

Abstract
Objective
Osteopontin (OPN) is a pro-inflammatory cytokine important in rheumatoid arthritis (RA). OPN can be cleaved by thrombin, leading to OPN-Arg (OPN-R) and exposing the cryptic C-terminal α4β1 and α9β1 integrin-binding motif (SVVYGLR). Thrombin-activatable carboxypeptidase B (CPB), also termed thrombin-activatable fibrinolysis inhibitor (TAFI), removes the C-terminal arginine from OPN-R, generating OPN-Leu (OPN-L) and abrogating its enhanced cell binding. We investigated the roles of OPN-R and OPN-L in: (i) synoviocyte adhesion, which contributes to formation of invasive pannus, and (ii) neutrophil survival, which affects inflammatory infiltrates, in RA.
Methods and Results
We developed ELISAs specific for OPN-R and OPN-L, and demonstrate elevations of OPN-R and OPN-L in RA, but not in osteoarthritis or psoriatic arthritis, synovial fluid samples. OPN-R and OPN-L levels correlated with multiple inflammatory cytokines including TNFα and IL-6. Immunohistochemical analyses demonstrated robust expression of OPN-FL, but minimal OPN-R, in RA synovium, suggesting that cleaved OPN is released into the synovial fluid. In cellular assays, OPN-FL, and to a lesser extent OPN-R and OPN-L, had an anti-apoptotic effect on neutrophils. OPN-R, but not OPN-L, augmented RA fibroblast-like synoviocyte binding mediated by SVVYGLR binding to α4β1.
Conclusion
Thrombin activation of OPN (OPN-R) and its subsequent inactivation by thrombin-activatable CPB (OPN-L) occurs locally within inflamed joints in RA. Our data suggest that thrombin-activatable CPB plays a central homeostatic role in RA, by regulating neutrophil viability and reducing synoviocyte adhesion.
Keywords: osteopontin, thrombin-activatable carboxypeptidase B, thrombin-activatable fibrinolysis inhibitor, rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic, disabling systemic inflammatory joint disease, characterized by the propagation of inflammatory cells within the joint space, leading to the invasive growth of synovial tissues and destruction of cartilage and bone (1). Recent evidence suggests that osteopontin (OPN) may play an important role in its pathogenesis (2–4). OPN is a glycosylated phosphoprotein that functions as a plasma pro-inflammatory cytokine or as an extracellular matrix protein in bone remodeling (5, 6). OPN is expressed by many different cell types including activated immune cells such as macrophages, natural killer cells, and activated T lymphocytes (7–10). There is increased local production of OPN in human rheumatoid joints and it is expressed in invading synoviocytes, chondrocytes and CD4+ synovial T cells (2, 4, 11, 12).
OPN contains a RGD site that mediates its attachment to osteoclast αvβ3 integrin and a CD44-binding domain for bone matrix interaction, both of which are involved in bone resorption (13). In addition to αvβ3, the RGD site can interact with multiple other integrins, including αvβ1, αvβ5, α5β1, and α8β1, conferring OPN with many cytokine-like functions including chemotaxis and signal transduction (5, 6). Of note, OPN has a conserved thrombin cleavage site at 168R/S169 immediately downstream of the 159RGD161 site. Thrombin cleavage exposes a new carboxyl terminus 162SVVYGLR168 in the cleaved OPN (OPN-Arg, or OPN-R) that interacts with α4β1 and α9β1 integrins in a non-RGD-dependent manner (14–19). Both integrin α4β1, expressed on lymphocytes, smooth muscle cells, and integrin α9β1, expressed on neutrophils, epithelial cells and smooth muscle cells, represent a defined subset within the broad integrin family. The essential role of this thrombin cleavage-dependent, cryptic integrin-binding site in OPN has been recently demonstrated in a murine model of RA (3). A monoclonal antibody directed against this site inhibits proliferation of synovium, infiltration of inflammatory cells, and development of bone erosions in vivo. The presence of cleaved OPN fragments has been detected in synovial fluids of RA patients, although the precise fragments were not defined (12).
We have previously shown that the thrombin-activatable procarboxypeptidase B (pCPB, also termed thrombin-activatable fibrinolysis inhibitor or TAFI), upon activation to CPB, cleaves the C-terminal arginine from SVVYGLR, thereby converting OPN-Arg (OPN-R) to OPN-Leu (OPN-L), and inhibits the enhanced OPN-R adhesion to Jurkat cells (20). Prothrombin fragments F1+2, and thrombin-antithrombin complexes, both indices of thrombin generation, are found at much higher levels in RA synovial fluid than in plasma (21), suggesting that a prothrombotic environment exists within the inflammatory joint compartment, capable of generating thrombin locally for hydrolysis of OPN. Thrombomodulin, an essential cofactor for the activation of pCPB to CPB by thrombin that is normally present on the vascular endothelial cell surface, is also present on synovial lining cells (22). Both soluble thrombomodulin (22) and pCPB (21) are greatly increased in RA synovial fluid. OPN cleavage by thrombin is dependent on both of thrombin’s anion-binding exosites that determine rates and specificity of thrombin proteolysis, establishing OPN as a physiological substrate for thrombin (23).
To further investigate the importance of OPN-R and OPN-L in vivo, we developed specific ELISAs for these cleaved OPN forms. We demonstrate that elevated levels of OPN-R and OPN-L are present in RA synovial fluid, and that OPN-R and OPN-L differentially regulate synoviocyte adhesion and neutrophil survival, two cellular responses known to play central roles in the pathogenesis of RA.
MATERIALS AND METHODS
Reagents
Rabbit polyclonal antibody sera were raised against the peptides SVVYGL (anti-OPN-L), SLAYGLR (anti-OPN-R), and VDTYDRGDSVVYGLR (anti-OPN) conjugated to KLH by the standard protocol of Covance (Denver, PA). Antibodies cross-reactive to OPN-L in anti-OPN-R were removed by adsorption on sepharose-linked SLAYGL. The human OPN Quantikine ELISA Kit, MAB14331 (anti-human OPN monoclonal antibody), AF1433 (goat anti-human OPN antibody), and purified recombinant eukaryotic human and mouse OPN were purchased from R&D Systems (Minneapolis, MN). 10A16 anti-OPN antibody was from IBL-America (Minneapolis, MN). Peroxidase- or FITC-conjugated goat anti-rabbit antibody, FITC-conjugated donkey anti-mouse antibody, and Cy3-conjugated goat anti-mouse IgG antibody, were from Jackson Immunological (West Grove, PA). The AminoLink Plus Immobilization Kit was from Pierce (Rockford, IL). Human thrombin and pCPB were from Haematologic Technologies (Essex Junction, VA). The ACTICHROME CPB activity kit and anti-thrombomodulin and anti-pCPB monoclonal antibodies were from American Diagnostica (Stamford, CT). Potato carboxypeptidase inhibitor (CPI) and all other reagents and chemicals, unless stated, were from Sigma (Atlanta, GA). The OPN peptides were synthesized by the peptide synthesis facility at Beckman Center, Stanford University School of Medicine. High protein-binding 96-well ELISA plates were from Granier Bio-One (Monroe, NC).
Preparation of OPN-R and OPN-L from human milk-derived OPN and recombinant OPN
Full length human milk (23, 24) or recombinant human OPN (OPN-FL, 10 μg each), quantitated by extinction at 280 nm, were digested with 100 nM thrombin for OPN-R or thrombin then 100 nM CPB for OPN-L in 20 mM HEPES pH 7.6, 1 mM CaCl2, 150 mM NaCl, 0.1% PEG8000. Thrombin was quenched by PPACK (5 μM); no further treatment of CPB was required due to its thermal instability. OPN-R and OPN-L were used without further purification.
Development of specific ELISAs for human OPN-R and OPN-L
A commercial antibody (MAB14331, 500 ng/well) was used for the capture of OPN-FL, OPN-R and OPN-L. OPN-FL was measured using the R&D Systems Quantikine OPN ELISA kit. Captured OPN-R and OPN-L were detected by anti-OPN-R or anti-OPN-L in conjunction with peroxidase-conjugated goat-anti-rabbit antibody and TMB substrate (Alpha Diagnostic, San Antonio, TX). Recombinant OPN-FL, OPN-R and OPN-L (0.023 – 1.5 nM; 0.625 ng/mL – 50 ng/mL) were used to construct the calibration curves.
Epitope mapping of anti-OPN-R and anti-OPN-L antibodies
The sandwich ELISA system using milk-derived OPN-R or OPN-L at 10 ng/mL was used for epitope mapping. Molar excesses of peptides to antibody (50 fold) were pre-incubated for 2 h then added to the wells. Peptides SAAYGLR, SLGYGLR, SLAAGLR, SLAYALR, SLAYGAR, SLAYGLA, SLAYGLR-NH2 and RLGYALS were incubated with anti- OPN-R antibody and SAVYGL, SVAYGL, SVVAGL, SVVYAL, SVVYGA, SVVYGL-NH2 and RLGYALS with anti-OPN-L antibody (substituted residue in bold font). The decrease in binding of the antibody to the immobilized OPN-R or OPN-L by the cognate unsubstituted peptide was defined as 100% inhibition, and results from the substituted peptides were normalized to this.
Detection of OPN-FL and its cleaved forms in RA, OA, and PSA synovial fluid
Synovial fluid specimens were obtained from 26 RA, 18 osteoarthritis (OA), and 10 psoriatic arthritis (PsA) patients following approved human subjects protocols at Stanford University Medical Center and Brigham and Women’s Hospital, and stored frozen at −80°C until analysis. Samples were thawed on ice, and clarified by centrifugation at 400x g for 10 min at 4°C. The various forms of OPN were measured using the specific ELISAs. Wilcoxon Rank test was performed, and P values < 0.05 were considered significant.
Multiplex cytokine analysis of synovial fluid
A 12-cytokine Beadlyte kit (Millipore, Billerica, MA) and the Luminex xMAP 100IS platform (Austin, TX) were used. To block non-specific cross-linking by rheumatoid factor, synovial fluid samples were pre-incubated with 3 μg/ml HeteroBlock (Omega Biologicals Inc, Bozeman, MT). The Wilcoxon rank test was used to compare the median cytokine levels in RA vs. OA. The correlation between cytokines and OPN was performed using Spearman correlation analysis, and all reported values have a Spearman’s rho P value < 0.01.
Immunofluorescence labeling of fibroblast-like synoviocytes
Fibroblast-like synoviocytes (FLS) obtained from human synovial fluid samples were cultured in DME with 10% FBS. Cells at passages 6–8 were used in immunofluorescence studies using standard procedures.
RT-PCR for pCPB detection in FLS
Total RNA (~1 μg) prepared from FLS was converted to cDNA using an oligo dT primer and superscript II (Invitrogen). The specific primers used for amplifying a 454bp pCPB fragment were CGTTTCAGAGTGGCCAAGTT (forward) and GGCATTTTTGGCTGTTTGTT (reverse). Annealing temperature used in the PCR reaction was 55°C and 35 cycles applied.
Activation of pCPB by thrombin in the presence of cultured FLS
The functional activity of thrombomodulin on the surface of FLS was determined by adding pCPB (40 nM) and thrombin (10 nM) in 100 μL PBS and incubating at room temperature (RT) for 30 min. The reactions were stopped by PPACK (10 μM). CPB activity was assessed using a chromogenic assay (Actichrome CPB kit). CPI (10 μg/mL) was added to inhibit CPB activity in some assays.
Direct ELISA of pCPB, OPN-R and OPN-L
Synoviocytes were cultured in a 96-well plate, washed, and agonists added at 37°C for 30 min. Aliquots of supernatants were transferred to a new 96-well plate and coated at RT for 2 h. Non-specific binding sites were blocked by incubation with BSA (2%) for 1 h, followed by anti-pCPB, anti-OPN-R or anti-OPN-L antibodies for 1 h and then developed as described in the OPN ELISAs.
Immunohistochemical detection of OPN-FL and OPN-R in RA synovium
Synovial tissue samples were obtained with informed consent from RA patients during total knee replacement surgery under human subjects protocols approved at Stanford University Medical Center. The tissue specimens were snap-frozen then embedded. For immunofluorescence analyses, cryosections were stained with anti-OPN-R or preimmune rabbit IgG. All cryosections were co-stained with monoclonal anti-OPN antibody (10A16). FITC-conjugated goat anti-rabbit IgG antibody was used to detect anti-OPN-R staining, and Cy3-conjugated goat anti-mouse IgG antibody to detect 10A16 staining. Some cryosections were pre-incubated with thrombin (100 nM) for 30 min before fixation to generate OPN-R in situ. Peptide quenching was performed by pre-incubation of anti-OPN-R with the peptide SLAYGLR in a 100:1 peptide:antibody molar ratio for 1.5 h. H&E staining was performed according to standard procedures.
Neutrophil apoptosis assay
Neutrophils were isolated from buffy coats (Stanford Blood Center) using density gradient ficoll separation, washed with RPMI 1640 with 10% FBS, and seeded into a 24-well plate at 106 cells/mL. Cells were incubated with various concentrations of recombinant WT and RGD-mutated human OPN-FL, OPN-R, OPN-L for 22 h at 37°C in the presence of 5% CO2. Cells were then washed and labeled with annexin-FITC and PI (BD Apoptosis Assay kit II), and analyzed by FACS.
Adhesion Assay of FLS to OPN-FL, OPN-R and OPN-L
Recombinant WT and RAA-substituted OPN-FL, OPN-R and OPN-L (100nM) as well as RGDS, GRGES, SVVYGLR, and SVVYGL peptides (0.1–1000 μM) were coated onto 96-well micro black fluorescence plates (Thermo Scientific, Waltham, MA). The recombinant proteins were prepared as previously described (20). Wells were washed and blocked using 2% BSA for 1 h. FLS were labeled with CFDA cell tracer dye (Invitrogen, Carlsbad, CA) and 5000 cells/well were incubated for 1 h at 37°C. The wells were washed, and the plate read at excitation wavelength of 488 nm and emission of 538 nm using Fluoroskan Ascent (Thermo Scientific, Waltham, MA); data were recorded as relative fluorescence unit (RFU).
RESULTS
Generation and characterization of specific antibodies for OPN-R and OPN-L
Specificity of purified rabbit IgG for various OPN forms was tested by western blot analysis (Figure 1A). The antibodies anti-OPN (raised against VDTYDRGDSVVYGLR) recognized all three forms of human OPN, and anti-OPN-R (raised against the mouse sequence SLAYGLR) recognized both mouse and human OPN-R. After removal of antibodies cross-reactive to OPN-L in anti-OPN-R by adsorption on sepharose-linked SLAYGL, the immune-adsorbed anti-OPN-R was then specific for both human and mouse OPN-R. The anti-OPN-L antibodies (raised against the human sequence SVVYGL) recognized human OPN-L specifically. The specificity of the antibodies was confirmed using blocking peptides. The peptide SLAYGLR blocked the interaction of anti-OPN-R with either human or mouse OPN-R but had no effect on anti-OPN-L recognition of human OPN-L. Conversely, the peptides SVVYGL and SLAYGL (data not shown) blocked anti-OPN-L recognition of human OPN-L, but had no effect on anti-OPN-R recognition of OPN-R.
Figure 1. Characterization of specific antibodies against OPN-FL, OPN-R and OPN-L and development of specific ELISAs.

Recombinant human OPN-FL was digested with thrombin (IIa) or thrombin (IIa)/CPB to produce OPN-R and OPN-L respectively. A. Western blot analysis of human OPN-FL, OPN-R and OPN-L with the specific anti-peptide antibodies anti-OPN, anti-OPN-R and anti-OPN-L. OPN-R (SLAYGLR) and OPN-L (SVVYGL) peptides were used for competitive inhibition of antibody binding. B, ELISAs for OPN-FL (commercial), OPN-R and OPN-L ELISA. C. Competition of substituted peptides for binding of anti-OPN-R and anti-OPN-L to immobilized purified OPN-R and OPN-L respectively. Inhibition of binding by the cognate unsubstituted peptide was defined as 100%.
Development of ELISA specific for OPN-R and OPN-L
Anti-OPN-R and anti-OPN-L antibodies were used to develop an ELISA specific for human OPN-R and OPN-L respectively. Purified recombinant human OPN was cleaved with thrombin to yield OPN-R and further cleaved with CPB to yield OPN-L which were then used to construct the calibration curves for the ELISAs. A commercially available monoclonal antibody (MAB14331) that recognized all three forms of human OPN was used as the capturing antibody in these ELISAs. A commercial OPN ELISA (Quantikine ELISA system, R&D systems) was able to detect all three forms of OPN, but with much greater sensitivity towards the OPN-FL than OPN-R or OPN-L (Figure 1B). When anti-OPN-R was used as the detecting antibody, OPN-R, but not OPN-FL or OPN-L, was detected in a dose-dependent manner with a lower limit of detection of 0.023 nM (0.625 ng/mL; Figure 1B). Likewise, anti-OPN-L specifically recognized OPN-L and not OPN-FL or OPN-R with similar sensitivity (Figure 1B). A 20-fold molar excess of OPN-FL to either OPN-R or OPN-L did not affect the readout of either of these two ELISAs (data not shown), indicating that these two ELISAs are applicable for the measurement of OPN-R and OPN-L in biological fluid samples. To further demonstrate the specificity of the ELISAs, soluble human OPN-R peptide (SVVYGLR), but not OPN-L peptide (SVVYGL), competitively inhibited the measurement of OPN-R, while soluble OPN-L peptide, but not OPN-R peptide, inhibited OPN-L measurement (data not shown).
To define the epitopes in OPN-R and OPN-L recognized by the respective antibodies, a series of peptides based on the mouse OPN-R (SLAYGLR) and human OPN-L (SVVYGL) carboxyl terminal sequences were synthesized and used to competitively inhibit antibody binding to immobilized OPN-R and OPN-L in the ELISA system. Peptides that had key residues substituted were unable to compete effectively. For recognition by either antibody the C-terminal arginine or leucine and its carboxyl group as well as the adjacent amino acid were critical (Figure 1C). In addition, the anti-OPN-R antibody recognition depended on tyrosine while the anti-OPN-L antibody depended on the glycine, tyrosine and N-terminal valine.
Determination of OPN-FL, OPN-R and OPN-L levels in synovial fluid of patients with rheumatoid arthritis, osteoarthritis and psoriatic arthritis
Increased OPN levels in RA synovial fluid samples have been reported (4, 12). We tested whether OPN-R and OPN-L were detectable in RA synovial fluid using our specific ELISAs. While OPN-FL was significantly elevated in RA synovial fluid (n = 26, median level 352.5 ng/mL), as compared to its normal plasma level (~50 ng/mL), it was not significantly higher when compared to OA (n = 18, 157.9 ng/mL, P value 0.142) or PsA (n = 10, 143.4 ng/mL, P value 0.074) synovial fluid samples (Figure 2A). On the other hand, a highly significant elevation of OPN-R and OPN-L levels was detected in the RA synovial fluid, as compared to OA and PsA. The median values of OPN-R and OPN-L in RA, OA and PSA were 69.7 ng/mL, 5.3 ng/mL and 1.1 ng/mL (for OPN-R, P < 0.003) and 102.2 ng/mL, 12.9 ng/mL and undetectable respectively (for OPN-L, P < 0.006). There was a wide range of intact and cleaved OPN levels in the synovial fluid samples in RA patients, from undetectable to greater than 50,000 ng/mL, suggesting significant disease heterogeneity. The median value for the ratio of cleaved OPN (OPN-R plus OPN-L) to total OPN was approximately 0.35, indicating extensive enzymatic cleavage activity within the RA inflammatory joint space (Figure 2B), whereas in samples from OA patients it was approximately 0.1 suggesting that there was much less proteolytic activity present.
Figure 2. Determination of OPN-FL, OPN-R and OPN-L levels in synovial fluid of patients with osteoarthritis, psoriatic arthritis and rheumatoid arthritis by ELISA.
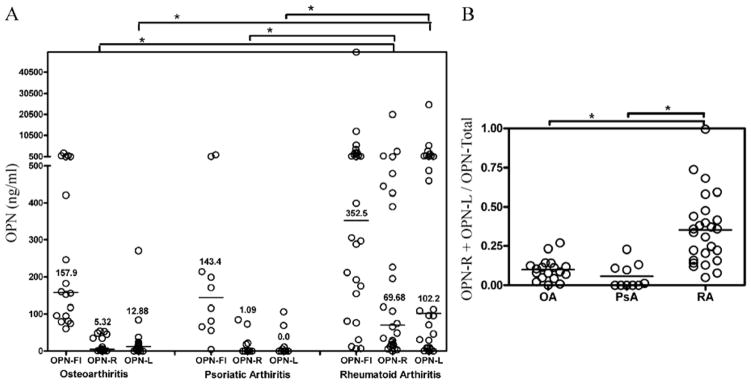
A, Osteoarthritis (OA) n = 18, psoriatic arthritis (PsA) n = 10, rheumatoid arthritis (RA) n = 26. Bars are median values of each disease group. *P < 0.003 by Wilcoxon rank test. B, The ratio of cleaved OPN levels (OPN-R plus OPN-L) to total OPN (sum of the three different OPN forms) for RA, OA and PsA. *P < 0.003.
Cytokine levels in RA and OA synovial fluid
Bead array multiplex cytokine analysis was performed on synovial fluid from both RA (n = 26) and OA (n = 13) patients. Significantly increased levels of the pro-inflammatory cytokines TNFα, FGF, GM-CSF, IP-10, IL-12p40, IL-6, and eotaxin in RA synovial fluid were noted (Table 1) that correlated with OPN-FL, OPN-R and OPN-L. It is notable that while OPN-FL levels only exhibited a statistical correlation with IL-6, increased OPN-R level was statistically correlated with multiple pro-inflammatory cytokines including IL-6, IL-12p40, FGF-2, IP-10 and TNFα. The same holds true for OPN-L, which was significantly correlated with FGF-2, IL-12p40, IL-6 and eotaxin.
Table 1.
Inflammatory cytokines in synovial fluid of OA and RA.
| RA (n=26) | OA (n=13) | p-valuea | OPN-FL | OPN-R | OPN-L | |
|---|---|---|---|---|---|---|
| FGF-2 (pg/ml) | 66.6 (51.2–124.8)b | 15.0 (8.6–37.5) | <.001 | - | 0.463 | 0.485 |
| IP-10 (μg/ml) | 19.2 (14.9–20.0) | 11.5 (8.1–12.7) | <.001 | - | 0.446 | - |
| IL-12p40 (pg/ml) | 158.0 (19.2–235.3) | 0 (0–35.6) | 0.001 | - | 0.619 | 0.54 |
| IL-6 (pg/ml) | 2375.0 (999.5–3502.5) | 253.0 (27.5–2185.0) | 0.003 | 0.676 | 0.524 | 0.437 |
| TNFa (pg/ml) | 81.6 (49.3–151.75) | 17.2 (14.4–17.2) | 0.005 | - | 0.434 | - |
| Eotaxin (pg/ml) | 165.0 (88.1–388.5) | 0 (0–147.5) | 0.02 | - | - | 0.469 |
| GM-CSF (pg/ml) | 14.0 (3.8–54.6) | 0 (0–16.4) | 0.05 | - | - | - |
| MCP-1 (pg/ml) | 174.0 (70.1–517.0) | 209.0 (50.0–393.0) | 0.697 | - | - | - |
Reported P values were calculated using the Wilcoxon rank test to compare the levels of cytokines measured in synovial fluid samples derived from RA as compared to OA.
Median (interquartile range; 25%–75%)
Last three columns represent the correlations between OPN-FL, OPN-R and OPN-L and inflammatory cytokines in synovial fluids derived from both patients with RA and OA. The correlations between the levels of cytokines and different forms of OPN were analyzed using the data from all synovial samples and the Spearman’s rank correlation test. Spearman’s rho values from significant correlations with p < 0.01 are shown while dashes indicate that the correlation was not significant.
pCPB is produced by synoviocytes in RA and OA, released into synovial fluid, and activated by thrombin in the presence of synovial thrombomodulin
A ~65 kDa protein similar to plasma pCPB was identified in RA and OA synovial tissue lysates by Western blot analysis (Figure 3A). A similar protein in cultured fibroblast-like synoviocytes (FLS) lysates from RA and OA patients was noted (data not shown). The anti-pCPB antibody also strongly labeled the cytoplasm of FLS (Figure 3B). RT-PCR using specific primers for pCPB showed the expected 454 bp amplification product using FLS RNA whose identity was confirmed by direct sequencing. pCPB was also detected in RA and OA synovial fluid by Western blot analysis (data not shown). Collectively the data indicate FLS synthesize and release pCPB into synovial fluid. Functional assays demonstrated CPB activity in synovial fluid from OA and RA patients (median value of 2.2 μg/mL in OA (n = 8) and 4.56 μg/mL in RA (n = 9) samples, P < 0.05). The presence of thrombomodulin in FLS was demonstrated by immunofluorescence, consistent with published data (22) (Figure 3B). When pCPB was added to cultured FLS in the presence of thrombin, significant levels of CPB activity were generated, which was inhibited by anti-thrombomodulin antibody, indicating the synovial FLS express functional thrombomodulin that can support thrombin activation of pCPB (Figure 3C).
Figure 3. Identification of thrombin-activatable pCPB and its activity in fibroblast-like synoviocytes.

A, Detection of pCPB in OA and RA synovial tissue lysates from two patients each by a monoclonal anti-pCPB antibody in Western blot. B, Immunofluoresence cell labeling of pCPB, thrombomodulin and OPN in FLS. In anti-pCPB panel, green color indicates pCPB and blue color stands for nuclear labeling by DAPI. C, Thrombomodulin expressed on FLS enhanced pCPB activation by thrombin. pCPB activation was blocked in the presence of anti-thrombomodulin antibody. Results represent the mean ± SEM (n = 6). D, Generation of endogenous OPN-R and OPN-L by incubation of thrombin with FLS, with or without exogenous pCPB. Thrombin was used at either 1 U/mL or 10 U/mL; OPN-R or OPN-L in FLS supernatant was measured by direct ELISA as described in Methods. Potato carboxypeptidase inhibitor (CPI) is a specific inhibitor of CPB. Results represent mean ± SEM (n = 6) P < 0.05.
Generation of OPN-R and OPN-L by thrombin and pCPB in the presence of FLS
By immunofluorescence, cultured FLS was strongly stained by anti-OPN mAb (Figure 3B), confirming increased local production of OPN in RA synovial tissues (12). When thrombin was added to the FLS, generation of OPN-R in the supernatant was observed, demonstrating thrombin cleavage of the endogenously produced OPN (Figure 3D). Of note, OPN-L was also detectable in the supernatant indicating that thrombin, in the presence of FLS-expressed thrombomodulin, was capable of activating the endogenous pCPB into active CPB and converted OPN-R to OPN-L in vitro (Figure 3D). The OPN-L concentration was further enhanced by the addition of exogenous pCPB, and the carboxypeptidase activity was completely abolished by potato carboxypeptidase inhibitor (CPI), a specific CPB inhibitor, indicating that the carboxypeptidase activity was derived from CPB and not CPN.
Detection of OPN-FL but not OPN-R in RA synovium
There was abundant expression of OPN-FL (Figure 4B) but OPN-R was barely detectable in the synovial lining of RA patients (Figure 4C). Thrombin treatment of synovial tissue sections, cleaving OPN-FL to OPN-R in situ, resulted in robust staining of the synovial lining by the anti-OPN-R antibody (Figure 4D) in a pattern similar to that observed for OPN-FL. This suggests that the low level of OPN-R detection in the synovial lining reflected its paucity rather than inability of the antibody to detect OPN-R in tissues. Pre-incubation of the OPN-R antibody with the OPN-R peptide SLAYGLR blocked its ability to stain thrombin-treated tissue sections (data not shown), confirming its specificity. Taken together, our ELISA and immunohistological data suggest that OPN-FL is highly expressed at the inflamed synovial surface, where it is cleaved by thrombin and CPB, and the cleaved OPN products are then released into the synovial fluid.
Figure 4. Detection of OPN-FL and OPN-R in RA synovium.

Immunostaining of RA synovial tissue: A, H&E stain. B, anti-OPN (10A16). C, anti-OPN-R. D, anti-OPN-R following in situ treatment with thrombin, magnification 400X.
Effect of OPN and its cleaved forms on neutrophil apoptosis and cultured synoviocyte adhesion
OPN-FL, and to a lesser extent OPN-R and OPN-L, significantly protected neutrophils from apoptosis (Figure 5A). The anti-apoptotic effect is dependent on the RGD site of OPN, since mutation of the RGD site in either the intact or cleaved OPN produced an almost complete loss of protection against apoptosis.
Figure 5. OPN protected neutrophils from apoptosis and fibroblast-like synoviocytes binding to OPN was enhanced in OPN-R but not in OPN-L.

A, Effect of recombinant WT OPN-FL, OPN-R, OPN-L and their RAA substituted counterparts (RAA-FL, RAA-R, and RAA-L) on neutrophil apoptosis. Each data point represents mean ± SEM (n = 3), and the results are representative of one of three separate experiments. B, FLS adhesion to immobilized WT and RGD-substituted OPN-FL, OPN-R, OPN-L. Data are presented as mean ± SEM (n = 8), and results are representative of one of four separate experiments. C, Schematic model of OPN-R and OPN-L in RA: While OPN and pCPB within the inflammatory joint space can be derived from plasma, synoviocytes also produce and release these molecules locally. Following initial insult, thrombin is generated and cleaves OPN to OPN-R, which enhances tissue inflammation. 27 On the other hand, thrombin also binds to thrombomodulin (TM) on the synovial cell surface, which then activates pCPB locally to CPB and converts OPN-R to OPN-L, thereby dampening inflammation.
Adhesion of FLS to immobilized OPN and its cleaved forms was studied in vitro. OPN-FL supported FLS cell binding compared to BSA control (Figure 5B); this was significantly enhanced in OPN-R (>2-fold, P < 0.05) but not in OPN-L, suggesting that it required a functional SVVYGLR domain. Similar to the protective effect on neutrophil apoptosis, it also required an intact RGD site since mutation of the RGD site in either the intact or cleaved OPN abolished cell-binding activity. This was further demonstrated by binding of FLS to immobilized RGD but not RGE peptides, and to SVVYGLR but not to SVVYGL peptides in a dose-dependent manner (data not shown), confirming that an intact SVVYGLR domain is necessary to support the enhanced cell binding to OPN-R. FLS expressed both α4 and β1 integrin subunits as confirmed by flow cytometry, which may mediate the enhanced cell binding to OPN-R (data not shown).
DISCUSSION
We have recently shown that OPN is a physiological substrate for thrombin (23). Thrombin cleavage of OPN, leading to the exposure of the cryptic α4β1 or α9β1 integrin-binding site in the C-terminal SVVYGLR domain in OPN-R, plays an important role in tissue inflammation (3, 25, 26). Our basic premise is that the subsequent cleavage of the C-terminal arginine by the thrombin/thrombomodulin-activated CPB, thereby abolishing its integrin-binding function, represents a homeostatic feedback response in modulating this inflammatory process (20). In this study we provide the first direct evidence that thrombin-cleaved OPN-R, and thrombin and CPB-cleaved OPN-L, are markedly elevated in RA synovial fluid (Fig. 2). We performed cellular assays to investigate the roles of OPN-R and OPN-L in the pathogenesis of RA, and show differential regulation of fibroblast-like synoviocyte adhesion and neutrophil survival. The adhesion of fibroblast-like synoviocytes plays a critical role in the formation and growth of pannus, a tumor-like growth of the synovial lining that invades cartilage and bone to contribute to joint destruction. Neutrophils are recruited in large numbers into the pannus and synovial fluid, and we demonstrate that the cleaved forms of OPN regulate neutrophil survival and apoptosis (Fig. 5).
It is now well established that thrombin has intrinsic proinflammatory properties in addition to its recognized procoagulant role in fibrin clot formation (28). The source of thrombin in RA may in part originate from the blood compartment, with leakage of thrombin into the joint space as part of the inflammatory vascular response. On the other hand, thrombin may also be generated locally. Multiple clotting factors, including factor VII, factor X, and prothrombin, are present in synovial fluid of RA patients (29, 30). Thrombin-AT levels were markedly elevated in synovial fluid, at 50–500 fold its plasma levels (29, 30). Activated neutrophils and monocytes, infiltrating the inflamed joint, are known to express tissue factor, which will lead to activation of the clotting cascade and fibrin deposition locally (31). In this study, we showed that synoviocytes, in addition to their known production of OPN, also synthesize and release pCPB (Figure 3A and 3B). Synovial cell surface thrombomodulin supports thrombin activation of pCPB to CPB, which cleaves OPN-R to OPN-L (Figure 3C and 3D). Thus the whole process of synthesis and release of OPN and pCPB, thrombin activation of OPN and its subsequent inactivation by CPB, can all occur within the inflamed joint space and be regulated locally (Figure 5C).
OPN confers an anti-apoptotic effect on many different cell types, mediated through its binding to CD44 and/or αvβ3, which may be cell-type specific (32–34). Consistent with this, OPN confers protection against apoptosis in neutrophils (Figure 5A), and it is absolutely dependent on an intact RGD site, with the RAA substitution completely abolishing its anti-apoptotic activity, supporting the importance of signaling through the αvβ3 integrin pathway. However it is somewhat surprising that OPN-R and OPN-L, while still quite active, have less protective effect since the major CD44-binding site is located at amino acid 121–140 (34) and thus still retained within OPN-R and OPN-L. It is possible that thrombin cleavage at 168R/S169 alters the conformation of the adjacent 159RGD161 motif, thereby affecting its affinity for the αvβ3 integrin receptor. The enhanced synoviocyte binding to OPN-R, but not OPN-L (Figure 5B), and its direct binding to SVVYGLR, but not SVVYGL, is consistent with binding via the α4β1 receptor on synoviocytes, and is similar to our previous observation on enhanced Jurkat cell binding to OPN-R but not OPN-L (20). Whether OPN-R and OPN-L will have distinct effects on other aspects of neutrophil and synoviocyte biology is being studied.
RA is a complex disease and a number of different pathophysiological mechanisms could give rise to inflammatory polyarthritis that is clinically classified as RA. In this study, marked heterogeneity was observed in the elevated levels of OPN-FL, OPN-R and OPN-L in RA synovial fluid (Figure 2). The ratio of cleaved OPN to OPN-FL, with a median value of ~0.5, suggests that in a significant subset of RA patients, almost 50% of the OPN within the joint space underwent proteolytic cleavage(s). This heterogeneity may reflect different clinical severity at the time the synovial fluid samples were obtained, or intrinsic heterogeneity in the pathophysiological mechanisms in RA. It is notable that elevated levels of OPN-R and OPN-L correlated with increased levels of multiple pro-inflammatory cytokines including IL-6, IL-12p40, TNFα, and the chemokines IP-10 and eotaxin, while the levels of OPN-FL did not correlate well with pro-inflammatory cytokines other than IL-6 (Table 1). It is possible that elevation of OPN-FL is part of a general and less-specific inflammatory response, and the elevated OPN-R and OPN-L levels in a subset of RA patients suggest that thrombin and CPB-regulated pathways may play a major role in mediating synovitis in this patient subgroup. It is interesting that hirudin, a specific thrombin inhibitor, has been shown to be efficacious in an experimental RA model (35), suggesting that inhibition of thrombin may have therapeutic potential in these patients.
Acknowledgments
Funding: National Institutes of Health (grant RO1 HL57530 (L.L)), NHLBI contract N01-HV-28183 (W.H.R), and an American College of Rheumatology Research and Education Foundation Physician Scientist Development Award (J.S)
Nonstandard abbreviations used
- pCPB
procarboxypeptidase B
- CPB
carboxypeptidase B
- TAFI
thrombin-activatable fibrinolysis inhibitor
- FLS
fibroblast-like synoviocytes
- PsA
psoriatic arthritis
- OPN
osteopontin
Footnotes
Conflict of interest: None
References
- 1.Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–61. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 2.Yumoto K, Ishijima M, Rittling SR, Tsuji K, Tsuchiya Y, Kon S, et al. Osteopontin deficiency protects joints against destruction in anti-type II collagen antibody-induced arthritis in mice. Proc Natl Acad Sci USA. 2002;99:4556–61. doi: 10.1073/pnas.052523599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yamamoto N, Sakai F, Kon S, Morimoto J, Kimura C, Yamazaki H, et al. Essential role of the cryptic epitope SLAYGLR within osteopontin in a murine model of rheumatoid arthritis. J Clin Invest. 2003;112:181–8. doi: 10.1172/JCI17778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu G, Nie H, Li N, Zheng W, Zhang D, Feng G, et al. Role of osteopontin in amplification and perpetuation of rheumatoid synovitis. J Clin Invest. 2005;115:1060–7. doi: 10.1172/JCI23273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Denhardt DT, Noda M, O’Regan AW, Pavlin D, Berman JS. Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J Clin Invest. 2001;107:1055–61. doi: 10.1172/JCI12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wai PY, Kuo PC. The role of Osteopontin in tumor metastasis. J Surg Res. 2004;121:228–41. doi: 10.1016/j.jss.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 7.Patarca R, Saavedra RA, Cantor H. Molecular and cellular basis of genetic resistance to bacterial infection: the role of the early T-lymphocyte activation-1/osteopontin gene. Crit Rev Immunol. 1993;13:225–46. [PubMed] [Google Scholar]
- 8.Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Jansson M, Zawaideh S, et al. Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science. 2000;287:860–4. doi: 10.1126/science.287.5454.860. [DOI] [PubMed] [Google Scholar]
- 9.O’Regan AW, Nau GJ, Chupp GL, Berman JS. Osteopontin (Eta-1) in cell-mediated immunity: teaching an old dog new tricks. Immunol Today. 2000;21:475–8. doi: 10.1016/s0167-5699(00)01715-1. [DOI] [PubMed] [Google Scholar]
- 10.Chabas D, Baranzini SE, Mitchell D, Bernard CC, Rittling SR, Denhardt DT, et al. The influence of the proinflammatory cytokine, osteopontin, on autoimmune demyelinating disease. Science. 2001;294:1731–5. doi: 10.1126/science.1062960. [DOI] [PubMed] [Google Scholar]
- 11.Petrow PK, Hummel KM, Schedel J, Franz JK, Klein CL, Müller-Ladner U, et al. Expression of osteopontin messenger RNA and protein in rheumatoid arthritis: effects of osteopontin on the release of collagenase 1 from articular chondrocytes and synovial fibroblasts. Arthritis Rheum. 2000;43:1597–605. doi: 10.1002/1529-0131(200007)43:7<1597::AID-ANR25>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 12.Ohshima S, Yamaguchi N, Nishioka K, Mima T, Ishii T, Umeshita-Sasai M, et al. Enhanced local production of osteopontin in rheumatoid joints. J Rheumatol. 2002;29:2061–7. [PubMed] [Google Scholar]
- 13.Chellaiah MA, Kizer N, Biswas R, Alvarez U, Strauss-Schoenberger J, Rifas L, et al. Osteopontin deficiency produces osteoclast dysfunction due to reduced CD44 surface expression. Mol Biol Cell. 2003;14:173–89. doi: 10.1091/mbc.E02-06-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith LL, Giachelli CM. Structural requirements for alpha 9 beta 1-mediated adhesion and migration to thrombin-cleaved osteopontin. Exp Cell Res. 1998;242:351–60. doi: 10.1006/excr.1998.4108. [DOI] [PubMed] [Google Scholar]
- 15.Taooka Y, Chen J, Yednock T, Sheppard D. The integrin alpha9beta1 mediates adhesion to activated endothelial cells and transendothelial neutrophil migration through interaction with vascular cell adhesion molecule-1. J Cell Biol. 1999;145:413–20. doi: 10.1083/jcb.145.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yokosaki Y, Matsuura N, Sasaki T, Murakami I, Schneider H, Higashiyama S, et al. The integrin alpha(9)beta(1) binds to a novel recognition sequence (SVVYGLR) in the thrombin-cleaved amino-terminal fragment of osteopontin. J Biol Chem. 1999;274:36328–34. doi: 10.1074/jbc.274.51.36328. [DOI] [PubMed] [Google Scholar]
- 17.Barry ST, Ludbrook SB, Murrison E, Horgan CM. Analysis of the alpha4beta1 integrin-osteopontin interaction. Exp Cell Res. 2000;258:342–51. doi: 10.1006/excr.2000.4941. [DOI] [PubMed] [Google Scholar]
- 18.Bayless KJ, Davis GE. Identification of dual alpha 4beta1 integrin binding sites within a 38 amino acid domain in the N-terminal thrombin fragment of human osteopontin. J Biol Chem. 2001;276:13483–9. doi: 10.1074/jbc.M011392200. [DOI] [PubMed] [Google Scholar]
- 19.Green PM, Ludbrook SB, Miller DD, Horgan CM, Barry ST. Structural elements of the osteopontin SVVYGLR motif important for the interaction with alpha(4) integrins. FEBS Lett. 2001;503:75–9. doi: 10.1016/s0014-5793(01)02690-4. [DOI] [PubMed] [Google Scholar]
- 20.Myles T, Nishimura T, Yun TH, Nagashima M, Morser J, Patterson AJ, et al. Thrombin activatable fibrinolysis inhibitor, a potential regulator of vascular inflammation. J Biol Chem. 2003;278:51059–67. doi: 10.1074/jbc.M306977200. [DOI] [PubMed] [Google Scholar]
- 21.So AK, Varisco PA, Kemkes-Matthes B, Herkenne-Morard C, Chobaz-Peclat V, Gerster JC, et al. Arthritis is linked to local and systemic activation of coagulation and fibrinolysis pathways. J Thromb Haemost. 2003;1:2510–5. doi: 10.1111/j.1538-7836.2003.00462.x. [DOI] [PubMed] [Google Scholar]
- 22.Conway EM, Nowakowski B. Biologically active thrombomodulin is synthesized by adherent synovial fluid cells and is elevated in synovial fluid of patients with rheumatoid arthritis. Blood. 1993;81:726–33. [PubMed] [Google Scholar]
- 23.Myles T, Leung LL. Thrombin Hydrolysis of Human Osteopontin Is Dependent on Thrombin Anion-binding Exosites. J Biol Chem. 2008;283:17789–96. doi: 10.1074/jbc.M708629200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bayless KJ, Davis GE, Meininger GA. Isolation and biological properties of osteopontin from bovine milk. Protein Expr Purif. 1997;9:309–14. doi: 10.1006/prep.1996.0699. [DOI] [PubMed] [Google Scholar]
- 25.Diao H, Kon S, Iwabuchi K, Kimura C, Morimoto J, Ito D, et al. Osteopontin as a mediator of NKT cell function in T cell-mediated liver diseases. Immunity. 2004;21:539–50. doi: 10.1016/j.immuni.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 26.Kon S, Ikesue M, Kimura C, Aoki M, Nakayama Y, Saito Y, et al. Syndecan-4 protects against osteopontin-mediated acute hepatic injury by masking functional domains of osteopontin. J Exp Med. 2008;205:25–33. doi: 10.1084/jem.20071324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agnihotri R, Crawford HC, Haro H, Matrisian LM, Havrda MC, Liaw L. Osteopontin, a novel substrate for matrix metalloproteinase-3 (stromelysin-1) and matrix metalloproteinase-7 (matrilysin) J Biol Chem. 2001;276:28261–7. doi: 10.1074/jbc.M103608200. [DOI] [PubMed] [Google Scholar]
- 28.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–64. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 29.Carmassi F, de Negri F, Morale M, Song KY, Chung SI. Fibrin degradation in the synovial fluid of rheumatoid arthritis patients: a model for extravascular fibrinolysis. Semin Thromb Hemost. 1996;22:489–96. doi: 10.1055/s-2007-999049. [DOI] [PubMed] [Google Scholar]
- 30.Furmaniak-Kazmierczak E, Cooke TD, Manuel R, Scudamore A, Hoogendorn H, Giles AR, et al. Studies of thrombin-induced proteoglycan release in the degradation of human and bovine cartilage. J Clin Invest. 1994;94:472–80. doi: 10.1172/JCI117358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berckmans RJ, Nieuwland R, Tak PP, Boing AN, Romijn FP, Kraan MC, et al. Cell-derived microparticles in synovial fluid from inflamed arthritic joints support coagulation exclusively via a factor VII-dependent mechanism. Arthritis Rheum. 2002;46:2857–66. doi: 10.1002/art.10587. [DOI] [PubMed] [Google Scholar]
- 32.Burdo TH, Wood MR, Fox HS. Osteopontin prevents monocyte recirculation and apoptosis. J Leukoc Biol. 2007;81:1504–11. doi: 10.1189/jlb.1106711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scatena M, Almeida M, Chaisson ML, Fausto N, Nicosia RF, Giachelli CM. NF-kappaB mediates alphavbeta3 integrin-induced endothelial cell survival. J Cell Biol. 1998;141:1083–93. doi: 10.1083/jcb.141.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin YH, Yang-Yen HF. The osteopontin-CD44 survival signal involves activation of the phosphatidylinositol 3-kinase/Akt signaling pathway. J Biol Chem. 2001;276:46024–30. doi: 10.1074/jbc.M105132200. [DOI] [PubMed] [Google Scholar]
- 35.Marty I, Peclat V, Kirdaite G, Salvi R, So A, Busso N. Amelioration of collagen-induced arthritis by thrombin inhibition. J Clin Invest. 2001;107:631–40. doi: 10.1172/JCI11064. [DOI] [PMC free article] [PubMed] [Google Scholar]