absence of p21 in p53-null cells, suggesting that the lack of a functional p53 protein could lead to loss of p21 protein levels in response to noscapine exposure. This was also manifested in a pronounced reduction of apoptosis in p53-null cells on noscapine treatment. Although our data showed that G2-M arrest was a major characteristic of almost all cell types before they succumb to apoptosis, the inactivation of p21 in HCT116 cells resulted in a significant polyploid population on noscapine exposure at 48 h. Moreover, p21-null cells that had intact p53 offered a proapoptotic role of p21 because these cells not only showed a higher IC50 value but also escaped apoptosis on noscapine treatment. The current study thus suggests that p21 plays an important role in exerting the apoptotic activity of noscapine in a p53-dependent manner.
Chemotherapeutic regimens including Adriamycin, etoposide, cisplatinum, colcemid, and resveratrol induce distinct cellular responses; however, absence of p21 favors apoptosis induction by Adriamycin, etoposide, and colcemid. In our context, in the p53-wt cells, noscapine stimulates p21 expression and induces apoptosis in a p53-dependent manner, and the absence of p21 decreases apoptosis. The apoptotic response is mediated, perhaps, through the mitochondria and is accompanied by an alteration of the Bcl-2/BAX ratio. An increasing ratio of BAX/Bcl-2 proteins thus plays an important role in the induction of apoptosis by noscapine in p53-wt cells. Furthermore, it is suggestive of involvement of a mitochondrial-mediated apoptotic process on noscapine treatment of p53-wt cells. This was, however, not true in the case of BAX-null cells because they showed a good apoptotic response, thereby suggesting BAX-independent cell death mechanisms. Although p53 status alone may not be a predictor of chemosensitivity to noscapine, it is apparent from this study that wild-type p53 in tumor tissues could be at least one factor in predicting drug efficacy. The apoptotic response, however, was greatly decreased in HCT116 lines with homozygous knockout of p53. The ability of wild-type p53 to induce apoptosis was restored in HCT116 cells that are null for p53 by infection with adenoviral p53. Intriguingly, despite an increase in p53 levels on noscapine treatment over time in p21-null cells, the apoptotic response was significantly reduced. It was restored on reintroduction of the p21 gene, thus showing that p53 is a necessary but not sufficient condition for noscapine-induced apoptosis. Nevertheless, the role of p21 in apoptosis is controversial. There is much evidence that implicates p21 as an antiapoptotic factor (42-46). On the other hand, its role as a proapoptotic factor is also well documented (47-49). In summary, we have clearly shown, using isogenic cell lines, that noscapine activates growth arrest and apoptosis primarily via a p53-dependent pathway that necessarily involves the function of p21 such that the abrogation of either molecule confers a growth advantage to the cells.
Acknowledgments
Grant support: NIH grants CA095317 (H.C. Joshi) and CA84197 and DK64399 (V.W. Yang).
We thank Dr. Bert Vogelstein for the HCT116 p53-wt, p53-null, p21-null, and BAX-null cells; Binfei Zhou for the superb technical assistance; and members of the Joshi and Yang laboratory for discussion.
References
- 1.Joshi HC. Microtubule dynamics in living cells. Curr Opin Cell Biol. 1998;10:35–44. doi: 10.1016/s0955-0674(98)80084-7. [DOI] [PubMed] [Google Scholar]
- 2.Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by Taxol at low concentrations. Proc Natl Acad Sci U S A. 1993;90:9552–6. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L. Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 1996;56:816–25. [PubMed] [Google Scholar]
- 4.Gundersen GG, Cook TA. Microtubules and signal transduction. Curr Opin Cell Biol. 1999;11:81–94. doi: 10.1016/s0955-0674(99)80010-6. [DOI] [PubMed] [Google Scholar]
- 5.Meek DW. Multisite phosphorylation and the integration of stress signals at p53. Cell Signal. 1998;10:159–66. doi: 10.1016/s0898-6568(97)00119-8. [DOI] [PubMed] [Google Scholar]
- 6.Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001;13:332–7. doi: 10.1016/s0955-0674(00)00216-7. [DOI] [PubMed] [Google Scholar]
- 7.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 8.el-Deiry WS. p21/p53, cellular growth control and genomic integrity. Curr Top Microbiol Immunol. 1998;227:121–37. doi: 10.1007/978-3-642-71941-7_6. [DOI] [PubMed] [Google Scholar]
- 9.Gupta M, Fan S, Zhan Q, Kohn KW, O’Connor PM, Pommier Y. Inactivation of p53 increases the cytotoxicity of camptothecin in human colon HCT116 and breast MCF-7 cancer cells. Clin Cancer Res. 1997;3:1653–60. [PubMed] [Google Scholar]
- 10.Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B. 14-3-3σ is required to prevent mitotic catastrophe after DNA damage. Nature. 1999;401:616–20. doi: 10.1038/44188. [DOI] [PubMed] [Google Scholar]
- 11.Herr I, Debatin KM. Cellular stress response and apoptosis in cancer therapy. Blood. 2001;98:2603–14. doi: 10.1182/blood.v98.9.2603. [DOI] [PubMed] [Google Scholar]
- 12.Bunz F, Hwang PM, Torrance C, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–9. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace-Brodeur RR, Lowe SW. Clinical implications of p53 mutations. Cell Mol Life Sci. 1999;55:64–75. doi: 10.1007/s000180050270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu S, Bishop WR, Liu M. Differential effects of cell cycle regulatory protein p21WAF/Cip1 on apoptosis and sensitivity to cancer chemotherapy. Drug Resist Updat. 2003;6:183–95. doi: 10.1016/s1368-7646(03)00044-x. [DOI] [PubMed] [Google Scholar]
- 15.Ye K, Ke Y, Keshava N, et al. Opium alkaloid noscapine is an antitumor agent that arrests metaphase and induces apoptosis in dividing cells. Proc Natl Acad Sci U S A. 1998;95:1601–6. doi: 10.1073/pnas.95.4.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aneja R, Vangapandu SN, Lopus M, et al. Synthesis of microtubule-interfering halogenated noscapine analogs that perturb mitosis in cancer cells followed by cell death. Biochem Pharmacol. 2006;72:415–26. doi: 10.1016/j.bcp.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 17.Zhou J, Gupta K, Yao J, et al. Paclitaxel-resistant human ovarian cancer cells undergo c-Jun NH2-terminal kinase-mediated apoptosis in response to noscapine. J Biol Chem. 2002;277:39777–85. doi: 10.1074/jbc.M203927200. [DOI] [PubMed] [Google Scholar]
- 18.Hu MC, Qiu WR, Wang YP. JNK1, JNK2 and JNK3 are p53 N-terminal serine 34 kinases. Oncogene. 1997;15:2277–87. doi: 10.1038/sj.onc.1201401. [DOI] [PubMed] [Google Scholar]
- 19.Buschmann T, Potapova O, Bar-Shira A, et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol Cell Biol. 2001;21:2743–54. doi: 10.1128/MCB.21.8.2743-2754.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW. Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene. 2004;23:2934–49. doi: 10.1038/sj.onc.1207515. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290:989–92. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- 22.Bunz F, Dutriaux A, Lengauer C, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 23.Zhou J, Gupta K, Aggarwal S, et al. Brominated derivatives of noscapine are potent microtubule-interfering agents that perturb mitosis and inhibit cell proliferation. Mol Pharmacol. 2003;63:799–807. doi: 10.1124/mol.63.4.799. [DOI] [PubMed] [Google Scholar]
- 24.Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–12. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 25.Landen JW, Lang R, McMahon SJ, et al. Noscapine alters microtubule dynamics in living cells and inhibits the progression of melanoma. Cancer Res. 2002;62:4109–14. [PubMed] [Google Scholar]
- 26.Brown JM, Wilson G. Apoptosis genes and resistance to cancer therapy: what does the experimental and clinical data tell us? Cancer Biol Ther. 2003;2:477–90. doi: 10.4161/cbt.2.5.450. [DOI] [PubMed] [Google Scholar]
- 27.Gong J, Ammanamanchi S, Ko TC, Brattain MG. Transforming growth factor β1 increases the stability of p21/WAF1/CIP1 protein and inhibits CDK2 kinase activity in human colon carcinoma FET cell. Cancer Res. 2003;63:3340–6. [PubMed] [Google Scholar]
- 28.Zhang W, Grasso L, McClain CD, et al. p53-independent induction of WAF1/CIP1 in human leukemia cells is correlated with growth arrest accompanying monocyte/macrophage differentiation. Cancer Res. 1995;55:668–74. [PubMed] [Google Scholar]
- 29.Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M. Tumor susceptibility of p21Waf1/Cip1-deficient mice. Cancer Res. 2001;61:6234–8. [PubMed] [Google Scholar]
- 30.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–84. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 31.Blijham GH. Chemotherapy of colorectal cancer. Anticancer Drugs. 1991;2:233–45. doi: 10.1097/00001813-199106000-00003. [DOI] [PubMed] [Google Scholar]
- 32.Shah MA, Schwartz GK. Cell cycle-mediated drug resistance: an emerging concept in cancer therapy. Clin Cancer Res. 2001;7:2168–81. [PubMed] [Google Scholar]
- 33.Hollstein M, Rice K, Greenblatt MS, et al. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994;22:3551–5. [PMC free article] [PubMed] [Google Scholar]
- 34.Weller M. Predicting response to cancer chemotherapy: the role of p53. Cell Tissue Res. 1998;292:435–45. doi: 10.1007/s004410051072. [DOI] [PubMed] [Google Scholar]
- 35.Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–3. doi: 10.1038/358080a0. [DOI] [PubMed] [Google Scholar]
- 36.Kastan MB, Zhan Q, el-Deiry WS, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–97. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 37.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–4. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 38.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75:805–16. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 39.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–90. [PubMed] [Google Scholar]
- 40.Agrawal S, Agarwal ML, Chatterjee-Kishore M, Stark GR, Chisolm GM. Stat1-dependent, p53-independent expression of p21 (waf1) modulates oxysterol-induced apoptosis. Mol Cell Biol. 2002;22:1981–92. doi: 10.1128/MCB.22.7.1981-1992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haldar S, Negrini M, Monne M, Sabbioni S, Croce CM. Down-regulation of bcl-2 by p53 in breast cancer cells. Cancer Res. 1994;54:2095–7. [PubMed] [Google Scholar]
- 42.Asada M, Yamada T, Ichijo H, et al. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J. 1999;18:1223–34. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bissonnette N, Hunting DJ. p21-induced cycle arrest in G1 protects cells from apoptosis induced by UV-irradiation or RNA polymerase II blockage. Oncogene. 1998;16:3461–9. doi: 10.1038/sj.onc.1201899. [DOI] [PubMed] [Google Scholar]
- 44.Gorospe M, Wang X, Guyton KZ, Holbrook NJ. Protective role of p21 (Waf1/Cip1) against prostaglandin A2-mediated apoptosis of human colorectal carcinoma cells. Mol Cell Biol. 1996;16:6654–60. doi: 10.1128/mcb.16.12.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosato RR, Wang Z, Gopalkrishnan RV, Fisher PB, Grant S. Evidence of a functional role for the cyclin-dependent kinase-inhibitor p21WAF1/CIP1/MDA6 in promoting differentiation and preventing mitochondrial dysfunction and apoptosis induced by sodium butyrate in human myelomonocytic leukemia cells (U937) Int J Oncol. 2001;19:181–91. doi: 10.3892/ijo.19.1.181. [DOI] [PubMed] [Google Scholar]
- 46.Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–49. [PubMed] [Google Scholar]
- 47.Chen T, Turner J, McCarthy S, Scaltriti M, Bettuzzi S, Yeatman TJ. Clusterin-mediated apoptosis is regulated by adenomatous polyposis coli and is p21 dependent but p53 independent. Cancer Res. 2004;64:7412–9. doi: 10.1158/0008-5472.CAN-04-2077. [DOI] [PubMed] [Google Scholar]
- 48.Dong C, Li Q, Lyu SC, Krensky AM, Clayberger C. A novel apoptosis pathway activated by the carboxyl terminus of p21. Blood. 2005;105:1187–94. doi: 10.1182/blood-2004-06-2188. [DOI] [PubMed] [Google Scholar]
- 49.Yang HL, Pan JX, Sun L, Yeung SC. p21 Waf-1 (Cip-1) enhances apoptosis induced by manumycin and paclitaxel in anaplastic thyroid cancer cells. J Clin Endocrinol Metab. 2003;88:763–72. doi: 10.1210/jc.2002-020992. [DOI] [PubMed] [Google Scholar]