

Abstract
Primary plasma cell leukemia (pPCL) is a rare and aggressive plasma cell proliferative disorder with a very poor prognosis and with distinct biologic, clinical, and laboratory features. Compared with multiple myeloma, pPCL presents more often with extramedullary involvement, anemia, thrombocytopenia, hypercalcemia, elevated serum β2-microglobulin and lactate dehydrogenase levels, as well as impaired renal function. Many of the genetic aberrations observed in newly diagnosed pPCL are typically found in advanced multiple myeloma. These cytogenetic abnormalities and mutations lead to increased proliferation, enhanced inhibition of apoptosis, escape from immune surveillance, and independence from the BM microenvironment, with changes in expression of adhesion molecules or chemokine receptors. The outcome of pPCL has improved with the introduction of autologous stem cell transplantation and combination approaches with novel agents, including bortezomib and immunomodulatory drugs, such as lenalidomide. In this review, we provide an overview of currently available therapeutic options with recommendations of how these treatment modalities can best be used to improve outcome for plasma cell leukemia patients.
Introduction
Primary plasma cell leukemia (pPCL) is the most aggressive form of the plasma cell dyscrasias. It is defined by the presence of > 2 × 109/L peripheral blood plasma cells or plasmacytosis accounting for > 20% of the differential white cell count, and does not arise from preexisting multiple myeloma (MM).1,2 Secondary PCL (sPCL), however, is a leukemic transformation of end-stage MM. The first case of pPCL was described by Gluzinski and Reichenstein in 1906 (Figure 1).3 In contrast to the clonal plasma cells in PCL, reactive plasmacytosis associated with bacterial or viral infections, autoimmune disorders, and serum sickness is polyclonal in nature. pPCL is rare, with only 1%-4% of MM patients presenting as pPCL.4–10 In addition, < 1% of patients presenting with extreme leucocytosis (> 50 × 109/L) are diagnosed with PCL.11 Compared with classic MM, pPCL has both a different biologic background as well as distinct clinical and laboratory features. The prognosis of pPCL is very poor, with a median overall survival (OS) of only 7 months with standard chemotherapy, and therefore requires innovative treatment approaches incorporating various modalities to improve outcome.
Figure 1.
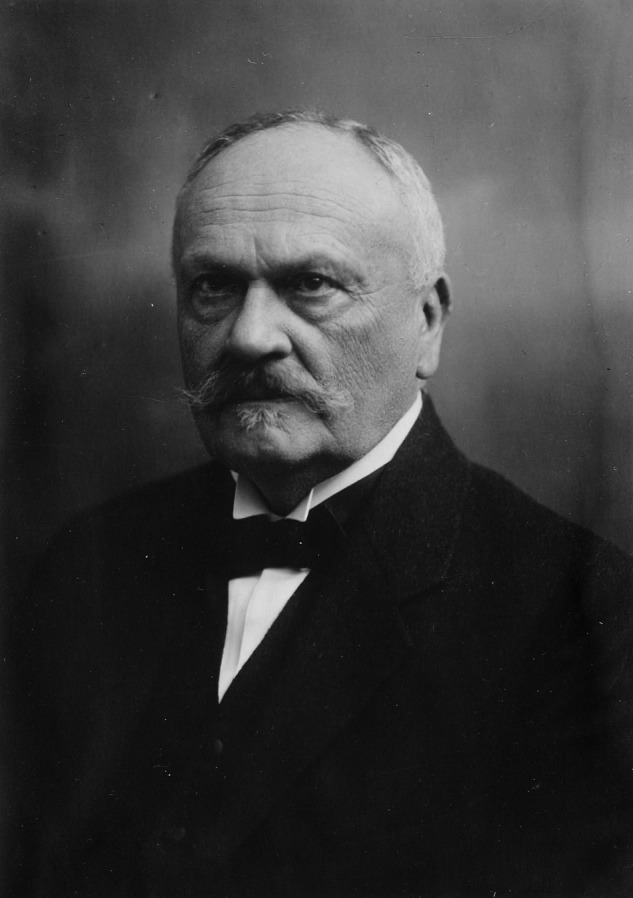
Wladyslaw Antoni Gluzinski around 1930. In 1906, Professor A. Gluzinski and Dr M. Reichenstein described the first case of pPCL in the “Wiener Klinische Wochenschrift.”3,7 At that time, the authors worked in the University Hospital of Lemberg (Lvov), which was then part of the Austrian Empire, but is now one of the main cities in western Ukraine. Professor Gluzinski was born in Wloclawek (Poland) in 1856 and died in Warsaw in 1935.100 The authors describe a 47-year-old train ticket inspector who presented with bone pain, a palpable mass over one of his ribs, rib fractures, anemia, and splenomegaly. Urine contained large amounts of a proteinaceous substance. In the blood smear, the authors observed erythroblasts and immature plasma cells, sometimes with 2 or 3 nuclei. The absolute number of circulating tumor cells increased significantly during further follow-up (one week before patient died, leucocytes were estimated at 39.4 × 109/L, containing 91% plasma cells). The diagnosis of MM and “leucaemia lymphatica plasmocellularis” was made. Treatment consisted of the organoarsenic compounds Atoxyl and Neoarsicodile. This was followed by a brief period with reduced levels of pain and reduction of the size of the rib tumor, enabling him to resume working. However, the pain and rib tumors recurred and the patient died 6 months after his first presentation of pneumonia. Autopsy confirmed the premortem diagnosis. Interestingly, much later, several groups reported activity from arsenic in MM and related disorders.
Case presentation
A 51-year-old woman presented to our hospital with easy bruising for 1 week, combined with heavy menses, weight loss, and increasing fatigue over 2 months. Physical examination was noteworthy for pallor and splenomegaly. Laboratory evaluation showed a white blood cell count of 56 × 109/L, hemoglobin of 8.8 g/dL, platelet count of 30 × 109/L, lactate dehydrogenase (LDH) of 832 U/L (reference range < 250 U/L), a creatinine of 1.21 mg/dL, and serum calcium of 11.5 mg/dL. In the peripheral blood smear, the white blood cells had a very atypical appearance and immunophenotypic analysis demonstrated the presence of circulating clonal plasma cells (CD38+, CD138+, CD56−, kappa+). Additional investigation revealed the presence of an IgA-κ M-protein of 1.1 g/dL, albumin of 2.9 g/dL, and β2-microglobulin of 3.1 mg/L. The BM biopsy demonstrated diffuse infiltration with almost 100% atypical plasma cells. Cytogenetic analysis was remarkable for the presence of multiple deletions and t(14;20). Several small osteolytic bone lesions were noted on skeletal survey, and overall her findings were considered consistent with a diagnosis of pPCL.
Treatment with bortezomib combined with doxorubicin and dexamethasone (PAD) was initiated. Supportive therapy consisted of hydration, allopurinol, pamidronate, antimicrobial prophylaxis, and hormonal therapy to prevent menorraghia. After cycle 1, circulating plasma cells were no longer detectable; and after 3 cycles of PAD, she proceeded to protocol-directed autologous stem cell transplantation followed by nonmyeloablative, fully matched unrelated donor, allogeneic stem cell transplantation; she achieved complete remission (CR). Her post-transplantation course was uncomplicated. Unfortunately, 12 months later, she developed oculomotor nerve palsy, caused by leptomeningeal myelomatosis. Staging also showed extensive BM involvement. Intrathecal chemotherapy was administered combined with systemic therapy (lenalidomide/dexamethasone and donor lymphocyte infusions), and she obtained a second CR. Ultimately, 24 months after diagnosis the patient died of systemic and neurologic disease progression despite salvage therapy with a 4-drug regimen (bortezomib, dexamethasone, lenalidomide, and cyclophosphamide).
This case illustrates the aggressive clinical course of pPCL. In this review, we discuss the pathogenesis and presentation of pPCL and provide recommendations for how to treat patients with this high-risk variant of MM. We also draw comparisons between pPCL and sPCL to illustrate key distinctions between these 2 entities.
Biology of PCL
During initial stages of MM, tumor cells are mainly localized in BM where they depend on the BM microenvironment for growth, survival, and protection against drug-induced apoptosis. In PCL, tumor cells accumulate in the BM but also have an increased capacity to recirculate in blood, with subsequent egression, and formation of extramedullary disease. The dissemination of tumor cells out of the BM is not only related to changes in expression of adhesion molecules and chemokine receptors but also to the presence of several molecular aberrations, which contribute to BM microenvironment-independent tumor growth, inhibition of apoptosis, and escape from immune surveillance. In pPCL, these genetic abnormalities are already present at diagnosis, whereas during the process of progression of monoclonal gammopathy of undetermined significance (MGUS) to MM and finally to sPCL, there is a gradual accumulation of genetic events, resulting in the acquisition of a more aggressive phenotype of the clonal plasma cells (Figure 2). Interestingly, gene expression profiling identifies pPCL as a distinct molecular entity among myeloma samples.10
Figure 2.

Model for the mechanisms that contribute to the development of primary and secondary plasma cell leukemia. Primary immunoglobulin heavy chain (IgH) translocations with 5 recurrent chromosomal partners (4p16, 6p21, 11q13, 16q23, and 20q11) and hyperdiploidy (typically with trisomies of chromosomes 3, 5, 7, 9, 11, 15, 19, and 21) are early events and associated with the initiation of limited clonal plasma cell proliferation in MGUS. Acquisition of secondary chromosomal abnormalities (such as deletions of [parts of] chromosomes or secondary chromosomal translocations) and mutations involving individual genes results in the stepwise progression from MGUS to newly diagnosed symptomatic MM and, finally, aggressive forms of MM, such as sPCL or extramedullary MM. During this process, there is a progressive replacement of normal/polyclonal plasma cells (red) by clonal plasma cells (green). Progression of the plasma cell disorder is also accompanied by altered interactions of the tumor cells with various components of their microenvironment, such as osteoclasts, endothelial cells, and cells of the immune system. On the other hand, the immortalized cell may accumulate various genetic aberrations in a short period of time, resulting in de novo pPCL or rapid progression of MGUS to PCL, without preceding MM. Importantly, in pPCL, hyperdiploidy is very uncommon, whereas primary IgH translocations have a higher incidence compared with the incidence of these primary genetic events in newly diagnosed MM.
MM is preceded by MGUS in the majority of patients, and some case series report PCL patients with a preexisting MGUS.12 However, it is currently unknown to what extent such a protracted premalignant phase precedes pPCL or whether indeed pPCL predominantly arises de novo.
Initial immortalizing events
Primary IgH translocations and hyperdiploidy are considered to be early oncogenic events necessary for the immortalization of the plasma cell clone but insufficient for the development of symptomatic disease. In pPCL, hyperdiploidy is observed in only 0%-8.8% of the cases,5,13 whereas it is observed in ∼ 50% of newly diagnosed MM.14 Conversely, the incidence of hypodiploidy5,6,8,10,13,15,16 and IgH translocations is significantly increased,5,13,17 which is consistent with the association of 14q32 translocations with nonhyperdiploidy in MM.18 A similar pattern is observed in sPCL, with the exception that hyperdiploidy is slightly more prevalent in sPCL (∼ 17%) compared with pPCL.5,15,16 Altogether, this suggests that nonhyperdiploid tumors are less dependent on the BM microenvironment than hyperdiploid tumors.
The most prevalent IgH translocation in pPCL is t(11;14) at a frequency of 25%-65%,4,5,13,16,17,19–22 which is higher than observed in MM (∼ 15%23,24; Table 1). The poor prognosis of t(11;14) bearing pPCL is in contrast with the more favorable prognosis associated with this abnormality in MM.23,24 One study showed a significantly higher incidence of t(11;14) in pPCL compared with sPCL,5 whereas another study observed no difference.16 The frequencies of t(4;14) and t(14;16) in pPCL are higher compared with newly diagnosed MM but comparable with the prevalence in sPCL,4,5,13,16,17,22 and these translocations are clearly associated with poor prognosis in newly diagnosed MM.23,25
Table 1.
Presence of cytogenetic abnormalities in pPCL by FISH
| Reference | N | del(13q); NDMM: ∼ 50% | 14q32 translocations; NDMM: 50%-60% | t(4;14); NDMM: ∼ 15% | t(11;14); NDMM: ∼ 15% | t(14;16); NDMM: ∼ 5% | del(17p); NDMM: ∼ 10% | amp(1q21); NDMM: ∼ 30%-43% | del(1p21); NDMM: ∼ 20% |
|---|---|---|---|---|---|---|---|---|---|
| 5 | 18 | 85 | 87 | 0 | 65 | 0 | 50 | NA | NA |
| 4 | 70 | 65 | NA | 21 | 25 | 17 | 20 | NA | NA |
| 6 | 13 | 86 | NA | NA | NA | NA | NA | NA | NA |
| 13 | 40 | 68 | 80 | 12 | 33 | 13 | NA | NA | NA |
| 16 | 15 | 57 | NA | 25 | 50 | NA | 29 | 57 | 21 |
| 22 | 22 | 73 | NA | 14 | 32 | 36* | 32 | 46 | 41 |
| 17 | 10 | 60 | 70 | 0 | 40 | 30 | 20 | 67† | 44† |
Values are percentages.
NDMM indicates newly diagnosed myeloma; and NA, not available.
Both t(14;16) and t(14;20).
Determined by aCGH in 9 patients.
Secondary (epi)genetic events
In both pPCL and sPCL, the prevalence of poor-risk chromosomal abnormalities, such as del(17p),4,5,16,17,21 del(13q),4–6,13,16,17,22,26,27 del(1p21),16,17,21,28 ampl(1q21),16,17,21,27,28 and MYC translocations or amplifications,5,17,29,30 is markedly higher compared with newly diagnosed MM (Table 1). Coding mutations in TP53 are common in both pPCL and sPCL,5,31 whereas these mutations are rare in newly diagnosed MM.31 Activating K-RAS and N-RAS mutations are frequently observed in advanced MM and more prevalent in pPCL5,32,33 compared with newly diagnosed MM.33,34 In addition to genetic lesions, epigenetic modifications, such as increased methylation of CpG dinucleotides in the promoters of genes, appear to play an important role in the pathogenesis of PCL.5,35,36
Adhesion molecules and chemokine receptors
Compared with MM, tumor cells from pPCL and sPCL patients have reduced expression of the adhesion molecules NCAM (neural cell adhesion molecule/CD56) and LFA-1 (leukocyte function-associated antigen-1), which may contribute to the extramedullary accumulation of tumor cells in PCL.6,37–39 The absence of CD56 or LFA-1 is associated with reduced binding of tumor cells to BM stromal cells.40,41 Furthermore, loss of CD56 results in increased production of matrix metalloproteinase-9, which leads to destruction of the basal membrane and extracellular matrix.42 Extramedullary MM or PCL is also associated with loss of very late antigen-5 (VLA-5),39,43 LFA-3,37,44 and tetraspanin proteins, such as CD9 and CD82, which are cell surface molecules involved in adhesion, motility, and invasion.6,45 Down-regulation of chemokine receptors, such as CXCR4 on tumor cells, may also lead to impaired retention of PCL in the BM.45
Immune evasion
Various abnormalities may result in impaired antitumor T cell and NK cell-mediated immune responses in PCL. One of these aberrations is reduced expression of antigen-presenting HLA class I molecules in pPCL compared with MM or MGUS.46 Loss of adhesion molecules, such as LFA-1, may also facilitate escape of tumor cells from immune surveillance.47
Clinical and laboratory features of PCL
pPCL versus newly diagnosed MM
pPCL patients have a younger age at presentation compared with MM or sPCL patients (Table 2).4,5,7,48,49 However, their performance status at diagnosis is usually worse,6 which may be related to the more advanced stage of disease (Durie-Salmon stage III: ∼ 80%-96%; International Staging System stage III: ∼ 63%-80%).4,6,50–54 Extramedullary involvement, such as hepatomegaly, splenomegaly, lymphadenopathy, leptomeningeal infiltration, or extramedullary plasmacytomas, is more frequent in pPCL,1,5–10,49,51,53–55 with extensive bone disease being more common in patients with MM.5,6,53
Table 2.
Clinical and laboratory characteristics at diagnosis of newly diagnosed pPCL patients, as reported in selected larger informative series of patients
| Characteristic | Index | Values in NDMM | Newly diagnosed pPCL |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Vela-Ojeda et al9 | Kraj et al52 | Tiedemann et al5 | Pagano et al51 | Garcia-Sanz et al6 | Costello et al49 | Colovic et al54 | Noel and Kyle7 | Avet-Loiseau et al13 | Woodruff et al53 | Dimopoulos et al8 | |||
| No. of patients | 24 | 23 | 41 | 73 | 26 | 18 | 30 | 25 | 40 | 15 | 27 | ||
| Clinical characteristics | |||||||||||||
| Age, y | Median | 66-68 | 64 | 63 | 55 | — | 66 | 52 | 60 | 53 | 62 | 59 | 57 |
| Male | % | 53-59 | 38 | 43 | 59 | 59 | 46 | 61 | 73 | 60 | — | 47 | — |
| Durie-Salmon stage III | % | 50-60 | — | 85 | — | — | 92 | — | 87 | — | — | 87 | 96 |
| ISS stage III | % | 26-34 | — | 80 | — | 78 | — | — | — | — | — | — | — |
| Extramedullary plasmacytomas | % | 5-15 | — | — | 22 | 14 | 23 | 22 | 23 | — | — | — | — |
| Splenomegaly | % | 1 | 21 | — | 18 | 18 | — | 28* | 53 | 44 | — | 53 | 26 |
| Hepatomegaly | % | 4 | 25 | — | 32 | 23 | — | 28* | 57 | 52 | — | 73 | 7 |
| Lymphadenopathy | % | 1 | 17 | — | 6 | 4 | — | 0 | 3 | 12 | — | 40 | 11 |
| Bone disease | % | 79-81 | — | 67 | 35 | 64 | 48 | 61 | 60 | 44 | 59 | 67 | — |
| Laboratory characteristics | |||||||||||||
| PB plasmacytosis, × 109/L | Median | 0.003-0.0044 | 3.9 | — | 7.2 | 3.7 | — | 7.5 | 4.0 | 7.9 | 9.2 | 1.9 | — |
| Range | 0-2.0 | 1.6-19.5 | — | 1.5-164 | 0.4-49.9 | — | 2.1-42 | 1.7-105 | 2.1-145.8 | 2.0-64 | 0.6-76.7 | — | |
| White blood cells, × 109/L | Median | 5.8 | — | 15 | 21.5 | 13.7 | — | 24 | 16 | 19.7 | — | 14.5 | — |
| Hemoglobin, g/dL | Median | 10.1-10.9 | — | — | 9.4 | 9.1 | 8.2 | 9 | 7.8 | 8.4 | — | 7.4 | — |
| Platelets, × 109/L | Median | 209-242 | — | — | 98 | 116 | 123 | 62 | — | 94 | — | 112 | — |
| Serum creatinine, mg/dL | Median | 1.2 | — | — | 1.9 | 1.7 | — | 1.5 | — | 2.6 | — | — | — |
| ≥ 2 mg/dL, % | 19-21 | 50 | 33 | — | 44 | 44 | 50† | 43‡ | — | — | 53 | 37 | |
| Serum calcium, mg/dL | Median | 9.5-9.6 | — | — | 10 | 9.8 | — | 10 | — | 10.1 | — | 11.3 | — |
| ≥ 11 mg/dL, % | 13-20 | 37 | 52 | — | 27 | 48 | — | 60 | — | — | 60 | 44§ | |
| β2-Microglobulin, mg/L | median | 3.6-3.9 | 10.4 | 8.4 | 6.8 | 8.5 | — | 6.0 | — | — | — | — | — |
| ≥ 6 mg/dL, % | 26-27 | — | — | — | — | 65 | 50 | 64 | — | — | — | 91 | |
| Serum albumin, g/dL | median | 3.6 | — | 3.9 | — | 3.7 | — | — | 2.8 | — | — | — | — |
| < 3.5 g/dL, % | 40 | — | 33 | — | 46 | 52 | — | — | — | — | — | — | |
| Light-chain only | % | 10-20 | 21 | 19 | 41 | 41 | 31 | 28 | 20 | 50 | — | 40 | 26 |
| BM plasmacytosis | Median | 40-50 | 90 | 80 | 78 | 80 | — | 76 | 80 | 84 | — | ≥ 70 | — |
Presenting features of patients with newly diagnosed myeloma (NDMM) are mainly derived from 3 large series of patients.5,6,99
— indicates not available; ISS, International Staging System; and PB, peripheral blood.
Splenomegaly and/or hepatomegaly.
Creatinine ≥ 1.4 mg/dL.
Creatinine ≥ 2.8 mg/dL.
Calcium > 11.5 mg/dL.
Various laboratory characteristics reflect a high tumor load. For example, the median percentage of BM plasma cells is significantly higher in pPCL than in MM.5,6,54 In addition, renal failure is more common in pPCL, which can be partly explained by the higher incidence of light-chain disease.5,6,10,49 Furthermore, hypercalcemia, anemia, thrombocytopenia, elevated plasma cell labeling index, increased LDH, and GEP-defined high-risk disease are more frequent at presentation in pPCL compared with MM.5,6,8,10,50,51,53,55 Consistent with higher tumor burden and an increased incidence of renal impairment, significantly elevated β2-microglobulin levels in pPCL are seen compared with MM.5,6,10,50,55
pPCL versus secondary PCL
Secondary PCL is typically the terminal stage of preexistent MM, and this in part explains the higher prevalence of advanced bone disease compared with pPCL.5,7 In contrast, extramedullary involvement may be less common in sPCL.5,7 Patients with sPCL present more often with renal failure than MM patients, but renal dysfunction is more common in pPCL.5
Blood and BM findings
Peripheral blood examination in pPCL shows circulating tumor cells and typically a leuko-erythroblastic blood picture in up to 67% of patients (Figure 3).5,53 BM biopsy typically demonstrates extensive BM involvement, disrupting normal hematopoiesis. In some cases, tumor resembles normal plasma cells, whereas in others, lymphoplasmacytoid or immature plasma cells predominate.19,56,57 Sometimes the circulating plasma cells are difficult to classify by light microscopy alone and differentiation from other conditions, such as chronic lymphocytic leukemia, hairy cell leukemia, or marginal zone lymphoma with circulating lymphocytes requires immunophenotypic analysis, which can also be useful to differentiate reactive from clonal plasma cells.
Figure 3.

Peripheral blood and BM features of pPCL. (A) May-Grünwald-Giemsa staining of peripheral blood smears from 3 different patients with pPCL. (i) The peripheral blood smear of patient 1 is characterized by the presence of circulating plasma cells, including binucleated cells. Some plasma cells have a lymphoplasmacytoid appearance. There is also rouleaux formation and leuko-erythroblastosis (myelocyte in Ai). Cytogenetic analysis demonstrated the presence of an IgH translocation with an unknown partner chromosome as well as deletion of the Y chromosome. (ii) Peripheral blood of patient 2 contains circulating plasma cells, some of which have 2 nuclei or a lymphoplasmacytoid aspect. FISH analysis showed the presence of t(14;16) and ampl(1q21). (iii) The staining of patient 3 shows very atypical circulating plasma cells often with a monocytic appearance. Numerous cells contain cytoplasmic (Russell bodies) and intranuclear inclusions (Dutcher bodies). Cytogenetic analysis revealed multiple deletions, including chromosomes 6q, 12p, 13, 16, and 20, and the presence of t(14;20). (B) May-Grünwald-Giemsa-stained BM smears from the same 3 pPCL (i-iii) patients. (C) Immunophenotypic analysis of the leukemic plasma cells from the patient shown in subpanels Aii and Bii. The tumor cells are positive for CD138 and CD27, weakly positive for CD38 and CD45, and negative for CD56 and CD19. Cells were also negative for CD20 (not shown).
The most striking immunophenotypic difference between newly diagnosed MM and pPCL is that pPCL tumor cells are less often positive for CD56, CD71, CD117, and HLA-DR, but more likely express CD20, CD45, CD19, CD27, and CD23.6,20,37–39,49,58,59 Both increased expression of CD20 and CD23 and down-regulation of CD56 may be related to the high incidence of t(11;14) in pPCL.6,20,58,60 Tumor cells are positive for CD38 and CD138 in both PCL and MM6,19,37 (Figure 3C). Interestingly, there is a progressive decrease in expression levels of CD38 from normal plasma cells to MGUS plasma cells, MGUS to MM, and finally MM to PCL,46 which probably reflects dedifferentiation into a more immature phenotype. Kraj et al compared pPCL and sPCL and found that their immunophenotypic profiles are comparable, except for CD56 expression, which was more often present in sPCL.37 In contrast, in another comparative study, both types of PCL had no or weak CD56 expression, whereas CD28 expression was more frequent in sPCL compared with pPCL.38
Diagnostic workup of pPCL
At the time of pPCL diagnosis, laboratory studies, such as complete blood count with differential, peripheral blood smear, blood chemistry including tumor lysis parameters, β2-microglobulin, serum and urine protein electrophoresis with immunofixation, and measurement of free light chains, are essential (Table 3). Staging procedures should also include skeletal survey and BM biopsy, as well as aspiration for morphology, immunophenotyping, and cytogenetic analysis by FISH, which is focused on del(17p13), del(13q), del(1p21), ampl(1q21), and the presence of 14q32 abnormalities (t(11;14)(q13;q32), t(4;14)(p16;q32), and t(14;16)(q32;q23)). Because pPCL is a clearly defined high-risk plasma cell disorder, the value of additional risk stratification by cytogenetics to guide type of therapy or predict outcome is currently limited. Lumbar puncture, MRI, or CT (which can be combined with PET) should be performed when extramedullary involvement is suspected (eg, if cranial nerve palsies, cord compression, obstructive icterus, or palpable masses are noted on examination).
Table 3.
Diagnostic evaluation for pPCL
| Medical history and physical examination |
|---|
| Blood |
| Complete blood count with differential, peripheral blood smear |
| BUN, creatinine, liver enzymes, bilirubin, alkaline phosphatase, total protein, CRP |
| Tumor lysis parameters (LDH, uric acid, calcium, phosphate, potassium) |
| β2-microglobulin and albumin |
| Serum protein electrophoresis, immunofixation, serum free light chain analysis |
| Immunophenotyping |
| Urine |
| 24-hour urine collection for electrophoresis and immunofixation |
| 24-hour urine for total protein |
| BM |
| Biopsy for histology |
| Aspirate for: |
| Morphology |
| Immunophenotyping |
| Cytogenetic analysis by FISH* focused on del(17p13), del(13q), del(1p21), ampl(1q21), t(11;14), t(4;14), and t(14;16) |
| Radiographic skeletal survey, including skull, pelvis, vertebral column, and long bones |
| Additional investigations, which may be useful under certain circumstances |
| Lumbar puncture (cell counts, chemistry, cytology, immunophenotyping): suspicion of leptomeningeal involvement |
| MRI: evaluation of cord compression or painful area of the skeleton (suspicion of soft tissue plasmacytomas arising from bone) |
| CT or 18F-FDG-PET/CT: suspicion of extramedullary plasmacytomas |
| Survey for evaluation of AL amyloidosis |
| Bleeding time, APTT, PT |
| Cryoglobulins, cold agglutinins |
| Serum viscosity, fundoscopy: symptoms of hyperviscosity |
| HLA typing: in case allo-SCT is considered |
BUN indicates blood urea nitrogen; CRP, C-reactive protein; MRI, magnetic resonance imaging; CT, computed tomography; 18F-FDG-PET, 18F-fluorodeoxyglucose positron emission tomography; AL, amyloid light chain; APTT, activated partial thromboplastin time; and PT, prothrombin time.
FISH is preferably performed on purified tumor cells or with simultaneous staining of cytoplasmic immunoglobulins (cIg-FISH).
Prognostic factors
Various unfavorable prognostic factors for newly diagnosed MM also have prognostic value in pPCL. However, the prevalence of these risk factors in pPCL is significantly higher. Prognostic parameters include low serum albumin,51 elevated β2-microglobulin,6 hypercalcemia,49,51 elevated serum LDH,9,54 advanced age,9,48 worse performance status,9,54 and increased percentage of S-phase plasma cells.6 Response to treatment is also of great prognostic value in pPCL. Patients presenting with disease that is resistant to initial therapy have a very poor prognosis, with survival estimates of a few months.9,49,51 The failure of blood plasma cells to decline by 50% within 10 days or to be cleared within 4 weeks has been proposed as a criterion identifying patients with unresponsive disease.8
Because most studies performed in pPCL are small and retrospective in nature with heterogeneous treatments, the value of the cytogenetic abnormalities with prognostic impact in MM remains unclear in pPCL. The presence of hypodiploidy, complex karyoptype, del(13q), del(17p), del(1p), or ampl(1q) was associated with reduced OS in a retrospective study performed in Italy.51 Survival in a cohort of both primary and secondary PCL patients was negatively affected by the presence of t(4;14) and del(1p21).16 Avet-Loiseau et al showed that pPCL patients with t(11;14) had a longer OS,13 and Tiedemann et al found that MYC rearrangements predicted for shorter OS.5 Larger prospective studies with standardized treatments are needed to establish the prognostic value of the diverse cytogenetic abnormalities in PCL overall.
Treatment modalities in pPCL
There is a paucity of literature on the treatment of pPCL, and no randomized trials have been reported exclusively for patients with pPCL.
Conventional chemotherapy
The prognosis of pPCL after conventional chemotherapy without novel agents is poor, with median OS of ∼ 7 months.6–8,49,55,57,61 There appears to be limited benefit in terms of survival for multiagent conventional chemotherapy, such as vincristine, adriamycin, and dexamethasone (VAD)-based regimens, compared with regimens containing only an alkylating agent plus a corticosteroid.5–8,51,55
Novel agents
The introduction of immunomodulatory drugs and proteasome inhibitors has significantly improved survival of MM patients.62,63 Increasing evidence suggests that these agents also improve outcome of pPCL, but the benefit may be less pronounced compared with classic MM. A retrospective analysis performed by the Intergroupe Francophone du Myélome showed that pPCL patients treated with novel agents had a survival of 15 months compared with 8 months for patients who did not receive novel agents as part of their treatment.64 In addition, a retrospective analysis performed by GIMEMA showed improved survival for those patients who received bortezomib and/or thalidomide at any stage of their treatment.51 In contrast, a SEER database analysis failed to show enhanced survival of pPCL patients in the period 1973-2004, but information on treatment changes over time were lacking in this study.48 There remain a limited number of prospective studies evaluating novel agents in pPCL, with several retrospective studies providing additional information on the efficacy of these drugs.
Bortezomib
Bortezomib is probably the most important drug in pPCL because bortezomib-based therapy rapidly reduces tumor load and reverses complications, including renal failure and hypercalcemia. Bortezomib also overcomes the poor prognosis conferred by del(13q) or t(4;14) and mitigates the adverse outcome associated with del(17p).65–67 Several case reports and small case series suggest that bortezomib, alone or in combination with other agents, is effective in newly diagnosed pPLC and may also be active in refractory pPCL or sPCL.61,68–74
The largest retrospective analysis of newly diagnosed pPCL patients treated with bortezomib-based regimens (n = 29) comes from the Italian GIMEMA MM Working Party, with an overall response rate of 79%, and 38% ≥ very good partial response (VGPR).71 Importantly, there was improvement or normalization of renal function in 10 of 11 patients presenting with renal failure. Two-year progression-free survival (PFS) was 40% and 2-year OS 55%, with the best long-term results achieved in patients who received stem cell transplantation (SCT) after bortezomib induction.71
In another retrospective analysis, Musto et al showed that response rate to bortezomib or bortezomib-based combinations in 8 newly diagnosed or relapsed pPCL patients was 100%, with median PFS and OS not reached after 21 months.61
A single-center retrospective analysis of all pPCL and sPCL patients has shown a survival advantage for bortezomib-treated patients compared with a non–bortezomib-treated group.75 This is in line with a retrospective analysis of 73 pPCL patients treated with different regimens, which showed best results for patients treated with bortezomib followed by autologous SCT (auto-SCT).51 In contrast, a retrospective analysis performed by the Intergroupe Francophone du Myélome showed no improvement in OS for patients treated with relatively short courses of bortezomib.4 Although the number of patients is small, the Arkansas group showed that Total Therapy 3 (which incorporates both bortezomib and thalidomide) did not result in an improved survival compared with the preceding Total Therapy regimens, which incorporated thalidomide alone.10
Thalidomide
Efficacy of single-agent thalidomide is limited in pPCL compared with the activity of this agent in MM.76,77 Although some reports with small numbers of patients showed that thalidomide may result in durable responses in sPCL or pPCL, its decreased activity in extramedullary MM makes its use less attractive.78,79 Conversely, addition of thalidomide to dexamethasone, conventional chemotherapy, or bortezomib may result in enhanced activity in pPCL.64,80,81
Lenalidomide
Lenalidomide is less toxic and more potent than thalidomide. The combination of lenalidomide with dexamethasone has been effective in newly diagnosed pPCL.22 In a prospective phase 2 study, 23 newly diagnosed pPCL patients were treated with lenalidomide and dexamethasone: 14 patients completed the initial 4 planned cycles and partial response (PR) was achieved in 61%, with ≥ VGPR in 35%. Five patients underwent auto-SCT and 1 received tandem auto-SCT/allogeneic SCT (allo-SCT) after lenalidomide plus dexamethasone treatment. After a median follow-up of 15 months, OS and PFS were 63% and 52%, respectively.22 Lenalidomide-based therapies also appear promising in the setting of relapsed/refractory disease, especially in combination with bortezomib.64,82,83
Combinations of novel agents
In pPCL, the efficacy of combinations of novel agents, such as lenalidomide, bortezomib, and dexamethasone (RVD),84 bortezomib, thalidomide, and dexamethasone (VTD),61,64,71 or melphalan, prednisone, bortezomib, and thalidomide (VMPT),71 appears very promising. Studies describing these regimens invoke only a small numbers of patients but are based on the biologic and clinical features seen.
Auto-SCT
McElwain and Powles were the first to describe the efficacy of high-dose melphalan in a pPCL patient, who survived more than 30 months after melphalan 140 mg/m2.85 Since then, other case reports and small case series suggest that high-dose therapy with hematopoietic stem cell support improves OS.4,5,9,51,56
The largest retrospective analysis to date was performed by the European Group for Blood and Marrow Transplantation,50 who compared 272 pPCL patients with 20 844 MM patients undergoing auto-SCT between 1980 and 2006.50 Although CR rates before and after autologous SCT were higher in pPCL patients, median PFS (14.3 vs 27.4 months) and OS (25.7 vs 62.3 months) were significantly longer in MM patients. Conversion to CR after auto-SCT was associated with improved PFS and OS.50 Treatment-related mortality (TRM) was higher in the pPCL group. Importantly, this study lacked information regarding type of induction regimen, which is probably critical based on current data.
Another large retrospective analysis of 97 pPCL patients who received upfront auto-SCT between 1995 and 2006 was generated by the Center for International Blood and Marrow Transplant Research (CIBMTR).86 In contrast to the European Group for Blood and Marrow Transplantation results, 3-year PFS (34%) and OS (64%) were similar to that observed in MM, and there was a trend toward superior OS in patients who received a tandem auto-SCT compared with those receiving a single transplant. Nonrelapse mortality at 3 years was 5%. The use of novel agents as part of the induction therapy was very low, which may explain the absence of any major difference in outcome.
Moreover, when comparing both studies, PFS is almost identical, whereas OS was significantly longer in the CIBMTR study (Table 4). This outcome may be related to greater availability of novel agents for relapse treatment in the CIBMTR study (> 60% transplanted after 2000). Altogether, these studies show an encouraging survival after auto-SCT with acceptable toxicity. However, in both series, it is unclear which proportion of patients planned to undergo auto-SCT did not receive this treatment because of either early progression or death. This selection bias may lead to an overestimation of the effectiveness of auto-SCT in pPCL; nonetheless, integration of novel therapies into induction, consolidation, and maintenance around auto-SCT would seem justified.
Table 4.
Results from the 2 largest retrospective series of pPCL patients treated with auto-SCT
| Characteristics of patients | Drake et al50 | Mahindra et al86 |
|---|---|---|
| N | 272 | 97 |
| Age at SCT, y | 56 | 56 |
| SCT received | 1980-2006 | 1995-2006 |
| β2-Microglobulin, median (mg/L) | 6.8 | NA |
| Induction regimen, % | ||
| Thalidomide-based | NA | 25 |
| Bortezomib-based | NA | 5 |
| Lenalidomide-based | NA | 0 |
| Graft type | ||
| BM | 0.8 | NA |
| BM/PBSC | 1.5 | NA |
| PBSC | 97.7 | NA |
| Conditioning regimen, % | ||
| Melphalan alone | NA | 56 |
| Melphalan + TBI ± others | NA | 11 |
| Melphalan-based, no TBI | NA | 24 |
| TBI included | 9.1 | NA |
| No TBI included | 90.9 | NA |
| Second transplant, % | ||
| Auto-auto | NA | 26 |
| Auto-allo | 10.3 | 4 |
| Disease status at transplantation, % | ||
| CR | 25.5 | 20 |
| PR | 58.7 | 56 |
| ≥ PR | 84.2 | 76 |
| Response post-SCT, % | ||
| CR | 41.2 | NA |
| PR | NA | NA |
| ≥ PR | NA | NA |
| PFS at 3 years | 38 | 34% |
| OS at 3 years | 39.5 | 64% |
| NRM at 3 years | NA | 5% |
NA indicates not available; PBSC, peripheral blood stem cells; TBI, total body irradiation; and NRM, nonrelapse mortality.
Allo-SCT
Because response duration after auto-SCT is relatively short, other strategies are needed as a consolidation treatment, including allo-SCT, under the auspices of a clinical trial. Although TRM is high, several case reports and case series reported successful results and long-term survival after allo-SCT in pPCL.56,81,87
The retrospective CIBMTR analysis also compared the outcome of the 97 patients who received auto-SCT with 50 patients who received allo-SCT between 1995 and 2006.86 Most patients (68%) received a myeloablative conditioning regimen before allo-SCT, whereas 32% received a nonmyeloablative or reduced-intensity conditioning regimen. Only 4 patients received tandem auto-allo-SCT. Although the cumulative incidence of relapse at 3 years was significantly lower in the allogeneic group (allo-SCT vs auto-SCT, 38% vs 61%), TRM at 3 years was considerably higher in patients who received an allogeneic transplant (allo-SCT vs auto-SCT, 41% vs 5%). This resulted in a 3-year OS of 64% and 39% for the auto-SCT and allo-SCT group, respectively.86 Relapse risk was lower and PFS was superior in patients who were transplanted within 6 months of diagnosis. Importantly, the use of novel agents as part of the induction regimen remained low. Furthermore, this analysis covers a long time span, during which TRM of allo-SCT has decreased because of better supportive care and use of other reduced intensity conditioning regimens.
The European Myeloma Network is initiating a prospective study in which pPCL patients will be treated with a novel agent-containing induction regimen, followed by tandem auto-SCT and allo-SCT, the latter involving semi-intensive conditioning with melphalan 140 mg/m2 + fludarabine, as well as novel agent consolidation and maintenance. Such strategies would appear most likely to generate improved outcome in this setting.
Treatment recommendations for pPCL
pPCL requires urgent control of clinical manifestations to prevent early death because of irreversible disease complications.8 In patients presenting with renal failure, hypercalcemia, or elevated LDH, treatment with a bortezomib-based regimen should be started as soon as possible to rapidly reduce the high tumor load to reverse and/or prevent complications. Although participation in clinical trials is strongly recommended, this is not always feasible. Our recommendations of how to treat patients not eligible for studies are based on the increasing evidence that autologous SCT and novel agents improve outcome in pPCL (Figure 4). Because no specific response and relapse criteria have been defined in pPCL, we also propose to use definitions as formulated in Table 5.
Figure 4.

Treatment of pPCL. Flow chart with suggestions for how to treat patients with pPCL. *Consolidation for non–transplant-eligible patients is given once best response to induction therapy is achieved. Varicella zoster virus prophylaxis with acyclovir or valacyclovir is recommended for all patients receiving bortezomib-based therapy. Thromboprophylaxis (aspirin for patients at standard risk for thromboembolic events and low-molecular-weight heparin or adjusted-dose warfarin in high-risk patients) is indicated when either thalidomide or lenalidomide is combined with dexamethasone or chemotherapy. MUD indicates matched unrelated donor; DLI, donor lymphocyte infusion; PAD, bortezomib, adriamycin, and dexamethasone; CVD, cyclophosphamide, bortezomib, and dexamethasone; VMP, bortezomib, melphalan, and prednisone; and RIC, reduced intensity conditioning.
Table 5.
Proposed response and relapse criteria for pPCL
| Response/relapse subcategory | Criteria |
|---|---|
| MRD-negative CR* | sCR as defined below plus |
| MRD-negative BM† by multicolor flow cytometry or allele-specific oligonucleotide PCR‡ and | |
| MRD-negative peripheral blood by multicolor flow cytometry or allele-specific oligonucleotide PCR‡ | |
| sCR* | CR as defined below plus |
| Normal FLC ratio and | |
| Absence of clonal cells in BM† by immunohistochemistry or immunofluorescence§ and | |
| Absence of clonal cells in peripheral blood by immunofluorescence | |
| CR* | Negative immunofixation on the serum and urine and |
| Disappearance of any soft tissue plasmacytomas and | |
| ≤ 5% plasma cells in BM† and | |
| No plasma cells in blood smear | |
| VGPR* | Serum and urine M-protein detectable by immunofixation but not on electrophoresis or ≥ 90% reduction in serum M-protein plus urine M-protein level < 100 mg per 24 h and |
| No plasma cells in blood smear | |
| PR* | ≥ 50% reduction of serum M-protein and reduction in 24-h urinary M-protein by ≥ 90% or to < 200 mg per 24 h |
| If the serum and urine M-protein are immeasurable,‖ a ≥ 50% decrease in the difference between involved and uninvolved FLC levels is required in place of the M-protein criteria | |
| If serum and urine M-protein are immeasurable, and serum free light assay is also immeasurable, ≥ 50% reduction in BM plasma cells is required in place of M-protein, provided baseline BM plasma cell percentage was ≥ 30% | |
| In addition to the aforementioned criteria, a ≥ 90% reduction of peripheral blood plasma cells is required and peripheral blood plasma cells must be ≤ 5% of the differential white cell count, and if present at baseline, a ≥ 50% reduction in the size of soft tissue plasmacytomas is also required | |
| SD* | Not meeting criteria for CR, VGPR, PR, or progressive disease |
| PD¶ (to be used for calculation of TTP and PFS endpoints for all patients, including those in CR (includes primary progressive disease and disease progression on or off therapy) | Progressive disease: requires any one or more of the following: |
| Increase of ≥ 25% from baseline/nadir in | |
| Serum M-component and/or (the absolute increase must be ≥ 0.5 g/dL)# | |
| Urine M-component and/or (the absolute increase must be ≥ 200 mg/24 h) | |
| Peripheral blood plasma cells (with at least 2 × 109 cells per L or > 20% of the differential white cell count) | |
| Only in patients without measurable serum and urine M-protein levels: the difference between involved and uninvolved FLC levels; the absolute increase must be > 10 mg/dL | |
| BM plasma cell percentage: the absolute % must be ≥ 10%** | |
| Definite development of new bone lesions or soft tissue plasmacytomas or definite increase in the size of existing bone lesions or soft tissue plasmacytomas | |
| Development of hypercalcemia (corrected serum calcium > 11.5 mg/dL or 2.65mM) that can be attributed solely to the plasma cell proliferative disorder | |
| Relapse from CR¶ (to be used only if the endpoint studied is DFS) | Any one or more of the following: |
| Reappearance of serum or urine M-protein by immunofixation or electrophoresis | |
| Reappearance of peripheral blood plasma cells in blood smear | |
| Development of ≥ 5% plasma cells in the BM** | |
| Appearance of any other sign of progression (ie, new plasmacytoma, lytic bone lesion, or hypercalcemia) |
Criteria are based on the International Myeloma Working Group (IMWG) criteria with some modifications and inclusion of the response subcategory MRD-negative CR.
MRD indicates minimal residual disease; FLC, free light chain; sCR, stringent complete response; SD, stable disease; PD, progressive disease; TTP, time to progression; and DFS, disease-free survival.
All response categories require 2 consecutive assessments made at any time before the institution of any new therapy; all categories also require no known evidence of progressive or new bone lesions if radiographic studies were performed. Radiographic studies are not required to satisfy these response requirements.
Confirmation with repeat BM biopsy is not needed.
Sensitivity attainable with 8-color multiparameter flow cytometry and allele-specific oligonucleotide PCR is 10−6.
The presence or absence of clonal cells is based on the κ/λ ratio. An abnormal κ/λ ratio by immunohistochemistry and/or immunofluorescence requires a minimum of 100 plasma cells for analysis. An abnormal ratio reflecting the presence of an abnormal clone is κ/λ of > 4:1 or < 1:2.
For measurable disease, serum M-protein ≥ 1 g/dL (≥ 10 g/L); urine M-protein ≥ 200 mg/24 h; involved FLC level ≥ 10 mg/dL (≥ 100 mg/L) provided serum FLC ratio is abnormal.
All relapse categories require 2 consecutive assessments made at any time before classification as relapse or disease progression and/or the institution of any new therapy.
For progressive disease, serum M-component increases of ≥ 1 g/dL (10 g/L) are sufficient to define relapse if starting M-component is ≥ 5 g/dL (50 g/L).
Relapse from CR has the 5% cut-off versus 10% for other categories of relapse.
Younger patient with pPCL
In the transplantation-eligible patient, high-dose therapy with autologous stem cell rescue is currently the most effective therapeutic modality to achieve long-term remission. Because several studies report a survival benefit of bortezomib, our current practice is to treat patients with a 3-drug bortezomib-based induction regimen, such as RVD, PAD, CVD (cyclophosphamide, bortezomib, and dexamethasone), or VTD. This is followed by auto-SCT with high-dose melphalan as conditioning regimen. In the absence of randomized trials, no conclusions can be drawn regarding the superiority of one bortezomib-based triplet over another.
Post-transplantation consolidation and/or maintenance strategies with novel agents have not been extensively studied in pPCL, with only case reports and small case series describing prolonged remissions after maintenance treatment with thalidomide,51,83 lenalidomide,22,73 and bortezomib.69,71,72 Nonetheless, we typically offer consolidation and maintenance therapy to patients after transplantation because of increasing evidence that consolidation and/or maintenance treatments increase the quality of response,88–90 which is associated with improved OS and PFS in MM,91 and this may be especially important in high-risk disease, such as pPCL.50 In particular, achievement of a minimal residual disease-negative status is predictive of improved outcome, especially in the presence of high-risk cytogenetics.88,91 The generally short PFS after autologous-SCT (also for patients achieving CR)50,86 seen in pPCL is indicative of the persistence of a substantial burden of (minimal) residual disease. Altogether, this provides strong rationale for the use of post-transplantation therapies in pPCL to improve depth of response, maintain remission, and prolong survival. Furthermore, future studies incorporating detection of persistent minimal residual disease by multiparameter flow cytometry are warranted to identify which patients benefit most from consolidation/maintenance strategies.91
Consolidation regimens that can be used after transplantation include 2-4 cycles of RVD or VTD. This treatment can be followed by maintenance therapy with lenalidomide and/or bortezomib until progression. Maintenance treatment with bortezomib and/or lenalidomide also seems to improve outcome in MM patients with high-risk chromosomal aberrations,65,90,92 whereas data suggest that thalidomide maintenance is only beneficial in patients with standard-risk cytogenetics.93,94 This outcome, combined with the cumulative toxicity associated with thalidomide, favors the use of lenalidomide and/or bortezomib as maintenance therapy in pPCL.
Given the lower risk of relapse after allo-SCT compared with auto-SCT,86 consolidation with allo-SCT can also be considered as part of frontline treatment in pPCL in younger patients (< 65 years) with an available fully matched sibling or unrelated donor. Because the benefits of allo-SCT in this disease are still unclear and to improve efficacy and safety of allo-SCT, it should be performed in the setting of a clinical trial, in which new conditioning regimens may be explored and in which novel agents as part of induction and maintenance therapy are incorporated. Other methods, such as vaccination approaches, hold promise for improving outcome of allo-SCT but may be problematic in the setting of aggressive disease, such as pPCL. Treatment decisions for allo-SCT should be individually discussed with the patient. Thus, patients with pPCL can be treated with auto-SCT, followed 3-6 months later by allo-SCT with semi-intensive conditioning consisting of busulfan + fludarabine or melphalan 140 mg/m2 + fludarabine. Myeloablative conditioning should only be considered in fit young patients without comorbidities and on protocol, given its high TRM. In both semi-intensive and myeloablative conditioning protocols, the chemotherapy preceding allo-SCT is important for the prevention of early relapse before the graft-versus-myeloma effect becomes established. Moreover, the integration of combination therapy with novel therapies as part of induction, consolidation, and maintenance is vital.
Older patient with pPCL
Patients who are not transplant eligible because of advanced age and/or comorbidities should be treated with a bortezomib-based regimen, such as VMP or RVD. In addition, in older pPCL patients, the importance of consolidation and maintenance therapy after induction treatment is highlighted. In the very elderly population and in patients with multiple comorbidities, treatment tolerance is reduced, necessitating dose modifications to reduce toxic side effects and avoid early discontinuations, which eventually can lead to decreased efficacy.
Treatment of relapsed pPCL
When patients with pPCL relapse, outcome is generally poor. Based on various factors, including age, performance status, time from prior therapy, response to prior therapies, BM reserve, and presence of comorbidities (such as polyneuropathy or renal dysfunction), selection of a multiagent salvage chemotherapy regimen with several novel agents is recommended.95
Supportive care
Early mortality through complications is high,8 indicating that adequate supportive care is important in this vulnerable patient group. Varicella zoster virus prophylaxis is recommended for all patients receiving bortezomib-based therapy, and antibacterial and antifungal prophylaxis is given dependent on type of treatment regimen, especially with high-dose steroids. Intravenous immunoglobulin may be useful in case of recurrent bacterial infections in the presence of hypogammaglobulinemia. Other measures to prevent serious infection include the use of granulocyte colony-stimulating factor.
Thromboprophylaxis should be initiated when either thalidomide or lenalidomide is combined with dexamethasone or chemotherapeutic agents. Low-molecular-weight heparin or adjusted-dose warfarin should be selected for high-risk patients, whereas aspirin can be considered in patients at standard risk for venous thromboembolism.
Tumor lysis syndrome (TLS) is rare in myeloma (∼ 1%-2%) and typically occurs soon after initiation of therapy with an increased incidence in patients with high tumor burden (BM plasmacytosis > 70%), rapidly proliferating disease (labeling index > 3.5%, high LDH), immature plasma cell morphology, adverse cytogenetics, and compromised renal function. Because many of these characteristics are present in pPCL patients, the incidence of TLS is expected to be higher in this disorder.96 Therefore, patients should receive TLS prophylaxis (allopurinol and increased hydration for standard risk patients, and rasburicase plus increased hydration for patients with multiple risk factors or presenting with renal dysfunction or hyperuricemia), with careful monitoring for TLS and prompt intervention in case TLS develops to minimize any permanent renal dysfunction.
Patients with extramedullary disease may also benefit from locoregional therapy, including radiotherapy in case of plasmacytomas and/or intrathecal chemotherapy for leptomeningeal involvement. Bisphosphonates are recommended after correction of factors predisposing to renal deterioration, such as hypovolemia, except in patients with severe renal impairment, in which reversibility is anticipated. Finally, the use of erythropoiesis-stimulating agents is appropriate for Hb ≤ 10 g/dL.
Treatment recommendations for sPCL
Survival of patients with sPCL is shorter than survival in pPCL.5,7,57 As for other forms of relapsed and refractory MM, treatment depends on both patient- and disease-specific factors. Available data suggest that bortezomib-based treatment is the most effective treatment modality in sPCL.61,68,73,75 In case of refractory disease, combination approaches, such as lenalidomide combined with bortezomib,97 or participation in a clinical trial, if feasible, can be considered. High-dose methylprednisolone may be a useful adjunct together with other chemotherapeutics and novel agents. However, because sPCL represents heavily pretreated end-stage MM, patients are generally refractory to all available treatment modalities or have a response of short duration, with the emphasis of best supportive care and effective palliation being very important considerations once therapeutic failure occurs.
In conclusion, patients with pPCL have an aggressive clinical presentation and poor prognosis, resulting from a different biologic background compared with classic MM. Various studies demonstrate that the introduction of auto-SCT and bortezomib-based therapy, as well as other novel agents such as lenalidomide, have markedly improved survival of pPCL patients. However, survival of most pPCL patients is still inferior compared with outcome in newly diagnosed MM, indicating the need for novel treatment strategies.
Examples of newer novel agents include second-generation proteasome inhibitors and immunomodulatory drugs, monoclonal antibodies such as rituximab, elotuzumab, and daratumumab,77,98 as well as inhibitors of histone deacetylase, Akt, and mTOR (Figure 5). The introduction of these next-generation novel agents, combined with the identification of biomarkers that are predictive of therapeutic response, will eventually result in a more personalized targeted treatment to simultaneously augment efficacy and minimize toxicity. Because PCL patients do not benefit as much from the currently available treatment strategies compared with other MM patients, they should be considered primary candidates for these newer treatment options, and enrollment in clinical trials should be encouraged. Given the rarity of pPCL, collaboration will be essential in developing clinical trials to effectively evaluate new drug combinations and continued modality approaches (eg, allo-SCT) to further improve outcome.
Figure 5.

Novel agents in plasma cell leukemia. Immunomodulatory drugs, bortezomib, and next-generation novel agents have direct antitumor effects but also affect the BM microenvironment (ie, down-regulation of adhesion molecules, reduction of osteoclastogenesis, promotion of new bone formation, inhibition of angiogenesis, and stimulation of immune effector cells).
Acknowledgments
The authors thank Victor Muñoz Sanz (Harvard School of Design, Boston, MA) for creating Figures 2 and 5; Jeanette Kimmel, Gerda Engelsman, and Dr Andries C. Bloem (Department of Immunology, University Medical Center Utrecht, Utrecht, The Netherlands) for help with the selection of morphologic and flow cytometric images; Professor Eugene J. Kucharz (Department of Internal Medicine and Rheumatology, Medical University of Silesia, Katowice, Poland) for providing the image shown in Figure 1; and Dr Monique C. Minnema (Department of Hematology, University Medical Center Utrecht, Utrecht, The Netherlands), Dr Constantine S. Mitsiades (Dana-Farber Cancer Institute, Boston, MA), Dr Catriona A. Hayes (Dana-Farber Cancer Institute, Boston, MA), and Dr Jacob P. Laubach (Dana-Farber Cancer Institute, Boston, MA) for useful discussions and comments on the manuscript.
N.W.C.J.v.d.D. was supported by the Dutch Cancer Society and Catharijne Stichting Utrecht.
Authorship
Contribution: N.W.C.J.v.d.D. performed literature searches and prepared the first draft of the manuscript; and H.M.L., K.C.A., and P.G.R. reviewed and edited the draft of the report and approved the final manuscript.
Conflict-of-interest disclosure: N.W.C.J.v.d.D. has received research support from Celgene and honoraria as speaking fee from Johnson & Johnson and Celgene. H.M.L. has received research support from Celgene, Genmab, and Ortho-Biotech. K.C.A. is a scientific founder of Acetylon and is on the advisory boards of Celgene, Millennium, Onyx, BMS, and Merck. P.G.R. has served on the advisory board of Millennium Pharmaceuticals, Celgene Corporation, Novartis, Johnson & Johnson, and Bristol-Myers Squibb.
Correspondence: Niels W. C. J. van de Donk, Department of Hematology, University Medical Center Utrecht, Heidelberglaan 100, 3584CX Utrecht, The Netherlands; e-mail: n.w.c.j.vandedonk@umcutrecht.nl.
References
- 1.Kyle RA, Maldonado JE, Bayrd ED. Plasma cell leukemia: report on 17 cases. Arch Intern Med. 1974;133(5):813–818. doi: 10.1001/archinte.133.5.813. [DOI] [PubMed] [Google Scholar]
- 2.International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121(5):749–757. [PubMed] [Google Scholar]
- 3.Gluzinski A, Reichenstein M. Myeloma und leucaemia lymphatica plasmocellularis. Wien Klin Wochenschr. 1906;12:336–339. [Google Scholar]
- 4.Avet-Loiseau H, Roussel M, Campion L, et al. Cytogenetic and therapeutic characterization of primary plasma cell leukemia: the IFM experience. Leukemia. 2012;26(1):158–159. doi: 10.1038/leu.2011.176. [DOI] [PubMed] [Google Scholar]
- 5.Tiedemann RE, Gonzalez-Paz N, Kyle RA, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22(5):1044–1052. doi: 10.1038/leu.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia-Sanz R, Orfao A, Gonzalez M, et al. Primary plasma cell leukemia: clinical, immunophenotypic, DNA ploidy, and cytogenetic characteristics. Blood. 1999;93(3):1032–1037. [PubMed] [Google Scholar]
- 7.Noel P, Kyle RA. Plasma cell leukemia: an evaluation of response to therapy. Am J Med. 1987;83(6):1062–1068. doi: 10.1016/0002-9343(87)90942-9. [DOI] [PubMed] [Google Scholar]
- 8.Dimopoulos MA, Palumbo A, Delasalle KB, Alexanian R. Primary plasma cell leukaemia. Br J Haematol. 1994;88(4):754–759. doi: 10.1111/j.1365-2141.1994.tb05114.x. [DOI] [PubMed] [Google Scholar]
- 9.Vela-Ojeda J, Garcia-Ruiz Esparza MA, Rosas-Cabral A, et al. Intermediate doses of melphalan and dexamethasone are better than vincristine, adriamycin, and dexamethasone (VAD) and polychemotherapy for the treatment of primary plasma cell leukemia. Ann Hematol. 2002;81(7):362–367. doi: 10.1007/s00277-002-0480-5. [DOI] [PubMed] [Google Scholar]
- 10.Usmani SZ, Nair B, Qu P, et al. Primary plasma cell leukemia: clinical and laboratory presentation, gene-expression profiling, and clinical outcome with Total Therapy protocols [published online ahead of print April 17, 2012]. Leukemia. doi: 10.1038/leu.2012.107. doi: 10.1038/leu.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hugo ZP, Perry MC, Steensma DP. Extreme leukocytosis in the 21st Century is often iatrogenic [abstract]. Blood (ASH Annual Meeting Abstracts) 2008;112(11):5401. [Google Scholar]
- 12.Pasqualetti P, Festuccia V, Collacciani A, Acitelli P, Casale R. Plasma cell leukemia: a report on 11 patients and review of the literature. Panminerva Med. 1996;38(3):179–184. [PubMed] [Google Scholar]
- 13.Avet-Loiseau H, Daviet A, Brigaudeau C, et al. Cytogenetic, interphase, and multicolor fluorescence in situ hybridization analyses in primary plasma cell leukemia: a study of 40 patients at diagnosis, on behalf of the Intergroupe Francophone du Myelome and the Groupe Francais de Cytogenetique Hematologique. Blood. 2001;97(3):822–825. doi: 10.1182/blood.v97.3.822. [DOI] [PubMed] [Google Scholar]
- 14.Avet-Loiseau H, Li C, Magrangeas F, et al. Prognostic significance of copy-number alterations in multiple myeloma. J Clin Oncol. 2009;27(27):4585–4590. doi: 10.1200/JCO.2008.20.6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jimenez-Zepeda VH, Neme-Yunes Y, Braggio E. Chromosome abnormalities defined by conventional cytogenetics in plasma cell leukemia: what have we learned about its biology? Eur J Haematol. 2011;87(1):20–27. doi: 10.1111/j.1600-0609.2011.01629.x. [DOI] [PubMed] [Google Scholar]
- 16.Chang H, Qi X, Yeung J, Reece D, Xu W, Patterson B. Genetic aberrations including chromosome 1 abnormalities and clinical features of plasma cell leukemia. Leuk Res. 2009;33(2):259–262. doi: 10.1016/j.leukres.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 17.Chiecchio L, Dagrada GP, White HE, et al. Frequent upregulation of MYC in plasma cell leukemia. Genes Chromosomes Cancer. 2009;48(7):624–636. doi: 10.1002/gcc.20670. [DOI] [PubMed] [Google Scholar]
- 18.Fonseca R, Debes-Marun CS, Picken EB, et al. The recurrent IgH translocations are highly associated with nonhyperdiploid variant multiple myeloma. Blood. 2003;102(7):2562–2567. doi: 10.1182/blood-2003-02-0493. [DOI] [PubMed] [Google Scholar]
- 19.Johnson MR, Del Carpio-Jayo D, Lin P, et al. Primary plasma cell leukemia: morphologic, immunophenotypic, and cytogenetic features of 4 cases treated with chemotherapy and stem cell transplantation. Ann Diagn Pathol. 2006;10(5):263–268. doi: 10.1016/j.anndiagpath.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 20.Walters M, Olteanu H, Van Tuinen P, Kroft SH. CD23 expression in plasma cell myeloma is specific for abnormalities of chromosome 11, and is associated with primary plasma cell leukaemia in this cytogenetic sub-group. Br J Haematol. 2010;149(2):292–293. doi: 10.1111/j.1365-2141.2009.08042.x. [DOI] [PubMed] [Google Scholar]
- 21.Mosca L, Musto P, Fabris S, et al. Integrative genomic analysis of primary plasma cell leukemia revealed strong gene and microRNA dosage effect [abstract]. Blood (ASH Annual Meeting Abstracts) 2010;116(21):4040. [Google Scholar]
- 22.Musto P, D'Auria F, Petrucci MT, et al. Final results of a phase II study evaluating lenalidomide in combination with low dose dexamethasone as first line therapy for primary plasma cell leukemia [abstract]. Blood (ASH Annual Meeting Abstracts) 2011;118(21):2925. [Google Scholar]
- 23.Avet-Loiseau H, Attal M, Moreau P, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood. 2007;109(8):3489–3495. doi: 10.1182/blood-2006-08-040410. [DOI] [PubMed] [Google Scholar]
- 24.Fonseca R, Blood EA, Oken MM, et al. Myeloma and the t(11;14)(q13;q32); evidence for a biologically defined unique subset of patients. Blood. 2002;99(10):3735–3741. doi: 10.1182/blood.v99.10.3735. [DOI] [PubMed] [Google Scholar]
- 25.Fonseca R, Blood E, Rue M, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101(11):4569–4575. doi: 10.1182/blood-2002-10-3017. [DOI] [PubMed] [Google Scholar]
- 26.Lloveras E, Granada I, Zamora L, et al. Cytogenetic and fluorescence in situ hybridization studies in 60 patients with multiple myeloma and plasma cell leukemia. Cancer Genet Cytogenet. 2004;148(1):71–76. doi: 10.1016/s0165-4608(03)00233-4. [DOI] [PubMed] [Google Scholar]
- 27.Gutierrez NC, Hernandez JM, Garcia JL, et al. Differences in genetic changes between multiple myeloma and plasma cell leukemia demonstrated by comparative genomic hybridization. Leukemia. 2001;15(5):840–845. doi: 10.1038/sj.leu.2402116. [DOI] [PubMed] [Google Scholar]
- 28.Chang H, Qi X, Jiang A, Xu W, Young T, Reece D. 1p21 deletions are strongly associated with 1q21 gains and are an independent adverse prognostic factor for the outcome of high-dose chemotherapy in patients with multiple myeloma. Bone Marrow Transplant. 2010;45(1):117–121. doi: 10.1038/bmt.2009.107. [DOI] [PubMed] [Google Scholar]
- 29.Sumegi J, Hedberg T, Bjorkholm M, et al. Amplification of the c-myc oncogene in human plasma-cell leukemia. Int J Cancer. 1985;36(3):367–371. [PubMed] [Google Scholar]
- 30.Fabris S, Storlazzi CT, Baldini L, et al. Heterogeneous pattern of chromosomal breakpoints involving the MYC locus in multiple myeloma. Genes Chromosomes Cancer. 2003;37(3):261–269. doi: 10.1002/gcc.10211. [DOI] [PubMed] [Google Scholar]
- 31.Neri A, Baldini L, Trecca D, Cro L, Polli E, Maiolo AT. p53 gene mutations in multiple myeloma are associated with advanced forms of malignancy. Blood. 1993;81(1):128–135. [PubMed] [Google Scholar]
- 32.Bezieau S, Devilder MC, Avet-Loiseau H, et al. High incidence of N and K-Ras activating mutations in multiple myeloma and primary plasma cell leukemia at diagnosis. Hum Mutat. 2001;18(3):212–224. doi: 10.1002/humu.1177. [DOI] [PubMed] [Google Scholar]
- 33.Corradini P, Ladetto M, Voena C, et al. Mutational activation of N- and K-ras oncogenes in plasma cell dyscrasias. Blood. 1993;81(10):2708–2713. [PubMed] [Google Scholar]
- 34.Ortega MM, Faria RM, Shitara ES, et al. N-RAS and K-RAS gene mutations in Brazilian patients with multiple myeloma. Leuk Lymphoma. 2006;47(2):285–289. doi: 10.1080/10428190500300969. [DOI] [PubMed] [Google Scholar]
- 35.Walker BA, Wardell CP, Boyd KD, et al. Hypermethylation is a key feature of the transition of multiple myeloma to plasma cell leukemia [abstract]. Blood (ASH Annual Meeting Abstracts) 2010;116(21):535. [Google Scholar]
- 36.Walker BA, Wardell CP, Chiecchio L, et al. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood. 2011;117(2):553–562. doi: 10.1182/blood-2010-04-279539. [DOI] [PubMed] [Google Scholar]
- 37.Kraj M, Kopec-Szlezak J, Poglod R, Kruk B. Flow cytometric immunophenotypic characteristics of 36 cases of plasma cell leukemia. Leuk Res. 2011;35(2):169–176. doi: 10.1016/j.leukres.2010.04.021. [DOI] [PubMed] [Google Scholar]
- 38.Pellat-Deceunynck C, Barille S, Jego G, et al. The absence of CD56 (NCAM) on malignant plasma cells is a hallmark of plasma cell leukemia and of a special subset of multiple myeloma. Leukemia. 1998;12(12):1977–1982. doi: 10.1038/sj.leu.2401211. [DOI] [PubMed] [Google Scholar]
- 39.Pellat-Deceunynck C, Barille S, Puthier D, et al. Adhesion molecules on human myeloma cells: significant changes in expression related to malignancy, tumor spreading, and immortalization. Cancer Res. 1995;55(16):3647–3653. [PubMed] [Google Scholar]
- 40.Barille S, Collette M, Bataille R, Amiot M. Myeloma cells upregulate interleukin-6 secretion in osteoblastic cells through cell-to-cell contact but downregulate osteocalcin. Blood. 1995;86(8):3151–3159. [PubMed] [Google Scholar]
- 41.Schmidmaier R, Mandl-Weber S, Gaul L, et al. Inhibition of lymphocyte function associated antigen 1 by LFA878 induces apoptosis in multiple myeloma cells and is associated with downregulation of the focal adhesion kinase/phosphatidylinositol 3 kinase/Akt pathway. Int J Oncol. 2007;31(4):969–976. [PubMed] [Google Scholar]
- 42.Edvardsen K, Chen W, Rucklidge G, Walsh FS, Obrink B, Bock E. Transmembrane neural cell-adhesion molecule (NCAM), but not glycosyl-phosphatidylinositol-anchored NCAM, down-regulates secretion of matrix metalloproteinases. Proc Natl Acad Sci U S A. 1993;90(24):11463–11467. doi: 10.1073/pnas.90.24.11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohtake K, Takeuchi T, Saito K, et al. Integrin VLA-5 negative primary plasma cell leukemia. Intern Med. 1993;32(7):565–568. doi: 10.2169/internalmedicine.32.565. [DOI] [PubMed] [Google Scholar]
- 44.Barker HF, Hamilton MS, Ball J, Drew M, Franklin IM. Expression of adhesion molecules LFA-3 and N-CAM on normal and malignant human plasma cells. Br J Haematol. 1992;81(3):331–335. doi: 10.1111/j.1365-2141.1992.tb08236.x. [DOI] [PubMed] [Google Scholar]
- 45.Singh ZN, Owens RB, Nair B, Shaughnessy J, Jr., Barlogie B. Loss of chemokine receptor expression and tetraspanins is correlated with extramedullary disease in multiple myeloma [abstract]. Blood (ASH Annual Meeting Abstracts) 2010;116(21):2983. [Google Scholar]
- 46.Perez-Andres M, Almeida J, Martin-Ayuso M, et al. Clonal plasma cells from monoclonal gammopathy of undetermined significance, multiple myeloma and plasma cell leukemia show different expression profiles of molecules involved in the interaction with the immunological bone marrow microenvironment. Leukemia. 2005;19(3):449–455. doi: 10.1038/sj.leu.2403647. [DOI] [PubMed] [Google Scholar]
- 47.Clayberger C, Wright A, Medeiros LJ, et al. Absence of cell surface LFA-1 as a mechanism of escape from immunosurveillance. Lancet. 1987;2(8558):533–536. doi: 10.1016/s0140-6736(87)92924-2. [DOI] [PubMed] [Google Scholar]
- 48.Ramsingh G, Mehan P, Luo J, Vij R, Morgensztern D. Primary plasma cell leukemia: a Surveillance, Epidemiology, and End Results database analysis between 1973 and 2004. Cancer. 2009;115(24):5734–5739. doi: 10.1002/cncr.24700. [DOI] [PubMed] [Google Scholar]
- 49.Costello R, Sainty D, Bouabdallah R, et al. Primary plasma cell leukaemia: a report of 18 cases. Leuk Res. 2001;25(2):103–107. doi: 10.1016/s0145-2126(00)00102-8. [DOI] [PubMed] [Google Scholar]
- 50.Drake MB, Iacobelli S, van Biezen A, et al. Primary plasma cell leukemia and autologous stem cell transplantation. Haematologica. 2010;95(5):804–809. doi: 10.3324/haematol.2009.013334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pagano L, Valentini CG, De Stefano V, et al. Primary plasma cell leukemia: a retrospective multicenter study of 73 patients. Ann Oncol. 2011;22(7):1628–1635. doi: 10.1093/annonc/mdq646. [DOI] [PubMed] [Google Scholar]
- 52.Kraj M, Kopec-Szlezak J, Poglod R, Kruk B. Flow cytometric immunophenotypic characteristics of plasma cell leukemia. Folia Histochem Cytobiol. 2011;49(1):168–182. doi: 10.5603/fhc.2011.0024. [DOI] [PubMed] [Google Scholar]
- 53.Woodruff RK, Malpas JS, Paxton AM, Lister TA. Plasma cell leukemia (PCL): a report on 15 patients. Blood. 1978;52(4):839–845. [PubMed] [Google Scholar]
- 54.Colovic M, Jankovic G, Suvajdzic N, Milic N, Dordevic V, Jankovic S. Thirty patients with primary plasma cell leukemia: a single center experience. Med Oncol. 2008;25(2):154–160. doi: 10.1007/s12032-007-9011-5. [DOI] [PubMed] [Google Scholar]
- 55.Jimenez-Zepeda VH, Dominguez VJ. Plasma cell leukemia: a rare condition. Ann Hematol. 2006;85(4):263–267. doi: 10.1007/s00277-005-0054-4. [DOI] [PubMed] [Google Scholar]
- 56.Saccaro S, Fonseca R, Veillon DM, et al. Primary plasma cell leukemia: report of 17 new cases treated with autologous or allogeneic stem-cell transplantation and review of the literature. Am J Hematol. 2005;78(4):288–294. doi: 10.1002/ajh.20272. [DOI] [PubMed] [Google Scholar]
- 57.Bernasconi C, Castelli G, Pagnucco G, Brusamolino E. Plasma cell leukemia: a report on 15 patients. Eur J Haematol Suppl. 1989;51:76–83. doi: 10.1111/j.1600-0609.1989.tb01497.x. [DOI] [PubMed] [Google Scholar]
- 58.Buda G, Carulli G, Orciuolo E, et al. CD23 expression in plasma cell leukaemia. Br J Haematol. 2010;150(6):724–725. doi: 10.1111/j.1365-2141.2010.08277.x. [DOI] [PubMed] [Google Scholar]
- 59.Guikema JE, Hovenga S, Vellenga E, et al. CD27 is heterogeneously expressed in multiple myeloma: low CD27 expression in patients with high-risk disease. Br J Haematol. 2003;121(1):36–43. doi: 10.1046/j.1365-2141.2003.04260.x. [DOI] [PubMed] [Google Scholar]
- 60.Hundemer M, Klein U, Hose D, et al. Lack of CD56 expression on myeloma cells is not a marker for poor prognosis in patients treated by high-dose chemotherapy and is associated with translocation t(11;14). Bone Marrow Transplant. 2007;40(11):1033–1037. doi: 10.1038/sj.bmt.1705857. [DOI] [PubMed] [Google Scholar]
- 61.Musto P, Rossini F, Gay F, et al. Efficacy and safety of bortezomib in patients with plasma cell leukemia. Cancer. 2007;109(11):2285–2290. doi: 10.1002/cncr.22700. [DOI] [PubMed] [Google Scholar]
- 62.Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111(5):2516–2520. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Richardson PG, Mitsiades C, Schlossman R, Munshi N, Anderson K. New drugs for myeloma. Oncologist. 2007;12(6):664–689. doi: 10.1634/theoncologist.12-6-664. [DOI] [PubMed] [Google Scholar]
- 64.Chaoui D, Leleu X, Roussel M, et al. Has the prognostic of primary plasma cell leukemia improved with new drugs? [abstract]. Blood (ASH Annual Meeting Abstracts) 2009;114(22):3869. [Google Scholar]
- 65.Neben K, Lokhorst HM, Jauch A, et al. Administration of bortezomib before and after autologous stem-cell transplantation improves outcome in multiple myeloma patients with deletion 17p. Blood. 2012;119(4):940–948. doi: 10.1182/blood-2011-09-379164. [DOI] [PubMed] [Google Scholar]
- 66.Shaughnessy JD, Zhou Y, Haessler J, et al. TP53 deletion is not an adverse feature in multiple myeloma treated with total therapy 3. Br J Haematol. 2009;147(3):347–351. doi: 10.1111/j.1365-2141.2009.07864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Avet-Loiseau H, Leleu X, Roussel M, et al. Bortezomib plus dexamethasone induction improves outcome of patients with t(4;14) myeloma but not outcome of patients with del(17p). J Clin Oncol. 2010;28(30):4630–4634. doi: 10.1200/JCO.2010.28.3945. [DOI] [PubMed] [Google Scholar]
- 68.Esparis-Ogando A, Alegre A, Aguado B, et al. Bortezomib is an efficient agent in plasma cell leukemias. Int J Cancer. 2005;114(4):665–667. doi: 10.1002/ijc.20793. [DOI] [PubMed] [Google Scholar]
- 69.Katodritou E, Verrou E, Gastari V, Hadjiaggelidou C, Terpos E, Zervas K. Response of primary plasma cell leukemia to the combination of bortezomib and dexamethasone: do specific cytogenetic and immunophenotypic characteristics influence treatment outcome? Leuk Res. 2008;32(7):1153–1156. doi: 10.1016/j.leukres.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 70.Grassinger J, Sudhoff T, Andreesen R, Hennemann B. Complete remission and successful stem cell mobilization after treatment of refractory plasma cell leukemia with bortezomib. Ann Hematol. 2006;85(2):132–133. doi: 10.1007/s00277-005-0027-7. [DOI] [PubMed] [Google Scholar]
- 71.D'Arena G, Valentini CG, Pietrantuono G, et al. Frontline chemotherapy with bortezomib-containing combinations improves response rate and survival in primary plasma cell leukemia: a retrospective study from GIMEMA Multiple Myeloma Working Party. Ann Oncol. 2012;23(6):1499–1502. doi: 10.1093/annonc/mdr480. [DOI] [PubMed] [Google Scholar]
- 72.Libby E, Candelaria-Quintana D, Moualla H, Abdul-Jaleel M, Rabinowitz I. Durable complete remission of primary plasma cell leukemia with the bortezomib plus melphalan and prednisone (VMP) regimen. Am J Hematol. 2010;85(9):733–734. doi: 10.1002/ajh.21790. [DOI] [PubMed] [Google Scholar]
- 73.Al-Nawakil C, Tamburini J, Bardet V, et al. Bortezomib, doxorubicin and dexamethasone association is an effective option for plasma cell leukemia induction therapy. Leuk Lymphoma. 2008;49(10):2012–2014. doi: 10.1080/10428190802290660. [DOI] [PubMed] [Google Scholar]
- 74.Lebon D, Gruson B, Garidi R, et al. Bortezomib, doxorubicin and dexamethasone (PAD) prior to high-dose melphalan/autologous stem cell transplantation for newly diagnosed plasma cell dycrasias [abstract]. Blood (ASH Annual Meeting Abstracts) 2009;114(22):4960. [Google Scholar]
- 75.Lebovic D, Zhang L, Alsina M, et al. Clinical outcomes of patients with plasma cell leukemia in the era of novel therapies and hematopoietic stem cell transplantation strategies: a single-institution experience. Clin Lymphoma Myeloma Leuk. 2011;11(6):507–511. doi: 10.1016/j.clml.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 76.Petrucci MT, Martini V, Levi A, et al. Thalidomide does not modify the prognosis of plasma cell leukemia patients: experience of a single center. Leuk Lymphoma. 2007;48(1):180–182. doi: 10.1080/10428190601007570. [DOI] [PubMed] [Google Scholar]
- 77.Gemmel C, Cremer FW, Weis M, et al. Anti-CD20 antibody as consolidation therapy in a patient with primary plasma cell leukemia after high-dose therapy and autologous stem cell transplantation. Ann Hematol. 2002;81(2):119–123. doi: 10.1007/s00277-001-0397-4. [DOI] [PubMed] [Google Scholar]
- 78.Johnston RE, Abdalla SH. Thalidomide in low doses is effective for the treatment of resistant or relapsed multiple myeloma and for plasma cell leukaemia. Leuk Lymphoma. 2002;43(2):351–354. doi: 10.1080/10428190290006143. [DOI] [PubMed] [Google Scholar]
- 79.Bauduer F. Efficacy of thalidomide in the treatment of VAD-refractory plasma cell leukaemia appearing after autologous stem cell transplantation for multiple myeloma. Br J Haematol. 2002;117(4):996–997. doi: 10.1046/j.1365-2141.2002.03537_4.x. [DOI] [PubMed] [Google Scholar]
- 80.Wohrer S, Ackermann J, Baldia C, et al. Effective treatment of primary plasma cell leukemia with thalidomide and dexamethasone: a case report. Hematol J. 2004;5(4):361–363. doi: 10.1038/sj.thj.6200375. [DOI] [PubMed] [Google Scholar]
- 81.Murthy V, Mwirigi A, Ward S, Rassam SMB. Rapid and excellent response to hyper CVAD, particularly with thalidomide, in plasma cell leukaemia and long term remissions following allogeneic stem cell transplantation [abstract]. Blood (ASH Annual Meeting Abstracts) 2009;114(22):4963. [Google Scholar]
- 82.Musto P, Pietrantuono G, Guariglia R, et al. Salvage therapy with lenalidomide and dexamethasone in relapsed primary plasma cell leukemia. Leuk Res. 2008;32(10):1637–1638. doi: 10.1016/j.leukres.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 83.Guglielmelli T, Merlini R, Giugliano E, Saglio G. Lenalidomide, melphalan, and prednisone association is an effective salvage therapy in relapsed plasma cell leukaemia. J Oncol. 2009;2009:867380. doi: 10.1155/2009/867380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goyal M, Mohammad N, Palanki SD, Vaniawala SN. Primary plasma cell leukemia with light chain secretion and multiple chromosomal abnormalities: how successfully treated? A case report with review of literature. Indian J Med Paediatr Oncol. 2010;31(3):96–100. doi: 10.4103/0971-5851.73603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McElwain TJ, Powles RL. High-dose intravenous melphalan for plasma-cell leukaemia and myeloma. Lancet. 1983;2(8354):822–824. doi: 10.1016/s0140-6736(83)90739-0. [DOI] [PubMed] [Google Scholar]
- 86.Mahindra A, Kalaycio ME, Vela-Ojeda J, et al. Hematopoietic cell transplantation for primary plasma cell leukemia: results from the Center for International Blood and Marrow Transplant Research. Leukemia. 2012;26(5):1091–1097. doi: 10.1038/leu.2011.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lebovic DJ, Baz R, Alsina M, et al. Cytogenetics and clinical outcomes of plasma cell leukemia patients: a single institution experience [abstract]. Blood (ASH Annual Meeting Abstracts) 2009;114(22):4932. [Google Scholar]
- 88.Ladetto M, Pagliano G, Ferrero S, et al. Major tumor shrinking and persistent molecular remissions after consolidation with bortezomib, thalidomide, and dexamethasone in patients with autografted myeloma. J Clin Oncol. 2010;28(12):2077–2084. doi: 10.1200/JCO.2009.23.7172. [DOI] [PubMed] [Google Scholar]
- 89.Cavo M, Pantani L, Patriarca F, et al. Superior complete response rate (CR) and progression-free survival (PFS) with bortezomib-thalidomide-dexamethasone (VTD) versus thalidomide-dexamethasone (TD) as consolidation therapy after autologous stem-cell transplantation (ASCT) in multiple myeloma (MM) [abstract]. Blood (ASH Annual Meeting Abstracts) 2011;118(21):1871. [Google Scholar]
- 90.Attal M, Lauwers Vc Marit G, et al. Maintenance treatment with lenalidomide after transplantation for MYELOMA: final analysis of the IFM 2005-02 [abstract]. Blood (ASH Annual Meeting Abstracts) 2010;116(21):310. [Google Scholar]
- 91.Paiva B, Gutierrez NC, Rosinol L, et al. High-risk cytogenetics and persistent minimal residual disease by multiparameter flow cytometry predict unsustained complete response after autologous stem cell transplantation in multiple myeloma. Blood. 2012;119(3):687–691. doi: 10.1182/blood-2011-07-370460. [DOI] [PubMed] [Google Scholar]
- 92.Avet-Loiseau H, Caillot D, Marit G, et al. Long-term maintenance with lenalidomide improves progression free survival in myeloma patients with high-risk cytogenetics: an IFM study [abstract]. Blood (ASH Annual Meeting Abstracts) 2010;116(21):1944. [Google Scholar]
- 93.Morgan GJ, Gregory WM, Davies FE, et al. The role of maintenance thalidomide therapy in multiple myeloma: MRC Myeloma IX results and meta-analysis. Blood. 2012;119(1):7–15. doi: 10.1182/blood-2011-06-357038. [DOI] [PubMed] [Google Scholar]
- 94.Ludwig H, Durie BG, McCarthy P, et al. IMWG consensus on maintenance therapy in multiple myeloma. Blood. 2012;119(3):3003–3015. doi: 10.1182/blood-2011-11-374249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van de Donk NW, Lokhorst HM, Dimopoulos M, et al. Treatment of relapsed and refractory multiple myeloma in the era of novel agents. Cancer Treat Rev. 2011;37(4):266–283. doi: 10.1016/j.ctrv.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 96.Jaskiewicz AD, Herrington JD, Wong L. Tumor lysis syndrome after bortezomib therapy for plasma cell leukemia. Pharmacotherapy. 2005;25(12):1820–1825. doi: 10.1592/phco.2005.25.12.1820. [DOI] [PubMed] [Google Scholar]
- 97.Benson DM, Jr, Smith MK. Effectiveness of lenalidomide (Revlimid) for the treatment of plasma cell leukemia. Leuk Lymphoma. 2007;48(7):1423–1425. doi: 10.1080/10428190701361851. [DOI] [PubMed] [Google Scholar]
- 98.Oka S, Yokote T, Akioka T, et al. Successful treatment of multi-agent chemotherapy with rituximab for IgM plasma cell leukemia. Leuk Res. 2006;30(12):1581–1583. doi: 10.1016/j.leukres.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 99.Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21–33. doi: 10.4065/78.1.21. [DOI] [PubMed] [Google Scholar]
- 100.Kucharz EJ. [Wladyslaw Antoni Gluzinski: an eminent Polish internist, founder of the Polish Society of Internal Medicine]. Pol Arch Med Wewn. 2007;117(5):270–273. [PubMed] [Google Scholar]