

Abstract
Long-term humoral immunity elicited by pathogens and vaccines alike relies upon the generation of both memory B cells (Bmem) and long-lived plasma cells (PCs). Virtually all vaccine formulations induce the concomitant emergence of both Bmem and PCs, suggesting that the emergence of these two differentiated B cells subsets is commonly controlled. Evidence presented shows specific Toll-like Receptor (TLR) agonists coupled with soluble protein antigen (sAg) can selectively induce the expansion of antigen specific Bmem in the absence of PC generation. The co-administration of either TLR 3 or 9 agonists with sAg induced germinal centre (GC) formation, antigen-specific Bmem, but failed to substantively induce the generation of long-lived bone marrow (BM) PCs. Upon re-challenge, high levels of PCs were induced with concomitant high titres of antigen-specific serum IgG. Hence, vaccines can be developed that can prime and protect the host to subsequent infectious agents without initial, high levels of antibody production. Furthermore, these studies suggest that the signals that govern the expansion and differentiation of Bmem can be uncoupled from those that induce long-lived BM PCs.
Keywords: Memory B cell, Vaccine, Adjuvant, TLRs, B Cell Differentiation
1. Introduction
Memory B cells (Bmem) play a central role in protecting the host against pathogens. The generation and maintenance of this subset of B cells has long been considered an exclusive property of the acquired immune system and is dependent upon CD4 T cell help, B cell clonal expansion, germinal center (GC) formation and differentiation to Bmem [1,2]. Recent findings have clearly shown that the innate immune system, through direct and indirect effects on T cells and B cells, can exert profound influence on the generation of Bmem and plasma cells (PCs). [3-6]. A number of groups have reported TLR-induced activation and differentiation of B cells in vitro. Ligation of TLRs 3, 4, 7 and 9 on B cells have all been shown to induce polyclonal activation and differentiation in vitro [7-11]. In vivo, TLR agonists have been shown to enhance humoral responses [7,8]. Furthermore, experiments with mice lacking MyD88 have shown that this effect is B cell-intrinsic and is independent of dendritic cells (DCs) [4]. As many vaccine formulations used in both the clinic and laboratory setting utilize complex combinations of antigen coupled with adjuvant that often contain multiple TLR signals in the presence of a depot effect ( which enhances the pharmacokinetics of the antigen), we investigated whether select TLR agonists often present within these vaccine formulations can differentially induce the generation of Bmem and PCs when co-administered with soluble antigen (sAg). We systematically evaluated a wide range of TLR agonists and their ability to induce long-lived humoral immunity in the absence of any overt depot effect. Surprisingly, co-injection of TLR3 and 9 agonists with sAg induced generation of Bmem, but failed to generate high numbers of long-lived BM PCs. This result could be modulated by injection of additional TLR agonists or emulsifying the TLR agonists and sAg in IFA. Data presented here demonstrate a novel means to selectively induce Bmem in vivo by controlling TLR signals during immunization and provide an exciting new strategy for the formulation of humoral immunity-specific vaccines.
2. Materials & Methods
2.1. Mice & Reagents
C57BL/6 and BALB/c (Jackson Laboratory, Bar Harbor, ME) mice, 10-12 weeks old, were maintained in the specific pathogen-free animal facility at Dartmouth Medical School, Lebanon, New Hampshire.
The following monoclonal antibodies were utilized for these studies: APC-Cy7-Rat IgG2a anti-mouse B220 (clone RA3-6B2), APC- Rat IgG2a anti-mouse B220 (clone RA3-6B2), APC-Rat IgG2a anti-mouse CD38 (clone 90) (Ebiosciences, San Diego, CA); FITC- Rat IgM anti-mouse GL7 (clone GL7) (BD PharMingen, San Diego, CA); Biotin-Peanut Agglutinin (PNA; Vector Laboratories, Burlingame, CA); Rat IgG1 anti-mouse CD40 (Clone FGK 45) and Armenian Hamster IgG1 anti-mouse CD154 (Clone MR1) (Bioexpress, Lebanon, NH); Rat IgG2a anti-mouse IgD (clone 11-26) antibody (generously provided by Dr. Thomas Waldschmidt) was fluorescinated and biotinylated in the lab. Incubation with biotinylated antibodies was followed by incubation with streptavidin- peridinine chlorophyll protein (PerCP; BD PharMingen, San Deigo, CA) or streptavidin-alexa Flour488 (Molecular Probes, Eugene, OR). In order to identify antigen-specificity, cells were stained with Phycoerythrin (at 10μg/ml; Cyanotech Corp., Kailua-Kona, HI).
2.2. Immunization
All mice were challenged s.c (in the footpad and flank) with 10μg of Phycoerythrin (PE; Cyanotech Corp., Kailua-Kona, HI). PE was given emulsified in complete Freund's adjuvant, or incomplete Freund's adjuvant (CFA and IFA; Sigma-Aldrich; St.Louis, MO) or in 1XPBS containing either 25μgs Peptidoglycan (PGN; TLR2 agonist), or 50μgs Poly I:C (pIC; TLR3 agonist), or 2μgs Lipopolysachharide (LPS;TLR4 agonist) (Sigma-Aldrich; St.Louis, MO ); or 100μgs CpG 1826 (CpG;TLR9 agonist; Eurogentec, San Diego, CA); or 100μgs S27609 (TLR7 agonist) which was kindly provided by 3M Pharmaceuticals (St.Paul, MN) [12]. In case of in vivo recall experiments, mice that had been immunized for 4-6 weeks were re-challenged i.p. with 100μg of PE in 1XPBS, and analyzed for memory responses 5 days after re-challenge.
2.3. Flow cytometry
Cells from draining LNs (popliteal, inguinal, and axillary) were washed in 5% FCS in BSS and resuspended in low-pH (pH 4.0) acetate buffer containing 0.05 M Na acetate, 0.085 M NaCl, and 0.005 M KCl and 2% FCS in distilled H2O, for removal of cytophilic Ig. Samples were incubated on ice for 1 minute followed by the addition of an equal volume of 0.1 M Tris buffer, pH 8.0, containing 2% FCS. Samples were washed twice in 5% FCS BSS and then stained. Chromatographically purified rat IgG was used as isotype control. For all studies, nonspecific staining was further reduced by the addition of heat-inactivated rat serum. Antibody incubations were for 20 min at 4°C followed by washing in BCS/BSS. A minimum of 500,000 events per sample was collected on a FACScan (BDBiosciences, San Deigo, CA) with 4th and 5th color upgrade by Cytek (Gordonsville, VA). Dead cells were excluded based on forward and 90° light scatter. Data were analyzed with FlowJo software (Treestar, CA).
2.4. Immunohistochemistry
On day 21 after immunization, intact lymph nodes were harvested and 4- to 5-μm frozen sections were fixed in acetone at 4°C for 15 minutes and allowed to air dry at 4°C prior to staining. Sections were stained in 5% BCS- PBS for germinal centre formation, identified by PNA+B220+PE+ cells. To block non-specific binding, 10% normal rat serum was included in the staining process. Samples were analyzed on a Zeiss LSM 510 confocal microscope using Zeiss LSM software (Carl Zeiss, Oberkochen, Germany).
2.5. ELISA & ELISPOT Assay
Antigen-specific IgG-secreting cells were enumerated by an antigen-specific ELISPOT assay as previously described [13]. Briefly, PE -specific Plasma cells (PCs) were enumerated by an IgG ELISPOT assay. After isolation of cells, 2.4 × 106 cells per well were apportioned to PE-coated Multi screen® Millipore 96-well plates (Fisher scientific; USA) and threefold serial dilutions were made before incubation. Plates were incubated for 5 h at 37°C within a CO2 incubator. After the incubation, plates were washed in 0.05% Tween and in nano-pure water. PCs were detected by HRP-conjugated anti–mouse IgG (H+L) (Southern Biotech, Birmingham, AL). ELISPOTS were developed by a SIGMA FAST BCIP/NBT (Sigma, St. Louis, MO) chromagen substrate. ELISPOTS were enumerated by direct visual counting using a dual-axis light-dissecting microscope.
Circulating antibodies in sera was measured by an antigen-specific ELISA. PE was used to capture all antigen-specific Ig and captured antibodies were then detected with an enzyme-conjugated goat ant-mouse IgG (H+L) (Southern biotech, Birmingham, AL).
3. Results
3.1. Generation of a robust antigen-specific B cell response upon immunization with soluble antigen and TLR agonist
Immunization with sAg rarely elicits robust, humoral immune responses. Most immune responses that are generated against soluble proteins rely either on an oil or alum adjuvant in combination with multiple TLR agonists, like CFA. CFA contains many TLR agonists of which some or all may contribute to the robust humoral immune responses that it induces. To begin to decipher the contribution of distinct TLRs in inducing humoral immunity, different TLR agonists were used to vaccinate mice to sAg. As such, mice were immunized with soluble PE (10μg) along with a range of TLR agonists. As a positive control, a group of mice were also immunized with PE emulsified in CFA (sAg/CFA). In mice immunized with CFA or sAg/CpG (TLR 9 agonist), enhanced polyclonal B cell expansion was evident (Fig.1B). In contrast, a TLR3 agonist, PolyI:C (pIC) administered in conjunction with sAg induced expansion of antigen-specific B cells in the absence of generalized B cell expansion (Fig.1B & C). Like pIC, CpG induced an increase in the frequency of antigen-specific B cells but commensurate with an increase in total B cell number. TLRs 3, 4, 7 and 9 have all been implicated in B cell activation in vitro and in vivo [7,8,10,11,14-18]. Surprisingly, LPS, a TLR4 agonist, did not induce an increase in the frequency of antigen-specific response in our experiments [8]. The same was observed with TLR7, which has been shown to induce polyclonal B cell activation both in vitro and in vivo [11,16].
Figure I.
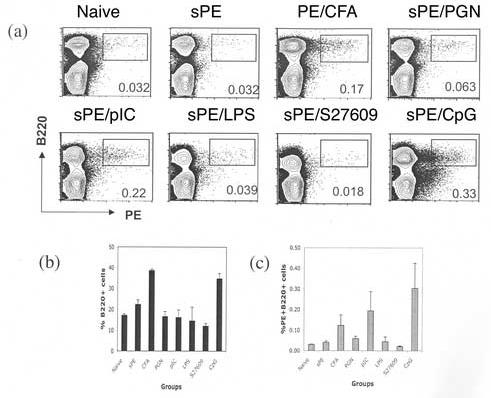
Induction of an Antigen-specific B cell response on immunization with sAg and TLR agonist. (a) C57BL/6 mice were immunized with sAg/TLR agonist or sAg/CFA. Draining Lymphnodes were isolated 7 days post- immunization and stained for B220 and PE. Percent B220+ (b) and B220+PE+ (c) cells in draining LN were calculated. Standard error was calculated between values from three independent mice for each group. Data is representative of three independent experiment. Students T- test was used to calculate P values between sPE and PE/CFA immune mice (0.04); sPE and sPE/pIC immune mice (0.06); and sPE and PE/CpG (0.009).
3.2. Analysis of antigen-specific GC formation induced by TLR agonists and antigen
Most of the literature on TLR agonists suggest that they polyclonally activate B cells in a T-independent manner. It has been shown that CpG induces B cell proliferation and differentiation into PCs [10,18]. The TLR 3 agonist pIC has been shown to activate B cells either directly or indirectly through the activation of dendritic cells and macrophages [7,9]. To gain further insights into the fate of TLR-activated B cells, the differentiation of Ag-specific B cells to GC B cells was investigated. Following the administration of sAg/TLR agonist, PE-specific GCs were formed by 7 days post-priming, with well over 50% PE-specific B cells expressing GL7 and losing expression of CD38, a GC phenotype, by three weeks post-immunization (Fig 2A,S1). Evaluation over a longer time course revealed that the kinetics and magnitude of the number of antigen-specific GC cells induced by CFA/Ag vs TLR/sAg were similar from weeks 1 thru 5. (Fig S1). In addition, immunohistochemical analysis confirmed bona fide GC formation induced by sAg/TLR agonist by staining for PNA and PE-binding (Fig. 2C). Interestingly, CpG induced far more PNA positive B cells that were Ag-nonspecific compared to pIC, as was suggested by the ability of CpG to be an overt B cell mitogen.
Figure II.
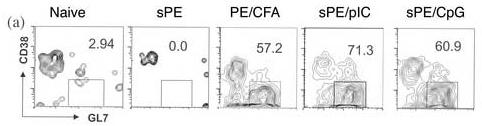
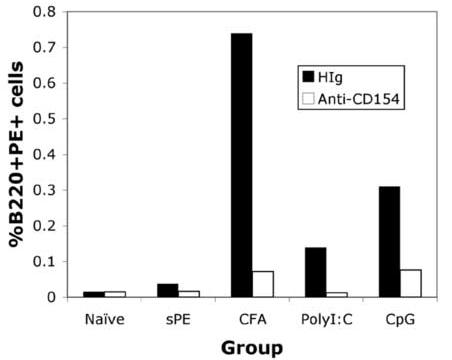
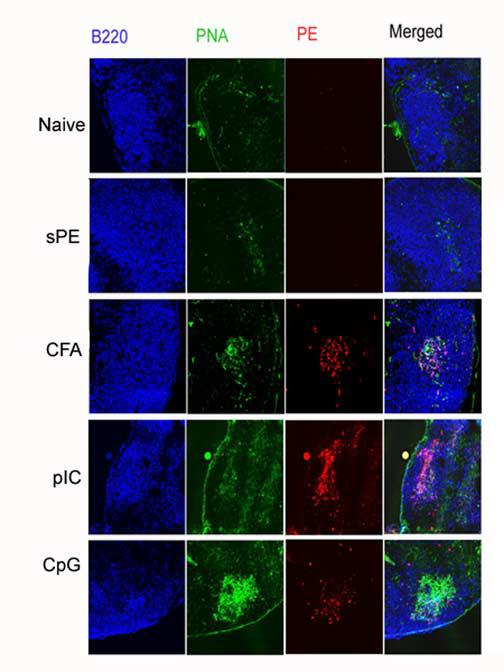
GC Formation in response to soluble PE/TLR agonist immunization. (a) Draining LNs were isolated 3 weeks post- immunization with sAg/TLR agonist and stained for B220, PE, CD38 and GL7. B220+PE+ cells were gated and analysed for down-regulation of CD38 and up-regulation of GL7. These data are representative of four independent experiments. (b) Mice were immunized with sAg/TLR agonist and treated with 250μg of anti-CD154 or Hamster Ig for every three days for a period of three weeks. At three weeks, draining LNs were isolated and stained for B220 and PE. Percent B220+PE+ cells were calculated. Data is representative of two independent experiments. (c) Draining LNs were frozen at week3 for immuno-histology. Frozen tissue sections were stained for B220, PE and PNA. Data is representative of two independent experiments.
Since recent reports suggest that GC-formation can also be induced in response to T-independent antigens, mice were treated with αCD154 antibody to block CD154-dependent B cell activation [19-21]. Mice treated with αCD154 failed to generate any PE-specific B cells, clearly indicating that sAg/TLR agonist- induced GC-formation was a T-dependent phenomenon (Fig.2B).
3.3. Failure to generate Long-lived BM PCs in response to sAg/TLR agonist immunization
Robust, GC formation to TD antigens using alum or CFA inevitably leads to the generation of long-lived BM PCs, and an increase in titre of Ag-specific antibody in the serum of immune mice. In general, the fate of Ag-specific GC B cells is to differentiate to long-lived PCs, PC pre-cursors and/or Bmem [22-25]. The data show that either pIC or CpG and sAg is sufficient to induce the expansion of Ag-specific B cells and GC formation. Initial insight into the long-lived fate of B cells following immunization with sAg/ TLR agonist was provided by analysis of Ag-specific IgG levels in the serum of immune mice. As seen in Fig. 3A, mice immunized with sAg/ TLR agonists, had low levels of PE-specific Ig in sera 3 weeks post-immunization which failed to increase with time. In contrast, mice immunized with sAg /CFA produced high titres of antigen-specific serum Ig. Additionally, the number of Ag-specific BM PCs were also quantified (Fig 3B). While high numbers of ELISPOT positive cells (PCs) were seen in mice immunized with sAg/CFA, substantially lower numbers of PCs were present in mice immunized with sAg/TLR agonist in mice immunized both 3 and 4-6 weeks prior, indicating the absence of PC generation.
Figure III.
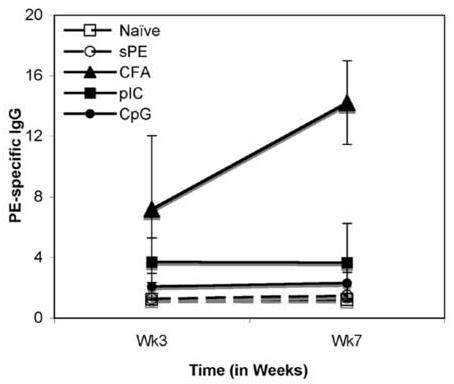
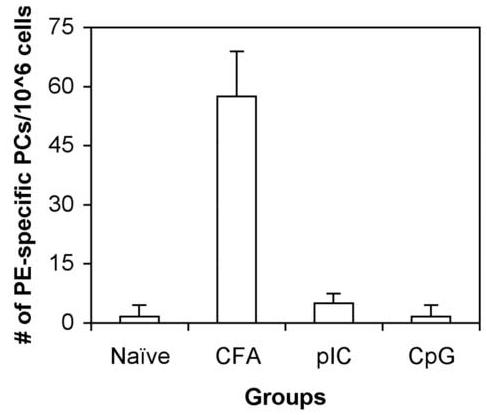
Lack of long-lived PCs in the BM with sAg/TLR immunization. (a) C57BL/6 mice immunized with sAg/TLR agonist were bled at Weeks, 3 and 7 post-immunization. PE-specific Ab in sera was quantitated by ELISA. Data is representative of two independent experiments. (b) Bone marrow (BM) was isolated 4-6 weeks post-immunization and number of PE-specific Plasma cells (PCs) were enumerated by ELISPOT analysis. Data is representative of 5 independent experiments.
3.4. Generation of Bmem cells in response to sAg/TLR stimulus
Immunization with sAg/TLR agonist produced a robust immune response that induced GC formation but failed to generate long-lived BM PCs. Lack of long-lived immunity in these mice may suggest that the generation of Bmem in these mice may also be absent. To evaluate if sAg/TLR administration induced B cell memory, immunized mice were re-challenged with soluble antigen and the recall response was evaluated 5 days later. As seen in Table 1, upon recall, mice immunized with sAg/CFA produced high levels of PCs in LN, SPL and BM, as anticipated. However, high numbers of PCs were now observed in mice initially primed with sAg/TLR agonist upon re-challenge with sAg.
Table I.
In vivo-recall response in C57BL/6 mice immunized with sAg/TLR agonist. C57BL/6 mice immunized with sAg/TLR agonist were re-challenged with 100μg of sAg at 4 weeks i.p. 4 weeks post primary immunization. Draining LNs, Spleen (SPL) and BM was isolated on day 5 post-challenge and number of PE–specific PCs were enumerated by ELISPOT analysis.
| Group | LN Primary |
LN Secondary |
SPL Primary |
SPL Secondary |
BM Primary |
BM Secondary |
|---|---|---|---|---|---|---|
| Naïve | 1± 1 | 0± 0 | 0± 0 | 0± 0 | 0± 0 | 0± 0 |
| sPE | 0± 0 | 0± 0 | 0± 0 | 0± 0 | 1±1 | 0± 0 |
| CFA | 50±1 | 4866±157 | 18±7 | 5505±272 | 57±8 | 467±2 |
| pIC | 0 ± 0 | 167±19 | 0 ± 0 | 1401±239 | 0 ± 0 | 122±1 |
| CpG | 2±1 | 321±19 | 1 ± 0 | 2171±304 | 3 ± 0 | 134±54 |
Phenotypic analysis of differentiated B cells in immune mice also revealed the induction of Bmem by sAg/TLR immunization. As seen in Fig.4A, on day 10 post secondary challenge, there was expansion of IgDlo PE-binding cells in the LNs of sAg/TLR agonist immunized mice. Re-immunization of mice initially immunized with sAg/CFA did not increase the frequency of IgDlo PE-binding B cells even though it induced PC formation, likely due to the depot of long-lived antigen in these mice (Data not shown).
Figure IV.
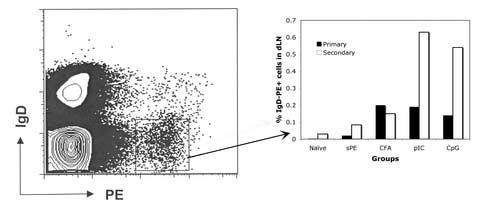
Antigen-specific expansion of Bmem cells. (a) Balb/C mice immunized with sAg/TLR agonist or sAg/CFA were re-challenged with 100μg of sAg i.p. at 4 weeks. Draining LNs were isolated and analysed for antigen-specific Bmem expansion 10 days after re-challenge by staining for IgD and PE. Data is representative of two independent experiments.
3.5. Failure to generate long-lived PCs can be partially remedied by provision of additional TLR agonists or thru emulsification in IFA
The lack of generation of long-lived PCs in sAg/TLR agonist immunization is likely due to insufficient signals. Mice were immunized with sAg and different combinations of three TLR agonists (LPS, pIC, and CpG) or agonistic α-CD40 injected i.p. As seen in Fig.5, mice immunized with sAg plus a combination of TLR agonists showed an additive increase in long-lived BM PC numbers, with no impact on PC numbers afforded by agonistic α-CD40. Additionally, immunization with sAg/IFA alone or with the addition of TLR3 or TLR9 agonists rescued the generation of long-lived PCs in the BM, with no discernable difference between the groups. Our results clearly show that by adding layers of TLR agonists, one can control the output of the humoral response.
Figure V.
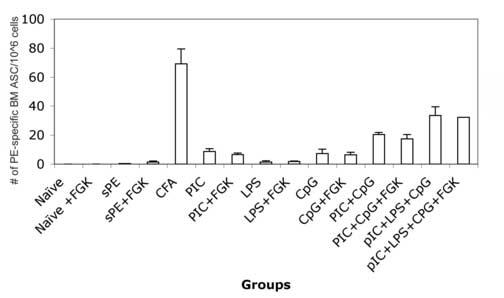
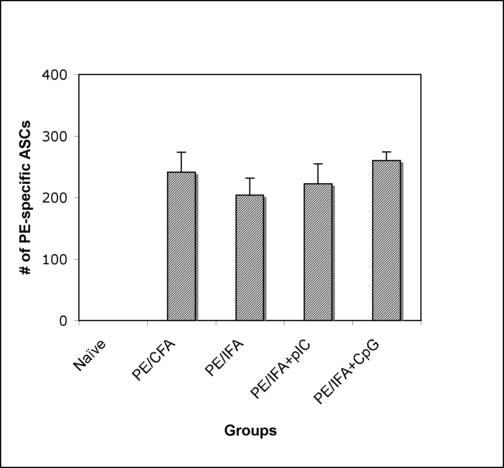
Defect in the generation of long-lived PCs can be remedied by provision of additional signals. (a) Balb/C mice were immunized with PE and a combination of TLR agonists –pIC, CpG, and LPS, in the presence or absence of anti-CD40. BM was isolated 4 weeks post-immunization and analysed for PE-specific long-lived PCs in the BM. Data is representative of two independent experiments. (b) Balb/C mice were immunized with PE either emulsified in CFA or IFA alone or IFA containing pIC or IFA containing CpG. BM was isolated 4-6 weeks post-immunization and analysed for PE-specific long-lived PCs in the BM. Data is representative of three independent experiments.
4. Discussion
Historically, immunologists have used complex concoctions of activators to elicit humoral immunity. Most of the published data generated for assessing Bmem and PC development have been generated using CFA or IFA, killed B. Pertussis, Ribi and Alum, some of which contain overt TLR agonists [24,26]. This report shows that molecularly defined vaccine compositions can restrict the extent of humoral immune responses in vivo. Mice immunized with soluble antigen and a defined TLR agonist showed GC formation and Bmem generation. In comparison to CFA, both CpG and pIC are similar in their capacity to induce the expansion of antigen specific B cells and GC formation (Fig.1&2) over an extended time course (Fig S1). While the memory response observed in mice immunized with CFA is 2-3-fold more robust then with defined TLR agonists, it is clear that TLR agonists can indeed induce Bmem. However, these B cells failed to generate long-lived PCs in the BM (Figs.1, 2 &3). Proof that Bmem were induced by this vaccine approach was afforded by the fact that, on recall with sAg, there was a 50-100 fold increase in the number of BM PCs as early as day 5 post-challenge (Table1) in immune mice. Further, we could modulate the primary response induced by sAg/TLR agonist and increase long-lived BM PC numbers by providing additional TLR agonists, suggesting this effect is quantitative in regard to the number of TLR signals required for humoral priming (Fig.5a). Thus, our data clearly show that defined TLRs can trigger restricted patterns of B cell differentiation in vivo.
A number of studies have shown that TLR agonists can exert direct and indirect effects on B cell function in vivo. TLR 3 and TLR9 have been shown to induce isotype switching in B cells in either IFN-dependent or IFN-independent manner [7,15]. Recent work by Bernasconi et al. showed that TLR signals play an important role in both maintaining Bmem and inducing their differentiation into PCs [27,28]. In vivo studies by Tough and colleagues showed that immunization in the presence of pIC or IFN-α augments humoral responses in a DC-dependent manner [7]. Pertinent to our study, a report from Medzhitov and colleagues suggest that B cells lacking MyD88, an adaptor protein that associates with the cytoplasmic domain of TLRs, respond poorly to sAg/TLR agonists in IFA and alum [4]. However, the requirement for TLR agonists in driving the primary humoral response has recently come under scrutiny with the finding that a number of commonly used vaccine adjuvants such as alum, CFA and RIBI adjuvants were still able to drive considerable humoral responses in mice lacking the components of TLR signalling [29]. It appears that the depot effect provided by such adjuvants is dominant over their ability to agonize TLR receptors during induction of a humoral immune response. In the study presented here, we eliminate the depot effect of adjuvants by using TLR agonists coupled to sAg to induce a humoral response, enabling us to examine the qualitative and quantitative impact of different TLR agonists during priming. It is indeed surprising that individual TLR agonists, when segregated and administered with antigen, can selectively drive the generation of Bmem in the virtual absence of antigen-specific PC. This suggests that the multiple TLR agonists inherent within a pathogen may play a quantitative role (and potentially a qualitative role as indicated by the failure of TLR4 and TLR7 agonists to induce a primary humoral response) in the induction of humoral immunity: TLR3 or TLR9 ligation alone functions to induce Bmem generation, when these TLR agonists are coupled together, both Bmem and long-lived PCs are generated. Our data indicates that the constituents of our vaccines were equally able to generate GCs as shown by Figure S1, thus the defect in long-lived PC generation observed with sAg/TLR3 and TLR9 agonists likely lies in the development of PCs from GC B cells.
Another report in the literature that describes the generation of Bmem in the absence of long-lived PCs such as that reported here is that by Bachmann and colleagues, who found that the immunization of mice lacking the complement receptors CD21/35 resulted in the absence of long-lived PCs despite normal GC and Bmem formation [30]. This impairment of PC generation was found to be due to an increase in the B-cell receptor (BCR) signalling threshold because of the absence of CD21/35 from the BCR. As a result of attenuated BCR signalling throughout the course of the primary immune response, the PC transcriptional pathway was never induced, suggesting that high BCR signalling strength is required for PC generation, with moderate signal strength is sufficient for Bmem generation. Our findings are significant in this context as they implicate TLR signals as able to modulate the PC transcriptional pathway. Likely, the sum signal strength induced by sAg/TLR agonist on the B cell did not reach the threshold required to induce the PC transcriptional pathway within responding antigen-specific B cells. The addition of further signals such as that afforded by additional TLR agonists or a depot effect overcame this threshold and rescued PC development. This suggests that we modify our view on which signals drive the PC transcriptional pathway within responding B cells. Current work indicates that high BCR affinity to antigen and subsequent BCR signalling is the primary variable mediating the PC transcriptional pathway.[30,31] Our work suggests that insufficient inflammatory signals such as those initiated by a single TLR agonist either fail to induce the PC transcriptional pathway irrespective of BCR affinity to antigen, or fail to generate antigen-specific clones with a high enough affinity to initiate the PC transcriptional pathway within the GC. It appears that the addition of two TLR agonists during priming crossed the threshold required for the responding B cell to initiate the PC transcriptional pathway within the GC. It is apparent that signals independent of BCR signalling, such as TLR agonists, modulate PC development, with these signals able to be exploited to generate memory in the absence PCs. Thus, the ‘sum signal strength’ on the responding B cell constituted by BCR affinity to antigen as well as signals driven by TLR agonists mediates PC generation.
The findings of this study have significant implications in the context of vaccine development. A safety concern with vaccines is that the high-titered primary humoral immune response against the vaccine may cross-react to self and cause autoimmunity [32,33]. Several groups have shown an association between vaccination and autoimmunity, such as Hepatitis B vaccine (HBV) inducing anti-phospho lipid antibodies [34]. Additionally, strong primary responses to vaccines can have maltoward symptoms other then autoimmunity: an inappropriate antibody response to the measles component of the Measles-Mumps-Rubella vaccine has been correlated to pathogenesis of autism [35]. Hence, strategies to limit the intensity of the primary immune response to vaccination may improve the safety profile of our vaccines.
Our studies clearly show that adjuvants can be formulated such that high levels of Bmem can be induced in the absence of high titres of antigen-specific Ig. Furthermore, we show that upon challenge, the immune host makes a robust secondary antibody response. As such, by carefully choosing the appropriate TLR agonist, safer vaccines can be formulated.
Supplementary Material
{kind=link}
Acknowledgements
This work was supported by NIH AI26296 to RJN
We would like to acknowledge Kathryn Bennet for help with animal care and housing. We would also like to thank Shinichiro Fuse for discussion.
Abbreviations
- sAg
Soluble Antigen
- PE
Phycoerythrin
- Bmem
Memory B cell
- PC
Plasma cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McHeyzer-Williams MG, Ahmed R. Current Opinion in Immunology. 1999;11:172–9. doi: 10.1016/s0952-7915(99)80029-6. [DOI] [PubMed] [Google Scholar]
- 2.McHeyzer-Williams LJ, McHeyzer-Williams MG. Annual Review of Immunology. 2005;23:487–513. doi: 10.1146/annurev.immunol.23.021704.115732. [DOI] [PubMed] [Google Scholar]
- 3.Janeway CA, Jr., Medzhitov R. Annual Review of Immunology. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 4.Pasare C, Medzhitov R. Nature. 2005;438:364–8. doi: 10.1038/nature04267. [DOI] [PubMed] [Google Scholar]
- 5.Pasare C, Medzhitov R. Immunity. 2004;21:733–41. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 6.Pasare C, Medzhitov R. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 7.Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, Tough DF. Immunity. 2001;14:461–70. doi: 10.1016/s1074-7613(01)00126-1. [DOI] [PubMed] [Google Scholar]
- 8.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S. Journal of Immunology. 1999;162:3749–52. [PubMed] [Google Scholar]
- 9.Finkelman FD, Svetic A, Gresser I, Snapper C, Holmes J, Trotta PP, Katona IM, Gause WC. Journal of Experimental Medicine. 1991;174:1179–88. doi: 10.1084/jem.174.5.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. Nature. 1995;374:546–9. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 11.Bekeredjian-Ding IB, Wagner M, Hornung V, Giese T, Schnurr M, Endres S, Hartmann G. Journal of Immunology. 2005;174:4043–50. doi: 10.4049/jimmunol.174.7.4043. [DOI] [PubMed] [Google Scholar]
- 12.Ballas ZK, Krieg AM, Warren T, Rasmussen W, Davis HL, Waldschmidt M, Weiner GJ. Journal of Immunology. 2001;167:4878–86. doi: 10.4049/jimmunol.167.9.4878. [DOI] [PubMed] [Google Scholar]
- 13.Erickson LD, Durell BG, Vogel LA, O'Connor BP, Cascalho M, Yasui T, Kikutani H, Noelle RJ. Journal of Clinical Investigation. 2002;109:613–20. doi: 10.1172/JCI14110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin L, Gerth AJ, Peng SL. European Journal of Immunology. 2004;34:1483–7. doi: 10.1002/eji.200324736. [DOI] [PubMed] [Google Scholar]
- 15.Liu N, Ohnishi N, Ni L, Akira S, Bacon KB. Nature Immunology. 2003;4:687–93. doi: 10.1038/ni941. [DOI] [PubMed] [Google Scholar]
- 16.Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:5598–603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogata H, Su I, Miyake K, Nagai Y, Akashi S, Mecklenbrauker I, Rajewsky K, Kimoto M, Tarakhovsky A. Journal of Experimental Medicine. 2000;192:23–9. doi: 10.1084/jem.192.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poeck H, Wagner M, Battiany J, Rothenfusser S, Wellisch D, Hornung V, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Blood. 2004;103:3058–64. doi: 10.1182/blood-2003-08-2972. [DOI] [PubMed] [Google Scholar]
- 19.Foy TM, Laman JD, Ledbetter JA, Aruffo A, Claassen E, Noelle RJ. Journal of Experimental Medicine. 1994;180:157–63. doi: 10.1084/jem.180.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lentz VM, Manser T. Journal of Immunology. 2001;167:15–20. doi: 10.4049/jimmunol.167.1.15. [DOI] [PubMed] [Google Scholar]
- 21.Toellner KM, Jenkinson WE, Taylor DR, Khan M, Sze DM, Sansom DM, Vinuesa CG, MacLennan IC. Journal of Experimental Medicine. 2002;195:383–9. doi: 10.1084/jem.20011112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McHeyzer-Williams LJ, Cool M, McHeyzer-Williams MG. Journal of Experimental Medicine. 2000;191:1149–66. doi: 10.1084/jem.191.7.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McHeyzer-Williams MG, McLean MJ, Lalor PA, Nossal GJ. Journal of Experimental Medicine. 1993;178:295–307. doi: 10.1084/jem.178.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Connor BP, Cascalho M, Noelle RJ. Journal of Experimental Medicine. 2002;195:737–45. doi: 10.1084/jem.20011626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slifka MK, Antia R, Whitmire JK, Ahmed R. Immunity. 1998;8:363–72. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]
- 26.McHeyzer-Williams LJ, McHeyzer-Williams MG. Methods in Molecular Biology. 2004;271:173–88. doi: 10.1385/1-59259-796-3:173. [DOI] [PubMed] [Google Scholar]
- 27.Bernasconi NL, Onai N, Lanzavecchia A. Blood. 2003;101:4500–4. doi: 10.1182/blood-2002-11-3569. [DOI] [PubMed] [Google Scholar]
- 28.Bernasconi NL, Traggiai E, Lanzavecchia A. Science. 2002;298:2199–202. doi: 10.1126/science.1076071. [DOI] [PubMed] [Google Scholar]
- 29.Gavin AL, Hoebe K, Duong B, Ota T, Martin C, Beutler B, Nemazee D. Science. 2006;314:1936–8. doi: 10.1126/science.1135299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gatto D, Pfister T, Jegerlehner A, Martin SW, Kopf M, Bachmann MF. J Exp Med. 2005;201:993–1005. doi: 10.1084/jem.20042239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phan TG, Paus D, Chan TD, Turner ML, Nutt SL, Basten A, Brink R. J Exp Med. 2006;203:2419–24. doi: 10.1084/jem.20061254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Molina V, Shoenfeld Y. Autoimmunity. 2005;38:235–45. doi: 10.1080/08916930500050277. [DOI] [PubMed] [Google Scholar]
- 33.Cohen AD, Shoenfeld Y. J Autoimmun. 1996;9:699–703. doi: 10.1006/jaut.1996.0091. [DOI] [PubMed] [Google Scholar]
- 34.Martinuc Porobic J, Avcin T, Bozic B, Kuhar M, Cucnik S, Zupancic M, Prosenc K, Kveder T, Rozman B. Clin Exp Immunol. 2005;142:377–80. doi: 10.1111/j.1365-2249.2005.02923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh VK, Lin SX, Newell E, Nelson C. J Biomed Sci. 2002;9:359–64. doi: 10.1007/BF02256592. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.