

Abstract
PA-U2, an engineered anthrax protective antigen that is activated by urokinase was combined with wild-type lethal factor in the treatment of Colo205 colon adenocarcinoma in vitro and B16-BL6 mouse melanoma in vitro and in vivo. This therapy was also tested in combination with the small molecule paclitaxel, based on prior reports suggesting synergy between ERK1/2 inhibition and chemotherapeutics. Colo205 was sensitive to PA-U2/LF while B16-BL6 was not. For the combination treatment of B16-BL6, paclitaxel showed a dose response in vitro, but cells remained resistant to PA-U2/LF even in the presence of paclitaxel. In vivo, each therapy slowed tumor progression, and an additive effect between the two was observed. Since LF targets tumor vasculature while paclitaxel is an anti-mitotic, it is possible the agents were acting against different cells in the stroma, precluding a synergistic effect. The engineered anthrax toxin PA-U2/LF warrants further development and testing, possibly in combination with an anti-angiogenesis therapy such as sunitinib or sorafinib.
Keywords: anthrax toxin, B16 melanoma, urokinase, paclitaxel, combination therapy
Introduction
Despite the declaration of the War on Cancer by Richard Nixon in 1971, cancer remains the second leading cause of death in the United States, accounting for 23.3 % of deaths in 2009 (http://www.cdc.gov/nchs/data/dvs/deaths_2009_release.pdf). Huge strides have been made in the past forty years in both prevention and treatment of disease, but locally advanced and metastatic cancers remain deadly. Chemotherapeutics such as cyclophosphamide [1,2], cisplatin [3,4], doxorubicin [5], and paclitaxel [6,4,7] now serve as backbone agents for combination therapies that have lengthened survival times for many forms of cancer; however, these drugs are limited in their efficacy by toxicities, often cardiac [8], hematopoietic [9], neurological [10], and nephritic [11] in nature. They also cause nausea, vomiting, and alopecia, decreasing a patient’s quality of life [12].
Recent advances in treatment include tyrosine kinase inhibitors (TKIs) and biological agents, which have much better toxicity profiles compared to standard chemotherapeutics. TKIs include erlotinib, imatinib, and crizotinib, and patients whose tumors have sensitizing mutations show marked responses within the first few months of treatment, but are prone to relapse within 18 months due to the evolution of various resistance mechanisms [13–15]. Biologics include several different categories of agents such as interferons, interleukins, and colony stimulating factors, but the most prevalent are monoclonal antibodies such as bevacizumab and trastuzumab. These agents neutralize growth factors or block signaling of important receptor tyrosine kinases and can be employed as an add-on to a standard chemotherapy backbone or as single agents [16–18]. Another subset of biologic therapies is the immunotoxins, which consist of cell-targeting ligands such as growth factors or antibodies that are conjugated to toxic drugs, peptides, or proteins [19]. Toxic moieties include the ADP-ribosyltransferase Pseudomonas exotoxin A [20], diphtheria toxin [21], the small molecule DM1 [22,23], deglycosylated ricin A-chain [24], and the plant toxin gelonin [25]. These immunotoxins bind to cancer-specific cell surface markers and are endocytosed into the cytoplasm where they kill the cancer cell.
An alternate system of employing bacterial toxins as therapeutic agents involves specific activation of the toxins by proteases overexpressed in tumor stroma, such as matrix metalloproteases (MMPs), urokinase plasminogen activator (uPA), and in the case of prostate cancer, prostate specific antigen (PSA). These toxins are administered systemically but show activity only in tissues that express the selected protease. To date, only two toxins have been so altered: B. anthracis protective antigen [26,27] and A. hydrophila proaerolysin [28]. Protective antigen (PA) is the cellular binding component of lethal toxin, an AB toxin that utilizes PA and a separate catalytic protein, lethal factor (LF), while proaerolysin is a single-part, pore forming toxin [29,30]. Proaerolysin is secreted as a dimer that binds to glycophosphatidylinositol-anchored proteins on the cell surface, while PA is secreted as a monomer and binds to one of two transmembrane proteins, capillary morphogenesis protein 2 or tumor endothelial marker 8. Both PA and proaerolysin are cleaved by cell surface furin or related proteases to release an inhibitory fragment. The activated proteins then oligomerize to form 7- (PA and aerolysin) and 8- (PA only) membered rings [31]. At this point, the mechanism of the two toxins diverges. Aerolysin forms pores in the cellular membranes, allowing for loss of small molecules and ions, ultimately leading to apoptosis. In contrast, PA oligomers bind LF and the entire complex is endocytosed. A pH change within the vesicle causes a pore to form, and LF is translocated into the cytosol where it cleaves mitogen-activated protein kinase kinases 1, 2, 3, 4, 6, and 7, shutting down the ERK, p38, and JNK pathways [32–35]. This leads to death in many cell types. A recent report found that anthrax lethal factor also cleaves rat Nlrp1b, activating the inflammasome and leading to release of IL-1β [36].
PRX302, an engineered version of proaerolysin with activating protease specificity altered to PSA, is currently in phase II trials for benign prostatic hyperplasia (NCT01454349) [28]. Several labs have been investigating the use of engineered anthrax lethal toxin for various tumors and different combinations of PA mutants combined with natural and engineered catalytic moieties have been published. Tumor-specific mutants include PA-L1, which is activated by matrix metalloproteases, combined with either wild-type lethal factor or FP59, and PA-U2, a urokinase plasminogen activator-activated PA, in combination with FP59 [27,26,37–39]. FP59 is a fusion protein containing the PA binding domain of LF fused to the catalytic domain of Pseudomonas exotoxin A. Another version of lethal toxin requires both MMP and uPA proteases for full cytotoxicity by mutating the LF binding site to prevent binding except when the differentially activated PAs are next to each other in the oligomer [40]. These four combinations have shown activity in vitro and in vivo; however, it is important to note that ERK-addiction is necessary for in vitro sensitivity to lethal factor-containing regimens [39]. PA-L1/LF shrank tumors in vivo regardless of BRAF mutational status, indicating that the mechanism of action in vivo is not related to ERK-addiction in the cancer cell. Histology found that tumor vasculature cross-sectional area was significantly reduced compared to PBS-treated tumors, suggesting that angiogenesis was the key target of this regimen. Based on these findings, we set out to evaluate the efficacy of PA-U2/LF on a sensitive and resistant line in vitro, and a resistant line in vivo.
In addition to determining the efficacy of PA-U2/LF alone, we believe that combining the toxin with currently used therapies might be of benefit. Several previous reports have found evidence suggesting benefit in combining chemotherapeutics with ERK blockage. ERK inhibition has been found to downregulate P-glycoprotein, the product of the MDR1 gene [41] and to show increased activity when combined with irinotecan or taxanes [42]. In addition, while the allosteric ERK1/2 inhibitor AZD6244 was found to be cytostatic in several melanoma lines, the combination of AZD6244 and a taxane proved to be cytotoxic [43]. Other work has examined the effectiveness of combination therapies involving immunotoxins, which showed synergy, albeit by a mechanism unrelated to ERK inhibition [44–46]. Since lethal factor inhibits ERK signaling, we desired to examine the effect of combination therapy with the urokinase-activated protective antigen, PA-U2, in combination with wild-type lethal factor and paclitaxel. The results from published studies involving AZD6244 suggest that cells which are not sensitive to LF in vitro may be sensitized by taxane treatment, allowing for direct targeting of the tumor cells.
Materials and methods
General methods
PA, PA-U2, and LF were purified as previously described [47]. Colo205 was obtained from the NCI-60 cell panel and B16-BL6 mouse melanoma was kindly provided by Judah Folkman, Harvard Medical School, Boston, MA. Both were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented to 10 % fetal bovine serum, 50 μg/mL gentamycin (both from Life Technologies, Grand Island, NY), and 10 mM Hepes, pH 7.3 (Quality Biological, Gaithersburg, MD) in a humidified incubator at 37 °C and 5 % CO2. Paclitaxel was purchased from Teva Pharmaceuticals (Washington, DC). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma (St. Louis, MO). Female C57BL6/J mice, aged 6–8 weeks, were purchased from Jackson Labs (Bar Harbor, ME). The mice were housed in a pathogen-free facility certified by the Association for Assessment and Accreditation of Laboratory Animal Care International, and the study was carried out in accordance with institutional guidelines.
In vitro toxicity assays
Colo205 and B16-BL6 cells were plated at 50,000 cells/well and 5,000 cells/well, respectively, in a 96-well plate in supplemented DMEM. The next day, PA or PA-U2 was added at 7 different concentrations (1–1000 ng/mL) with a constant concentration of LF at 500 ng/mL in triplicate and the cells were incubated for an additional 48 h. MTT was added to a concentration of 0.5 mg/mL and the cells were incubated for an additional hour. The medium was aspirated and MTT was solubilized in 91% isopropanol containing 0.5% sodium dodecylsulfate and 0.038 M hydrochloric acid and read using a SpectraMax 190 plate reader and Softmax Pro 4.7.1 software (Molecular Devices, Sunnyvale, CA) at 570 and 490 nm. The difference of the absorbances was used to determine percent survival as compared to a no-treatment control.
In vitro combination cytotoxicity assay
B16-BL6 cells were seeded in 96-well plates at 5,000 cells/well in supplemented DMEM. The following day, paclitaxel was added to a final concentration of 0, 0.75, 2.5 or 7.5 ng/mL. The next day, PA or PA-U2 was added at 7 different concentrations (1–1000 ng/mL) with a constant concentration of LF at 500 ng/mL in triplicate and the cells were incubated for an additional 48 h. MTT was added for the last hour of incubation and the plates were read as above. Mouse tumor models
B16-BL6 cells (5 ×105) were injected into the right flank of 40 C57BL/6J mice in 0.1 mL unsupplemented DMEM. On day 5 after inoculation, mice received intraparitoneally (i.p.) either paclitaxel at 50 mg/kg or blank vehicle in 0.5 mL PBS. On days 6, 9, and 12, mice received i.p. either 30 μg PA-U2 and 10 μg LF in 0.5 mL PBS or 0.5 mL PBS alone. Mice were weighed before injection and tumors were measured on days 5, 6, 9, 12, and 14 and volumes were calculated by the formula, (length × width × height)/2.
Statistical analysis
Tumor sizes were analyzed by repeated measures ANOVA. To account for differences in the initial tumor volume between mice, the volume measurements were normalized to their values on the first day of treatment. In order to approximate the assumptions for the distributions within groups, the volumes were transformed to the spherical diameter. Treatments were assessed for their individual efficacy in reducing tumor growth as compared to vehicle control, as well as their synergistic relationship over days five to fourteen. Synergy was defined as the combination treatment having a significantly greater effect than the sum of the two individual treatments.
Results
In vitro activity on tumor cell lines
Two cell lines, Colo205 colon adenocarcinoma and B16-BL6 melanoma were treated with PA/LF and urokinase-activated PA-U2/LF to evaluate the sensitivity of these cells to the toxin as a single agent. Wild-type PA was used to control for the unknown expression levels of urokinase in the cell lines. The cells were treated with 7 different concentrations of the PA variants (1–1000 ng/mL) in the presence of a constant amount of LF. While Colo205 showed sensitivity to both toxin combinations, with an EC50 of less than 1 ng/mL, B16-BL6 proved to be only partially sensitive to even the highest amount of lethal toxin (Figure 1). To study the ability of the combination therapy of paclitaxel and PA-U2/LF to kill the resistant line B16-BL6, the cells were exposed to a 24-h pretreatment of four different doses of paclitaxel (0, 0.75, 2.5, or 7.5 ng/mL) followed by addition of PA/LF or the PA-U2/LF in varying doses of PA (1–1000 ng/mL). While the paclitaxel treatment showed some cytotoxic effect in a dose-dependent manner, neither PA variant showed an effect alone, or in combination with paclitaxel (Figure 2).
Fig. 1.
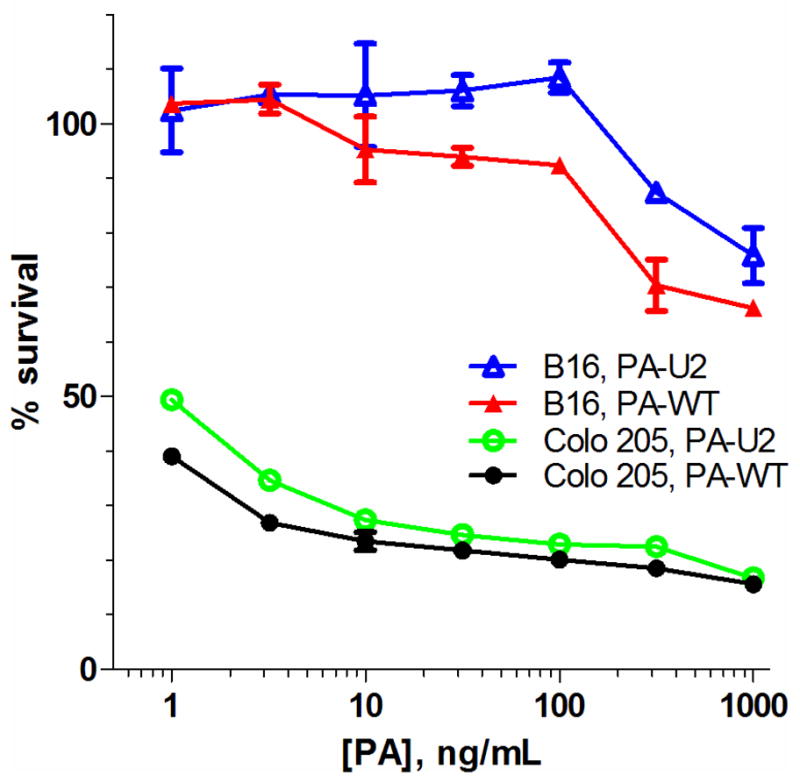
Sensitivity of Colo205 and B16-BL6 cells to PA/LF or PA-U2/LF. PA (solid markers) or PA-U2 (open markers) was added at varying concentrations of PA and 500 ng/mL LF. The cells were allowed to grow 48 h before the addition of 0.5 mg/mL MTT for an additional 1 h before lysis. Percent survival is reported as the absorbance of treatment wells compared to a vehicle-only control. Data is presented as the mean of three independent experiments and error bars are standard error. For many points, error bars are too small to see
Fig. 2.
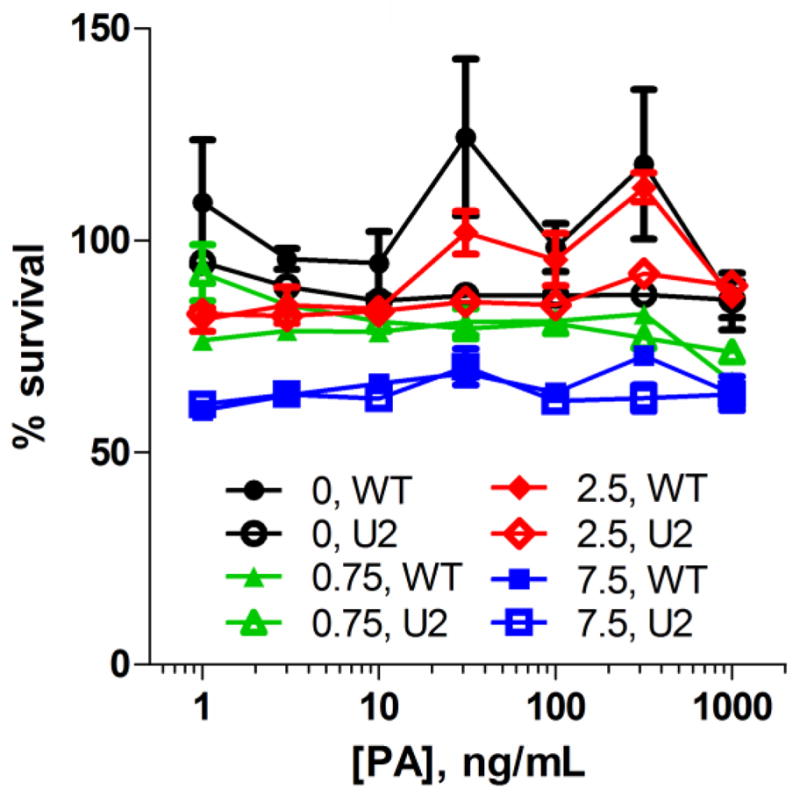
B16-BL6 mouse melanoma cells are not sensitized to PA toxins by prior paclitaxel treatment. Cells were seeded in 96-well plates, allowed to grow overnight, and treated with paclitaxel at 0–7.5 ng/mL for 24 h before the addition varying concentrations of PA (solid markers) or PA-U2 (open markers) with 500 ng/mL LF. The cells were allowed to grow an additional 48 h before the addition of 0.5 mg/mL MTT for an additional 1 h before lysis. Percent survival is reported as the absorbance of treatment wells compared to a vehicle-only control. Data is presented as the mean of three independent experiments and error bars are standard error. For many points, error bars are too small to see
In vivo treatment of B16-BL6 tumors
To study the effectiveness of the combination of paclitaxel and PA-U2/LF on a resistant cell line in vivo, B16-BL6 tumors were inoculated into the right flank of 40 C57BL/6J mice. At day 5 after inoculation, the mice received paclitaxel at 50 mg/kg or vehicle, and on days 6, 9, and 12, mice received 30 μg PA-U2 and 10 μg LF or vehicle. Tumors in the control group of mice grew quickly and reached an average size of 992 mm2 by day 14 (Figure 3a). Mice that had received a sub-therapeutic dose of paclitaxel had less growth, reaching 878 mm2 by day 14. The PA-U2/LF and combination groups showed much slower tumor growth compared to either the vehicle or paclitaxel groups, with the combination group showing the smallest tumors. Although one mouse in both the vehicle and PA-U2/LF treatment groups died of disease progression at day 12, these were included in the statistical analysis. The individual treatment with either paclitaxel or PA-U2/LF, as well as the combination, led to significantly reduced tumor volumes compared to the vehicle treatment arm (p = 0.0002, p < 0.0001, and p < 0.0001, respectively). The combination arm was also more effective than PA-U2/LF alone (p=0.0037). There was no evidence of synergy between the treatments (p = 0.69). The paclitaxel group had a faster effect on tumor volumes, probably due to its earlier administration. None of the treatments caused a significant effect on body weight (Figure 3b).
Fig. 3.
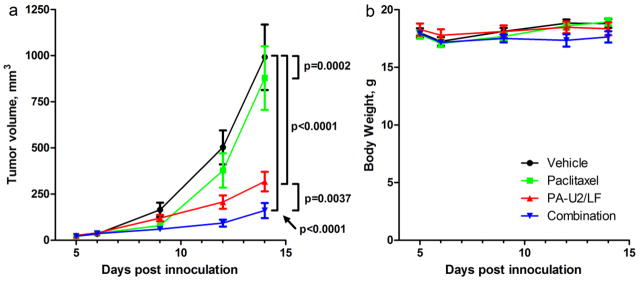
Paclitaxel and anthrax toxin have additive but not synergistic anti-tumor activities. Average tumor sizes (a) and body weights (b) of mice receiving vehicle (circles), paclitaxel only (squares), PA-U2/LF only (upward triangles), or the combination (downward triangles) for the duration of the study. B16-BL6 tumors were implanted in the right flank of C57BL6 mice. The mice were treated with 50 mg/kg paclitaxel or vehicle on day 5. Half of each of these groups then received 30 μg PA-U2 and 10 μg LF or vehicle on days 6, 9, and 12. The final group sizes were 10 mice per treatment. Measurements were taken before each injection and at day 14. Data are presented as mean tumor volume and error bars are standard error
Discussion
To establish the effectiveness of the engineered bacterial toxin PA-U2/LF, Colo205 colon adenocarcinoma and B16-BL6 mouse melanoma were treated with wild-type PA/LF or PA-U2/LF over the range of 1–1000 ng/mL in vitro. Colo205 showed a dose response to the treatment with an EC50 of <1 ng/mL while B16-BL6 was resistant at all doses tested. Previous tests of the matrix metalloprotease-activated PA (PA-L1) in combination with LF found that Colo205 was sensitive to the toxin with an EC50 of 10 ng/mL, which is 10-fold higher than the EC50 of the PA-U2/LF tested here. It was previously noted that BRAF mutational status was predictive of an in vitro response to lethal toxin treatment [48,39]. This is presumably because cells harboring the B-RAF V600E mutation depend on ERK activation for survival and growth, whereas other cell types are able to survive in the presence of ERK inhibition. The resistance of B16-BL6 melanoma to both PA/LF and PA-U2/LF in vitro is expected since this line does not harbor tumorigenic mutations in H-, N-, and K-RAS, or B-RAF [49].
Despite the cell line’s resistance to lethal toxin in vitro, the B16-BL6 tumors implanted in mice were susceptible to PA-U2/LF as a single agent, and also in combination with paclitaxel. This is in agreement with previous data for PA-L1/LF, which has been shown to slow the growth of both LF-sensitive and LF-resistant tumors implanted in mice. It is thought that the toxin is not targeting the tumor cells directly in these situations, but is instead impairing another component of the tumor stroma, leading to shrinkage or retardation of growth, depending on the tumor type. Based on the massive reduction in vasculature cross section seen in histological sections of tumors treated with PA-L1/LF, a component of tumor blood vessels is thought to be the main target, but studies to identify the cell type involved continue [39].
While the treatments with PA-U2/LF, paclitaxel, and the combination each reduced B16-BL6 tumor growth, synergy was not found between PA-U2/LF and paclitaxel. The absence of synergy could be due to the high efficacy of the individual treatments, as the combination arm would have had to completely block tumor in order to demonstrate synergy. Such a response to therapy is very difficult to achieve with this highly aggressive tumor type. Despite the lack of evident synergy between PA-U2/LF and paclitaxel, it is possible that other drugs which work via alternate mechanisms may show a greater-than-additive effect when combined with PA-U2/LF. Since PA-U2/LF is thought to work by targeting tumor vasculature and other components of tumor stroma, it is possible that cytotoxic agents that are designed to kill the cancer cells directly may not be capable of showing synergy with PA-U2/LF since there is no overlap in target. Alternatively, a drug which also targets vasculature, such as sorafinib or sunitinib, may show synergy when combined with PA-U2/LF.
The majority of previous work in evaluating synergy between protein and small molecule drugs has either been suggestive of a mechanism or did not examine the mechanism. The only work to elucidate the mechanism of synergy between biological agents and chemotherapeutics found that paclitaxel pretreatment decreased mesothelin shedding, thereby decreasing the decoy-mediated inhibition of the anti-mesothelin immunotoxin SS1P, and producing synergy [46]. Suggested effects of ERK1/2 inhibition on the activity of paclitaxel, such as down-regulation of P-glycoprotein, could not have been seen in the animal work in this study since the drug was administered before PA-U2/LF and not given a second time. However, in the in vitro cytotoxicity experiment, while the cells were also pretreated with paclitaxel, the drug was left in the culture medium when PA-U2/LF was added, allowing for ERK inhibition to begin to down-regulate P-glycoprotein. If this mechanism were operative in B16-BL6 cells, then the combination treatments should have shown some measure of a dose-response to PA-U2/LF, which was not observed.
This study was the first to examine the combination of the urokinase-activated anthrax protective antigen PA-U2 and lethal factor for the treatment of tumors. Colo205, which harbors B-RAF V600E, was sensitive to toxin treatment in vitro, while B16-BL6 melanoma was not. However, when implanted in mice, the growth of B16-BL6 tumors was slowed by PA-U2/LF as a single agent and in combination with paclitaxel. In addition to the merit shown by PA-U2/LF monotherapy, the safety and additive effect of combination therapy with PA-U2/LF and paclitaxel suggest combination therapies with bacterial toxins deserve further exploration.
Acknowledgments
The authors thank Rasem Fattah for protein purification, Devorah Crown and Mahtab Moayeri for assistance with animal protocols, David J. Liewehr for contributing to the statistical analysis, and Diane E. Peters for advice regarding the manuscript. This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH, HHS.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Hallek M, Pflug N. Chronic lymphocytic leukemia. Ann Oncol. 2010;21(Suppl 7):vii154–vii164. doi: 10.1093/annonc/mdq373. [DOI] [PubMed] [Google Scholar]
- 2.White L. Chemotherapy in retinoblastoma: current status and future directions. Am J Pediatr Hematol Oncol. 1991;13 (2):189–201. [PubMed] [Google Scholar]
- 3.Kawai K, Akaza H. Current status of chemotherapy in risk-adapted management for metastatic testicular germ cell cancer. Cancer Sci. 2010;101 (1):22–28. doi: 10.1111/j.1349-7006.2009.01373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grossi F, Kubota K, Cappuzzo F, de Marinis F, Gridelli C, Aita M, Douillard JY. Future scenarios for the treatment of advanced non-small cell lung cancer: focus on taxane-containing regimens. Oncologist. 2010;15 (10):1102–1112. doi: 10.1634/theoncologist.2010-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thigpen T, Vance R, Puneky L, Khansur T. Chemotherapy in advanced ovarian carcinoma: current standards of care based on randomized trials. Gynecol Oncol. 1994;55 (3 Pt 2):S97–107. doi: 10.1006/gyno.1994.1347. [DOI] [PubMed] [Google Scholar]
- 6.Sparano JA. Taxanes for breast cancer: an evidence-based review of randomized phase II and phase III trials. Clin Breast Cancer. 2000;1 (1):32–40. doi: 10.3816/CBC.2000.n.002. [DOI] [PubMed] [Google Scholar]
- 7.Baird RD, Tan DS, Kaye SB. Weekly paclitaxel in the treatment of recurrent ovarian cancer. Nat Rev Clin Oncol. 2010;7 (10):575–582. doi: 10.1038/nrclinonc.2010.120. [DOI] [PubMed] [Google Scholar]
- 8.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339 (13):900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 9.Prevention and Treatment of Cancer-Related Infections . J Natl Compr Cancer Netw. 2008;6 (2):122. doi: 10.6004/jnccn.2008.0013. [DOI] [PubMed] [Google Scholar]
- 10.Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst. 2008;13 (1):27–46. doi: 10.1111/j.1529-8027.2008.00156.x. [DOI] [PubMed] [Google Scholar]
- 11.de Jonge MJ, Verweij J. Renal toxicities of chemotherapy. Semin Oncol. 2006;33 (1):68–73. doi: 10.1053/j.seminoncol.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 12.Hesketh PJ. Understanding the pathobiology of chemotherapy-induced nausea and vomiting. Providing a basis for therapeutic progress. Oncology. 2004;18 (10 Suppl 6):9–14. [PubMed] [Google Scholar]
- 13.Roychowdhury S, Talpaz M. Managing resistance in chronic myeloid leukemia. Blood Rev. 2011;25 (6):279–290. doi: 10.1016/j.blre.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Chen FL, Xia W, Spector NL. Acquired resistance to small molecule ErbB2 tyrosine kinase inhibitors. Clin Cancer Res. 2008;14 (21):6730–6734. doi: 10.1158/1078-0432.CCR-08-0581. [DOI] [PubMed] [Google Scholar]
- 15.Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat Rev Clin Oncol. 2010;7 (9):493–507. doi: 10.1038/nrclinonc.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koukourakis GV, Sotiropoulou-Lontou A. Targeted therapy with bevacizumab (Avastin) for metastatic colorectal cancer. Clin Transl Oncol. 2011;13 (10):710–714. doi: 10.1007/s12094-011-0720-z. [DOI] [PubMed] [Google Scholar]
- 17.Croom KF, Dhillon S. Bevacizumab: a review of its use in combination with paclitaxel or capecitabine as first-line therapy for HER2-negative metastatic breast cancer. Drugs. 2011;71 (16):2213–2229. doi: 10.2165/11207720-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 18.Okines A, Cunningham D, Chau I. Targeting the human EGFR family in esophagogastric cancer. Nat Rev Clin Oncol. 2011;8 (8):492–503. doi: 10.1038/nrclinonc.2011.45. [DOI] [PubMed] [Google Scholar]
- 19.Choudhary S, Mathew M, Verma RS. Therapeutic potential of anticancer immunotoxins. Drug Discov Today. 2011;16 (11–12):495–503. doi: 10.1016/j.drudis.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 20.Weldon JE, Pastan I. A guide to taming a toxin--recombinant immunotoxins constructed from Pseudomonas exotoxin A for the treatment of cancer. FEBS J. 2011;278 (23):4683–4700. doi: 10.1111/j.1742-4658.2011.08182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manoukian G, Hagemeister F. Denileukin diftitox: a novel immunotoxin. ExpertOpinBiolTher. 2009;9 (11):1445–1451. doi: 10.1517/14712590903348135. [DOI] [PubMed] [Google Scholar]
- 22.LoRusso PM, Weiss D, Guardino E, Girish S, Sliwkowski MX. Trastuzumab emtansine: a unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin Cancer Res. 2011;17 (20):6437–6447. doi: 10.1158/1078-0432.CCR-11-0762. [DOI] [PubMed] [Google Scholar]
- 23.Mathew J, Perez EA. Trastuzumab emtansine in human epidermal growth factor receptor 2-positive breast cancer: a review. Curr Opin Oncol. 2011;23 (6):594–600. doi: 10.1097/CCO.0b013e32834b895c. [DOI] [PubMed] [Google Scholar]
- 24.Messmann RA, Vitetta ES, Headlee D, Senderowicz AM, Figg WD, Schindler J, Michiel DF, Creekmore S, Steinberg SM, Kohler D, Jaffe ES, Stetler-Stevenson M, Chen H, Ghetie V, Sausville EA. A phase I study of combination therapy with immunotoxins IgG-HD37-deglycosylated ricin A chain (dgA) and IgG-RFB4-dgA (Combotox) in patients with refractory CD19(+), CD22(+) B cell lymphoma. Clin Cancer Res. 2000;6 (4):1302–1313. [PubMed] [Google Scholar]
- 25.Dean A, Talpaz M, Kantarjian H. Phase I clinical trial of the anti-CD33 immunotoxin HuM195/rgel in patients (pts) with advanced myeloid malignancies. J Clin Oncol. 2010;28(15 SUPPL) [Google Scholar]
- 26.Liu S, Bugge TH, Leppla SH. Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. J Biol Chem. 2001;276 (21):17976–17984. doi: 10.1074/jbc.M011085200. [DOI] [PubMed] [Google Scholar]
- 27.Liu S, Netzel-Arnett S, Birkedal-Hansen H, Leppla SH. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 2000;60 (21):6061–6067. [PubMed] [Google Scholar]
- 28.Williams SA, Merchant RF, Garrett-Mayer E, Isaacs JT, Buckley JT, Denmeade SR. A prostate-specific antigen-activated channel-forming toxin as therapy for prostatic disease. J Natl Cancer Inst. 2007;99 (5):376–385. doi: 10.1093/jnci/djk065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abrami L, Fivaz M, van der Goot FG. Adventures of a pore-forming toxin at the target cell surface. Trends Microbiol. 2000;8 (4):168–172. doi: 10.1016/s0966-842x(00)01722-4. [DOI] [PubMed] [Google Scholar]
- 30.Collier RJ, Young JAT. Anthrax toxin. Ann Rev Cell Dev Biol. 2003;19:45–70. doi: 10.1146/annurev.cellbio.19.111301.140655. [DOI] [PubMed] [Google Scholar]
- 31.Kintzer AF, Thoren KL, Sterling HJ, Dong KC, Feld GK, Tang II, Zhang TT, Williams ER, Berger JM, Krantz BA. The protective antigen component of anthrax toxin forms functional octameric complexes. J Mol Biol. 2009;392:614–629. doi: 10.1016/j.jmb.2009.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collier RJ. Membrane translocation by anthrax toxin. Mol Aspects Med. 2009;30 (6):413–422. doi: 10.1016/j.mam.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pellizzari R, Guidi-Rontani C, Vitale G, Mock M, Montecucco C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNgamma-induced release of NO and TNFalpha. FEBS Lett. 1999;462 (1–2):199–204. doi: 10.1016/s0014-5793(99)01502-1. [DOI] [PubMed] [Google Scholar]
- 34.Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, Montecucco C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem Biophys Res Commun. 1998;248:706–711. doi: 10.1006/bbrc.1998.9040. [DOI] [PubMed] [Google Scholar]
- 35.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280 (5364):734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 36.Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, Moayeri M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012;8 (3):e1002638. doi: 10.1371/journal.ppat.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu S, Aaronson H, Mitola DJ, Leppla SH, Bugge TH. Potent antitumor activity of a urokinase-activated engineered anthrax toxin. P Natl Acad Sci USA. 2003;100 (2):657–662. doi: 10.1073/pnas.0236849100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abi-Habib RJ, Singh R, Liu S, Bugge TH, Leppla SH, Frankel AE. A urokinase-activated recombinant anthrax toxin is selectively cytotoxic to many human tumor cell types. Mol Cancer Ther. 2006;5 (10):2556–2562. doi: 10.1158/1535-7163.MCT-06-0315. [DOI] [PubMed] [Google Scholar]
- 39.Liu S, Wang H, Currie BM, Molinolo A, Leung HJ, Moayeri M, Basile JR, Alfano RW, Gutkind JS, Frankel AE, Bugge TH, Leppla SH. Matrix metalloproteinase-activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J Biol Chem. 2008;283 (1):529–540. doi: 10.1074/jbc.M707419200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu S, Redeye V, Kuremsky JG, Kuhnen M, Molinolo A, Bugge TH, Leppla SH. Intermolecular complementation achieves high-specificity tumor targeting by anthrax toxin. Nat Biotechnol. 2005;23 (6):725–730. doi: 10.1038/nbt1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Katayama K, Yoshioka S, Tsukahara S, Mitsuhashi J, Sugimoto Y. Inhibition of the mitogen-activated protein kinase pathway results in the down-regulation of P-glycoprotein. Mol Cancer Ther. 2007;6 (7):2092–2102. doi: 10.1158/1535-7163.MCT-07-0148. [DOI] [PubMed] [Google Scholar]
- 42.Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, Cockerill M, Cartlidge S, Smith PD. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007;6 (8):2209–2219. doi: 10.1158/1535-7163.MCT-07-0231. [DOI] [PubMed] [Google Scholar]
- 43.Haass NK, Sproesser K, Nguyen TK, Contractor R, Medina CA, Nathanson KL, Herlyn M, Smalley KS. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14 (1):230–239. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 44.Hassan R, Broaddus VC, Wilson S, Liewehr DJ, Zhang J. Anti-mesothelin immunotoxin SS1P in combination with gemcitabine results in increased activity against mesothelin-expressing tumor xenografts. Clin Cancer Res. 2007;13 (23):7166–7171. doi: 10.1158/1078-0432.CCR-07-1592. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Xiang L, Hassan R, Paik CH, Carrasquillo JA, Jang BS, Le N, Ho M, Pastan I. Synergistic antitumor activity of taxol and immunotoxin SS1P in tumor-bearing mice. Clin Cancer Res. 2006;12 (15):4695–4701. doi: 10.1158/1078-0432.CCR-06-0346. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, Xiang L, Hassan R, Pastan I. Immunotoxin and Taxol synergy results from a decrease in shed mesothelin levels in the extracellular space of tumors. P Natl Acad Sci USA. 2007;104 (43):17099–17104. doi: 10.1073/pnas.0708101104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pomerantsev AP, Pomerantseva OM, Moayeri M, Fattah R, Tallant C, Leppla SH. A Bacillus anthracis strain deleted for six proteases serves as an effective host for production of recombinant proteins. Protein Expr Purif. 2011;80 (1):80–90. doi: 10.1016/j.pep.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alfano RW, Leppla SH, Liu S, Bugge TH, Herlyn M, Smalley KS, Bromberg-White JL, Duesbery NS, Frankel AE. Cytotoxicity of the matrix metalloproteinase-activated anthrax lethal toxin is dependent on gelatinase expression and B-RAF status in human melanoma cells. Mol Cancer Ther. 2008;7 (5):1218–1226. doi: 10.1158/1535-7163.MCT-08-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melnikova VO, Bolshakov SV, Walker C, Ananthaswamy HN. Genomic alterations in spontaneous and carcinogen-induced murine melanoma cell lines. Oncogene. 2004;23 (13):2347–2356. doi: 10.1038/sj.onc.1207405. [DOI] [PubMed] [Google Scholar]