

Abstract
Neuroimaging consistently reveals smaller hippocampal volume in recurrent or chronic major depressive disorder (MDD). The underlying cellular correlates of the smaller volume are not clearly known. Postmortem tissues from 17 pairs of depressed and control subjects were obtained at autopsy, and informant-based retrospective psychiatric assessment was performed. Formalin-fixed left temporal lobes were sectioned (40 μm), stained for Nissl substance, and every 60th section selected throughout the entire hippocampus. Total volume of the hippocampal formation was calculated, and total numbers of pyramidal neurons (in hippocampal fields CA1, CA2/3, hilus), dentate gyrus (DG) granule cells, and glial cells were estimated stereologically. While hippocampal volume in all MDD subjects was not significantly smaller versus control subjects, in recurrent/chronic MDD, total volume decreased with duration of depressive illness (r=−0.696, p<0.026). There was no significant difference between MDD and controls in total number or density of pyramidal neurons/granule cells or glial cells in CA1, CA2/3, hilus, or DG. However, CA1 pyramidal neuron density increased with duration of illness in recurrent/chronic MDD (r=0.840, p<0.002). Granule cell (r=0.971, p<0.002) and glial cell numbers (r=0.980, p<0.001) increased with age in those taking antidepressant medication (n=6). Increasing DG granule cell and glial cell numbers with age in antidepressant-treated subjects may reflect proliferative effects of antidepressant medications. Decreasing total volume and increasing CA1 pyramidal neuron density with duration of illness in recurrent/chronic MDD lends support to the neuropil hypothesis of MDD.
Keywords: Hippocampus, Depression, Postmortem, Stereology, Neuropil
1. INTRODUCTION
Major depressive disorder (MDD) is a debilitating illness affecting 121 million worldwide and the leading cause of disability and fourth greatest source of disease burden worldwide (WHO, 2010). Annual and lifetime prevalence is estimated at 6.6% and 16.2%, respectively (Kessler et al., 2003). Medications effective in treating MDD have been available for over half a century. However, despite improvements in side-effect profile and toxicity, efficacy has improved little (Gartlehner et al., 2011), with at least a third of patients responding partially or not at all (Rush et al., 2006). This suggests much remains unknown about the pathophysiology underlying the disorder.
Solid evidence of hippocampal involvement in depression comes from neuroimaging studies revealing smaller hippocampal volume in MDD (Bremner et al., 2000; Sheline et al., 1996), though the microscopic basis for smaller volume is not known. It is not clear whether smaller volume precedes or follows onset of MDD. Recent meta-analyses of magnetic resonance imaging (MRI) studies indicate smaller hippocampal volume in individuals with multiple depressive episodes or longer duration of depression (Kempton et al., 2011; McKinnon et al., 2009), suggesting the smaller volume is a consequence of depression, perhaps involving elevated cortisol (Conrad, 2008). However, another recent meta-analysis of MRI studies found smaller hippocampal volume in the first depressive episode (Cole et al., 2011), suggesting that smaller hippocampal volume precedes onset of depression and could instead indicate an etiological risk factor.
Apparent hippocampal atrophy in MDD has been proposed to reflect attenuated structural plasticity due to impaired neurotrophin signaling (Blugeot et al., 2011; Calabrese, 2009; Cooke & Bliss, 2006). Hence, it may be reversible through successful treatment. Antidepressant drugs are believed to exert their effects, at least in part, by enhancing neurotrophin signaling (Castrén & Rantamäki, 2008). Brain-derived neurotrophic factor (BDNF) is perhaps best characterized in this respect. BDNF exerts its effects through receptor tyrosine kinase B (TrkB) (Castrén & Rantamäki, 2010). In suicide, mRNA and protein expression of both BDNF and TrkB are reduced (Dwivedi et al., 2003).
Activation of TrkB by BDNF invokes the mitogen-activated protein (MAP) kinase pathway (Huang & Reichardt, 2003; Reichardt, 2006). Duric et al. (2010) reported elevated expression of mitogen-activated protein kinase phosphatase 1 (MKP-1), a negative regulator of MAP kinase signaling, in CA1 and dentate gyrus in postmortem tissue in MDD. Subsequent experiments revealed reduced sucrose preference and impaired active avoidance performance associated with upregulated MKP-1 mRNA transcription in hippocampus of rats exposed to chronic stress and that fluoxetine ameliorated these effects of stress (ibid.). Finally, viral-mediated MKP-1 overexpression in hippocampus of mice produced depressive-like behaviors in absence of stress, whereas constitutive deletion of the MKP-1 gene rendered mice unsusceptible to depressogenic effects of stress, indicating MKP-1 expression is necessary and sufficient for expression of depressive-like behavior.
Elevated MKP-1 expression may contribute to smaller hippocampal volume by disrupting propagative effects of neurotrophins on neuropil, thereby diminishing the volume of neuropil within hippocampus and resulting in tighter packing density of neurons and glia. Consistent with this hypothesis, we previously reported higher densities of pyramidal neurons, dentate granule cells, and glial cells and smaller pyramidal neuronal somata in postmortem hippocampus of MDD patients (Stockmeier et al., 2004). However, that study, examining only three sections from body of hippocampus, could not comment on total numbers of neurons and glia as the entire structure was not sampled. Until now, no one has completed a stereological estimation of total numbers of neurons and glia in hippocampus in MDD.
The aim of this study was to assess whether smaller hippocampal volume in MDD might reflect smaller numbers or sizes of hippocampal neurons or glia. Using formalin-fixed postmortem human brain tissue from subjects with MDD and psychiatrically-healthy controls matched for age and sex, total volume of hippocampal formation was calculated using serial tissue sections. Stereological methods were used to estimate total numbers of CA field pyramidal neurons, dentate granule cells, and glia, as well as average sizes of neuronal somata and glial cell nuclei. We hypothesized hippocampal volume would be smaller in MDD and that smaller volume would be associated with fewer neurons or glia.
2. MATERIALS AND METHODS
2.1 Subjects
The protocol for recruitment, tissue collection, and interviews was approved by the Institutional Review Boards of University Hospitals of Cleveland and University of Mississippi Medical Center. Written informed consent was obtained from legal next-of-kin for tissue collection and informant-based retrospective diagnostic interviews. Tissues were collected at autopsy at the Cuyahoga County Coroner’s Office and cause of death ruled by the coroner. Cases with history or evidence of neurological injury or disorder were excluded.
All subjects underwent retrospective assessment for Axis I diagnosis according to the Diagnostic and Statistical Manual of Mental Disorders (4th ed.) (DSM-IV) (APA, 1994) by a trained interviewer using the Structured Clinical Interview for DSM Axis I Disorders modified for third-person reporting (First et al., 1995). Interview notes and clinical histories were reviewed independently by two licensed mental health clinicians, who assigned consensus diagnoses in conference.
Each subject diagnosed with MDD (n = 17) was paired with a control subject matched for sex, age (± 5 years), postmortem interval (PMI) (± ~7.5 h), and tissue pH. There was no significant difference between MDD and Control in age, PMI, tissue pH, or fixation time in formalin (Table 1). Urine and blood collected at autopsy were examined by the coroner for presence of psychotropic medication or psychoactive substances. Laboratory personnel were unaware of individual diagnoses throughout the study. Clinical characteristics of MDD Cohort are summarized in Table 2. For individual Subject details, see Supplementary Tables 1 through 3.
Table 1.
Demographic and histological characteristics (mean ± SEM shown)
| Control n = 17 | Major depressive disorder n = 17 | ||
|---|---|---|---|
| Age (years) | 51.8 ± 3.4 | 51.5 ± 3.1 | p < 0.950 |
| Sex (M/F) | 13/4 | 12/5 | - |
| Postmortem interval (PMI; hours) | 23.4 ± 1.5 | 24.4 ± 2.4 | p < 0.811 |
| Tissue pH | 6.6 ± 0.1 | 6.4 ± 0.1 | p < 0.119 |
| Fixation time in formalin (weeks) | 155 ± 21 | 127 ± 15 | p < 0.290 |
Means ± SEM are shown here. Demographic and histological characteristics did not differ between Control and MDD subjects.
Table 2.
Clinical characteristics of the MDD Cohort (mean ± SEM shown)
| Major depressive disorder n = 17 | |
|---|---|
| Age of onset of depression (years) | 42.3 ± 4.2 |
| Duration of depression (years) | 9.0 ± 2.3 |
| Percent of life with depression (%) | 18.3 ± 4.6 |
| Single-episode v. Recurrent/Chronic MDD (S/RC) | 6/11 |
| Suicide (Y/N) | 10/7 (59%) |
| Antidepressant detected in postmortem toxicology (Y/N) | 7/10 (41%) |
| Antidepressant prescription in last month of life (Y/N) | 10/7 (59%) |
All MDD subjects met criteria for depression within last two weeks of life except one subject in remission. No Control subject met criteria for any current or past mood disorder or other psychiatric disorder. However, 1 Control subject did have a history of alcohol abuse, in full remission. Similarly, 1 MDD subject had a history of alcohol dependence, also in full remission. Three MDD subjects had active sedative and alcohol abuse, active cannabis abuse, and active cannabis dependence, respectively.
For measurement of total volume, one control subject was excluded because the caudal end of hippocampus was missing. As only a portion of subiculum was lost, the tissue loss did not affect CA fields or dentate gyrus so total cell counts were unaffected in this subject.
2.2. Tissue preparation and histology
Left temporal lobe was collected at autopsy and submerged in phosphate-buffered formalin (10%) for 21 to 369 weeks (141.1 ± 13.1 weeks, mean ± SEM). Temporal lobes were divided along the rostrocaudal axis into 6 mm-thick slabs, embedded in celloidin, and sectioned with a microtome (40 μm). Every tenth section was stained for Nissl substance using cresyl violet. Approximately every sixth Nissl-stained section throughout entire rostrocaudal length of hippocampal formation was selected for each subject, starting at anterior pole.
2.3. Stereological estimation of total volume and cell numbers
Total volume of hippocampal formation, consisting of Cornu Ammonis (CA), dentate gyrus, and subicular complex, was estimated (Uylings et al., 1986). A contour outlining the hippocampal formation in each tissue section was traced under 20× magnification via a Nikon Plan UW 2× objective (N.A. = 0.06, W.D. = 7.5 mm) (Nikon Instruments, Melville, NY) on a Nikon Eclipse 80i microscope using Stereo Investigator version 7.0 (MBF Bioscience, Williston, VT). Area within the contour was determined cartographically by Stereo Investigator. Partial volumes were calculated by multiplying contour areas by distance between each section and then summed to yield estimated total volume (Fig. 1. A.).
Fig. 1. Total hippocampal volume and stereology.
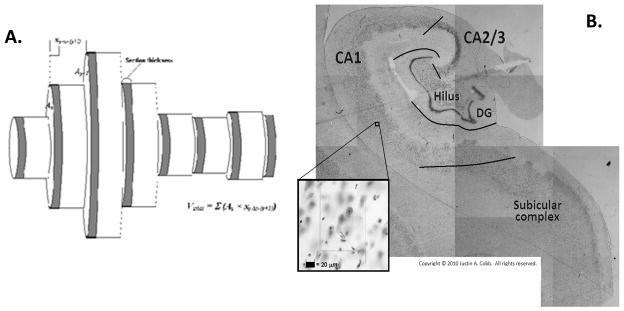
(A) Volume calculation. Each grey cylinder represents the cartographic area of region of interest traced in Stereo Investigator. Section thickness and distance between sections are indicated in the figure. (B) Cytoarchitectonic delineation of hippocampal fields by criteria of Amaral and Insausti (1990). Inset image depicts a single sampling site under 400× total magnification.
Total volumes of individual hippocampal fields were calculated similarly for the purpose of estimating total cell numbers. Total numbers of pyramidal neurons and glial cells were estimated in pyramidal cell layer of CA1, CA2/3, and hilus and granule cells and glial cells in granule cell layer of dentate gyrus. Hippocampal fields were delineated via cytoarchitectonic criteria (Amaral & Insausti, 1990) (Fig. 1. B.). Hilus was defined as the pyramidal cell layer located within the concavity of dentate gyrus. CA2/3 referred to the pyramidal cell layer interposed between its borders with hilus and CA1 and is characterized by relatively high cell density and large pyramidal neuron somata size. CA1 is interposed between CA2/3 and subicular complex and exhibits smaller pyramidal neuron somata and lower cell density versus CA2/3.
Estimation of cell number was carried out using the Optical Fractionator in Stereo Investigator. Stereological parameters are summarized in Table 3. Average volumes of neuronal somata and glial cell nuclei in each hippocampal field were estimated using the Isotropic Nucleator. Stereological procedures were carried out under 400× magnification using Nikon Plan Apo 40× oil-immersion objective (N.A. = 1.0, W.D. = 0.16 mm). Pyramidal neurons were identified by the pyramidal morphology of somata and discrete nucleoli; soma size varied by CA field. Dentate granule cells were small neuronal cells exclusive to DG granule cell layer. Glial cells were identified by the round morphology of their nuclei and distinct staining patterns that differ by cell type. No attempt was made to differentiate glial cell types. Unlike neurons, a Nissl stain makes observable only the nuclei of glial cells and not their somata.
Table 3.
Stereological parameters for cell quantification
| Disector dimensions (μm) | Grid dimensions (μm) | |
|---|---|---|
| CA1 | 100 (X) × 100 (Y) × 25 (Z) | 1000 (X) × 1000 (Y) |
| CA2/3 | 90 (X) × 90 (Y) × 25 (Z) | 650 (X) × 650 (Y) |
| Hilus | 100 (X) × 100 (Y) × 25 (Z) | 800 (X) × 800 (Y) |
| Dentate gyrus | 35 (X) × 35 (Y) × 25 (Z) | 450 (X) × 450 (Y) |
2.4. Statistical analyses
Sample size was determined by power analyses (Lenth, 2006–2009) based on previous results (Stockmeier et al., 2004). All subsequent statistical analyses were performed using SAS software, version 9.2 or 9.3 (SAS Institute, Cary, NC). Threshold for statistical significance was set at Type I error rate of α = 0.05, nonsignificant trends noted at 0.05 ≤ p ≤ 0.10.
Dependent variables were tested by Cohort for normality of distribution via Shapiro-Wilk W statistic. Those variables exhibiting non-normal distribution in both Cohorts underwent comparison by Cohort via Wilcoxon rank sum test. Otherwise, Cohort comparisons were made via t-tests. Pooled t-tests were used if folded F statistic indicated equality of variances between the two Cohorts, Satterthwaite t-tests if unequal variances.
To test for potential effects of suicide, antidepressant prescription (valid within last month of life), detection of antidepressant medication in postmortem toxicology, or recurrence/chronicity of depression, the MDD cohort was divided accordingly into two groups, yielding a total of three groups to compare. Subjects were considered to have suffered recurrent or chronic depression if they had multiple episodes or at least one continuous episode lasting >2 years. Eleven subjects were included in this group and had an average illness duration of 13.8 ± 2.7 years. Amongst the remainder who had only a single episode, average duration was 0.3 ± 0.1 years, approximately two to five months. Dependent variables were tested by these new groupings for normality of distribution, as done earlier by cohort, and comparisons amongst groups were made using either analyses of variance (ANOVA) or Kruskal-Wallis tests. Interactions of cohort with sex or age dichotomized into young (<50 years old) versus old (≥50 years old) were assessed using two-way factorial ANOVA (cohort × sex and cohort × young/old, respectively) or corresponding Kruskal-Wallis tests treating the interaction as sole factor in the model. Post hoc comparisons for all ANOVA were made using Tukey’s Honestly Significant Difference test, whereas those following Kruskal-Wallis tests were made via Wilcoxon rank sum tests across all possible comparisons, with Bonferroni correction.
Analyses of covariance (ANCOVA) were conducted to adjust for potential covariance due to age. Age was chosen for inclusion in the model through examination of Pearson’s correlations, across cohort, between dependent variables and continuous demographic variables. Variables assessed but ultimately not included were PMI, tissue pH, and fixation time in formalin. Post hoc comparisons utilized least-squares means.
Pearson’s correlations were examined by class variable for linear associations between dependent and continuous demographic variables.
3. RESULTS
3.1. Total hippocampal volume
There was no significant difference in total volume between MDD and control (Table 4). Moreover, contrary to expectation (Sheline et al., 1996), total volume was not significantly lower in recurrent/chronic MDD (Table 4). However, total volume did decrease with duration in recurrent/chronic MDD (r = −0.696, p < 0.026) (Fig. 2).
Table 4.
Total hippocampal volume
| Control | MDD | ||
|---|---|---|---|
| Total volume, mm3 | 2,535 ± 156 | 2,712 ± 90 | p < 0.340 |
|
Recur/Chronic: 2,586.2 ± 102.7 Single-Ep: 2,921.3 ± 153.3 |
p < 0.290 |
Means ± SEM are shown here. Total volume did not differ between MDD and Control subjects (p < 0.340), even after parsing MDD by recurrence or chronicity (p < 0.290).
Fig. 2. Total volume and pyramidal neuron density as a function of duration.

Total hippocampal volume decreased (r = −0.696, p < 0.026), and CA1 pyramidal neuron density increased (r = 0.840, p < 0.002), as a function of duration of depressive illness in those with recurrent or chronic MDD. Subjects who had had only a single episode are not shown in the figure, as these correlations were not statistically significant within that subset of depressed subjects.
Recent imaging studies suggest hippocampal subregions are differentially affected in MDD (Maller et al., 2007, 2011; Malykhin et al., 2010). However, subdivision of total volumes here into head, middle, and tail yielded no differences, even after considering recurrence/chronicity, suicide, or antidepressants (Supplementary Table 4).
3.2. Total hippocampal neuron and glial cell numbers
There was no significant difference in pyramidal neuron or glial cell number between MDD and control in CA1, CA2/3, or hilus (Table 5).
Table 5.
Stereological results
| Control | MDD | ||||
|---|---|---|---|---|---|
| Total cell numbers | Pyramidal neurons | CA1 | 4,836,111 ± 423,683 | 5,466,898 ± 364,068 | p < 0.268 |
| CA2/3 | 901,142 ± 92,245 | 969,506 ± 110,770 | p < 0.638 | ||
| Hilus | 1,432,024 ± 110,995 | 1,594,785 ± 131,514 | p < 0.350 | ||
|
| |||||
| Dentate granule cells | 8,028,600 ± 600,548 | 8,223,057 ± 699,064 | p < 0.835 | ||
|
| |||||
| Glial cells | CA1 | 14,346,097 ± 1,218,376 | 15,693,925 ± 1,806,176 | p < 0.541 | |
| CA2/3 | 2,189,443 ± 283,082 | 2,280,400 ± 337,407 | p < 0.838 | ||
| Hilus | 5,917,229 ± 771,941 | 6,064,486 ± 653,996 | p < 0.886 | ||
| DG | 1,884,273 ± 187,097 | 1,743,968 ± 154,256 | p < 0.567 | ||
|
| |||||
| Cell packing densities, cells/mm3 | Pyramidal neurons | CA1 | 13,672 ± 621 | 14,483 ± 615 | p < 0.361 |
| CA2/3 | 21,295 ± 1,630 | 21,405 ± 988 | p < 0.955 | ||
| Hilus | 11,733 ± 514 | 11,942 ± 515 | p < 0.777 | ||
|
| |||||
| Dentate granule cells | 245,581 ± 12,893 | 251,324 ± 10,353 | p < 0.731 | ||
|
| |||||
| Glial cells | CA1 | 40,793 ± 1,844 | 39,687 ± 1,929 | p < 0.708 | |
| CA2/3 | 48,900 ± 3,492 | 48,966 ± 3,035 | p < 0.989 | ||
| Hilus | 45,849 ± 3,901 | 44,317 ± 3,309 | p < 0.768 | ||
| DG | 55,113 ± 2,165 | 53,538 ± 2,671 | p < 0.650 | ||
|
| |||||
| Cellular volumes, μm3 | Pyramidal neuron somata | CA1 | 4,654 ± 166 | 4,755 ± 102 | p < 0.610 |
| CA2/3 | 3,433 ± 120 | 3,615 ± 143 | p < 0.334 | ||
| Hilus | 3,589 ± 111 | 3,674 ± 116 | p < 0.601 | ||
|
| |||||
| Dentate granule cell somata | 1,565 ± 52 | 1,494 ± 50 | p < 0.336 | ||
|
| |||||
| Glial nuclei | CA1 | 63.3 ± 2.4 | 72.2 ± 3.9 | ‡p < 0.062 | |
| CA2/3 | 54.3 ± 2.8 | 57.5 ± 2.9 | p < 0.434 | ||
| Hilus | 55.5 ± 2.2 | 61.7 ± 3.3 | p < 0.124 | ||
| DG | 82.3 ± 5.1 | 91.3 ± 5.2 | p < 0.225 | ||
There was no significant difference between MDD and control in DG granule cell or glial cell number (Table 5). However, granule cell number decreased with duration in MDD subjects with no current antidepressant prescription at time of death (r = −0.739, p < 0.037) and with percent of life with MDD in suicide (r = −0.632, p < 0.050). Moreover, both granule cell (r = 0.971, p < 0.002) and glial cell numbers (r = 0.980, p < 0.001) increased with age in MDD subjects with antidepressant detected in postmortem toxicology (Fig. 3).
Fig. 3. Dentate gyrus cell numbers, age, and antidepressants.

Total numbers of granule cells (r = 0.971, p < 0.002) and glial cells (r = 0.980, p < 0.001) in DG increased as a function of age in subjects with MDD in whom evidence of antidepressant intake was detected in postmortem toxicology. If the eldest subject (age 79 years) is excluded, the correlation remains statistically significant for granule cells (r = 0.814, p < 0.049) but is reduced to a nonsignificant trend for glial cells (r = 0.749, p < 0.087). However, please note that there is no empirical, a priori basis to exclude this subject. Please also note that neither total hippocampal volume nor volume of the dentate gyrus was significantly different amongst controls and MDD subjects with or without either antidepressant in postmortem toxicology.
3.3. Hippocampal neuron and glial cell densities
There was no significant difference in pyramidal neuron or glial cell density between MDD and control in CA1, CA2/3, or hilus (Table 5). In CA1, pyramidal neuron density increased with duration in recurrent/chronic MDD (r = 0.840, p < 0.002) (Fig. 2) and glial cell density with age in control (r = 0.499, p < 0.042) but not MDD. CA2/3 pyramidal neuron density increased with age in MDD subjects with no antidepressant detected postmortem (r = 0.620, p < 0.042).
In DG, neither granule cell nor glial cell density was significantly different between MDD and control (Table 5). However, granule cell density decreased with duration in MDD subjects with no current antidepressant prescription (r = −0.734, p < 0.039).
Like volume, cell densities were not significantly different in MDD in head, middle, or tail, even after accounting for recurrence/chronicity, suicide, or antidepressants (Supplementary Table 5).
3.4. Volumes of neuron somata and glial cell nuclei
There was no significant difference between MDD and control in pyramidal neuron soma volume or glial nuclear volume in CA1, CA2/3, or hilus (Table 5). However, glial nuclear volume in CA1 increased with age in MDD (r = 0.560, p < 0.020) but not control.
In DG, there was no significant difference in granule cell soma volume or glial nuclear volume between MDD and control (Table 5). However, granule cell soma volume decreased with age in MDD subjects with antidepressant detected postmortem (r = −0.977, p < 0.001), whereas glial nuclear volume increased with age in suicide (r = 0.744, p < 0.014).
4. DISCUSSION
Hippocampal volumes reported here in postmortem tissue were comparable to those observed via neuroimaging (Gur et al., 2002; Lupien et al., 2007; Pavić et al., 2007; Villarreal et al., 2002). Meta-analyses of neuroimaging studies generally report subtle but significant reduction in hippocampal volume in MDD (Cole et al., 2011; Kempton et al., 2011; McKinnon et al., 2009). Here, a significant decrease was noted in total hippocampal volume but only as a function of duration of depressive illness in recurrent or chronic MDD. Total numbers of pyramidal neurons and glial cells were estimated to determine whether cellular pathology is basis for reductions in hippocampal volume in depression. Total numbers of neurons and glia here did not differ in any hippocampal field examined. However, total number of dentate granule cells decreased as a function of illness duration in untreated MDD or suicide, and both granule cell and glial cell numbers in increased with age in MDD subjects treated with antidepressant drugs. Moreover, although neither neuronal nor glial cell density differed in MDD in any hippocampal field, pyramidal neuron density in CA1 increased with duration of depressive illness in recurrent/chronic MDD.
Neuronal and glial cell densities were previously estimated in replicate sections from body of right, frozen postmortem hippocampus in subjects with MDD and matched controls (Stockmeier et al., 2004). In that study, higher neuronal and glial cell density and smaller neuronal soma size in MDD were reported in all hippocampal fields examined. Minor methodological differences may account for the subtle differences between here and the 2004 study. For example, whereas fixed, celloidin-embedded tissues exhibiting no tissue shrinkage were used here, the 2004 study reported substantially (~18%) greater shrinkage in frozen sections from MDD subjects versus controls, closely matching reductions in neuron soma volume (17–21%). It was proposed that differential tissue shrinkage might reflect a difference in water content. Cellular volume is mostly water so the soma volume reduction reported in 2004 may have been an artifact of water loss during tissue processing, as cell densities but not soma sizes were corrected for tissue shrinkage. Additionally, the current study applied the Isotropic Nucleator to estimate soma volume, whereas “projected surface area measurements” were collected previously (Stockmeier et al., 2004). As for cell densities, only three sections were examined within the body of hippocampus in 2004, whereas the entire structure was sampled here. Cell densities cannot be assumed homogeneous throughout an entire structure so greater cell densities in MDD may be restricted to hippocampal body, yet this was not replicated here (Supplementary Table 5). Relative dearth of antidepressant-treated subjects could explain greater cell densities in MDD in the earlier study. Over a third of MDD subjects studied here had antidepressant medication detected in postmortem toxicology, versus only two subjects in 2004.
Decreasing total hippocampal volume as a function of duration of depression in recurrent or chronic MDD observed here was consistent with previous neuroimaging results (Sheline et al., 1996). This finding in conjunction with increasing CA1 pyramidal neuron density with duration of depression in the same subjects supports the hypothesis that smaller hippocampal volume in MDD reflects attenuated volume of neuropil. This putative neuropil deficit has been proposed to involve diminished neurotrophic factor signaling, particularly brain-derived neurotrophic factor (BDNF) (Castrén et al., 2007; Castrén & Rantamäki, 2010; Kuipers & Bramham, 2006). Indeed, using tissues from the same collection as here, Duric et al. (2010) reported reduced gene transcription for BDNF, as well as CREB and several other growth factors. BDNF is thought essential to efficacy of antidepressant medications (Kozisek et al., 2008; Lee & Kim, 2010; Masi & Brovedani, 2011). Cumulative evidence in depression and from suicide victims (Duric et al., 2010; Dwivedi et al., 2003) and stress-induced animal models related to depression (Yu & Chen, 2011) suggests mRNA and protein expression of BDNF and its receptor, tyrosine kinase B (TrkB), are reduced in hippocampus and prefrontal cortex and elevated in amygdala and nucleus accumbens, whereas antidepressant drugs restore normal BDNF expression in these areas. At least in rats, antidepressant compounds can restore normal TrkB expression in brain (Yu & Chen, 2011). We recently observed reduced TrkB mRNA density in CA2/3 in MDD (Stockmeier et al., 2009).
Activation of Trk receptors by neurotrophins invokes downstream intracellular signaling, including the mitogen-activated protein (MAP) kinase pathway (Huang & Reichardt, 2003; Reichardt, 2006). The MAP kinase pathway appears to be critical to etiology and treatment of MDD (Duric et al., 2010). Whole-genome assessment in postmortem hippocampal tissue revealed elevated expression of a negative regulator of MAP kinase signaling, mitogen-activated protein kinase phosphatase 1 (MKP-1), in CA1 and dentate gyrus in MDD. On this basis, Duric et al. (2010) exposed rats to chronic unpredictable stress, with or without fluoxetine. Stress resulted in decreased sucrose preference and impaired active-avoidance performance, with increased MKP-1 mRNA transcription in dentate gyrus, CA1, and CA3, whereas fluoxetine ameliorated these behaviors and opposed the upregulation of MKP-1. Finally, viral-mediated overexpression of MKP-1 in hippocampus of mice produced depressive-like behavior in absence of stress, whereas constitutive deletion of the MKP-1 gene rendered mice unsusceptible to depressogenic effects of stress, suggesting MKP-1 is necessary and sufficient for expression of depressive-like behavior. It is plausible MKP-1 expression would be increased in MDD subjects examined here. However, the neuropil reduction proposed here may not in fact reflect a direct consequence of enhanced MKP-1expression. BDNF via TrkB actually induces MKP-1 expression in vitro, and reduced MKP-1 expression is associated with reduced axonal arborization in vivo (Jeanneteau et al., 2010).
In addition to development and maintenance of neuropil, BDNF is implicated in neurogenic effects of antidepressant drugs (Castrén et al., 2007; Duman & Monteggia, 2006; Lee & Kim, 2010; Masi & Brovedani, 2011). Interventions effective against depressive symptoms generally enhance hippocampal neurogenesis (Bolwig, 2011; Malberg et al., 2000; Paizanis et al., 2007). Antidepressant efficacy may in fact require hippocampal neurogenesis (David et al., 2009; Malberg, 2004; Perera et al., 2011). Thus, the observation here that numbers of dentate granule cells and glial cells increased with age in MDD treated with antidepressant drugs requires further exploration.
Boldrini et al. (2009) reported greater numbers of nestin-immunoreactive neural progenitor cells (NPCs) in postmortem anterior hippocampal DG of subjects who had been treated with a selective serotonin reuptake inhibitor or tricyclic antidepressant versus untreated MDD subjects or controls. Moreover, numbers of NPCs decreased with age in treated MDD, though there was no relationship between age and numbers of Ki-67-immunoreactive mitotic cells in any group (Boldrini et al., 2009). We observed here increased numbers of presumably mature dentate granule cells and glial cells in antidepressant-treated MDD. Synthesizing the observations of these two studies, it is possible that while differentiation of proliferating cells toward neural fate, though not cell proliferation per se, decreases with age over course of treatment, antidepressants promote or prolong survival of fully-differentiated, mature granule cells over the course of treatment or in conjunction with age. Speculation vis-à-vis increased number of glial cells with age in antidepressant-treated MDD observed here is more difficult given that Nissl staining methods allow for identification of glial cells per se, whereas reliable identification of specific glial cell types requires immunohistochemical labeling. Boldrini et al. (2009) did examine GFAP-immunoreactivity but reported only qualitative observations. A forthcoming companion study to the one reported here may provide pertinent insights.
Increasing granule cell and glial cell numbers with age in antidepressant-positive subjects may not necessarily reflect effect of illness or treatment duration. Indeed, age did not correlate with illness duration, even when single-episode subjects are excluded. (Only 1 of 6 [17%] with single episode tested positive for antidepressants versus 6 of 11 [55%] recurrent/chronic.) Moreover, no direct information on treatment duration was available. Postmortem toxicology reveals only whether a drug was taken within a brief window of days to weeks. Long-term antidepressants may reduce likelihood of relapse (Kim et al., 2011; Nutt, 2010), but effects of treatment duration on hippocampal morphology or cell numbers are not well studied.
There are several limitations to the present study. Major limitations of any study of postmortem brain tissue are that subjects are examined at only a single point in time. Direct exploration of neural, biochemical, or genetic mechanisms involved in the etiopathology of depression is impossible in postmortem brain tissue. The fact that much of the information about our subjects was obtained second-hand via interviews with next-of-kin might appear prima facie to be a critical limitation. However, such informant-based methods have been validated as consistent with direct subject interviews or review of medical records (DeJong et al., 2010; Kelly & Mann, 1996). Another limitation includes the small number of subjects to support our conclusions regarding MDD after segregating subjects by recurrence or chronicity of MDD, duration of depressive illness, or toxicological evidence or documentation of treatment with antidepressant compounds. Moreover, conclusions about glia are limited by use of Nissl staining methods, as discussed in the preceding paragraph. Finally, comparison between imaging work and the present study is complicated by the majority of our subjects being male and many older than 50 years of age, whereas cohorts in imaging studies tend to be younger and better balanced by sex.
In conclusion, whether smaller hippocampal volume in recurrent or chronic MDD is due to fewer neurons or glia is only partially resolved by this study. However, decreasing total volume and increasing CA1 pyramidal neuron density with duration of depressive illness, taken together, are consistent with the neuropil hypothesis of depression. This, along with increasing numbers of dentate gyrus granule cells and glial cells with age in antidepressant-treated MDD, may reflect the action of neurotrophic factors.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amaral DG, Insausti R. 21. Hippocampal formation. In: Paxinos G, editor. The human nervous system. 1. San Diego, CA: Academic Press, Inc; 1990. pp. 711–755. [Google Scholar]
- American Psychiatric Association (APA) Diagnostic and statistical manual of mental disorders. 4. Washington, DC: Authors; 1994. [Google Scholar]
- Blugeot A, Rivat C, Bouvier E, Molet J, Mouchard A, Zeau B, Bernard C, Benoliel J-J, Becker C. Vulnerability to depression: From brain neuroplasticity to identification of biomarkers. The Journal of Neuroscience. 2011;31(36):12889–12899. doi: 10.1523/JNEUROSCI.1309-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, Mann JJ, Arango V. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology. 2009;34(11):2376–2389. doi: 10.1038/npp.2009.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolwig TG. How does electroconvulsive therapy work? Theories on its mechanism. Canadian Journal of Psychiatry. 2011;56(1):13–18. doi: 10.1177/070674371105600104. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Narayan M, Anderson ER, Staib LH, Miller HL, Charney DS. Hippocampal volume reduction in major depression. The American Journal of Psychiatry. 2000;157(1):115–118. doi: 10.1176/ajp.157.1.115. [DOI] [PubMed] [Google Scholar]
- Calabrese F, Molteni R, Racagni G, Riva MA. Neuronal plasticity: A link between stress and mood disorders. Psychoneuroendocrinology. 2009;34(Suppl 1):S206–S216. doi: 10.1016/j.psyneuen.2009.05.014. [DOI] [PubMed] [Google Scholar]
- Castrén E, Võikar V, Rantamäki T. Role of neurotrophic factors in depression. Current Opinion in Pharmacology. 2007;7(1):18–21. doi: 10.1016/j.coph.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Castrén E, Rantamäki T. Neurotrophins in depression and antidepressant effects. Novartis Foundation Symposium; 2008. pp. 43–59.pp. 87–93. [DOI] [PubMed] [Google Scholar]
- Castrén E, Rantamäki T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Developmental Neurobiology. 2010;70(5):289–297. doi: 10.1002/dneu.20758. [DOI] [PubMed] [Google Scholar]
- Cole J, Costafreda SG, McGuffin P, Fu CHY. Hippocampal atrophy in first episode depression: A meta-analysis of magnetic resonance imaging studies. Journal of Affective Disorders. 2011;134(1–3):483–487. doi: 10.1016/j.jad.2011.05.057. [DOI] [PubMed] [Google Scholar]
- Conrad CD. Chronic stress-induced hippocampal vulnerability: the glucocorticoid vulnerability hypothesis. Reviews in the Neurosciences. 2008;19(6):395–411. doi: 10.1515/revneuro.2008.19.6.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke SF, Bliss TVP. Plasticity in the human central nervous system. Brain. 2006;129(7):1659–1673. doi: 10.1093/brain/awl082. [DOI] [PubMed] [Google Scholar]
- David DJ, Samuels BA, Rainer Q, Wang J-W, Marsteller D, Mendez I, Drew M, Craig DA, Guiard BP, Guilloux J-P, Artymyshyn RP, Gardier AM, Gerald C, Antonijevic IA, Leonardo ED, Hen R. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009;62(4):479–493. doi: 10.1016/j.neuron.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJong TM, Overholser JC, Stockmeier CA. Apples to oranges? A direct comparison between suicide attempters and suicide completers. Journal of Affective Disorders. 2010;124(1–2):90–97. doi: 10.1016/j.jad.2009.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biological Psychiatry. 2006;59(12):1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biological Psychiatry. 2006;59(12):1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Duric V, Banasr M, Licznerski P, Schmidt HD, Stockmeier CA, Simen AA, Newton SS, Duman RS. A negative regulator of MAP kinase causes depressive behavior. Nature Medicine. 2010;16(11):1328–1332. doi: 10.1038/nm.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN. Altered gene expression of brain-derived neurotrophic factor and receptor tyrosine kinase B in postmortem brain of suicide subjects. Archives of General Psychiatry. 2003;60(8):804–815. doi: 10.1001/archpsyc.60.8.804. [DOI] [PubMed] [Google Scholar]
- First M, Spitzer R, Gibbon M, Williams J. Structured Clinical Interview for the DSM-IV Axis I Disorders (SCID Patient Edition), version 2.0. New York, NY: New York State Psychiatric Institute; 1995. [Google Scholar]
- Gartlehner G, Hansen RA, Morgan LC, Thaler K, Lux L, Van Noord M, Mager U, Thieda P, Gaynes BN, Wilkins T, Strobelberger M, Lloyd S, Reichenpfader U, Lohr KN. Comparative benefits and harms of second-generation antidepressants for treating major depressive disorder: An updated meta-analysis. Annals of Internal Medicine. 2011;155(11):772–785. doi: 10.7326/0003-4819-155-11-201112060-00009. [DOI] [PubMed] [Google Scholar]
- Gur RC, Gunning-Dixon F, Bilker WB, Gur RE. Sex differences in temporo-limbic and frontal brain volumes of healthy adults. Cerebral Cortex. 2002;12(9):998–1003. doi: 10.1093/cercor/12.9.998. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: Roles in neuronal signal transduction. Annual Review of Biochemistry. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Jeanneteau F, Deinhardt K, Miyoshi G, Bennett AM, Chao MV. The MAP kinase phosphatase MKP-1 regulates BDNF-induced axon branching. Nature Neuroscience. 2010;13(11):1373–1379. doi: 10.1038/nn.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly TM, Mann JJ. Validity of DSM-III-R diagnosis by psychological autopsy: a comparison with clinician ante-mortem diagnosis. Acta Psychiatrica Scandinavica. 1996;94(5):337–343. doi: 10.1111/j.1600-0447.1996.tb09869.x. [DOI] [PubMed] [Google Scholar]
- Kempton MJ, Salvador Z, Munafò MR, Geddes JR, Simmons A, Frangou S, Williams SC. Structural neuroimaging studies in major depressive disorder. Meta-analysis and comparison with bipolar disorder. Archives of General Psychiatry. 2011;68(7):675–690. doi: 10.1001/archgenpsychiatry.2011.60. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, Rush AJ, Walters EE, Wang PS. The epidemiology of major depressive disorder: Results from the National Comorbidity Survey Replication (NCS-R) The Journal of the American Medical Association. 2003;289(23):3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- Kim K-H, Lee S-M, Paik J-W, Kim N-S. The effects of continuous antidepressant treatment during the first 6 months on relapse or recurrence of depression. Journal of Affective Disorders. 2011;132(1–2):121–129. doi: 10.1016/j.jad.2011.02.016. [DOI] [PubMed] [Google Scholar]
- Kozisek ME, Middlemas D, Bylund DB. Brain-derived neurotrophic factor and its receptor tropomyosin-related kinase B in the mechanism of action of antidepressant therapies. Pharmacology & Therapeutics. 2008;117(1):30–51. doi: 10.1016/j.pharmthera.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Kuipers SD, Bramham CR. Brain-derived neurotrophic factor mechanisms and function in adult synaptic plasticity: New insights and implications for therapy. Current Opinion in Drug Discovery & Development. 2006;9(5):580–586. [PubMed] [Google Scholar]
- Lee B-H, Kim Y-K. The roles of BDNF in the pathophysiology of major depression and in antidepressant treatment. Psychiatry Investigation. 2010;7(4):231–235. doi: 10.4306/pi.2010.7.4.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenth RV. Java applets for power and sample size, 2006–2009. Retrieved 2010 Oct 28 from http://www.stat.uiowa.edu/~rlenth/Power.
- Lupien SJ, Evans A, Lord C, Miles J, Pruessner M, Pike B, Pruessner JC. Hippocampal volume is as variable in young as in older adults: Implications for the notion of hippocampal atrophy in humans. NeuroImage. 2007;34(2):479–485. doi: 10.1016/j.neuroimage.2006.09.041. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. The Journal of Neuroscience. 2000;20(24):9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malberg JE. Implications of adult hippocampal neurogenesis in antidepressant action. Journal of Psychiatry & Neuroscience. 2004;29(3):196–205. [PMC free article] [PubMed] [Google Scholar]
- Maller JJ, Daskalakis ZJ, Fitzgerald PB. Hippocampal volumetrics in depression: The importance of the posterior tail. Hippocampus. 2007;17(11):1023–1027. doi: 10.1002/hipo.20339. [DOI] [PubMed] [Google Scholar]
- Maller JJ, Réglade-Meslin C, Chan P, Daskalakis ZJ, Thomson RHS, Anstey KJ, Budge M, Sachdev P, Fitzgerald PB. Hippocampal sulcal cavities: Prevalence, risk factors and relationship to memory impairment. Brain Research. 2011;1368:222–230. doi: 10.1016/j.brainres.2010.10.089. [DOI] [PubMed] [Google Scholar]
- Malykhin NV, Carter R, Seres P, Coupland NJ. Structural changes in the hippocampus in major depressive disorder: Contributions of disease and treatment. Journal of Psychiatry & Neuroscience. 2010;35(5):337–343. doi: 10.1503/jpn.100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masi G, Brovedani P. The hippocampus, neurotrophic factors and depression: Possible implications for the pharmacotherapy of depression. CNS Drugs. 2011;25(11):913–931. doi: 10.2165/11595900-000000000-00000. [DOI] [PubMed] [Google Scholar]
- McKinnon MC, Yucel K, Nazarov A, MacQueen GM. A meta-analysis examining clinical predictors of hippocampal volume in patients with major depressive disorder. Journal of Psychiatry & Neuroscience. 2009;34(1):41–54. [PMC free article] [PubMed] [Google Scholar]
- Nutt DJ. Rationale for, barriers to, and appropriate medication for the long-term treatment of depression. The Journal of Clinical Psychiatry. 2010;71(Suppl E1):e02. doi: 10.4088/JCP.9058se1c.02gry. [DOI] [PubMed] [Google Scholar]
- Paizanis E, Hamon M, Lanfumey L. Hippocampal neurogenesis, depressive disorders, and antidepressant therapy. Neural Plasticity. 2007:Article 73754. doi: 10.1155/2007/73754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavić L, Gregurek R, Rados M, Brkljacić B, Brajković L, Simetin-Pavić I, Ivanac G, Pavlisa G, Kalousek V. Smaller right hippocampus in war veterans with posttraumatic stress disorder. Psychiatry Res. 2007;154(2):191–198. doi: 10.1016/j.pscychresns.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Perera TD, Dwork AJ, Keegan KA, Thirumangalakudi L, Lipira CM, Joyce N, Lange C, Higley JD, Rosoklija G, Hen R, Sackeim HA, Coplan JD. Necessity of hippocampal neurogenesis for the therapeutic action of antidepressants in adult nonhuman primates. PLoS One. 2011;6(4):e17600. doi: 10.1371/journal.pone.0017600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philosophical Transactions of the Royal Society of London: Series B, Biological Sciences. 2006;361(1473):1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, Niederehe G, Thase ME, Lavori PW, Lebowitz BD, McGrath PJ, Rosenbaum JF, Sackeim HA, Kupfer DJ, Luther J, Fava M. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. The American Journal of Psychiatry. 2006;163(11):1905–1917. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(9):3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY, Uylings HBM, Friedman L, Rajkowska G. Cellular changes in the postmortem hippocampus in major depression. Biological Psychiatry. 2004;56(9):640–650. doi: 10.1016/j.biopsych.2004.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockmeier CA, Mahajan G, Kooiman H, Austin MC, Szewczyk B, Iyo AH, May W, Rajkowska G. Cell loss and impaired neuroplasticity in prefrontal cortex and hippocampus during major depressive disorder. Biological Psychiatry. 2009;65:173S. (Prog. No. 568) [Google Scholar]
- Uylings HBM, van Eden CG, Hofman MA. Morphometry of size/volume variables and comparisons of their bivariate relations in the nervous system under different conditions. Journal of Neuroscience Methods. 1986;18(1–2):19–37. doi: 10.1016/0165-0270(86)90111-1. [DOI] [PubMed] [Google Scholar]
- Villarreal G, Hamilton DA, Petropoulos H, Driscoll I, Rowland LM, Griego JA, Kodituwakku PW, Hart BL, Escalona R, Brooks WM. Reduced hippocampal volume and total white matter volume in posttraumatic stress disorder. Biological Psychiatry. 2002;52(2):119–125. doi: 10.1016/s0006-3223(02)01359-8. [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO) Depression. 2010 Retrieved 2010 Jun 21 from http://www.who.int/mental_health/management/depression/definition/en/
- Yu H, Chen Z-Y. The role of BDNF in depression on the basis of its location in the neural circuitry. Acta Pharmacologica Sinica. 2011;32(1):3–11. doi: 10.1038/aps.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.