

Abstract
Tyrosinase is a multifunctional oxidase that is widely distributed in nature. It is a key enzyme in melanin biosynthesis and is involved in determining the color of mammalian skin and hair. In addition it is responsible for the undesirable enzymatic browning that occurs in plant-derived foods, limiting the shelf-life of fresh-cut products with the resultant economic loss. In recent years there has been considerable interest to study the inhibitory activity of tyrosinase and a number of inhibitory compounds derived from natural sources or partly/fully synthetic have been described. However, the current conventional methods to control tyrosinase action are inadequate. Considering the significant industrial and economic impact of the inhibitors of tyrosinase, this study was set to seek new potent inhibitors of this enzyme. A series of 3-hydroxypyridine-4-one derivatives were prepared in high yield and evaluated for their inhibitory activity on tyrosinase enzyme using dopachrome method. Our results show that all synthesized compounds have inhibitory effect on tyrosinase activity for the oxidation of L-DOPA. Among compounds studied those containing two free hydroxyl group (ie Va and V’a) were more potent than their analogues with one hydroxyl group (ie Vb and V’b). Also substitution of a methyl group on position N1 of the hydroxypyridinone ring seems to confer more inhibitory potency.
Keywords: Tyrosinase, Inhibition, Hyperpigmentation, Kojic acid, Bleeching
INTRODUCTION
Melanin is a dark pigment produced by the skin cells in the innermost layer of the epidermis. Melanin plays an important role in protecting human skin from the harmful effects of UV radiation from the sun. Melanin also determines our phenotypic appearance. Although melanin has mainly a photo-protective function in human skin, the accumulation of an abnormal amount of melanin in parts of the skin resulting in more pigmented patches might become an esthetic problem. In addition, the enzymatic browning that occurs on the cut surface of fresh fruits and vegetables can limit the shelf-life of the products and affect their quality which is undesirable. Hyperpigmentation in human skin and enzymatic browning in fruits is both undesirable (1). Melanogenesis has been defined as the entire process leading to the formation of dark macromolecular pigments, i.e., melanin (2). Melanogenesis is initiated with the first step of tyrosine oxidation by tyrosinase. When the skin is exposed to UV radiation, the formation of abnormal melanin pigment occurs, which constitutes a serious esthetic problem that is particularly prevalent in middle-aged and elderly individuals (3,4).
Tyrosinase (EC 1.14.18.1) is a copper-containing enzyme that catalyzes two distinct reactions of melanin biosynthesis : the hydroxylation of tyrosine to 3,4-dihydroxy-phenylalanine (L-DOPA) by monophenolase action and the oxidation of L-DOPA to o-dopaquinone by diphenolase action. However, if L-DOPA is an active cofactor, its formation as an intermediate during o-dopaquinone production is still controversial. o-Dopaquinone is unstable in aqueous solution and rapidly undergoes a non-enzymatic cyclization to leukodopachrome, which is further oxidized non-enzymatically by another molecule of o-dopaquinone to yield dopachrome and one molecule of regenerated L-DOPA (5–7). Tyrosinase exists widely in plants and animals tissues, and is involved in the formation of melanin pigments (8–10). Tyrosinase is also linked to Parkinson’s and other neurodegenerative diseases, oxidizing excess dopamine to produce dopamine quinones, highly reactive species which induce neural damage and cell death (11).
Many candidate inhibitors are examined in the presence of tyrosine or DOPA as the substrate. The inhibitory activities of these compounds are expressed in terms of dopachrome formation. Thus, experimentally observed inhibitors of tyrosinase activity can fall into six categories as described by Chang (12). Among these only two groups of compounds actually bind to the enzyme and inhibit its activity and therefore are regarded as specific or “true inhibitors,” of tyrosinase. These include:
1. Suicide substrates or specific tyrosinase inactivators such as mechanism-based inhibitors. These can be catalyzed by tyrosinase and form covalent bond with the enzyme, thus irreversibly inactivating the enzyme during catalytic reaction. They inhibit tyrosinase activity by inducing the enzyme to catalyze “suicide reaction”.
2. Specific tyrosinase inhibitors such as polyphenols, benzaldehyde and benzoate derivatives, long-chain lipids and steroids. These compounds reversibly bind to tyrosinase and reduce its catalytic capacity.
Inhibitory strength is the primary criterion of an inhibitor. The strength of an inhibitor is usually expressed as the inhibitory IC50 value, which is the concentration of an inhibitor needed to inhibit half of the enzyme activity in the tested condition. However, the IC50 values for the tyrosinase inhibitors in the literature are incomparable due to the varied assay conditions, including different substrate concentrations, incubation times and different batches of commercial tyrosinase. Fortunately, in most studies conducted to describe new tyrosinase inhibitors, a well-studied tyrosinase inhibitor such as kojic acid (KA) is often used as a positive standard at the same time (13).
KA, a fungal metabolite, acts as a good chelator of transition metal ions such as Cu+2 and Fe+3 and is a scavenger of free radicals (14). It is currently applied as a cosmetic skin-lightening agent and is used as a food additive to prevent enzymatic browning (15). KA shows a competitive inhibitory effect on the monophenolase activity and a mixed inhibitory effect on the diphenolase activity of mushroom tyrosinase (16,17). However, its use in cosmetics has been limited, because of the skin irritation caused by its cytotoxicity and also instability during storage.
Therefore, the development of novel, potent, non-toxic and stable tyrosinase inhibitors is of great importance in the medical, cosmetic and agricultural fields (18). Hence, this study was set to synthesize some derivatives of iron and copper chelating 3-hydroxypyridinone and to assess their inhibitory effect on the tyrosinase enzyme activity. We speculated that by increasing the chelating capacity of KA its inhibitory activity will increase accordingly, thus, selected the compounds described herein.
MATERIALS AND METHODS
Materials and instrumentation
All chemicals used for the synthesis of compounds were supplied by Merck or Sigma. Melting points were determined using a Mettler capillary melting point apparatus and are reported uncorrected. The FT-IR and IR spectra were recorded with a Latronics WQF-510 Ratio Recording FT-IR spectrometer and a PERKIN-ELMER 1420 Ratio Recording Infrared spectrometer as KBr disc (γ, cm-1) respectively. The1 H-NMR spectra (DMSO-d6 ) were recorded using a Bruker 400 MHz spectrometer. Chemical shifts (δ) are reported in ppm downfield from the internal standard tetramethylsilane (TMS). The tautomer ratio was calculated using the peak area ratio observed in NMR spectra. The inhibitory effect of compounds on tyrosinase was recorded by micro titer plate reader (Stat Fax 3200. USA). The purity of the compounds was checked by thin layer chromatography (TLC) on silica gel plate using chloroform and methanol as solvent. The procedure for the synthesis of the desired compounds is depicted in Fig. 1.
Fig. 1.
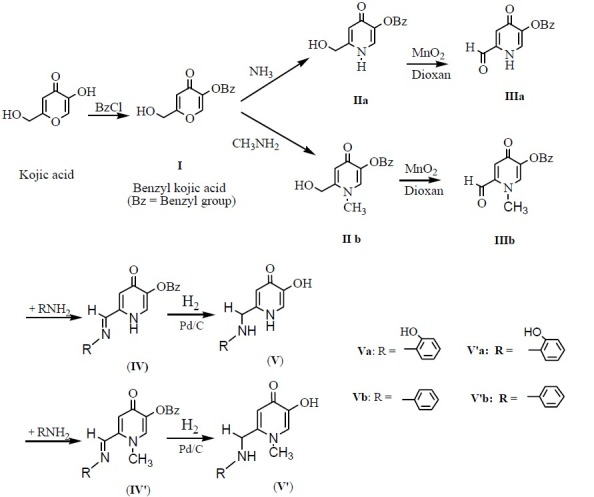
General procedure for the synthesis of the studied compounds
Synthesis of 5-(benzyloxy)-2-(hydroxymethyl)-4H-pyran-4-one (I)
KA (25.34 g, 0.178 mol) was dissolved in 200 ml of methanol. Then, 7.08 g (0.177mol) of sodium hydroxide (dissolved in 10 ml of water) and 20.3 ml (0.177 mol, 22.36 g) of benzyl chloride were added to the reaction mixture. The mixture was refluxed in an oil bath and the progress of the reaction was monitored by TLC (in 3:1 chloroform/ methanol solvent system) every 1 h. Reaction was completed after 6 h. The mixture was evaporated in vacuum and an oily dark brown substance was obtained. This was dissolved in 250 ml of dichloromethane and washed three times with 100 ml of water and once with 150 ml of 5% (W/V) Sodium hydroxide solution. The organic phase was collected, dried over Na2 SO4 and solvent evaporated in vacuo. The brown solid, remained after evaporation, was dissolved in ethanol and recrystallized as white and clear needles of compound I. The crystals were dried in vacuo at 40°C for 12 h (19).
Synthesis of 5-Benzyloxy-2-(hydroxymethyl)-pyridin-4(1H)-one (IIa)
25 g (0.108 mol) of compound I was dissolved in 100 ml of methanol and 150 ml of 30% aqueous solution of ammonia was added. The mixture was refluxed in an oil bath and the progress of the reaction was monitored by TLC (in 3:1 chloroform/methanol solvent system). Because ammonia is a volatile gas, 250 ml of 30 % ammonia solution was added by a dropping funnel during the reflux course. The reaction was completed after 18 h. The mixture was evaporated in a rotary evaporator to obtain a pale brown solid residue. The resulted solid was recrystallized from ethanol/ether to afford compound II. The crystals were dried in vacuo at 40°C for 12 h (20).
Synthesis of 5-(benzyloxy)-2-(hydroxymethyl)-1-methylpyridin-4(1H)-one (IIb)
Compound I (25 g, 0.108 mol) and methylamine (10.05 g, 0.324 mol) were reacted as described for IIa to give IIb as a pale yellowish crystalline solid.
Synthesis of 5-(benzyloxy)-4-oxo-1,4-dihydro pyridine-2-carbaldehyde (IIIa)
10 g (0.043 mol) of compound IIa was dissolved in 500 ml of 1,4-dioxan and the mixture was heated in an oil bath to boiling. Then 70 g of MnO2 was added and the reflux continued. Progress of the reaction was monitored by TLC (in 3:1 chloroform /methanol solvent system) every 30 min. The reaction was completed after 90 min. After cooling at room temperature the reaction mixture was filtered by sintered glass and MnO2 was separated. The separated MnO2 was washed 3 times with 100 ml of boiling 1,4-dioxan. The filtrate solution was evaporated in vacuo and a pale brown solid compound was obtained. This solid was recrystallized in methanol/ether and opaque and cream colored crystals were obtained. The resulted crystals were dried in vacuo at 40°C for 12 h (21).
Synthesis of 5-(Benzyloxy)-1-methyl-4-oxo-1,4 -dihydropyridine-2-carbaldehyde (IIIb)
Compound IIb (10 g, 0.041 mol) and 150 ml of 1,4-dioxan were reacted as described for IIIa to afford IIIb as a yellowish crystals.
Synthesis of 5-Benzyloxy-2-[2-hydroxy phenyl imino)methyl]pyridin-4(1H)-one (IVa)
1.5 g (6.5 mmol) of compound IIIa was dissolved in 30 ml of chloroform and 10 ml of ethanol-2-aminophenol (6.5 mmol, 0.71 g) was added. The mixture was then refluxed and the progress of the reaction was monitored every 30 min by TLC (in 3:1 chloroform/methanol solvent system). The reaction was completed within 90 min. The product was collected by filtration, washed with chloroform and then recrystallized in methanol/ether to give a yellowish crystals. The obtained compound was dried in vacuo at 40°C for 12 h.
Synthesis of 5- Benzyloxy -2 -[ phenylimino) methyl] pyridin-4(1H)-one (IVb)
Compound IIIa (1.5g, 6.5 mmol) and aniline (6.5 mmol, 0.60 ml) were reacted as described for IVa to obtain IVb as a white crystalline solid.
Synthesis of 5-Benzyloxy-2-[2-hydroxy phenyl imino)methyl]-1-methylpyridin-4(1H)-one (IV′a)
Compound IIIb (2 g, 8.2 mmol) and 2-aminophenol (8.2 mmol, 0.90 g) were reacted as described for IVa to give IV′a as yellow crystals.
Synthesis of 5-Benzyloxy -2-[pheny limino) methyl]-1-methylpyridin-4(1H)-one (IV′b)
Compound IIIb (2 g, 8.2 mmol) and aniline (8.2 mmol, 0.75 ml) were reacted as described for IVa to afford IV′b as white crystals.
Synthesis of 5-hydroxy-2-[2-hydroxyphenyl imino) methyl]pyridin-4(1H)-one (Va)
0.5 g (1.56 mmol) of compound IVa was dissolved in 100 ml of absolute ethanol. Then 5.00 g of 10 % palladium/charcoal was added. Hydrogen gas was bubbled into the reaction mixture using a balloon connected to the reaction vessel. The mixture was shaken vigorously at room temperature for 30-60 min. When the reaction was completed, palladium/ charcoal was collected by filtration and washed 3 times by ethanol. The solvent was evaporated under reduced pressure to obtain a brown solid. The resulted solid was re-crystallized from ethanol/ether to give yellowish crystals which were dried in vacuo at 40°C for 12 h.
Synthesis of 5 -hydroxy - 2-[phenylimino) methyl] pyridin-4(1H)-one (Vb)
(0.500 g, 1.65 mmol) of compound IVb and 0.500 g of 10% palladium/charcoal were reacted as described for Va to obtain Vb as a pale crystalline solid.
Synthesis of 5-hydroxy-2-[2-hydroxyphenyl imino)methyl]-1-methylpyridin-4(1H)-one (V′a)
(0.500 g, 1.50 mmol) of compound IV′a and 0.500 g of 10% palladium/charcoal were reacted as described for Va to give V′a as a yellowish crystal.
Synthesis of 5 -hydroxy -2 -[phenylimino) methyl]-1-methylpyridin-4(1H)-one (V′b)
(0.500 g, 1.58 mmol) of compound IV′b and 0.500 g of 10% palladium/charcoal were reacted as described for Va to give V′b as a white crystalline solid.
Enzymatic assay for measurement of tyrosinase inhibition
The dopachrome method described by Iida and coworkers (22), was employed with slight modifications. Briefly, 120 μl of phosphate buffer (20 mM, pH 6.8), 2 unit of mushroom tyrosinase (40 μl, 48 U/ml) and 20 μl of the inhibitor solution were placed in the wells of a 96-well micro titer plate. After pre-incubation for 10 min at 25°C, 20 μl of L-DOPA (0.85 mM) was added and the plate was further incubated at 25°C for 20 min. Subsequently the absorbance of dopachrome was measured at 492 nm using a micro titer plate reader (Stat Fax 3200, USA). KA and phosphate buffer were used instead of the inhibitor solution as positive and negative tyrosinase inhibitor control respectively. The extent of inhibition by the test compounds was expressed as the percentage of concentration necessary to achieve 50% inhibition (IC50 ). The percentage of tyrosinase inhibition was calculated as follow:
Inhibitory activity (%) = [Ac -At )/ Ac ] Χ 100
where, Ac is the absorbance of the negative control and At is the absorbance of the test compound.
RESULTS
Chemistry
The synthesis routes of 5-hydroxy pyridinone-4-one derivatives are summarized in Fig. 1. Treatment of KA (5-Hydroxy-2-(hydroxymethyl)-4H-pyran-4-one) with benzyl chloride resulted the protected form of this molecule (I) which was then reacted with ammonia or methyl amine to produce the corresponding pyridin-4(1H)-one derivatives (IIa,b). Oxidation of these alcohols with activated manganese (IV) oxide in dioxane afforded the desired aldehydes (IIIa,b). These prepared aldehydes were used as intermediates for the preparation of some 5-hydroxy pyridine-4-one derivatives containing appropriate moieties for stabilizing the radical species which would be formed from the reaction of these compounds with free radicals. Thus, IIIa and IIIb were reacted with aniline or 2-aminophenol to afford the desired final compounds after debenzylation of the 5-benzyloxy group of the obtained molecules.
Tautomerization occurs in compounds IIIa, IVa and IVb which posses a hydrogen atom attached to nitrogen ring of hydroxy pyridinone-4-one. In this case the electron lone pair of nitrogen takes part in hydrogen movement in pyridinone ring in that a hydrogen atom is transferred from nitrogen to the carbonyl group.
The existence of tautomerism in this type of compound was confirmed by1 H-NMR spectra. In such cases, each type of hydrogen in compounds IIIa, IVa and IVb appears as two (or more) different signals. Obviously,1 H-NMR spectra include two separate signals for any hydrogen belonging to each of the two types of tautomers. The calculated integral values of the two signals are not necessarily equal because of the unequal ratio of tautomers.
These compounds were revealed to be mixtures of two tautomers as shown in Fig. 2.
Fig. 2.
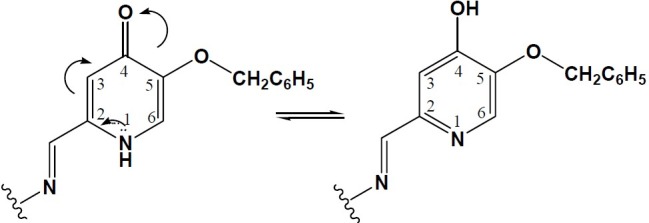
The observed tautomerism in compounds IVa andIVb
Interestingly, such tautomerism is not expected to occur in compounds Va and Vb. The hydrogen bonding between the ketone and phenol groups of the molecule, shown in Fig. 3 and Fig. 4 decreases the electron density on the ketone oxygen, diminishing the possibility of the tautomerism.
Fig. 3.
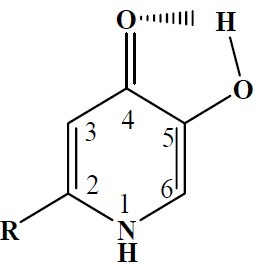
Hydrogen bonding in the unprotected 3-hydroxypyridine-4-ones
Fig. 4.
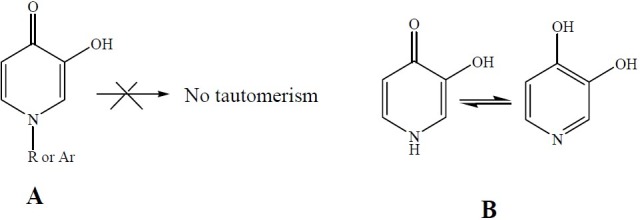
A: 1-alkyl (or aryl)-5-hydroxy-pyridine-4-one, B: Tautomerization in 5-hydroxy-pyridine-4-one
The structures of all compounds were confirmed by FTIR,1 HNMR.
5-(benzyloxy)-2-(hydroxymethyl)-4H-pyran-4-one (I)
White crystal; yield 89% ; m.p. 129-130°C; IR (KBr) cm-1 : 3138 (O-H, str.), 3091 (=C-H, aromatic, str.), 2882 and 2749 (C-H, aliphatic, str.), 1626 (C=O, str.), 1537 and 1501 (C=C, aromatic, str.);1 HNMR (DMSO-d6): δ 8.18 [s, 1H, C3-H], 7.30-7.45 [m, 5H, -C6 H5 ], 6.33 [s, 1H, C6-H], 5.71 [t, 1H, -CH2 -O H ], 4.94 [s, 2H, -C H2 -ph], 4.30 [d, 2H, -C H2 -OH]
5-Benzyloxy-2-(hydroxymethyl)-pyridin-4(1H)-one (IIa)
Colorless crystal; yield 87% ; m.p. 225-226°C; IR (KBr) cm-1 : 3768 (O-H, str.), 3319 (N-H, str.), 2908 and 2750 (-CH2, aliphatic, str.), 1652 (C=O, str.), 1601 and 1462 (C=C, aromatic, str.);1 HNMR (DMSO-d6); δ 11.18 [s, 1H, N1 -H], 7.30-7.60 [m, 6H, C3-H, -C6 H5], 6.24 [s, 1H, C6-H], 5.63 [s, 1H, -CH2 -O H ], 5.06 [s, 2H, -C H2 -ph], 4.39 [s, 2H, -C H2 -OH]
5- (benzyloxy) -2- (hydroxymethyl)-1- methyl pyridin-4(1H)-one (IIb)
Yellow crystal; yield 83% ; m.p. 212.5-213.8°C; IR (KBr) cm-1 : 3134 (O-H, str.), 3061 (=C-H, aromatic, str.), 2858 (CH3, str.), 2750 (CH2, aliphatic, str.), 1641 (C=O, str.), 1578 and 1564 (C=C, aromatic, str.);1 HNMR (DMSO-d6); δ 7.64 [s, 1H, C3-H], 7.32-7.70 [m, 5H, -C6 H5 ], 6.29 [s, 1H, C6-H], 5.65 [s, 1H, -CH2 -O H ], 5.05 [s, 2H, -C H2 -ph], 4.43 [s, 2H, -C H2 -OH], 3.65 [s, 3H, N1 -CH3 ]
5-(benzyloxy) -4 -oxo -1, 4-dihydropyridine-2-carbaldehyde (IIIa)
Opaque and cream colored crystal; yield 82.5% ; m.p. 163.5-164.4°C; a,b tautomers: a/b=100/30. IR (KBr)cm-1 : 3344 (N-H, srt.), 3055 (=C-H, aromatic), 2823 and 2730 (-CHO, str.), 1714 (C=O, aldehyde, str.), 1619 (C=O, ketone, srt.), 1537 and 1481 (C=C, aromatic, str.);1 HNMR (DMSO-d6 ): δ 11.16 [s, 1H, N1 -H], 9.76 [s, 1H, C4-OH], 8.24 [s, 1H, C3-H(b], 7.25-7.60 [m, 11H, C6 H5 (a,b), C3-H(a], 6.74 [s, 1H, C6-H(a], 6.35 [s, 1H, C6-H(b], 5.31 [s, 2H, -C H2 -ph(a], 5.02[s, 2H, -C H2 -ph(b]
5-(Benzyloxy)-1 -methyl -4- oxo- 1,4-dihydro pyridine-2-carbaldehyde (IIIb)
Opaque and cream colored crystal; Yield 77%; m.p. 167-168°C; IR (KBr) cm-1 : 2850 and 2729 (-CHO, str.), 1712 (C=O, aldehyde, str.), 1630 (C=O, ketone, str.), 1600 and 1462 (C=C, str.);1 HNMR (DMSO-d6): δ 7.59 [s, 1H, C3-H], 7.30-7.42 [m, 5H, -C6 H5 ], 6.39 [s, 1H, C6-H], 5.44 [d, 1H, -CHO], 5.00 [s, 2H, -C H2 -ph]
5- Benzyloxy- 2- [(2 -hydroxy phenyl imino) methyl]pyridin-4(1H)-one (IVa)
Pale yellow crystals; yield 86% ; m.p. 237-238°C [literature 235-236°C (23] ; a,b tautomers: a/b=100/30. IR (KBr) cm-1 : 3219 (N-H, str.), 3044 (=C-H, aromatic, str.), 2855 and 2690 (-CH2, aliphatic, str.), 1602 (C=O, str.), 1539 and 1487 (C=C, aromatic, str.);1 HNMR (DMSO-d6 ): δ 11.73 [s, 1H, N1 -H(a], 10.75 [s, 1H, C4-OH(b], 9.15-9.35 [m, 2H, disubstituted phenyl-OH (a,b] 8.72[s, 1H, -CH=N- (a] 8.53[s, 1H, -CH=N- (b] 8.35[s,1H, C3-H(b] 6.65-7.85[m, 20H, CH2 -C6 H5 (a,b), C6-H (b), C3-H (a,b), disubstituted phenyl ring hydrogens(a,b], 6.70 [s, 1H, C6-H(a], 5.34 [s, 2H, -C H2 -C6 H5(b]
5-Benzyloxy-2- [(phenylimino)methyl] pyridin-4 (1H)-one (IVb)
0Yellow crystals; yield 66% ; m.p. 203.5-204.7°C; a,b tautomers: a/b=100/53. IR (KBr) cm-1 : 3664 (N-H, str.), 3014 (=C-H, aromatic, str.), 1596 (C=O, str.), 1534 and 1486 (C=C, str.);1 HNMR (DMSO-d6 ): δ 11.68 [s, 1H, N1 -H(a], 10.80 [s, 1H, C4-OH(b], 8.48 [s,1H, -CH=N- (a], 8.31 [s, 1H, -CH=N- (b], 6.55-7.85 [m, 24H, (10H, -C6 H5 (a,b)) ,(10H, CH2 -C6 H5 (a,b)), (2H, C3-H (a,b)), (2H,C6-H (a,b], 4.90-5.45 [m, 4H, -C H2- C6 H5 (a,b]
5- Benzyloxy -2- [(2-hydroxy pheny limino) methyl]-1-methylpyridin-4(1H)-one (IV′a)
Yellow crystals; yield 82% ; m.p. 223 -2248°C [literature 228-229°C (23] ; IR (KBr) cm-1 : 3350 (O-H, str.), 3051 (=C-H, aromatic, str.), 2887 (-CH3, str.), 2750 and 2540 (-CH2, aliphatic, str.), 1622 (C=O, str.), 1552 and 1463 (C=C, str.),1 HNMR (DMSO-d6 ): δ 9.30 [s, 1H, C6 H4 -O H], 8.68 [s, 1H, -N=CH-], 7.72 [s, 1H, C6-H], 7.25-7.50 [m, 5H, -CH2 -C6 H5 ], 7.26-7.30 [d, 1H, C΄2-H], 7.12-7.18 [t, 1H, C΄4-H], 7.07 [s, 1H, C3-H], 6.92 [d, 1H, C΄3-H], 6.87 [t, 1H, C΄5-H], 5.07 [s, 2H, C H2 -C6 H5 ], 3.95 [s, 3H, -N1 C H3 ]
5-Benzyloxy-2- [(phenylimino)methyl]-1-methy lpyridin -4(1H)-one (IV′b)
0White crystal; yield 71% ; m.p. 184-185°C; IR (KBr) cm-1 : 3836 (N-H, str.), 3067 (=C-H, aromatic, str.), 3059 (-CH3, str.), 3047 and 2850 (-CH2, aliphatic, str.), 1634 (C=O, str.), 1571 and 1486 (C=C, aromatic, str.);1 HNMR (DMSO-d6 ): δ 8.61 [s, 1H, -CH=N-], 7.75 [s, 1H, C3-H], 7.25-7.60 [m, 10H, (5H, CH2 -C6 H5 ), (5H, =N-C6 H5none], 6.87 [s, 1H, C6-H], 5.07 [s, 2H, C H2 -ph], 3.96 [d, 3H, -N-C H3]
5- hydroxy- 2- [2-hydroxy phenyl amino) methyl]pyridin-4(1H)-one (Va)
Yellow crystal; yield 80% ; m.p. 249-250°C [literature 255-256°C (23 ] ; IR (KBr) cm-1 : 3414 (N-H, str.), 3280 (O-H, str.), 3071 (C-H, aromatic, str.), 2959 and 2854 (C-H, aliphatic, str.), 1607 (C=O, str.), 1514 and 1448 (C=C, aromatic, str.);1 HNMR (DMSO-d6): δ 11.16 [s, 1H, -N1 -H], 7.29 [s, 1H, C3-H], 671 [d, 1H, C΄6-H], 6.54-6.60 [t, 1H, C΄4-H], 6.41-6.46 [t, 1H, C΄5-H], 6.33-6.38 [d, 1H, C΄3-H], 6.18 [s, 1H, C6-H], 5.41 [s, 1H, -N H -C6 H4 OH], 4.16 [d, 2H, -C H2 -NH-]
5-hydroxy-2- [phenylamino)methyl]pyridin-4 (1H)-one (Vb)
Pale crystal; yield 68% ; m.p. 252-253°C; IR (KBr) cm-1 : 3416 (N-H, str.), 3241 (O-H, str.), 3050 (C-H, aromatic, str.), 2920 and 2762 (C-H, aliphtic, str.), 1641 (C=O, str.), 1598 and 1539 (C=C, str.);1 HNMR (DMSO-d6 ): δ 11.22 [s, 1H, N1 -H], 7.29 [s, 1H, C3-H], 6.51-7.30 [m, 6H, -C6 H5 ,C6-H], 6.15-6.28 [m, 2H, (1H, -OH), (1H, -N H -ph], 4.14 [d, 2H, -C H2 -NH-]
5-hydroxy-2- [(2-hydroxyphenylamino)methyl]-1-methylpyridin-4(1H)-one (V′a)
Yellow crystal; yield 81% ; m.p. 254-255°C [literature 244-245°C (23]; IR (KBr) cm-1 : 3254 (N-H, str.), 3148 (O-H, str.), 3027 (C-H, aromatic, str.), 2927 (C-H, aliphatic, str.), 1610 (C=O, str. ), 1523 and 1475 (C=C, str.);1 HNMR (DMSO-d6): δ 7.42 [s, 1H, C3-H], 6.67-6.77 [d, 1H, C΄3-H], 6.55-6.65 [t, 1H, C΄5-H], 6.41-6.52 [m, 2H, (1H, C΄4-H), (1H, C΄6-H], 6.14 [s, 1H, C6-H], 5.24 [s, 1H, -N H -C6 H4 OH], 4.26 [d, 2H, -C H2 -NH-], 3.67 [s, 3H, =N-C H3 ]
5-hydroxy-2- [(phenylamino)methyl]-1-methyl pyridin-4(1H)-one (V′b)
White crystal; yield 78% ; m.p. 258-259°C; IR (KBr) cm-1 : 3396 (N-H, str.), 3341 (O-H, str.), 3080 (C-H, str.), 2948 and 2878 (C-H, str), 1640 (C=O, str.), 1603 and 1563 (C=C, str.);1 HNMR (DMSO-d6 ): δ 7.44 [s, 1H, C3-H], 6.55-7.15 [m, 5H, -NH-C6 H5 ], 6.22 [t, 1H, -N H -C6 H5 ], 6.16 [s, 1H, C6-H], 4.22 [d, 2H, -NH-C H2 ], 3.66 [s, 3H, =N-C H3 ]
Enzymatic assay for inhibition of tyrosinase activity
Among the many known inhibitors of tyrosinase, KA has been intensively studied. KA shows a competitive inhibitory effect on the monophenolase activity and a mixed inhibitory effect on diphenolase activity of mushroom tyrosinase (24). Therefore, KA was used as the standard inhibitor of tyrosinase for comparison purposes.
In this study, the effect of KA and its derivatives (Va, Vb, V′a, V′b) on the inhibition of tyrosinase activity was measured at inhibitor concentrations of 0.1, 0.2, 0.3, 0.4 and 0.5 mM and data are shown in Fig 5.
Fig. 5.
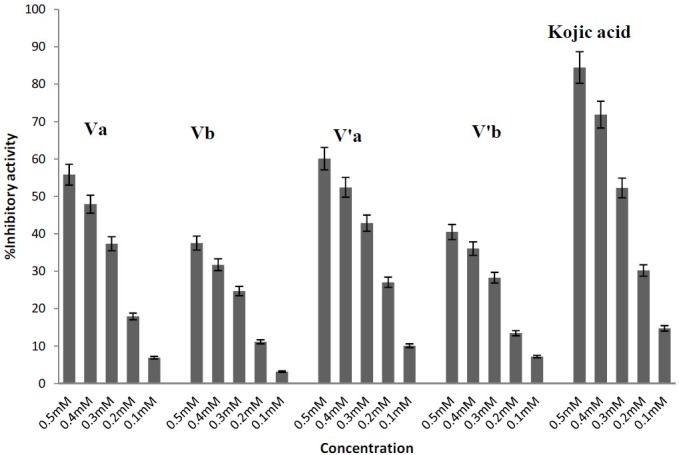
Tyrosinase inhibitory activity of compounds Va, b, V′a, b and Kojic acid
From the experimental data it appears that all of the synthesized compounds have inhibitory effect on tyrosinase activity for the oxidation of L-DOPA, although KA is stronger than other synthesized compounds.
The calculated IC50 values are reported in Table 1. It appears that the potency of inhibitory activity of compounds to be in the order of KA> V′a> Va> V′b> Vb
Table 1.
IC50 values for tyrosinase inhibition ability of kojic acid and its synthesized derivatives

When L-DOPA solution was incubated with tyrosinase at 25°C, its color turned to red-brown because of the formation of dopachrome. In the presence of KA and its derivatives the color intensity of the solution became faint as the result of enzyme activity inhibition.
DISCUSSION
Our results indicated that compounds with polarity lower than that of KA show lower inhibitory effect on tyrosinase activity. Compounds Va, Vb, Va′ and Vb′ contain 2-hydroxyphenylamino or phenylamino substituent on the C-2 position of the pyridinone ring that results in a reduction in the hydrophilicity and hence a decrease in the inhibitory activity compared to the original compound. This mean that a bulky moiety attached to KA may hinder its approach to the active site of tyrosinase.
In consideration of our results, free hydroxyl group at the C-3 and C′-2 positions of the inhibitor seems to be the most important factor for the inhibition of tyrosinase activity. Similarly it has been reported that a hydroxyl group at the C-3 and C-4 positions in flavonoids is the most important factor for their tyrosinase inhibitory activity (25). Miyazawa and coworkers reported that the presence of hydroxyl groups at the C-6 and C-7 positions of coumarin skeleton plays an important role in the expression of tyrosinase inhibition (26). Also, the additional hydroxyl group in quercetin caused it to be a more potent tyrosinase inhibitor than kaempferol (27). In addition the study on the potency of tyrosinase inhibition of chalcones has been shown that the number of hydroxyl groups and their location was the most important factor in their efficacy (24). From these results, we conclude that hydroxyl moiety seems to be an effective functional group for the inhibition of tyrosinase.
Hence, amongst the compounds synthesized in this study, Va and V′a were more potent than Vb and V′b because they had two free hydroxyl groups in their structure compared to the latter that have only one hydroxyl group.
Furthermore, the presence of a methyl group substitute at position N1 of the hydroxypyridinone ring (ie V′a and V′b) caused a stronger tyrosinase inhibitory effect. The reason for this inductive effect of methyl group is probably that it pushes more electrons to the oxygen atom attached to the C-4 position of the ring making it a better ligand for the copper chelating in the active site of the enzyme.
CONCLUSION
Our result showed that all synthesized compounds had inhibitory activity on tyrosinase. It seems that the presence of free hydroxyl group confers more inhibitory potency in that compounds Va and V′a containing two free hydroxyl groups were more potent than Vb and V′b containing one hydroxyl group. In addition compounds that had a methyl group substitute on position N1 of the hydroxypyridinone ring (ie V′a and V′b) were more potent than those without the substitute.
ACKNOWLEDGMENT
The authors wish to send their gratitude to all members of Isfahan Pharmaceutical Sciences Research Center for their support and encouragement. This research was performed by financial support from Isfahan University of Medical Sciences, Isfahan, Iran.
REFERENCES
- 1.Artés F, Castañer M, Gil MI. Review: enzymatic browning in minimally processed fruit and vegetables. Journal Agric Food Chem. 1998;4:377–389. [Google Scholar]
- 2.Schallreuter KU, Kothari S, Chavan B, Spencer JD. Regulation of melanogenesis-controversies and new concepts. Exp Dermatol. 2008;17:395–404. doi: 10.1111/j.1600-0625.2007.00675.x. [DOI] [PubMed] [Google Scholar]
- 3.Parvez S, Kang M, Chung HS, Cho C, Hong MC, Shin MK, Bae H. Survey and mechanism of skin depigmenting and lightening agent. Phytother Res. 2006;20:921. doi: 10.1002/ptr.1954. [DOI] [PubMed] [Google Scholar]
- 4.Parvez S, Kang M, Chung HS, Bae H. Naturally occurring tyrosinase inhibitors; Mechanism and application in skin health, cosmetics and agriculture industries. Phytother Res. 2007;21:8053. doi: 10.1002/ptr.2184. [DOI] [PubMed] [Google Scholar]
- 5.García-Cánovas F, García-Carmona F, Vera-Sánchez J, Iborra-Pastor JL, Lozano-Teruel JA. The role of pH in the melanin biosynthesis pathway. Journal Biol Chem. 1982;257:8738–8744. [PubMed] [Google Scholar]
- 6.Rodríguez-López JN, Tudela J, Varón R, García-Cánovas F. Kinetic study on the effect of pl-l on the melanin biosynthesis pathway. Biochim Biophys Acta. 1991;1076:379–386. doi: 10.1016/0167-4838(91)90480-n. [DOI] [PubMed] [Google Scholar]
- 7.Cooksey C J, Garratt PJ, Land EJ, Pavel S, Ramsden CA, Riley PA, et al. Evidence of the indirect formation of the catecholic intermediate substrate responsible for the autoactivation kinetics of tyrosinase. J Biol Chem. 1997;272:26226–26235. doi: 10.1074/jbc.272.42.26226. [DOI] [PubMed] [Google Scholar]
- 8.Pawelek JM, Körner AM. The biosynthesis of mammalian melanin. AMSCA. 1982;70:136–145. [PubMed] [Google Scholar]
- 9.Mayer AM. Polyphenol oxidases in plants: recent progress. Phytochemistry. 1987;26:11–20. [Google Scholar]
- 10.Whittaker JR. “Polyphenol oxidase” in: food enzymes: structure and mechanism. New York: Chapman and Hall; 1995. pp. 271–307. [Google Scholar]
- 11.Asanuma M, Miyazaki I, Ogawa N. Dopamine or L-DOPA induced neurotoxicity: the role of dopamine quinone formation and tyrosinase in a model of Parkinsonõs disease. Neurutox Res. 2003;5:165–176. doi: 10.1007/BF03033137. [DOI] [PubMed] [Google Scholar]
- 12.Chang TS. An Updated Review of Tyrosinase Inhibitors. Int J Mol Sci. 2009;10:2440–2475. doi: 10.3390/ijms10062440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen JS, Wei C, Marshall MR. Inhibition mechanism of kojic acid on polyphenol oxidase. Journal Agric Food Chem. 1991;39:1897–1901. [Google Scholar]
- 14.Kahn V, Ben-Shalom N, Zakin V. Effect of kojic acid on the oxidation of N-acetyldopamine by mushroom tyrosinase. Jounal Agric Food Chem. 1997;45:4460–4465. [Google Scholar]
- 15.Burdock GA, Soni MG, Carabin IG. Evaluation of health aspects of kojic acid in food. Regul Toxicol Pharmacol. 2001;33:80–101. doi: 10.1006/rtph.2000.1442. [DOI] [PubMed] [Google Scholar]
- 16.Schurink M, Van Berkel WJH, Wichers HJ, Boeriu CG. Novel peptides with tyrosinase inhibitory activity. Peptides. 2007;28:485–495. doi: 10.1016/j.peptides.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 17.Chen JS, Wei CI, Marshall MR. Inhibition mechanism of kojic acid on polyphenol oxidase. Journal Agric Food Chem. 1991;39:1897–1901. [Google Scholar]
- 18.Battaini G, Monzani E, Casella L, Lonardi E, Tepper AW, Canters GW, Bubacco L. Tyrosinase-catalyzed oxidation of fluorophenols. Journal Biol Chem. 2002;227:44606–44612. doi: 10.1074/jbc.M207829200. [DOI] [PubMed] [Google Scholar]
- 19.Streater M, Taylor PD, Hider RC, Porter J. Novel 3-hydroxy-2(1H)-pyridinones. Synthesis, iron (III)-chelating properties and biological activity. Journal of Medicinal Chemistry. 1990;33:1749–1755. doi: 10.1021/jm00168a033. [DOI] [PubMed] [Google Scholar]
- 20.Yongming M, Wei L, Quinn PJ, Zudong L, Hider RC. Design, synthesis, physicochemical properties, and evaluation of novel iron chelators with fluorescent sensors. Journal of Medicinal Chemistry. 2004;47:6349–6362. doi: 10.1021/jm049751s. [DOI] [PubMed] [Google Scholar]
- 21.Becker HD. The conversion of kojic acid into comenaldehyde and comenic acid. Acta Chemica Scandinavica. 1962;16:78–82. [Google Scholar]
- 22.Nithitanakool S, Pithayanukul P, Bavovada R, Saparakorn P. Molecular docking studies and anti-tyrosinase activity of Thai mango seed kernel extract. Molecules. 2009;14:257–265. doi: 10.3390/molecules14010257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mohammadpour M, Sadeghi A, Fassihi A, Saghaei L, Movahedian A, Rostami M. Synthesis and antioxidant evaluation of some novel ortho-hydroxypyridine-4-one iron chelators. J Research in Pharmaceutical Sciences. 2012;7:171–179. [PMC free article] [PubMed] [Google Scholar]
- 24.Noh JM, Kwak SY, Seo HS, Seo JH, Kim BG, Lee YS. Kojic acid-amino acid conjugates as tyrosinase inhibitors. Bioorganic & Medicinal Chemistry Letter. 2009;19:5586–5589. doi: 10.1016/j.bmcl.2009.08.041. [DOI] [PubMed] [Google Scholar]
- 25.Ohad N, Ramadan M, Soliman K, Snait T, Jacob V. Chalcones as potent inhibitors: the effect of hydroxyl positions and numbers. Phytochemistry. 2004;65:1389–1395. doi: 10.1016/j.phytochem.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 26.Miyazawa M, Tamura N. Inhibitory compound of tyrosinase activity from the sprout of polygonum hydropiper L(Benitade) Biol Pharm Bull. 2007;30(3):595–597. doi: 10.1248/bpb.30.595. [DOI] [PubMed] [Google Scholar]
- 27.Shimizo K, Kondo R, Sakai K. Inhibition of tyrosinase by flavonoids, stilbenes and 4-substituted resorsinols: structure-activity investigation. Planta Med. 2000;66:11–15. doi: 10.1055/s-2000-11113. [DOI] [PubMed] [Google Scholar]