

Abstract
Adult stem cells persist throughout the lifetime of the organism and may therefore require specific mechanisms to limit the effects of chronic oxidative stress. Recently, several instructive genetic mouse models have demonstrated the unique susceptibility of stem cells to perturbations in metabolic or redox homeostasis. These results have implications not only for stem cell biology but also suggest a mechanistic link between intracellular oxidants and the decline in regenerative function that occurs as a normal consequence of aging.
Introduction
In the adult mammalian organism, rare mostly quiescent tissue specific stem cells can give rise to an expanding population of committed progenitor cells. These progenitor cells can further divide and differentiate and thereby function as replacement for a range of mature cells that have been lost to injury, damage or disease. Besides the capacity to give rise to various progenitor cells, one unique property of stem cells is their capacity to divide and produce another stem cell, a process known as self-renewal. This property of self-renewal is essential for the stem cell pool to expand during fetal development, as well as to maintain and persist throughout the life of the organism. For instance, a single hematopoietic stem cell (HSC) can divide to give rise to a new HSC (e.g. self-renew), as well as giving rise to various committed progenitor cells. The progenitors can then further expand and divide and in doing so, maintain and replenish all of the blood cell lineages throughout the lifetime of the organism.
The observation that adult stem cells are the source of multiple different types of mature cells and that the stem cell persists over long periods of time suggests that these unique multipotent cells might require specific protection from the long term effects of reactive oxygen species (ROS) and oxidative damage. One adaptation may involve alterations of the intrinsic metabolism of the stem cell. For instance, studies with embryonic stem cells suggest that in the undifferentiated state, these cells rely predominantly on glycolysis to meet their energetic demand [1]. The decreased reliance on mitochondrial oxidative phosphorylation presumably results in a lower intracellular burden of ROS within the long lived stem cell. In contrast, as these pluripotent cells differentiate, there appears to be an increasing reliance on mitochondrial metabolism [1]. Similarly, it would appear that low ambient oxygen conditions are conducive for maintaining stem cell cultures in an uncommitted state [2].
In addition to reducing ROS levels by altering intracellular metabolism, there is some evidence that in vivo, adult stem cells preferentially reside in areas of low oxygen. For instance, previous observations suggest that HSCs are localized to the most hypoxic regions of the bone marrow [3]. The absolute level of oxygen is difficult to accurately assess in this or any niche, although mathematical models suggest that certain regions of the bone marrow may approach levels of oxygen of 1% or less [4]. In contrast, it is worth remembering that most routine ex vivo culturing of stem cells is done in 20% oxygen conditions. There is also evidence that activation of the cellular response to low oxygen is important for overall stem cell function. For instance, hypoxia-inducible factor 1α (HIF-1α) is a transcription factor whose protein level and activity increases in the setting of low oxygen [5]. In turn, HIF-1α regulates the transcription of a host of genes that provides the cell with a variety of adaptive functions including an increased capacity for glucose uptake and a switch towards glycolysis for ATP generation. Interestingly, there also appears to be important interactions between hypoxia, HIF-1α and critical regulators of stem cell activity including both Notch [6] and Wnt signaling [7]. These interactions may in part explain why in mouse models, HIF-1α conditional deletion can give rise to depletion of both neural [7] and hematopoietic stem cells [8]. Furthermore, there is a growing connection between intracellular metabolism and pathways such as Wnt signaling, that are known regulators of stem cell activity [9, 10]
ROS and stem cell function
The above observations suggest that the maintenance of the stem cell pool may require careful regulation of both intracellular metabolism and redox homeostasis. Recent work has solidified this notion and pointed to potentially interestingly links between oxidative stress and the known age-dependent decline in regenerative function. It has been over a half century since Denham Harman proposed what has been come to be known as the ‘Free Radical Theory of Aging’ [11]. The central tenet of this theory is that the accumulation of oxidative damage is the fundamental mechanism by which we age. Harman’s theory remains unproven, and while there is significant evidence to support a link between ROS and lifespan [12], some recent studies have discovered that this relationship is not entirely straightforward or predictable [13]. Leaving the notion of overall aging aside, there is increasing evidence that a rise in oxidants within the stem and progenitor compartment may be particularly deleterious and may in turn contribute to a number of the pathologies that are commonly associated with aging. Most of this evidence has come from mouse genetic models and therefore care must be taken in broadly applying the lessons learned to human aging. Nonetheless, these models are potentially instructive and suggest that redox homeostasis is central to the maintenance of stem cell function.
The first example of the intersection of ROS and stem cell function came from the analysis of mice deficient in the gene ataxia telangiectasia mutated (Atm). Atm encodes for a large serine/threonine protein kinase that is classically activated in the setting of DNA damage. Activation of the ATM kinase in this setting leads to phosphorylation of downstream effector molecules including the tumor suppressor p53, the cell cycle inhibitor Chk2 and the DNA damage response protein and histone variant H2AX [14]. Patients with Ataxia Telangiectasia (AT) have impaired ATM function and develop a constellation of symptoms including neurodegeneration, immune deficiency, small blood vessel dilatations (telangiectasias), hypersensitivity to radiation, increased incidence of cancer particularly lymphomas and leukemia’s, premature aging and death usually in the first or second decade[15].
Mouse knockout models of Atm mimic some but certainly not all aspects of the human disease. While most Atm-/- mice die in the first few months from an aggressive thymic lymphoma, some mice survive and are cancer free. Careful analysis of these surviving mice demonstrated that by six months of age, they all develop a progressive pancytopenia with a decline in red cells, white cells and platelets [16]. Such a phenotype is consistent with a defect within the Atm-/- hematopoietic stem cell (HSC). Further analysis allowed the authors to conclude that Atm-/- HSCs had a profound defect in self-renewal. Consistent with such a self-renewal defect, while the initial levels of HSCs were indistinguishable between wild type and Atm-/- mice, over time, there was a marked decline in the number of HSCs in the bone marrow of Atm-/- mice.
While ATM regulates a number of downstream effectors, the decline in HSCs self-renewal appears to be related to the role of ATM in maintaining the intracellular redox state. Using 2’-7’-dichlorofluorescene diacetate (DCFDA) fluorescence to assess intracellular ROS levels, it was noted that HSCs isolated from Atm-/- mice had a marked increase in ROS levels [16]. HSCs derived from Atm-/- mice also exhibited a rise in level of p16INK4a, a cell cycle inhibitor involved in G1 arrest. Interestingly, p16INK4a levels rise in tissues as a function of age [17], and this rise in p16INK4a had been previously shown to limit stem cell function [18-20]. Treatment of Atm-/- HSCs with the antioxidant N-acetylcysteine (NAC) was shown to reduce p16INK4a expression [16]. Moreover and quite surprisingly, when Atm-/- mice were given NAC supplementation in their drinking water, they did not develop bone marrow failure.
Subsequent studies demonstrated that the rise in ROS observed in HSCs derived from Atm-/- mice were sufficient to activate the p38 MAPK stress kinase pathway [21]. In other contexts, intracellular ROS were known to be potent activators of the p38 MAPK pathway [22]. Analogous to NAC treatment, pharmacological inhibition of the p38 pathway rescued the HSC defect seen in Atm-/- mice [21]. Interestingly, NAC treatment or pharmacological inhibition of p38 also extended the lifespan of wild-type HSC that had been subjected to the stress of serial bone marrow transplantation [21]. While the physiological relevance of being rescued from such serial transplantation can be debated, it should be noted that activation of the DNA damage response including the ATM target H2AX clearly increases in old versus young stem cells [23].
A second example of the intersection of ROS and stem cell function involves the FoxO family of transcription factors. Interest in this family of transcriptional regulators was spurred by observations in C. elegans where the FoxO family member Daf-16 was shown to regulate lifespan [24, 25]. In general, FoxO proteins are tethered in the cytoplasm and translocate to the nucleus following nutrient, oxidative or other stresses. Once in the nucleus, FoxO proteins such as Daf-16 bind to DNA and positively and negatively regulate a large subset of genes broadly involved in immunity, metabolism and stress resistance [26]. Evidence suggests that a modest increase in Daf-16 activity is associated with increased lifespan in the worm. In mammals there are multiple FoxO family members with FoxO3 being most homologous to Daf-16. Interestingly, FoxO3 appears to be able to activate the expression of the antioxidant superoxide dismutase [27], as well as the hydrogen peroxide scavenger catalase [28]. These targets are evolutionarily conserved as both antioxidant proteins are also regulated by Daf-16 in C. elegans[26].
Several studies have now demonstrated that deficiencies in FoxO activity lead to oxidative stress and subsequent defects in stem cell function. The first indication was using a model that led to the conditional deletion of FoxO1, FoxO3 and FoxO4 within the hematopoietic system [29]. Analysis of HSCs from these mice demonstrated a decline in long term repopulating activity. Interestingly this decline in stem cell function correlated with an increase in ROS levels. Consistent with its role in regulating antioxidant defenses, FoxO deficient stem cells exhibited reduced levels of MnSOD and catalase. As had been previously demonstrated for the Atm-/- mice, treatment with NAC rescued the self-renewal defect seen in FoxO deficient stem cells [29]. While these studies used a conditional deletion of three FoxO family members, a subsequent report noted that germline deletion of FoxO3 also resulted in a redox-dependent decline in HSC activity [30]. Similar to what was observed with Atm-/- mice, this FoxO3-/- mouse exhibited activation of the p38 MAPK pathway. Interestingly, within the hematopoietic system, FoxO proteins appear to regulate ROS levels predominantly within the stem cell compartment, as FoxO-deficient myeloid progenitors and wild type myeloid progenitors have similar ROS levels [29]. Indeed, the level of ROS in myeloid progenitors is significantly higher than seen in wild type or FoxO-deficient stem cells suggesting that long lived stem cells maybe much more susceptible to perturbations in redox homeostasis than shorter lived progenitor cells. Interestingly, the role of FoxO proteins in maintaining oxidative balance and stem cell function appears to extend to neural stem cells as well [31, 32].
Another recent example that again links ROS homeostasis with stem cell function came from analyzing mice deleted for the transcription factor Prdm16. Previous studies had identified this factor as an important regulator of the development of brown fat [33]. Surprisingly, Prdm16 appears to be highly expressed in HSCs and within neural stem cells but not in more mature progenitor cells [34]. Mice deficient in Prdm16 show defects in both their neural as well as hematopoietic stem cells. Interestingly, while the absence of Prdm16 leads to stem cell dysfunction in the bone marrow and the brain, only Prdm16 deficient neural stem cells show increased levels of ROS. Consistent with these observations, NAC rescues at least partially the defect observed in the neural stem cells derived from Prdm16 deficient mice as well as the reduced brain size seen in these animals. In contrast, antioxidant administration has no effect on the biology of HSCs from Prdm16 deficient mice. These results again support a connection between ROS homeostasis and the maintenance of stem cell function. Nonetheless, the observation that a single gene such as Prdm16 can have effects on ROS levels in one type of stem cell but not in another suggests that our understanding of redox homeostasis within the context of stem cell biology remains quite primitive at this point.
Energy metabolism and stem cells
In addition to redox homeostasis, there is a growing link between energy metabolism, mitochondrial function and stem cell function. One instructive example involves mice deficient in the Polycomb protein Bmi-1. The Polycomb proteins exist as complexes that function as important epigenetic regulators of gene expression [35]. Mice deleted for Bmi-1 have defects in transcriptional repression exhibiting increase expression of numerous Polycomb target genes. Perhaps the best studied locus for Bmi-1 mediated repression is INK4A. This locus was discussed briefly before and although a single genetic locus it actually encodes for two separate and important regulators, p16INK4A and p19INK4A. In the absence of Bmi-1, levels of both p16INK4A and p19INK4A rise. One well characterized physiological defect in Bmi-1-/- mice is a defect in self-renewal of both hematopoietic and neural stem cells [36-38]. Recently, a link between Bmi-1, mitochondrial function and oxidative stress has been made [39]. In particular, cells from Bmi-1-/- mice show altered mitochondrial function, reduced ATP, and increased ROS levels. The mechanism for all these effects are not completely understood although Bmi1 appears to be involved in the transcriptional repression of a number of intracellular oxidases and known regulators of mitochondrial function [39]. As was observed in the other models discussed, treatment of Bmi-1-/- mice with NAC rescued some of the observed defects.
The connection between a defect in energy metabolism and impaired stem cell biology first demonstrated in mice deficient in Bmi-1 has been reinforced by several subsequent observations in other models. For instance, several recent reports have examined the role of the tumor suppressor LKB1 in stem cell biology. LKB1 is activated under conditions of energetic stress and inactivation of LKB1 in humans leads to a familiar cancer syndrome known as Peutz-Jegher syndrome. Within the context of the hematopoietic stem cell, deletion of LKB1 resulted in a loss of stem cell quiescence [40-42]. In the niche, most HSCs appear to be quiescent with usually less than 10% of the population of stem cells entering the cell cycle at any time. In contrast, deletion of Lkb1 resulted in a significantly higher fraction of HSCs cycling, with a corresponding increase in the initial number of downstream progenitors. In each case this lack of stem cell quiescence and transient increase in progenitors leads to the eventual exhaustion of the stem cell population with a progressive decline in HSC number and circulating hematopoietic cells as the LKB1 deficient mice age. The exact mechanism for these effects is not entirely clear however some evidence suggests that the absence of LKB1 led to a decline in mitochondrial biogenesis and function and/or alteration in intracellular fatty acid metabolism [40, 41]. Interestingly, another recent report has also linked telomere dysfunction to mitochondrial biogenesis. In this report, shorten telomeres led to activation of the tumor suppressor p53. Activation of p53 in turn, led to repression of the transcriptional coactivator PGC-1α [43]. It is well known that PGC-1α is important regulator of mitochondrial number and in this recently described mouse model of telomere dysfunction, the absence of PGC-1α activity resulted in decreased mitochondrial biogenesis, impaired energetics and increased levels of ROS [43]. These changes appeared not only within HSCs but also within mature cells including hepatocytes and cardiomyocytes suggesting this important link between telomeres and mitochondria extends beyond stem cells and functions across a wide range of tissues.
This growing connection between energy metabolism and HSC maintenance was further underscored by analyzing mice lacking the essential autophagy gene Atg7 [44]. Autophagy is a process by which damaged cellular proteins and organelles can be shuttled to the lysosome for degradation and their basic constituents reused by the cell [45]. Autophagy is thought to play an essential role in energy homeostasis during nutrient deprivation and previous results have demonstrated that mice lacking Atg7 [46] or another essential autophagy gene Atg5 [47] die shortly after birth due to impaired energetics. Recent reports suggest that HSCs with a conditional deletion of Atg7 accumulate increased levels of presumably dysfunctional mitochondria, higher ROS levels and a corresponding profound defect in stem cell function [44]. In a separate but related study, mice that accumulate high levels of mitochondrial mutations due to a proofreading-defective mitochondrial DNA polymerase provided further demonstration of the need for intact mitochondrial function to maintain stem cell function [48].
Summary
Taken together these results suggest in adult stem cells, intracellular energetics and redox homeostasis is regulated by a diverse set of factors (Figure 1). Furthermore, in these instructive mouse models, dysregulation of energy or oxidative homeostasis results in profound impairment of stem cell quiescence or self-renewal capacity. It should be noted that most of the data has derived from the hematopoietic and nervous system and so it remains an open question as to how widely the lessons derived so far can be applied to other niches. Furthermore, while the preceding examples suggest multiple separate regulators of ROS and metabolism in stem cells, it is important to note that there is mounting evidence for significant crosstalk between these previously identified pathways. For instance, defects in HSCs from Atm-/- mice can be rescued by overexpression of Bmi1 [16]. Similarly, LKB1 is a known regulator of FoxO activity [49]. Finally, Prdm16 overexpression rescues defects seen in Bm1-/- HSCs [34]. Undoubtedly additional connections will be made as our understanding of the relative hierarchy of these various transcription factors, kinases and epigenetic regulators is refined.
Figure 1.
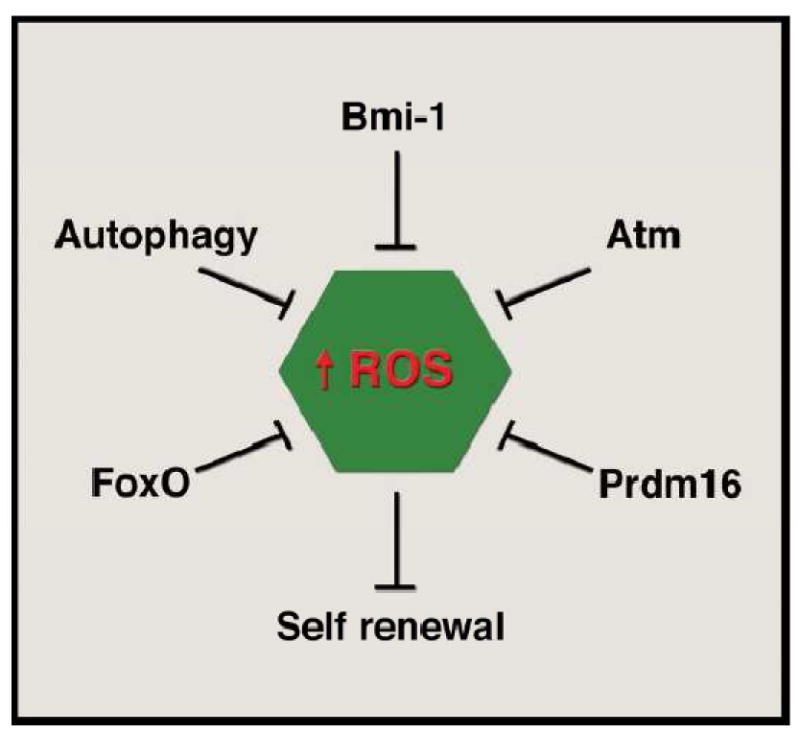
Multiple regulators of the intracellular redox state of the adult stem cell. Various transcription factors (FoxO family, Prdm16), epigenetic regulators (Bmi-1), kinases (ATM), or biological processes (autophagy) can coordinately regulate the levels of reactive oxygen species (ROS) within the stem cell. Dysregulation by genetic deletion of any of these factors in mice leads to alterations in stem cell properties, most notably the ability to undergo self-renewal. In the absence of self-renewal, levels of adult stem cells decline and there is ultimately an impaired capacity to meet the ongoing needs for tissue homeostasis. Although diagramed separately, there is a growing interconnection between the various factors thus far identified. See text for details.
While undoubtedly multifactorial, the age-dependent decline in regenerative and reparative function may reflect an underlying impairment in the biological activity of resident or circulating stem and progenitor cells. The emerging and intriguing links between oxidants, metabolism and stem cell function represents a potential window into this process. A more detailed understanding of those factors that regulate stem cell metabolism and redox homeostasis therefore promises to yield important insight into how we age. Furthermore, these insights may provide unique and potential valuable therapeutic targets to combat a wide range of age-related human diseases.
Highlights.
Recent evidence supports a unique sensitivity of stem cells to oxidative stress
Genes regulating stem cell energy balance and ROS levels recently characterized
ROS levels in stem cells may be important in age-dependent regenerative decline
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Facucho-Oliveira JM, St John JC. The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev. 2009;5:140–158. doi: 10.1007/s12015-009-9058-0. [DOI] [PubMed] [Google Scholar]
- 2.Ezashi T, Das P, Roberts RM. Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci U S A. 2005;102:4783–4788. doi: 10.1073/pnas.0501283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parmar K, Mauch P, Vergilio JA, Sackstein R, Down JD. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A. 2007;104:5431–5436. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow DC, Wenning LA, Miller WM, Papoutsakis ET. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. II. Modified Kroghian models. Biophys J. 2001;81:685–696. doi: 10.1016/S0006-3495(01)75733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 7.Mazumdar J, O'Brien WT, Johnson RS, LaManna JC, Chavez JC, Klein PS, Simon MC. O2 regulates stem cells through Wnt/beta-catenin signalling. Nat Cell Biol. 2010;12:1007–1013. doi: 10.1038/ncb2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, Shima H, Johnson RS, Hirao A, Suematsu M, Suda T. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7:391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Fergusson MM, Wu JJ, Rovira II, Liu J, Gavrilova O, Lu T, Bao J, Han D, Sack MN, Finkel T. Wnt signaling regulates hepatic metabolism. Sci Signal. 2011;4:ra6. doi: 10.1126/scisignal.2001249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoon JC, Ng A, Kim BH, Bianco A, Xavier RJ, Elledge SJ. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev. 2010;24:1507–1518. doi: 10.1101/gad.1924910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 12.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 13.Lee SJ, Hwang AB, Kenyon C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr Biol. 2010;20:2131–2136. doi: 10.1016/j.cub.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 15.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 16.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, Ikeda Y, Mak TW, Suda T. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 17.Sharpless NE. Ink4a/Arf links senescence and aging. Exp Gerontol. 2004;39:1751–1759. doi: 10.1016/j.exger.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 18.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 19.Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akala OO, Park IK, Qian D, Pihalja M, Becker MW, Clarke MF. Long-term haematopoietic reconstitution by Trp53-/-p16Ink4a-/-p19Arf-/- multipotent progenitors. Nature. 2008;453:228–232. doi: 10.1038/nature06869. [DOI] [PubMed] [Google Scholar]
- 21.Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, Ohmura M, Naka K, Hosokawa K, Ikeda Y, Suda T. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12:446–451. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 22.Torres M. Mitogen-activated protein kinase pathways in redox signaling. Front Biosci. 2003;8:d369–391. doi: 10.2741/999. [DOI] [PubMed] [Google Scholar]
- 23.Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447:725–729. doi: 10.1038/nature05862. [DOI] [PubMed] [Google Scholar]
- 24.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang RAC. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 25.Braeckman BP, Vanfleteren JR. Genetic control of longevity in C. elegans. Exp Gerontol. 2007;42:90–98. doi: 10.1016/j.exger.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 26.Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- 27.Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 28.Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- 29.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, Armstrong SA, Passegue E, DePinho RA, Gilliland DG. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, Matsuoka S, Miyamoto T, Ito K, Ohmura M, Chen C, Hosokawa K, Nakauchi H, Nakayama K, Nakayama KI, Harada M, Motoyama N, Suda T, Hirao A. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1:101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 31.Paik JH, Ding Z, Narurkar R, Ramkissoon S, Muller F, Kamoun WS, Chae SS, Zheng H, Ying H, Mahoney J, Hiller D, Jiang S, Protopopov A, Wong WH, Chin L, Ligon KL, DePinho RA. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell. 2009;5:540–553. doi: 10.1016/j.stem.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Renault VM, Rafalski VA, Morgan AA, Salih DA, Brett JO, Webb AE, Villeda SA, Thekkat PU, Guillerey C, Denko NC, Palmer TD, Butte AJ, Brunet A. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell. 2009;5:527–539. doi: 10.1016/j.stem.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scime A, Devarakonda S, Conroe HM, Erdjument-Bromage H, Tempst P, Rudnicki MA, Beier DR, Spiegelman BM. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chuikov S, Levi BP, Smith ML, Morrison SJ. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol. 2010;12:999–1006. doi: 10.1038/ncb2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beisel C, Paro R. Silencing chromatin: comparing modes and mechanisms. Nat Rev Genet. 2011;12:123–135. doi: 10.1038/nrg2932. [DOI] [PubMed] [Google Scholar]
- 36.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 37.Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 38.Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962–967. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, Ahn BH, Kumar NG, Rovira II, Xu XL, van Lohuizen M, Motoyama N, Deng CX, Finkel T. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–392. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, Fletcher-Sananikone E, Colla S, Wang YA, Chin L, Depinho RA. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468:701–704. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gurumurthy S, Xie SZ, Alagesan B, Kim J, Yusuf RZ, Saez B, Tzatsos A, Ozsolak F, Milos P, Ferrari F, Park PJ, Shirihai OS, Scadden DT, Bardeesy N. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468:659–663. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, Cooper M, Kotton D, Fabian AJ, Walkey C, Maser RS, Tonon G, Foerster F, Xiong R, Wang YA, Shukla SA, Jaskelioff M, Martin ES, Heffernan TP, Protopopov A, Ivanova E, Mahoney JE, Kost-Alimova M, Perry SR, Bronson R, Liao R, Mulligan R, Shirihai OS, Chin L, DePinho RA. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, Stranks AJ, Glanville J, Knight S, Jacobsen SE, Kranc KR, Simon AK. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med. 2011;208:455–467. doi: 10.1084/jem.20101145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 48.Norddahl GL, Pronk CJ, Wahlestedt M, Sten G, Nygren JM, Ugale A, Sigvardsson M, Bryder D. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell. 2011;8:499–510. doi: 10.1016/j.stem.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 49.Zhong D, Liu X, Khuri FR, Sun SY, Vertino PM, Zhou W. LKB1 is necessary for Akt-mediated phosphorylation of proapoptotic proteins. Cancer Res. 2008;68:7270–7277. doi: 10.1158/0008-5472.CAN-08-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]