

Abstract
Group B Streptococcus (GBS) is a major cause of morbidity and mortality among neonates. Though there have been tremendous advances in prevention of invasive neonatal GBS disease through prophylactic antibiotic treatment of pregnant women, the incidence of neonatal disease has not changed significantly over the past several years. Vaccination of pregnant women is an important strategy that has the potential to improve further on existing protocols. In this review, we explore the history of the design of maternal GBS vaccines. We also discuss how recent applications of genomics and immunology to vaccine design promise to further enhance our ability to develop more effective vaccines against this important disease.
Keywords: group B Streptococcus, capsular polysaccharide, maternal, glycoconjugate, reverse vaccinology, mechanism
Diseases Caused by Group B Streptococcus
Group B Streptococcus (GBS), also known as Streptococcus agalactiae, is a Gram-positive coccus that can act as either a commensal organism or a pathogen in humans [1]. It primarily colonizes the gastrointestinal and genital tracts but can also be found in the oropharynx [2]. While as many as 20–40% of healthy women are asymptomatically colonized [2], GBS can cause serious disease in neonates, pregnant women, and nonpregnant immunocompromised adults [3] (Table 1). Invasive GBS disease occurs in >0.1 pregnant women per 1000 live-born infants [4]. In pregnant women, the disease typically presents as infection of the genital tract, placenta, or amniotic sac or as bacteremia with either a genitourinary or an unknown source [4]. While GBS infections virtually never cause maternal death, ~60% of them result in miscarriage or stillbirth [4]. In nonpregnant adults, the annual incidence of invasive GBS disease is ~30 per 100,000 and has gradually been increasing [4]. Patients with immunocompromising conditions such as diabetes, cancer, cirrhosis, HIV infection, and an age >65 years are especially at risk [3]. In patients >65 years of age, the mortality rate for invasive GBS disease is >10% [4]. In nonpregnant adults, GBS most often presents as bacteremia with an unknown source but can also present as skin or soft tissue infection, pneumonia, or joint or bone infection [3].
Table 1.
Incidence and mortality of invasive GBS infections, based on patient age, 1999–2005.
| Population | Incidence | Syndrome | Mortality (%) | |
|---|---|---|---|---|
| Type | % of total | |||
| Early-onset neonatal (<7 days) | 0.29–0.37/1000 live births | Bacteremia without focus | 83 | 4–9 |
| Pneumonia | 9 | |||
| Meningitis | 7 | |||
| Late-onset neonatal (7–90 days) | 0.34–0.52/1000 live births | Bacteremia without focus | 65 | 4.7 |
| Meningitis | 27 | |||
| Cellulitis | 3 | |||
| Pneumonia | 3 | |||
| Children (90 days– 14 years) | 0.37–0.73/100,000 population | Bacteremia without focus | 58 | 90 days–1 year: 3 1–14 years: 13 |
| Meningitis | 19 | |||
| Pneumonia | 7 | |||
| Arthritis | 5 | |||
| Peritonitis | 4 | |||
| Adults (≥15 years) | 15–64 years: 3.5–5.0/100,000 population ≥65 years: 21.5–26.0/100,000 population |
Bacteremia without focus | 48 | 15–64 years: 6.1 ≥65 years: 11.5 |
| Cellulitis | 22 | |||
| Pneumonia | 11 | |||
| Osteomyelitis | 9 | |||
| Arthritis | 9 | |||
| Peritonitis | 3 | |||
| Abscess | 3 | |||
| Pregnant women | 0.11–0.14/1000 live births | Endometritis or chorioamnionitis with fetal death | 50 | Mother: <0.1 Fetus: 61 |
| Bacteremia without focus | 31 | |||
| Endometritis without fetal death | 8 | |||
| Chorioamnionitis without fetal death | 4 | |||
| Pneumonia | 2 | |||
Data are adapted from Phares et al. Epidemiology of invasive group B Streptococcal disease in the United States, 1999–2005. J Am Med Assoc 2008;299(17):2056–65.
Despite the increasing importance of invasive GBS disease in adults, GBS is perhaps best known as a cause of neonatal disease, which is traditionally classified as early-onset or late-onset [1]. Early-onset disease develops before 1 week of age, is often associated with obstetric complications, is typically acquired from the maternal genital tract, and generally consists of bacteremia without a focal source; pneumonia and meningitis are much less common [5]. Late-onset disease occurs after 1 week of age and is more often characterized by meningitis [5]. Late-onset disease probably has a different pathogenesis from early-onset disease. It is hypothesized that late-onset disease may be acquired from nosocomial sources, from breast milk, or from care providers (e.g., family and childcare workers [6]. While the risk factors for early- and late-onset GBS disease are largely the same, prematurity is a greater risk factor for late- than for early-onset disease: for every week of prematurity, the risk ratio for developing late-onset GBS disease is 1.34. This greater risk may be caused by decreased transplacental transmission of protective maternal antibodies [7, 8], as will be discussed in detail later.
Before the introduction of protocols to identify and treat colonized mothers prior to delivery, GBS was responsible for substantial perinatal morbidity and mortality, with as many as 1–3 in 1000 neonates affected [9]. When GBS first emerged in the 1970s as a common cause of invasive neonatal disease, the mortality rate from severe GBS infection (primarily sepsis and meningitis) was >50% [10]. Since then, there have been substantial advances in the prevention and treatment of neonatal GBS disease. A seminal 1986 paper demonstrated that the selective administration of prophylactic ampicillin treatment of pregnant women colonized with GBS resulted in decreased rates of neonatal GBS disease [11]. The Group B Strep Association was founded in 1990 to educate the public about GBS infections and promote the prevention and treatment of GBS disease [12]. In 1992 and 1993, the American Academy of Pediatrics [13] and the American Congress of Obstetricians and Gynecologists [14] published guidelines on the prevention and treatment of neonatal GBS disease. In 1996, the Centers for Disease Control and Prevention published consensus guidelines for GBS prevention and treatment [15]; these guidelines have since undergone further iterations that now include universal screening of pregnant women for GBS colonization [16–18].
Pregnant women at 35–37 weeks of gestation now undergo routine screening (vaginal and rectal cultures) for GBS carriage, and women who are colonized with GBS receive prophylaxis with ampicillin or other antibiotics to decrease the risk of vertical transmission [17]. Antibiotics are also given perinatally to GBS-colonized women, to women who have previously given birth to neonates with severe GBS disease, and certain other high-risk patients [17]. The incidence of early-onset invasive neonatal GBS disease has steadily declined (Fig. 1), and, with improvements in early detection of invasive GBS disease and supportive care, mortality rates decreased from >50% in the 1970s to 15–25% in the 1980s and <10% by the 1990s [10]. However, even in the era of antibiotic prophylaxis and sophisticated neonatal care in the United States, there are ~0.3 cases of early-onset disease and another 0.3 cases of late-onset disease per 1000 neonates, and GBS represents a major cause of neonatal bacterial meningitis and sepsis [19]. The mortality rate from early- and late-onset disease is ~4% overall but is far higher among premature neonates than among full-term neonates (10–30% vs. 2–3%) [4, 16, 20]. Survivors of GBS meningitis have substantial long-term deficits: >20% have moderate to severe deficits (cerebral palsy, learning difficulties, deafness, or global developmental delay), >5% have a seizure disorder, and fewer than two-thirds are neurologically intact [20, 21]. The worldwide burden of GBS infection is generally difficult to assess by continent; however, in areas of sub-Saharan Africa, the incidence of early-onset disease is at least 1.97 cases per 1000 neonates, and that of all neonatal GBS disease is >3.0 cases per 1000 [22].
Figure 1.
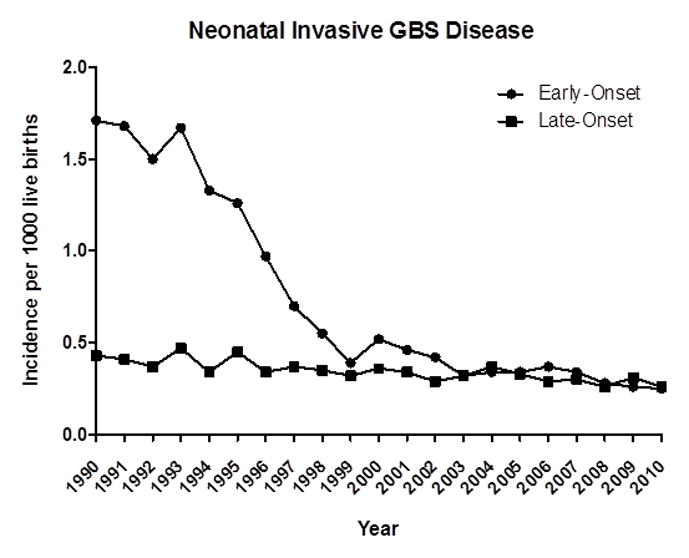
Incidence of early- and late-onset invasive neonatal GBS disease, 1990–2010. Adapted from (1) Verani et al. Prevention of perinatal group B Streptococcal disease. Revised guidelines from Centers for Disease Control and Prevention, MMWR Morb Mortal Wkly Rep 2010;59(RR10):1–31; (2) Centers for Disease Control and Prevention, 2010. Active bacterial core surveillance report, emerging infections program network, group B Streptococcus, 2009; and (3) Centers for Disease Control and Prevention. 2012. Active bacterial core surveillance report, emerging infections program network, group B Streptococcus, 2010.
GBS Virulence Factors
GBS makes use of a number of virulence factors, including pore-forming toxins that damage host cells; adhesion factors that increase binding to cells or extracellular matrix; evasion factors that decrease neutrophil recruitment and prevent complement binding; and factors that repel or otherwise induce resistance to antimicrobial peptides [23]. One of the most prominent and best-studied virulence factors is the capsular polysaccharide (CPS). CPSs play an important role in the pathogenesis of numerous disease-causing bacteria as well as in homeostasis with commensal bacteria [24–26]. GBS CPSs consist of various arrangements of monosaccharide building blocks, such as glucose, galactose, and/or N-acetylglucosamine, as well as a sialic acid residue on the branching terminus of each repeating unit [1] (Fig. 2). To date, nine distinct CPS structures (Ia, Ib, and II–VIII) have been identified [27], with a possible tenth polysaccharide, IX, reported as well [28]. In the 1970s, serotype III GBS (GBSIII) was the dominant cause of neonatal disease, but over time GBSIa, GBSII, and GBSV have increased in prominence as well [2, 4]. At the molecular level, CPS acts as a virulence factor by facilitating evasion of the host’s immune system. The sialic acid residues on the CPS accelerate dissociation of C3 convertase and thereby inhibit the complement cascade [29]. As a result, the CPS impairs opsonophagocytosis by host immune cells [29]. In addition, GBS sialic acid residues may act as ligands to sialic acid–recognizing immunoglobulin superfamily lectins and thus inhibit the phagocytosis of and oxidative burst from neutrophils and monocytes, enhancing bacterial survival [30].
Figure 2.
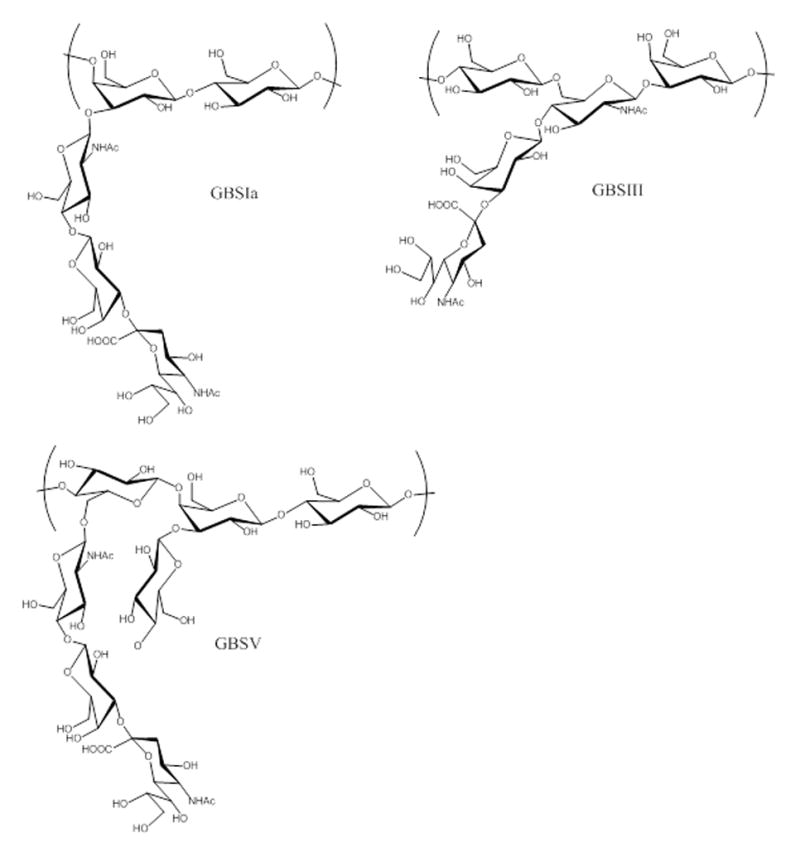
Repeating unit structures of selected CPSs.
GBSIa: (NeupNAcα2,3 - Galpβ1,4 - GlcpNAcβ1,3 -) - 4 - Galpβ1,4 - Glcpβ1 -.
GBSIII: - 4 - Glcpβ1,6 - (NeupNAcα2,3 - Galpβ1,4 -) - GlcpNAcβ1,3 - Galpβ1 -.
GBSV: (NeupNAcα2,3 - Galpβ1,4 - GlcpNAcβ1,6 -) - 4–Glcα1,4 - (Glcpβ1,3 -) Galpβ1,4 - Glcpβ1 -.
Why Develop a Vaccine against GBS?
Interest in the use of a CPS antigen for vaccination against GBS commenced when it was discovered that low levels of maternal antibodies to CPS antigens were correlated with neonatal susceptibility to serious GBS infection [31]. Baker and Kasper studied the ability of serum from pregnant women to bind to a type III CPS antigen. Among mothers of neonates without serious GBS disease, 76% had antibodies to type III CPS, as evidenced by a >40% antigen-binding capacity of maternal serum. In contrast, none of the mothers of neonates who developed serious GBS disease had antibodies to type III CPS. This observation was valid for both early-onset and late-onset GBS disease. Of note, this study also checked cord blood from some neonates for antibodies. All of the healthy neonates born to mothers with antibodies to type III CPS also had these antibodies. Among neonates who developed GBS disease, none had antibodies to type III CPS, nor did their mothers. This study demonstrated that maternal antibodies correlate with protection against serious GBS infection in neonates, possibly because of transplacental transmission of maternal immunoglobulin G (IgG). These findings also suggested that vaccination of pregnant women against CPS might be an effective strategy to protect neonates against GBS infection.
Since the advent of routine screening for maternal GBS colonization and antibiotic prophylaxis for GBS carriers, the incidence of early-onset neonatal GBS disease has declined dramatically—i.e., from 1.7 cases per 1000 neonates in 1990 to 0.34–0.37 case per 1000 in 2008 [17]. As discussed earlier, the development and implementation of guidelines (by organizations of pediatricians, obstetricians, microbiologists, and others) for universal screening for GBS and prophylactic treatment of pregnant mothers represent a tremendous advance in the prevention of neonatal GBS disease. However, these guidelines have not completely eliminated neonatal GBS disease. In fact, the incidence of early-onset GBS disease has plateaued for the past several years, and the incidence of late-onset disease has not decreased at all since the release of the Centers for Disease Control and Prevention guidelines [17].
Thus, there is still a role for vaccination in the prevention of GBS disease. The biologic or epidemiologic reasons for the plateau in levels of early-onset neonatal disease are unclear. Widespread vaccination may help decrease these levels. Since premature neonates have only 29–51% as much maternal antibody as full-term neonates [8], it may be critical for mothers of premature infants to have higher antibody levels that can compensate for prematurity and protect their infants from late-onset disease. Another issue is that, as a result of screening and treatment protocols, up to 30% of pregnant women ultimately receive antibiotics [16]. This practice imposes risks on the women and increases the possibility of selection for resistant organisms; for example, of clinical GBS isolates, 20% are resistant to clindamycin and 30–40% to erythromycin [32, 33]. Finally, rates of GBS infection in immunocompromised adults, including HIV-positive and older individuals, have been increasing [4], and vaccination may prevent morbidity and mortality in these high-risk populations.
This review recounts past attempts to develop vaccines that induce antibodies to CPS with the goal of preventing vertical transmission from mother to child. It also discusses emerging research that may influence the future of GBS vaccine design.
History of GBS Vaccine Development
The First Generation: Native Polysaccharide Vaccines
Initial attempts to develop a GBS vaccine focused on the use of purified native CPS as antigen [34]. These vaccines were found not to be toxic in a number of mammals [35–37]. More than 300 healthy adult volunteers were injected native type Ia, II, or III CPS in doses ranging from 10 to 150 μg [35–38]. Of these vaccine recipients, the vast majority had either mild local reactions, such as erythema or induration, or no reaction at all. The only possible systemic reaction reported involved a temperature elevation to 38°C, nausea, and myalgia with a total duration of <24 hours.
The native polysaccharide vaccines were immunogenic in humans. Recipients were stratified by previous exposure to the particular serotype of GBS in the vaccine, as evidenced by prevaccination antibody titers of >2–3 μg/mL [38]. Among previously exposed individuals, nearly all had antibody titer increases of ≥1 μg/mL. Among those who had not been previously exposed, responses were weaker and more variable. Type II CPS was the most immunogenic, with an 88% response rate, while type Ia CPS was the least, with 40% [38]. Moreover, these responses persisted for at least 2 years [38]. Notably, antibodies induced by vaccination had functional properties desirable for protection against perinatal GBS transmission: vaccination induced the production of IgG, which can cross the placenta; the antibodies had opsonic effects in vitro; and sera from vaccinated humans were protective in animal models of GBS infection [35–37].
While these early trials of native polysaccharide vaccines were promising, they notably revealed the reliance of protective effects on T cell–independent activation of antibody-producing B cells. As the first signal in B-cell activation, each B cell receptor on a B cell recognizes a component of a carbohydrate [39]. The second signal arises either from crosslinking of multiple B-cell receptors when they bind to a repeating unit of the carbohydrate or from binding of the antigen to another receptor—in certain cases, an innate receptor [39]. As a result, the B cells become activated and proliferate [39]. However, T cell–independent activation has its limitations. First, it does not induce B-cell memory. Indeed, when booster vaccine doses were administered, they did not result in an enhanced immune response relative to the initial vaccination, but rather brought antibody titers to a certain fixed level [39]. Second, T cells are not activated at all by these vaccines [24, 39]. Third, the response rate can be variable and sometimes rather low, especially in GBS-naïve individuals (as noted with the type Ia vaccine) [38]. Finally, the increases in titers of antibody to CPS were moderate (under 10-fold) compared with those elicited by later vaccine designs [34].
The Second Generation: Glycoconjugate Vaccines
Following the success of the Haemophilus influenzae type b CPS–protein conjugate vaccine [40], attention turned to glycoconjugate vaccines against GBS. These vaccines had the potential to overcome some of the difficulties encountered with polysaccharide-only vaccines described above. Covalent conjugation of a protein and a polysaccharide offers the potential for induction of B-cell memory against the polysaccharide [24]. In this process, B-cell receptors bind to the carbohydrate portion of the glycoconjugate vaccine; this binding acts as the first signal in B-cell activation [24, 39]. The B cells then endocytose the glycoconjugate, process both carbohydrate and protein in the endosomes, and present carbohydrate epitopes to T cells in a manner that requires the presence of peptide (to be described in greater detail in a later section) [24]. The T cell becomes activated and in turn activates the B cell to undergo class switching and proliferation [39]. Unlike T cell–independent B-cell activation, this process can induce both B- and T-cell memory and yield a more robust and highly functional IgG response through antibody class switching [24, 41].
CPS–protein coupling is typically achieved via reductive amination [42]. The first step in this technique is oxidation of sialic acid residues on the native CPS into aldehydes with sodium periodate; this oxidation retains antigenic epitopes on the CPS. The second step joins amine groups on protein to the aldehyde groups on the oxidized CPS, forming an imine. These imine groups are then reduced to secondary amines with sodium cyanoborohydride.
The first GBS glycoconjugate vaccine trial conducted in humans involved a GBSIII CPS–tetanus toxoid (III-TT) glycoconjugate [43]. This trial compared three doses of III-TT, type III CPS, and saline placebo. The vaccine was safe: no systemic responses were elicited, and only around one- quarter of recipients experienced localized redness and swelling (mostly <3 cm) lasting <72 hours. (One patient developed a temperature of 37.9°C that resolved in <24 hours; however, this reaction occurred in the setting of upper respiratory infection symptoms and was not thought to be related to vaccination.) Compared with type III CPS, the highest dose of III-TT produced higher levels of type III CPS–specific antibody measured 2 weeks after vaccination (4.89 μg/mL vs. 1.30 μg/mL). In addition, the proportion of recipients who responded to vaccination with III-TT was substantially greater than that responding to type III CPS; a fourfold increase in antibody titer was detected in 90% of those receiving III-TT but in only 50% of those receiving type III CPS. Together, these findings indicate that the response to glycoconjugate vaccination is both more robust and more reliable than the response to a carbohydrate-only vaccine.
Subsequent trials further expanded these results. While the original trial did not specifically investigate the effect in pregnant women and their babies, a later trial comparing III-TT with type III CPS in pregnant women showed that, after glycoconjugate vaccination, titers of protective IgG antibody to type III CPS were elevated in cord blood and persisted through at least 2 months of life; furthermore, levels of type III CPS–specific antibody in maternal serum correlated with those in cord serum [12]. Another important parameter considered in vaccine trials is whether vaccination decreases vaginal and rectal GBS carriage; this issue is significant because, if carriage is decreased, not only does the vaccine passively immunize the neonate, but the risk posed by direct inoculation of the neonate during delivery is also decreased. A trial addressing this issue found that women immunized with III-TT indeed had decreased levels of vaginal and rectal colonization with GBS [44].
Initially, type III CPS caused the majority of severe neonatal GBS infections. Over time, however, other serotypes (e.g., Ia and V) became increasingly prominent in human disease [4]. Thus it became increasingly important to develop vaccines against other serotypes of GBS CPS. Accordingly, a bivalent vaccine consisting of II-TT and III-TT glycoconjugates was tested in humans [12]. As with the III-TT monovalent vaccine, the response to vaccination was consistent: 80% and 90% of vaccine recipients had a fourfold or greater increase in type II CPS–specific antibodies and type III CPS–specific antibodies, respectively. These responses did not differ statistically from the antibody responses to monovalent vaccine; thus there was no significant interference between the two vaccines. The bivalent vaccine was associated with an incidence of adverse effects similar to that associated with each monovalent vaccine, even though the bivalent product contained more tetanus toxoid than either of the monovalent preparations. The opsonic abilities of the antibodies generated by the bivalent and monovalent vaccines were similar.
Although no trials have compared trivalent or higher-valence vaccines in humans, a tetravalent vaccine has been tested in mice [45]. This vaccine, which consists of Ia-TT, Ib-TT, II-TT, and III-TT, was compared with either tetanus toxoid alone or unconjugated CPS in a model in which pregnant mice were vaccinated and their newborn pups were challenged with one specific serotype of GBS. In this model, survival of pups was substantially increased after infection by all tested serotypes. This study demonstrates the potential value of human trials involving higher-valence GBS vaccines and suggests the feasibility of adding new GBS serotypes to a vaccine should they emerge as threats to human health.
The Third Generation: Application of Scientific Advances to GBS Vaccine Design
Reverse Vaccinology
Reverse vaccinology is a term coined by Rappuoli and describes a method for vaccine design that uses the information obtainable from whole-genome analysis [46]. In the conventional design of subunit vaccines, specific microbial antigens are identified and isolated by biochemical and other techniques. These antigens are then produced in quantities sufficient for use in in vivo vaccination testing; usually, production of such large quantities requires that the pathogen in question be grown in culture. The resulting products are tested for immunogenicity and protection against disease in animal experiments and human trials. This method has several shortcomings. First, identifying and isolating antigens are time-consuming tasks. Second, it is not always clear which antigens should be targeted, as all antigens are not equally immunogenic and antibodies to microbial antigens are not always protective. Finally, it is generally impossible to design vaccines against pathogens that cannot be grown in culture, including many bacteria and most viruses.
In reverse vaccinology, the starting point is the genome rather than the organism [46]. Using computer analysis, one identifies protein antigens that may be immunogenic and that therefore are potential vaccine targets. The antigens can be identified by sequencing the genome and pinpointing the open reading frames (i.e., potential DNA coding regions) that encode for surface-exposed proteins [47]. These surface-exposed proteins typically have certain features such as homology to known membrane proteins or transmembrane domains [47]. Next, one runs a high-throughput screen to determine whether immune responses to these antigens are protective. Such screens generally involve overexpression and purification of antigens from bacteria such as Escherichia coli, vaccination of mice or other animals with these antigens, and assessment of whether sera from the vaccinated animals kill bacteria in vitro [48]. Once these protective antigens, which generally make up only a small proportion of all surface antigens, are identified, they can be further investigated in animal protection models and then in human trials. In theory, this approach bypasses some of the problems of conventional vaccinology. For example, conventional vaccinology requires that antigens be plentiful and isolatable from the bacteria targeted by a vaccine; in contrast, reverse vaccinology simply depends on the overexpression of these antigens in other organisms, thus allowing the development of vaccines against pathogens that cannot be grown in culture [48]. The main limiting factor of reverse vaccinology is the high-throughput screen, which requires animal studies and is often the rate-limiting step of vaccine design [46]. In addition, reverse vaccinology cannot identify carbohydrates and other cellular structures that are not directly encoded by the genome.
Reverse vaccinology was first used against Neisseria meningitidis (meningococcus) [49], a Gram-negative coccus that is the second most common cause of bacterial meningitis in adults and the most common cause in 11- to 17-year-old children [50]. Like GBS, N. meningitidis is classified on the basis of its CPS structures; five serotypes—A, B, C, Y, and W135—cause virtually all meningococcal disease in humans [51]. Although serotype B accounts for >30% of cases of meningococcal disease in the United States [52], it was previously impossible to design a conventional vaccine against serotype B CPS, which is decorated with repeating units of α(2–8)N-acetylneuraminic acid residues that are identical to those found on many human polysaccharides [49]. Searching for novel antigens that might circumvent this problem, Rappuoli and colleagues identified five antigens that induced antibodies with bactericidal activity and could be produced in large quantities, then combined these into a single vaccine [47, 49]. When given with CpG oligonucleotides or an MF59 adjuvant, this combination vaccine induced protective antibodies to >90% of representative clinical isolates of serotype B meningococci [49].
Similar methods have been applied to vaccine design for GBS. Grandi and colleagues used the genome sequences from eight clinical GBS strains to select 589 surface proteins, 312 of which could be expressed in and purified from E. coli [53]. These purified proteins were used to immunize female mice, which were then bred and gave birth to pups that were challenged with a dose of one strain of GBS sufficient to cause a death rate of 80–90%. Of the 312 vaccine candidates, four increased the survival rate of infant mice. These candidates were further analyzed: immunizations were repeated, and pups were challenged with six GBS strains rather than just one. None of the four candidates individually conferred protection against all six strains. However, the four antigens were combined into a single vaccine and tested against 12 clinical GBS strains, and this combination provided 87% protection overall against GBS challenge. Further work examined the expression of three of these antigens, which were found to be pilus components [54], in a total of 289 GBS isolates. The results showed not only that all tested GBS strains express pili, but also that the structures of the pilus subunits are highly conserved [55]—a property that is promising in terms of the development of a universal GBS vaccine.
This work with GBS vaccination provides several interesting lessons about reverse vaccinology. Of the four antigens developed, none of them previously had a known function. Indeed, during the course of the vaccine design research, three of these four were discovered to be components of pili, which GBS had not previously been known to express [54]. These points highlight a major distinction between reverse and conventional vaccinology: in conventional vaccinology, targets are typically well-characterized antigens with a known function in pathogenesis, while in reverse vaccinology, it is possible to identify previously unknown antigens. Second, none of the four antigens was universally expressed in GBS, and only a combination of multiple antigens yielded a reasonably universal vaccine; this point underscores the importance of screening multiple strains for potential antigens. Finally, as expected, this research did not identify CPS (which had previously been the main target of vaccine design attempts) as a potential vaccine candidate. This point illustrates the inability of reverse vaccinology to discover antigens that are not directly encoded by the genome.
Mechanistic Insights
Although a number of vaccines have been developed against bacterial CPSs [40, 43], surprisingly little research has been conducted into the mechanisms that underlie protective immunity to a CPS [24]. In the traditional model for glycoconjugate vaccines, B-cell receptors specific to a vaccine’s carbohydrate components recognize and phagocytose the glycoconjugate [24]. The glycoconjugate is then degraded in B-cell endosomes, and the peptide (but not the carbohydrate) components are presented on major histocompatibility complex (MHC) II molecules. The MHC II–peptide complexes signal through T-cell receptors and thus induce T-cell maturation through interleukin 2 (IL-2) and IL-2 receptor production; this event, in turn, causes T cells to produce IL-4, which induces B-cell maturation, class switching, and memory. Thus the B cell can continue to produce antibodies specific to the carbohydrate components of these glycoconjugates, while the T cells recognize the peptide components.
Recent work from our laboratory has challenged this traditional model [41]. In a new model, endosomal degradation of glycoconjugates yields processed glycans conjugated to peptides (glycanp-peptides). It is glycanp-peptides rather than peptide-only components that are presented on MHC II molecules; the peptide component binds to the MHC II molecule, while the covalently attached glycanp component is recognized by the T-cell receptor. Our demonstration that memory T cells exposed to the III-TT glycoconjugate can be activated when exposed to III-ovalbumin (OVA) (a glycoconjugate with a different protein carrier) shows that T cells recognize the carbohydrate epitopes. These carbohydrate components are processed in the endosome and can be isolated from the cell surface on MHC II molecules, but only when they are covalently linked to peptides. In addition, T cells can distinguish III-OVA from OVA alone, which would not be the case if only peptides were recognized as the traditional model posits. Two T-cell clones that recognize MHC II–presented carbohydrate epitopes have been isolated and identified [41, 42].
These findings have significant implications for vaccine design. In particular, if the most immunogenic MHC II/T-cell receptor ligands are glycanp-peptides, then glycoconjugate vaccines should be designed to maximize the formation of glycanp-peptides upon endosomal processing—and thus to maximize the number of carbohydrate epitopes that can be presented by MHC II molecules. Conventional vaccine designs include polysaccharide chains with very few covalent bonds to proteins per polysaccharide [41]; the result is a relatively small number of glycanp-peptides. On the basis of our model, we designed a prototype new-generation glycoconjugate vaccine by conjugating polysaccharide with peptides (as opposed to proteins) to achieve an optimal number of glycan–MHCII-binding peptide bonds per glycoconjugate. Compared with a conventional glycoconjugate vaccine, this new-generation vaccine was 50–100 times more immunogenic and greatly enhanced the survival of challenged mouse pups in a maternal immunization model [41].
This novel vaccine design may have particularly important implications for late-onset neonatal GBS disease, which typically occurs in premature infants who have decreased levels of transplacentally transferred antibody [8]. However, even in premature infants, antibody levels appear to be proportional to maternal antibody levels; i.e., maternal–fetal antibody transfer is proportional to the maternal antibody concentration [7]. Thus, increasing maternal antibody levels with an improved vaccine will likely result in higher neonatal antibody levels. Admittedly, while the maternal antibody levels (and, by extrapolation, the fetal antibody levels) necessary to protect against early-onset GBS disease have been estimated [9], it remains to be determined what, if any, maternal/fetal antibody level is necessary for prevention of late-onset GBS disease. Certainly, however, maternal vaccination remains the most likely solution to the problem of late-onset GBS disease. With maternal–fetal transfer compromised as it is in premature infants, it will be critical to design as immunogenic a vaccine as possible in order to maximize the chances of successful disease prevention.
Summary.
The past 20 years have seen tremendous advances in the prevention and treatment of neonatal GBS disease. Despite these interventions, however, the incidence of such disease has plateaued, and invasive GBS disease still represents a major public health problem. Designing new-generation glycoconjugate vaccines against GBS has the potential to significantly improve on the prevention protocols already in practice. The efficacy of glycoconjugate vaccines is well established. The recent development of reverse vaccinology and elucidation of the mechanism of glycoconjugate vaccine–induced immunity offer promise in creating a new, more immunogenic generation of vaccines.
Acknowledgments
We thank Julie B. McCoy for her excellent editorial work.
Abbreviations
- CPS
capsular polysaccharide
- GBS
group B Streptococcus
- Ig
immunoglobulin
- MHC
major histocompatibility complex
- OVA
ovalbumin
- TT
tetanus toxoid
Footnotes
The authors report no conflicts of interest pertaining to the work described in this review.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Vincent L. Chen, Email: vincent_chen2@hms.harvard.edu.
Fikri Y. Avci, Email: fikri_avci@hms.harvard.edu.
Dennis L. Kasper, Email: dennis_kasper@hms.harvard.edu.
References
- 1.Paoletti LC, Kasper DL. Glycoconjugate vaccines to prevent group B streptococcal infections. Expert Opin Biol Ther. 2003;3(6):975–84. doi: 10.1517/14712598.3.6.975. [DOI] [PubMed] [Google Scholar]
- 2.Hickman ME, Rench MA, Ferrieri P, Baker CJ. Changing epidemiology of group B streptococcal colonization. Pediatrics. 1999;104(2):203–9. doi: 10.1542/peds.104.2.203. [DOI] [PubMed] [Google Scholar]
- 3.Sendi P, Johansson L, Norrby-Teglund A. Invasive group B streptococcal disease in non-pregnant adults - A review with emphasis on skin and soft-tissue infections. Infection. 2008;36(2):100–11. doi: 10.1007/s15010-007-7251-0. [DOI] [PubMed] [Google Scholar]
- 4.Phares CR, Lynfield R, Farley MM, Mohle-Boetani J, Harrison LH, Petit S, et al. Epidemiology of invasive group B streptococcal disease in the United States, 1999–2005. J Am Med Assoc. 2008;7;299(17):2056–65. doi: 10.1001/jama.299.17.2056. [DOI] [PubMed] [Google Scholar]
- 5.Stevens DL, Kaplan EL. Streptococcal infections: clinical aspects, microbiology, and molecular pathogenesis. United States: Oxford University Press; 2000. [Google Scholar]
- 6.Lin FYC, Weisman LE, Troendle J, Adams K. Prematurity is the major risk factor for late-onset group B streptococcus disease. J Infect Dis. 2003;15;188(2):267–71. doi: 10.1086/376457. [DOI] [PubMed] [Google Scholar]
- 7.Palmeira P, Quinello C, Silveira-Lessa AL, Zago CA, Carneiro-Sampaio M. IgG placental transfer in healthy and pathological pregnancies. Clin Devel Immunol. 2012 doi: 10.1155/2012/985646. (Epub 2011 Oct;2012:985646) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leineweber B, Grote V, Schaad UB, Heininger U. Transplacentally acquired immunoglobulin G antibodies against measles, mumps, rubella and varicella-zoster virus in preterm and full term newborns. Pediatr Infect Dis J. 2004;23(4):361–3. doi: 10.1097/00006454-200404000-00019. [DOI] [PubMed] [Google Scholar]
- 9.Lin FYC, Brenner RA, Johnson YR, Azimi PH, Philips JB, Regan JA, et al. The effectiveness of risk-based intrapartum chemoprophylaxis for the prevention of early-onset neonatal group B streptococcal disease. Am J Obstet Gynecol. 2001;184(6):1204–10. doi: 10.1067/mob.2001.113875. [DOI] [PubMed] [Google Scholar]
- 10.Schuchat A. Group B streptococcus. Lancet. 1999;353(9146):51–6. doi: 10.1016/S0140-6736(98)07128-1. [DOI] [PubMed] [Google Scholar]
- 11.Boyer KM, Gotoff SP. Prevention of early-onset neonatal group B streptococcal disease with selective intrapartum chemoprophylaxis. N Engl J Med. 1986;314(26):1665–9. doi: 10.1056/NEJM198606263142603. [DOI] [PubMed] [Google Scholar]
- 12.Baker CJ, Edwards MS. Group B streptococcal conjugate vaccines. Arch Dis Child 2003. 2003;88(5):375–8. doi: 10.1136/adc.88.5.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Committee on Infectious Diseases and Committee on Fetus and Newborn. Guidelines for prevention of group B streptococcal (GBS) in infection by chemoprophylaxis. Pediatrics. 1992;90(5):775–8. [PubMed] [Google Scholar]
- 14.ACOG. Group B streptococcal infections in pregnancy. ACOG technical bulletin number 170—July 1992. Int J Gynaecol Obstet. 1993;42(1):55–9. [PubMed] [Google Scholar]
- 15.Prevention of perinatal group B streptococcal disease: a public health perspective. Centers for Disease Control and Prevention. MMWR Recomm Rep. 1996;45(RR-7):1–24. [PubMed] [Google Scholar]
- 16.Schrag SJ, Zell ER, Lynfield R, Roome A, Arnold KE, Craig AS, et al. A population-based comparison of strategies to prevent early-onset group B streptococcal disease in neonates. New England Journal of Medicine. 2002 Jul 25;347(4):233–9. doi: 10.1056/NEJMoa020205. [DOI] [PubMed] [Google Scholar]
- 17.Verani JR, McGee L, Schrag SJ. Prevention of perinatal group B streptococcal disease. Revised guidelines from CDC. 2010 MMWR Morb Mortal Wkly Rep. 2010;59(RR10):1–31. [PubMed] [Google Scholar]
- 18.Schrag S, Gorwitz R, Fultz-Butts K, Schuchat A. Prevention of perinatal group B streptococcal disease. Revised guidelines from CDC. MMWR Recomm Rep. 2002 Aug 16;51(RR-11):1–22. [PubMed] [Google Scholar]
- 19.Gaschignard J, Levy C, Romain O, Cohen R, Bingen E, Aujard Y, et al. Neonatal bacterial meningitis 444 cases in 7 years. Pediatr Infect Dis J. 2011;30(3):212–7. doi: 10.1097/inf.0b013e3181fab1e7. [DOI] [PubMed] [Google Scholar]
- 20.Fluegge K, Siedler A, Heinrich B, Schulte-Moenting J, Moennig MJ, Bartels DB, et al. Incidence and clinical presentation of invasive neonatal group B streptococcal infections in Germany. Pediatrics. 2006 Jun;117(6):E1139–E45. doi: 10.1542/peds.2005-2481. [DOI] [PubMed] [Google Scholar]
- 21.Stevens JP, Eames M, Kent A, Halket S, Holt D, Harvey D. Long term outcome of neonatal meningitis. Arch Dis Child. 2003;88(3):179–84. doi: 10.1136/fn.88.3.F179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dagnew AF, Cunnington MC, Dube Q, Edwards MS, French N, Heyderman RS, et al. Variation in reported neonatal group B Streptococcal disease incidence in developing countries. Clin Infect Dis. 2012;55(1):91–102. doi: 10.1093/cid/cis395. [DOI] [PubMed] [Google Scholar]
- 23.Rajagopal L. Understanding the regulation of group B Streptococcal virulence factors. Future Microbiol. 2009;4(2):201–21. doi: 10.2217/17460913.4.2.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avci FY, Kasper DL. How bacterial carbohydrates influence the adaptive immune system. Ann Rev Immunol. 2010;28:107–30. doi: 10.1146/annurev-immunol-030409-101159. [DOI] [PubMed] [Google Scholar]
- 25.Duan J, Avci FY, Kasper DL. Microbial carbohydrate depolymerization by antigen-presenting cells: deamination prior to presentation by the MHCII pathway. Proc Nat Acad Sci USA. 2008;105(13):5183–8. doi: 10.1073/pnas.0800974105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008;6(4):288–301. doi: 10.1038/nrmicro1871. [DOI] [PubMed] [Google Scholar]
- 27.Cieslewicz MJ, Chaffin D, Glusman G, Kasper D, Madan A, Rodrigues S, et al. Structural and genetic diversity of group B Streptococcus capsular polysaccharides. Infect Immun. 2005;73(5):3096–103. doi: 10.1128/IAI.73.5.3096-3103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slotved H-C, Kong F, Lambertsen L, Sauer S, Gilbert GL. Serotype IX, a proposed new Streptococcus agalactiae serotype. J Clin Microbiol. 2007;45(9):2929–36. doi: 10.1128/JCM.00117-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wessels MR, Rubens CE, Benedi VJ, Kasper DL. Definition of a bacterial virulence factor—sialyation of the group B Streptococcal capsule. Proc Natl Acad USA. 1989;86(22):8983–7. doi: 10.1073/pnas.86.22.8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carlin AF, Chang Y-C, Areschoug T, Lindahl G, Hurtado-Ziola N, King CC, et al. Group B Streptococcus suppression of phagocyte functions by protein-mediated engagement of human Siglec-5. J Exp Med. 2009;206(8):1691–9. doi: 10.1084/jem.20090691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker CJ, Kasper DL. Correlation of maternal antibody deficiency with susceptibility to neonatal group-B Streptococcal infection. N Engl J Med. 1976;294(14):753–6. doi: 10.1056/NEJM197604012941404. [DOI] [PubMed] [Google Scholar]
- 32.Castor ML, Whitney CG, Como-Sabetti K, Facklam RR, Ferrieri P, Bartkus JM, et al. Antibiotic resistance patterns in invasive group B streptococcal isolates. Infect Dis Obstet Gynecol. 2008;2008:727505. doi: 10.1155/2008/727505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borchardt SM, DeBusscher JH, Tallman PA, Manning SD, Marrs CF, Kurzynski TA, et al. Frequency of antimicrobial resistance among invasive and colonizing Group B Streptococcal isolates. BMC Infect Dis. 2006;6:57. doi: 10.1186/1471-2334-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker CJ, Kasper DL. Group-B Streptococcal vaccines. Rev Infect Dis. 1985;7(4):458–67. doi: 10.1093/clinids/7.4.458. [DOI] [PubMed] [Google Scholar]
- 35.Baker CJ, Edwards MS, Kasper DL. Immunogenicity from polysaccharides from type-III, group-B Streptococcus. J Clin Invest. 1978;61(4):1107–10. doi: 10.1172/JCI109011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eisenstein TK, Decueninck BJ, Resavy D, Shockman GD, Carey RB, Swenson RM. Quantitative detemrination in human sera of vaccine-induced antibody to type-specific polysaccharides of group-B Streptococci using a enzyme-linked immunosorbent assay. J Infect Dis. 1983;147(5):847–56. doi: 10.1093/infdis/147.5.847. [DOI] [PubMed] [Google Scholar]
- 37.Kasper DL, Baker CJ, Galdes B, Katzenellenbogen E, Jennings HJ. Immunochemical analysis and immunogenicity of the type-II group-B Streptococcal capsular polysaccharide. J Clin Invest. 1983;72(1):260–9. doi: 10.1172/JCI110965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fischer G, Horton RE, Edelman R. Summary of the National Institutes of Health workshop on group-B Streptococcal infection. J Infect Dis. 1983;148(1):163–6. doi: 10.1093/infdis/148.1.163. [DOI] [PubMed] [Google Scholar]
- 39.Murphy K, Travers P, Walport M. Janeway’s immunobiology. New York: Garland Science; 2011. [Google Scholar]
- 40.Eskola J, Käyhty H, Takala AK, Peltola H, Rönnberg PR, Kela E, et al. A randomized, prospective field trial of a conjugate vaccine in the protection of infants and young children against invasive Haemophilus influenzae type b disease. N Engl J Med. 1990;323(20):1381–7. doi: 10.1056/NEJM199011153232004. [DOI] [PubMed] [Google Scholar]
- 41.Avci FY, Li X, Tsuji M, Kasper DL. A mechanism for glycoconjugate vaccine activation of the adaptive immune system and its implications for vaccine design. Nat Med. 2011;17(12):1602–U115. doi: 10.1038/nm.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avci FY, Li X, Tsuji M, Kasper DL. Isolation of carbohydrate-specific CD4+ T cell clones from mice after stimulation by two model glycoconjugate vaccines. Nat Protoc. 2012;7(12):2180–92. doi: 10.1038/nprot.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kasper DL, Paoletti LC, Wessels MR, Guttormsen HK, Carey VJ, Jennings HJ, et al. Immune response to type III group B streptococcal polysaccharide-tetanus toxoid conjugate vaccine. J Clin Invest. 1996;98(10):2308. doi: 10.1172/JCI119042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hillier S, Ferris D, Fine D, Ferrieri P, Edwards M, Carey V. Women receiving group B Streptococcus serotype III tetanus toxoid (GBS III-TT) vaccine have reduced vaginal and rectal acquisition of GBS type III [Presentation]. Annual meeting of the Infectious Diseases Society of America; Philadelphia, Pennsylvania. 2009. [Google Scholar]
- 45.Paoletti LC, Wessels MR, Rodewald AK, Shroff AA, Jennings HJ, Kasper DL. Neonatal mouse protection against infection with multiple group-B Streptococcal (GBS) serotypes by maternal immunization with a tetravalent GBS polysaccharide-tetanus toxoid conjugate vaccine. Infect Immun. 1994;62(8):3236–43. doi: 10.1128/iai.62.8.3236-3243.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rappuoli R. Reverse vaccinology. Curr Opin Microbiol. 2000;3(5):445–50. doi: 10.1016/s1369-5274(00)00119-3. [DOI] [PubMed] [Google Scholar]
- 47.Pizza M, Scarlato V, Masignani V, Giuliani MM, Arico B, Comanducci M, et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science. 2000;287(5459):1816–20. doi: 10.1126/science.287.5459.1816. [DOI] [PubMed] [Google Scholar]
- 48.Sette A, Rappuoli R. Reverse vaccinology: developing vaccines in the era of genomics. Immunity. 2010;33(4):530–41. doi: 10.1016/j.immuni.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Giuliani MM, Adu-Bobie J, Comanducci M, Arico B, Savino S, Santini L, et al. A universal vaccine for serogroup B meningococcus. Proc Natl Acad Sci USA. 2006;103(29):10834–9. doi: 10.1073/pnas.0603940103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thigpen MC, Whitney CG, Messonnier NE, Zell ER, Lynfield R, Hadler JL, et al. Bacterial meningitis in the United States, 1998–2007. N Engl J Med. 2011;364(21):2016–25. doi: 10.1056/NEJMoa1005384. [DOI] [PubMed] [Google Scholar]
- 51.Harrison LH, Trotter CL, Ramsay ME. Global epidemiology of meningococcal disease. Vaccine. 2009;27:B51–B63. doi: 10.1016/j.vaccine.2009.04.063. [DOI] [PubMed] [Google Scholar]
- 52.Rosenstein NE, Perkins BA, Stephens DS, Lefkowitz L, Cartter ML, Danila R, et al. The changing epidemiology of meningococcal disease in the United States 1992–1996. Journal of Infectious Diseases. 1999 Dec;180(6):1894–901. doi: 10.1086/315158. [DOI] [PubMed] [Google Scholar]
- 53.Maione D, Margarit I, Rinaudo CD, Masignani V, Mora M, Scarselli M, et al. Identification of a universal group B Streptococcus vaccine by multiple genome screen. Science. 2005;309(5731):148–50. doi: 10.1126/science.1109869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lauer P, Rinaudo CD, Soriani M, Margarit I, Maione D, Rosini R, et al. Genome analysis reveals pili in group B Streptococcus. Science. 2005;309(5731):105. doi: 10.1126/science.1111563. [DOI] [PubMed] [Google Scholar]
- 55.Margarit I, Rinaudo CD, Galeotti CL, Maione D, Ghezzo C, Buttazzoni E, et al. Preventing bacterial infections with pilus-based vaccines: the group B Streptococcus paradigm. J Infect Dis. 2009;199(1):108–15. doi: 10.1086/595564. [DOI] [PubMed] [Google Scholar]