

Abstract
Accurate quantum chemical computations based on density functional theory (DFT) were performed on the series of 2-(4-(naphthalen-2-yl)-1,2,3-thiadiazol-5-ylthio)-N-acetamide (TTA) derivatives. The local reactivity of the acetamide derivatives as anti-HIV drugs were studied in terms of Fukui functions in the framework of DFT. The results based on the basis set superposition error (BSSE) corrections showed that the mechanism of bond formation between the acetamide derivatives and tyrosine as a biological molecule occurs mainly through nitrogen atoms. The intramolecular interaction energies between the acetamide derivatives and tyrosine were calculated and the nature of the intermolecular interaction was revealed by natural bond orbital charge (NBO) analysis. The results suggest that acetamide derivatives with bromophenyl and nitrophenyl substitutions are the most potent as anti-HIV drugs.
Keywords: 2-(4-(naphthalen-2-yl)-1,2,3-thiadiazol-5-ylthio)-N-acetamides (TTAs); Anti-HIV drugs; Density functional theory (DFT); Fukui function; Softness indices
INTRODUCTION
Reversetranscriptase inhibitors (RTIs) are a class of antiretroviral drug used to treat HIV infection, tumors, and cancers. RTIs inhibit activity of reverse transcriptase, a viral DNA polymerase enzyme that retroviruses need to reproduce. Two kinds of the most important RTIs exist of nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs). The NNRTIs have a completely different mode of action. NNRTIs block reverse transcriptase by binding at a different site on the enzyme, compared to NRTIs. Therefore, NNRTIs are classified as non-competitive inhibitors of reverse transcriptase that have been developed over the past twenty years. However, the long-term usage of NNRTIs in HIV/AIDS patients causes virus-drug resistance. From the pharmacokinetic point of view, therefore, synthesis and design of novel NNRTIs are important(1,2). Despite of being useful and less toxic, NNRTIs like delavirdine, efavirenz, etravirine and nevirapine, cause drug- resistance and reduce the binding affinity of the inhibitors to the enzyme (3).
The novel oxindoles, quinolones and pyrrolidinones, and a novel series of sulphanyltriazoles and sulfanyltetrazoles have been reported as NNRTIs (4). Structure-activity relationship (SAR) studies have been performed on this scaffold. Molecular modelling studies have revealed that the N-substituted extension from the NNRTI binding pocket to the protein/solvent interface presents an attractive site (a tolerant region) for introducing structurally diverse moieties to generate novel molecules with reasonable anti-HIV activities (5,6). Moreover, there is a set of inhibitors referred to as 1,1,3-trioxo,1,2,,4-thiadiazine (TTD) derivatives showing anti-HIV activity within the NNRTI category (7,8). A three-dimensional quantitative SAR approach has been applied to this series of compounds.
Despite intensive experimental research, a detailed description of the enzyme-inhibitor interaction is a question of crucial importance, and the analysis of the activities of designed proteins will lead to a better understanding of inhibitors as pharmaceutical lead compounds. Therefore, theoretical investigation has been employed as an alternative method for studies on the enzyme-inhibitor interactions in details. Most theoretical investigations are limited by the required computational effort and the accuracy of the used method. There are some common molecular modellings in the molecular biology which have become more feasible and lead to new developments in computational chemistry (9).
The atomic charges, regioselective patterns of chemical reactivity, and other indices of biochemical activity may help us acquire a better understanding of how drugs work and the mechanism of drug resistance. Arissawal and Anthony (10) investigated a large number of drugs that inhibit HIV-1 reverse transcriptase using the ab initio Hartree-Fock method to obtain fully optimised geometric parameters. They calculated the vibration frequencies and also investigated the effects of solvents, the Mulliken population analysis approach and the natural bond orbital charge (NBO), as well as hydrogen bonding effects. They tried to correlate very low and very high anti-HIV activity with charges, vibration stretching frequencies, interatomic distances and the effect of solvents. Peeters and coworkers (11) reported the ab initio study of tetrahydroimidazo[4,5,1-jk][1,4]-benzodiazepin-2(H)-one and -thione derivatives R79882 and 2(H)-one and -thione derivatives R79882 and R82913. Conformational analysis of darunavir (TMC114), a novel HIV-1 protease inhibitor, and ab initio calculations were reported by Nivesanond and coworkers (12,13). Panicker and coworkers (14) reported the structural properties of 4-azatricyclo[5.2.2.02, 6]undecane-3, 5, 8-trione that were calculated by DFT and Hartree-Fock calculations.
Among representatives of the NNRTIs, 2-(4- (2, 4-dichlorophenyl)-1,2,3-thiadiazol-5-ylthio)-N-acetamide (TTA), shown in Fig. 1, there is an interesting structure which exhibits significant anti-HIV-1 activity (15,16). A series of TTA derivatives have been synthesised and evaluated as potent inhibitors of HIV-1. They possess improved or similar HIV-1 inhibition activity compared with nevirapine (EC50 =0.208 μM) and delavirdine (EC50 =0.320 μM). The preliminary SAR among the newly synthesised congeners have been discussed briefly and rationalised by docking studies (17). These new biologically active compounds of TTAs have a 4-(2-naphthoyl) moiety attached to 1, 2, 3-thiadiazole, with the aim of strengthening the π-π stacking interaction between the inhibitor and the aromatic residues (such as Tyr188 or Tyr181) of RT (17).
Fig. 1.
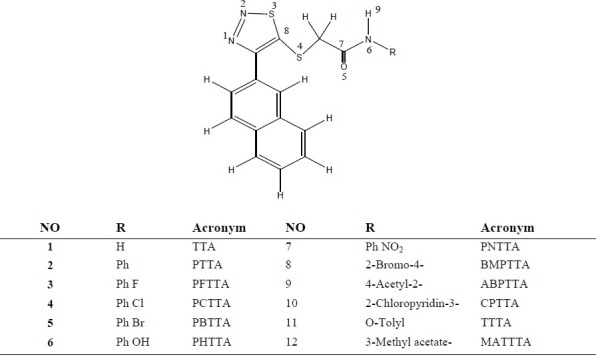
2-(4-(naphthalen-2-yl)-1,2,3-thiadiazol-5-ylthio) acetamide derivatives
The molecular structure and the electronic parameters, such as highest occupied molecular orbital (HOMO) energy, lowest unoccupied molecular orbital (LUMO) energy and the bond gap energy (ΔE=ELUMO - EHOMO ), can be obtained through theoretical calculations. These parameters are involved in the activity of the molecules, in addition to the reactive behaviour that can be assessed by means of the hard/soft acid−base (HSAB) theory (18–20). The objective of the present work was to calculate the more relevant molecular properties on the mechanism of action and reactivity of TTA and its derivatives as anti-HIV drugs. These properties include the molecular structure, the dipole moment, ELUMO, EHOMO, the bond gap energy (ΔE), and those parameters that provide valuable information on the reactive behaviours, such as electronegativity (χ) and global hardness (η). Local reactivity was analysed by means of Fukui indices (21), since they indicate the reactive regions in the form of the nucleophilic and electrophilic behaviour of each atom in the molecule. Also, second-order perturbation theory analysis of the Fock matrix at the B3LYP/ 6-31G level of theory and NBO analysis was carried out.
MATERIALS AND METHODS
Theory and computational details
Fukui functions (FF)
The hard/soft acid-base principle has long been known to be an excellent predictor of chemical reactivity (22–25). The Fukui functions can be defined in terms of the ionisation potential, I, and the electron affinity, A, which lead to:

where, ρN-1, ρN and ρN+1 are the electron density of cationic, neutral and anionic species, respectively. They are calculated under the frozen core approximation (26) which means a single calculation is done for the neutral species without any changes in the calculation method for the charged species, especially anions. Under the Mulliken population analysis approach, the condensed Fukui functions (27) at the atom k are (28,29):
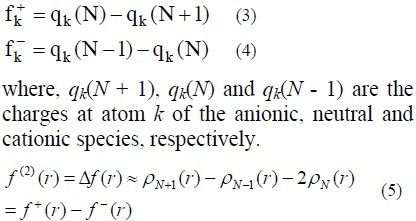
The reactivity descriptor Δf(r) provides useful information on both stabilising and destabilising interactions between a nucleophile and an electrophile and helps in identifying the electrophilic/ nucleophilic behaviour of a specific site within a molecule.
Since the dual descriptor has been very versatile for describing the regional stereoselectivity of a chemical reaction (29), it seems interesting to be used for assessing of the nucleophilicity of the nitrogen, oxygen and sulphur atoms in a biological system. Despite the most probable mechanism between two chemical species, the Fukui functions improve the study of the specific reactivity of TTA drug and some derivatives of TTA in terms of the HSAB principle through the DFT framework, the results of which would predict the nature of linkages in the drug and provide new insight into the development of synthetic drugs.
Interaction energy
Interaction energy (EintAB ) is defined as the difference between the energy of the complex and the sum of the energies of its fragments. It can be expressed as follows:
![]()
where, δABBSSE is the basis set superposition error (BSSE) correction. BSSE is calculated with the counterpoise procedure method advanced by Boys and Bernardi (30). BSSE is considered to be one of the major sources of error in calculation of the interaction energy of weakly bound (Van der Waals) and tightly bound covalent complexes (31,32).
The binding of the drug to the receptor will initially depend on the types of chemical bonds (covalent bonds, ionic bonds, hydrogen bonds and hydrophobic interactions) that can be established between the drug and its receptor. The overall strengths of these bonds will vary and will determine the degree of affinity between the drug and the receptor. The affinity of the compound to the receptor depends on its proper three-dimensional characteristics such as size, stereochemical orientation of functional groups, physical and electrochemical properties. The main amino acids of the NNRTI pocket are Val (Y187), Met (Y184) and Tyr (Y181 and Y188) (Fig. 2) (11). Val and Met are hydrophobic in nature (33), and they play a major role in hydrophobic interactions. The hydrophobic nature of the NNRTI pocket provides relatively few possibilities for polar interactions and hydrogen bonding which are investigated by searching the donor-acceptor sites and evaluating the related parameters in the system of TTAs derivatives and tyrosine.
Fig. 2.
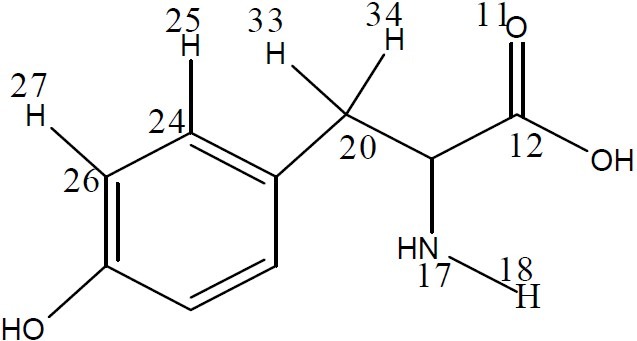
Tyrosine structure
Natural bond orbital (NBO) analysis
NBO analysis (34) stresses the role of intermolecular orbital interactions in the complex, particularly charge transfer. This is carried out by considering all possible interactions between filled donor and empty acceptor NBOs and estimating their energetic importance by second-order perturbation theory. For each donor NBO (i) and acceptor NBO (j), the stabilisation energy, E (2), associated with electron delocalisation between the donor and the acceptor is estimated as:
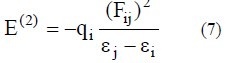
where, qi is the donor orbital occupancy, εi and εj are diagonal elements (orbital energies) and F(i,j) is the off-diagonal NBO Fock matrix element (35,36).
Computational methods
In the current work, a series of TTA derivatives as potent HIV-1 inhibitors were studied compuationally. All derivatives were selected based on the previous experimental work by Zhan and coworkers (11). Calculations were performed by resorting to density functional theory (DFT) of the three parameters of Becke (B3LYP) (23) by using the Gaussian 03W suite of programmes (37). The 6-31G(d) basis set was used for all calculations (38). Electron density ρ(r) for the N-1 and N+1 electron systems was calculated on the optimised geometry of the N electron system. Molecular geometries were fully optimised by Berny’s optimisation algorithm and harmonic vibrational wave numbers were calculated using analytic second derivatives in order to confirm the convergence to minima on the potential surface. At the optimised structure of the examined species, no imaginary wave number modes were obtained, proving that a true minimum on the potential surface was found. Also, the computation of the interaction energy at the optimised geometries for anti-HIV drug and tyrosine combined systems was carried out using the counterpoise procedure and NBO analysis (using by keywords POP=NBO COUNTERPOISE=2).
RESULTS
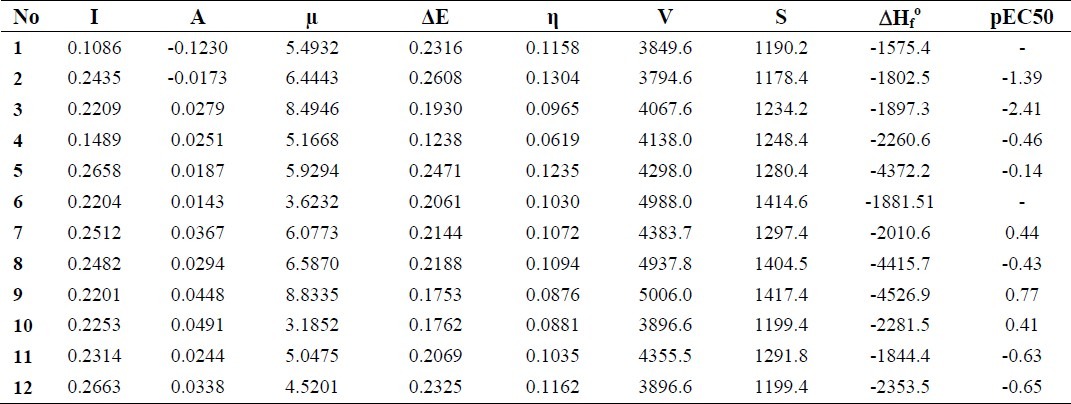
Table 1 reports the calculated quantum chemical parameters which are related to the molecular electronic structure of the molecules under consideration, including ionization energy electroaffinity energy, gap energy, hardness, dipole moment, molecular volume, molecular surface area and heat of formation for the gas phase at B3LYP/6-31G level of theory with experimental logarithm-based EC50 for TTAs molecules at B3LYP/6-31G level of theory.
Table 1.
Calculated ionization energy, I, electroaffinity energy, A, gap energy, ΔE, hardness, η (in Hartree), dipole moment, μ (in Debye), molecular volume, V (ino A3), molecular surface area, S (ino A2), and heat of formation, ΔHfo (in Hartree), for the gas phase at B3LYP/6-31G level of theory with experimental logarithm-based EC50 for TTAs molecules at B3LYP/6-31G level of theory
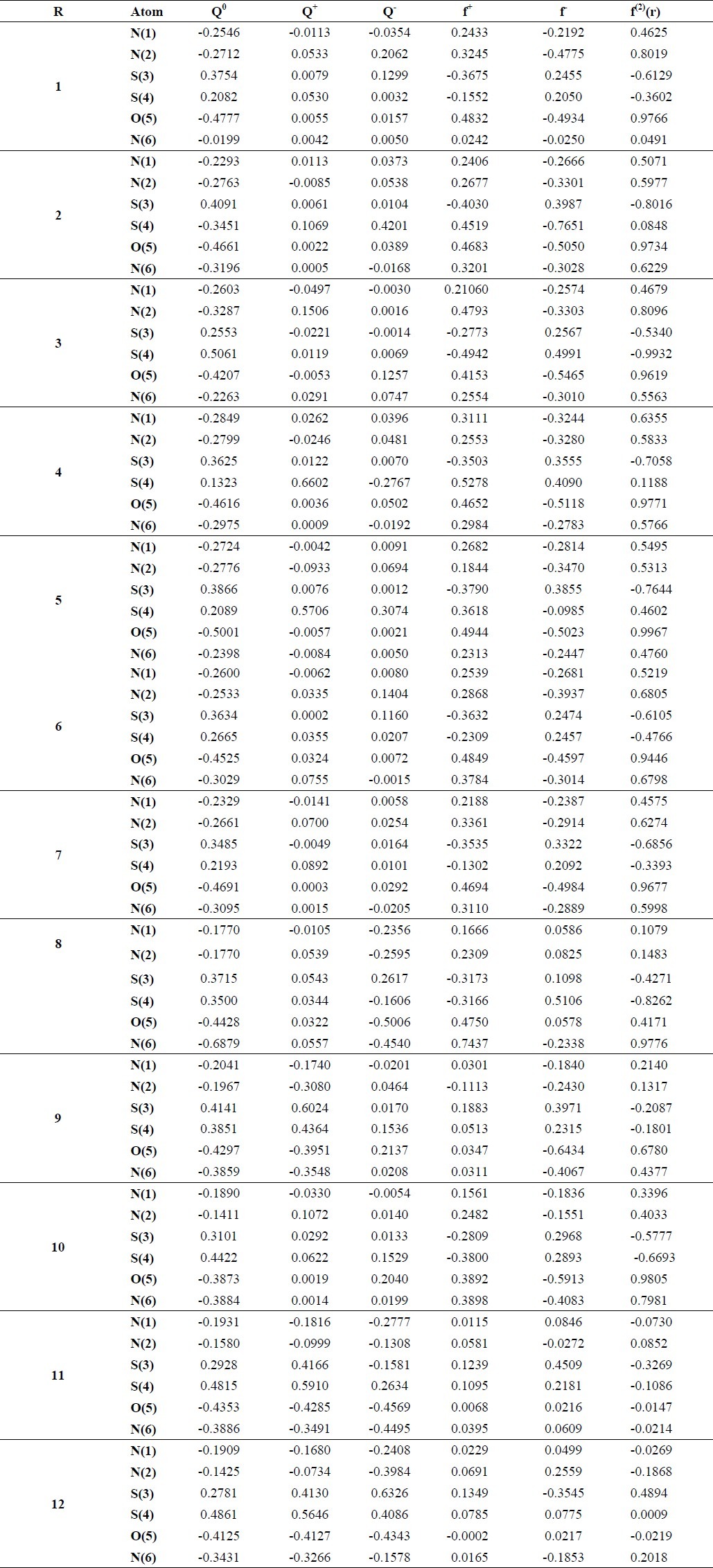
The results of Mulliken population analysis are reported in Table 2. The calculated Mulliken charges and Fukui indices have been presented only for those atoms that displayed an excess of charge according to the natural population analysis. This analysis was focused only on the N(1), N(2), S(3), S(4), O(5) and N(6) atoms, depicted in Fig. 1.
Table 2.
The calculated Mulliken charges and Fukui indices for some atoms in TTAs molecules at B3LYP/6-31G level of theory
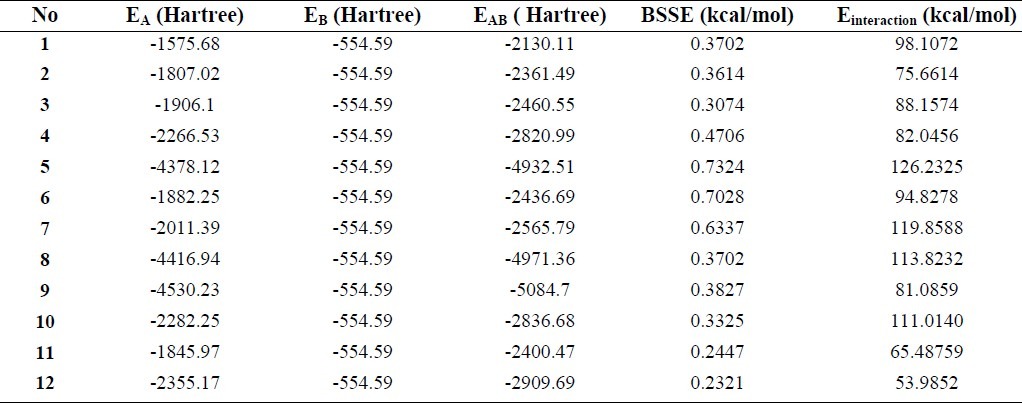
The interaction energies of the anti-HIV drugs (TTAs derivatives) and the tyrosine system, shown in Fig. 2, were calculated at the B3LYP/6-31G(d) level of theory with the functional counterpoise (CP) scheme and are presented in Table 3.
Table 3.
Total electronic energies for constituents, TTAs derivatives (EA) and tyrosine (EB), and combined systems, EAB, including BSSE and Einteraction, calculated by B3LYP/6-31G(d)
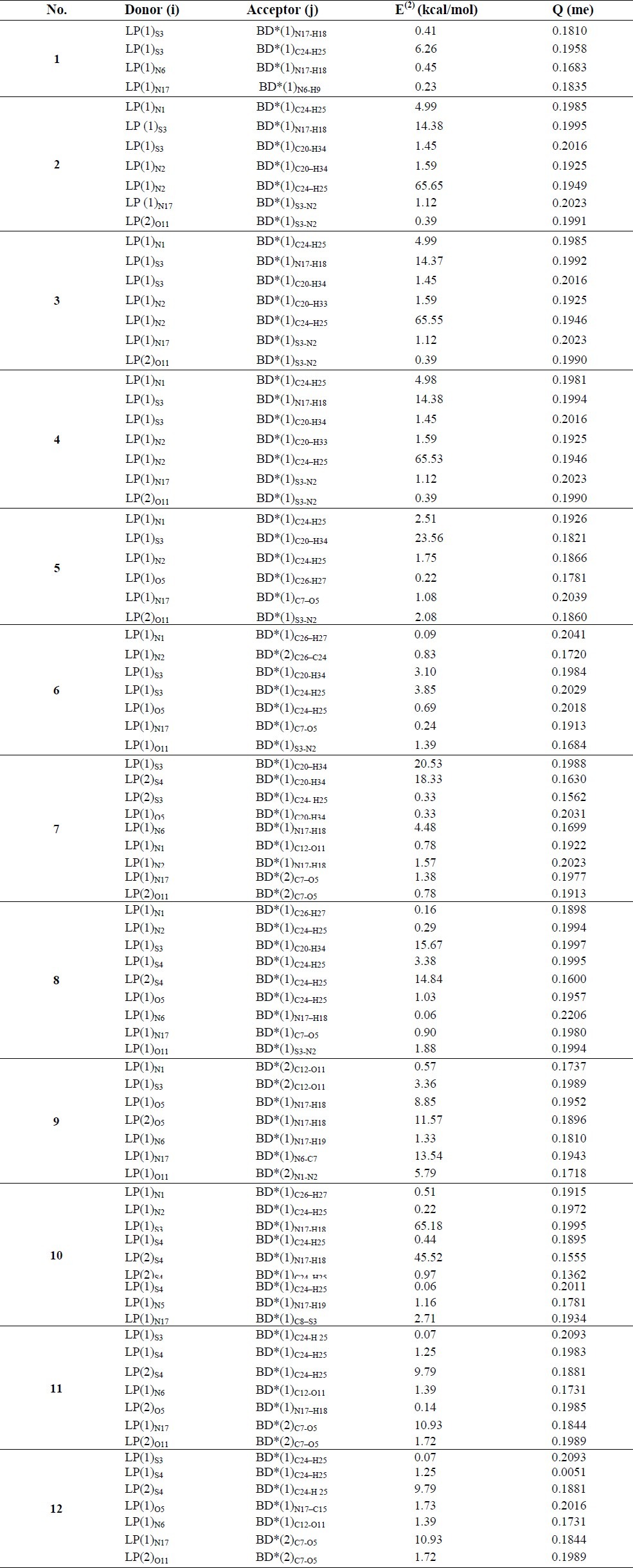
The selected results of some of the invaluable second-order interaction energies (E(2), kcal/mol) between donor and acceptor orbitals in the anti-HIV drugs (TTA derivatives) and tyrosine system have been collected in Table 4 (the results corresponding to the greatest E(2).
Table 4.
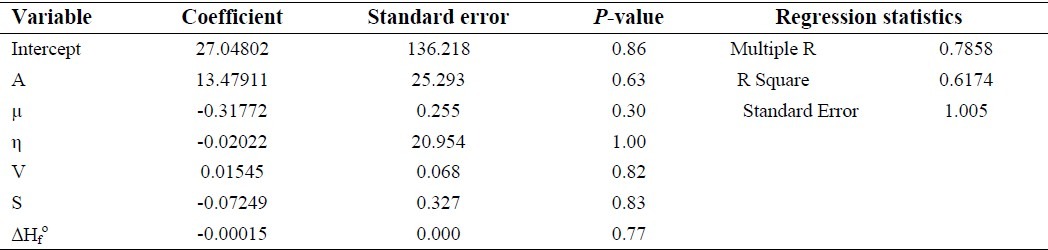
Statistical analysis and correlation coefficients between molecular descriptors as independent variables and pEC50 values for TTAs
DISCUSSION
Global hardness
Ionisation potential, I, and electron affinity, A, values were calculated by the application of Koopmans′ theorem (39–40). This theorem establishes a relationship between HOMO and LUMO with the ionisation potential and the electron affinity, respectively. Although there is not a formal proof of this theorem within DFT, its validity is generally accepted. The obtained values of I and A were considered for the calculation of the electronegativity χ and global hardness η in each of the molecules.
According to Table 1 the highest values of I=-EHOMO show the trend of 12~5>7>8>2>11. The calculations show that molecules 5, 7 and 12 had the highest HOMO levels up to 0.26583 Hartree. Higher values of EHOMO are likely to indicate a tendency of the molecule to donate electrons to the appropriate acceptor molecule of low empty molecular orbital energy. This is in good agreement with the experimental observations suggesting that molecules 5 and 7 were the most potent HIV-1 inhibitors among the investigated molecules (11).
The phenyl (2), flurophenyl (3), bromophenyl (5), nitrophenyl (7), 2-bromo-4-methylphenyl (8) and 4-acetyl-2-brophenyl (9) substitutions increased the dipole moment of molecule 1 (TTA) and others decreased it. So, these groups can promote the polarity and solubility of the TTA compound in the biological environment.
A comparison of the gap energies of these derivatives show that the phenyl (2) and cholorophenyl (4) substitutions caused the greatest increase and decrease in the gap energy related to TTA, respectively. The other substituted groups did not affect the gap energy. This trend was also observed for hardness. In accordance with the HSAB principle, the reactivity of molecules 1, 2, 5 and 12 against the high polarisability species and of molecules 3, 4, 6, 7, 9, 10 and 11 against of the low polarisability species was increased.
Fukui indices
Local reactivity was analysed by means of the Fukui indices (FI), since they indicated the reactive regions, as well as the nucleophilic and electrophilic behaviour of each molecule. This was done to obtain broader knowledge of the reactive site responsible for the activity and to show which atom is responsible for bond formation. The analysis of the Fukui indices along with the charge distribution and the global hardness provides a complete scheme of the reactivity of a molecule (41). The results of Mulliken population analysis, Table 2, show that the substituted groups changed the charge distribution of molecule 1, especially on the aforementioned atoms. For example, the charge on the N (1) and N (2) atoms in molecules 8-12 and 2-7 was generally more positive and negative than in molecule 1, respectively, and the charge on the N (6) atom in all TTAs was generally more negative than in molecule 1. Such a trend was observed for the O (5) atom, but there was no regular trend for the sulphur atoms. The charge on most of the atoms in the cations was more positive than in the neutral species, but this trend was not observed for sulphur atoms. Such a trend was not observed for the anions.
Analysing the dual descriptors for the aforementioned atoms revealed that the N(1), N(2), O(5) and N(6) atoms were the nucleophilic attack sites for most of the TTA molecules. The O(5) and N(6) atoms were most appropriate for nucleophilic attack. The substituted Ph, PhCl and PhBr groups in molecules 2, 4 and 5 changed the nucleophilic and electrophilic attack sites, and they generally increased the nucleophilic attack characterisation. These groups led to a change in the S (4) atom to an electrophilic attack site. The other substituted groups such as PhF, PhOH, PhNO2, 2-bromo-4-methylphenyl, 4-acetyl-2-bromophenyl and 2-chloropyridin-3-yl in molecules 3 and 6-10 did not change the sites with respect to TTA. For molecule 11, all of the atoms played role as electrophilic attack sites and represented good sites for electrophilic attack, except for the N (2) atom which acted as a nucleophilic attack site. In molecule 12, the S (3) and N (6) atoms were nucleophilic attack sites and the other atoms were electrophilic attack sites.
Interaction energy analysis
The obtained total electronic energy values were further corrected for the BSSE arising from the use of finite basis sets in the optimization procedure. Due to the different number of basic functions considered in the complex and monomer optimizations, BSSE is a very important computational issue in the proper treatment of intermolecular interactions and must definitely be taken into account. It is noteworthy that the BSSE is always large and cannot be neglected.
The results of the interaction energies of the anti-HIV drugs (TTAs derivatives) and the tyrosine system, Table 3, show that the high values of the computed interaction energies of the anti-HIV drugs (TTA derivatives) and tyrosine systems show high stability. These results demonstrate the ability of these drugs to bind to tyrosine as one of the amino acids constituting the NNRTI pocket. Molecules 5, 7, 8 and 10 had higher interaction energies than the others, which show that these compounds have stronger binding interactions. Since the interaction energy average is about 90 kcal/mol, the type of bonding during these interactions is covalent.
Correlating molecular properties
The biological activity evaluation of TTAs derivatives is done by pEC50 that indicates pharmacological potency. To investigate this quantity, quantum mechanical descriptors were included for these compounds (Table 1). Multiple linear regressions (MLR) of the dependent variable (pEC50) with these molecular descriptors such as electron affinity, dipole moment, softness, surface, volume and heat of formation were used to calculate a regression in SPSS software. The values of the regression coefficients have been estimated by applying the least square curve fitting method. The best regression equation is represented in the following equation:
pEC50=27.04802+13.47911(A)-0.31772(΅)-0.02022(η)+0.01545(Volume)-0.07249(Surface)-0.00015(Heat of formation) (8)
The statistical analysis for this correlation has been collected in Table 4. Our theoretical analysis suggested that the biological activity pEC50 detected in these compounds can be correlated with the molecular properties. The plot of predicted activity versus observed activity (Fig. 3) provides an idea about how well the model was trained and how well it predicts the activity of compounds. The positive coefficient of A (electron affinity) showed that increase in the values of this descriptor is useful for anti-HIV activity. Based on P-values of variables, softness descriptor has more contribution in biological activity. The next most important factors governing variation in the activity are volume and surface that directly proportional to the anti-HIV activity. The R square and P-values show the coefficients for selected independent variable may describe this relationship and other descriptors must be considered for this purpose.
Fig. 3.
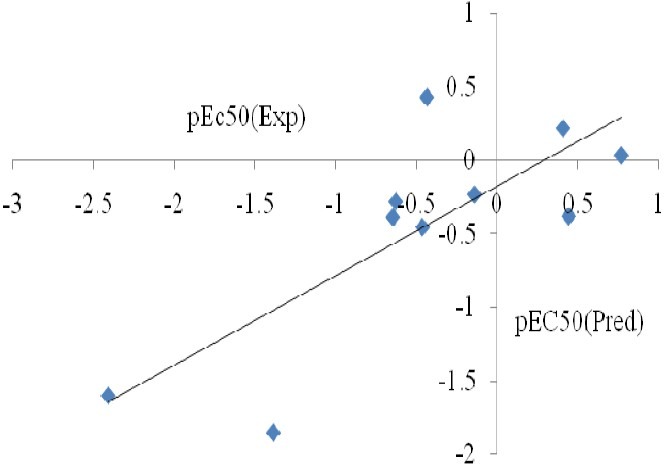
Relationship between predicted and experimental pEC50 for TTAs compounds. The symbols represent experimental pEC50 values.
NBO analysis
NBO calculations were performed using the Gaussian 03 package at the DFT/B3LYP level in order to understand various second-order interactions between the filled orbitals of one subsystem and the vacant orbitals of another subsystem, which is a measure of intermolecular delocalisation or hyper-conjugation. NBO analysis provides the most accurate possible “natural Lewis structure” picture of j, because all orbital details are mathematically chosen to include the highest possible percentage of the electron density. A useful aspect of the NBO method is that it provides information on interactions in both filled and virtual orbital spaces that could enhance the analysis of intra- and inter-molecular interactions. The second-order Fock matrix was carried out to evaluate the donor-acceptor interactions in the NBO basis set. The interactions result in a loss of occupancy from the localised NBO of the idealised Lewis structure into an empty non-Lewis orbital.
The stabilisation energy, E(2), provides a quantitative standard of the strength of the interaction between an electron donor and the receptor. With greater E(2), there is a stronger interaction between the electron donor orbital i and receptor orbital j, that is to say, i has a greater tendency to provide an electron to j and the degree of electron delocalisation is greater. NBO analysis stresses the role of intermolecular interactions in the drug-tyrosine system.
According to the data of Table 5, in most compounds, the antibonding orbitals of the acceptor can interact with the lone pair of N(1), N(2), S(3), S(4), O(5) and N(6) of the TTA drugs and N(17) and O(11) of tyrosine as the donor orbital. The data show that the calculated stabilisation energy, E(2), for the combined system of molecule 1-tyrosine is mainly due to the interaction of the antibonding orbitals of tyrosine with the lone pair electrons of S(3) and N(6). The greatest E(2) values appear in the LP(1)S4;σ*C24-H25) and LP(1) N6;σ*N17-H18 interactions. This indicates that the nature of this interaction is the N atom of the drug offering its lone pair electrons to σ*N17-H18. The calculated stabilisation energies of the 2, 3 and 4 systems are mainly due to the interaction of LP (1)S3→σ* (1)N17-H18, LP (1)N2→σ* (1)C24-H25 and LP (1)N1→σ* (1)C24-H25. E2 values for these compounds are almost identical. The 5 and 6 systems include the interaction of LP(1)O5→BD* (1)C26-H27 in addition to other interactions. The greatest E2 values appear in the LP (1)S3→σ* (1)C20-H34 interaction with a stabilisation energy of 23.56 kJ/mol, whereas in system 6, this value is 3.1 kJ/mol. The calculated stabilisation energies for system 7 are mainly due to LP(1)S3→σ*(1)C20-H34 with a stabilisation energy of 20.53 kJ/mol, as well as LP(1)O5 →BD* (1)C20-H34 and LP (1)N6→BD* (1)N17-H18. For system 8, the interactions LP(1)S3→BD*(1)C20-H34 and LP (1)S4→BD* (1)C24-H25 are effective. The greatest stabilisation energies of the combined system 9 are mainly due to the interaction LP (1)S3→BD* 2C12-O11, LP (1)O5→ BD* (1)N17-H18 and LP (1)N17→BD*(1)N6-C7, but in system 10, these interactions related to LP (1)S3→BD* (1)N17-H18, LP 2S4→BD* (1)N17-H18 and LP (1)N17→BD* (1)C8-S3. Finally, the stabilisation energies of systems 11 and 12 were the same and the greatest E2) values related to LP 2S4→BD* (1)C24-H25 and LP (1)N17→BD* 2C7-O5.
Table 5.
Some invaluable second-order interaction energies (E(2), kcal/mol) and partial electron transfer (q) between donor and acceptor orbitals in anti-HIV drug (TTAs derivatives) and tyrosine system calculated at B3LYP/6-31G(d) level of theory
CONCLUSION
Quantum mechanics was used to predict activity and reactivity of acetamide derivatives as anti-HIV drugs by studying the energy analysis and the Fukui functions. Also, the interactions of these drugs with individual active site residues of the reverse transcriptase enzyme (tyrosine) were investigated with BSSE corrections, and the interaction energies were calculated and NBO analysis was carried out. According to the obtained results, the TTA drugs and tyrosine act as acceptors and donors, respectively. The obtained results showed the C-N and C=0 bond lengths increased in the most systems (TTA-Tyr) compared to TTA, and C-N-H angle decreased significantly compared to drug (single) but N-H bond length decreased in the most systems. The substituted groups PhBr (PBTTA), PhNO2 (PNTTA), 2-bromo-4-methylphenyl (BMPTTA) and 2-chloropyridine-3-yl (CPTTA) had strong covalent bonding interactions. These derivatives could act as potent HIV-1 inhibitors. Finally the pEC50s for TTAs compounds correlate less significantly with the selected electronic descriptors.
ACKNOWLEDGMENT
The authors gratefully acknowledge the support of this work by Payame Noor University, Mashhad (Iran) and would like to thank Dr M. Shahlaei for his helpful discussions.
REFERENCES
- 1.Clercq ED. HIV-chemotherapy and -prophylaxis: New drugs leads and approaches. Int J Biochem Cell Biol. 2004;36:800–1822. doi: 10.1016/j.biocel.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Tan JJ, Cong XJ, L LH, Wang CX, Jia L, Liang XJ. Therapeutic strategies underpinning the develop-ment of novel techniques for the treatment of HIV infection. Drug Discov Today. 2010;15:185–197. doi: 10.1016/j.drudis.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuno M, Hannongbua S, Morokuma K. Theoretical investigation on nevirapine and HIV-1 reverse transcriptase binding site interaction, based on ONIOM method. Chem Phys Lett. 2003;380:456–463. [Google Scholar]
- 4.Wang Z, Wu B, Kuhen KL, Bursulaya B, Nguyen TN, Nguyen DG, et al. Synthesis and biological evaluations of sulfanyltriazoles as novel HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg & Med Chem Lett. 2006;16:4174–4177. doi: 10.1016/j.bmcl.2006.05.096. [DOI] [PubMed] [Google Scholar]
- 5.Omeara JA, Jakalian A, LaPlante S, Bonneau PR, Coulombe R, Faucher AM, et al. Scaffold hopping in the rational design of novel HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg & Med Chem Lett. 2007;17:3362–3366. doi: 10.1016/j.bmcl.2007.03.097. [DOI] [PubMed] [Google Scholar]
- 6.Gagnon A, Amad MH, Bonneau PR, Coulombe R, DeRoy PL, Doyon L, et al. Thiotetrazole alkynylacetanilides as potent and bioavailable non-nucleoside inhibitors of the HIV-1 wild type and K103N/Y181C double mutant reverse transcriptases. Bioorg Med Chem Lett. 2007;17:4437–4441. doi: 10.1016/j.bmcl.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 7.Arranz ME, Díaz JA, Ingate ST, Witvrouw M, Pannecouque C, Balzarini J, et al. Synthesis and anti-HIV activity of 1,1,3-trioxo-2H,4H-thieno[3,4-e1,2,4]thiadiazines (TTDs): a new family of HIV-1 specific non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem. 1999;7:2811–2822. doi: 10.1016/s0968-0896(99)00221-7. [DOI] [PubMed] [Google Scholar]
- 8.Ravichandran V, Ravanappa B, Kumar P, Sankar S, Agrawal RK. Predicting anti-HIV activity of 1,1,3-trioxo[1,2,4]-thiadiazine (TTD) derivatives: 3D QSAR approach. Med Chem Res. 2009;18:511–522. doi: 10.1016/j.ejmech.2008.05.036. [DOI] [PubMed] [Google Scholar]
- 9.Khandogin J, Musier-Forsyth K, York DM. Insights into the regioselectivity and RNA-binding affinity of HIV-1 nucleocapsid protein from linear-scaling quantum methods. J Mol Biol. 2003;330:993–1004. doi: 10.1016/s0022-2836(03)00658-2. [DOI] [PubMed] [Google Scholar]
- 10.Arissawa M, Anthony C. Investigation of nucleoside analogs with anti-HIV activity. Int J Quantum Chem. 2003;93:422–432. [Google Scholar]
- 11.Peeters A, Alsenoy CV, Dillen J, Geise HJ. Ab initio study of tetrahydro-imidazo[4,5,1-jk 1,4]-benzodiazepin- 2(1H)-one and -thione (TIBO) derivatives R79882 and R82913. J Mol Struct: Theochem. 1995;333:99–110. [Google Scholar]
- 12.Nivesanond K, Peeters A, Lamoen D, Alsenoy CV. Conformational analysis of TMC114, a novel HIV-1 protease inhibitor. J Chem Inf Model. 2008;48:99–108. doi: 10.1021/ci7001318. [DOI] [PubMed] [Google Scholar]
- 13.Nivesanond K, Peeters A, Lamoen D, Alsenoy CV. Ab initio calculation of the interaction Energy in the P2 binding pocket of HIV-1 protease Int. J Quantum Chem. 2005;105:292–299. [Google Scholar]
- 14.Panicker CY, Varghese HT, Pillai KM, Mary YS, Raju K, Manojkumar TK, et al. Quantum chemical DFT study of 4-azatricyclo [5220] undecane-3,5,8-trione. Spectrochim Acta A. 2010;75:1559–1565. doi: 10.1016/j.saa.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 15.Zhan P, Liu X, Cao Y, Wang Y, Pannecouque C, Clercq ED. 1,2,3-Thiadiazole thioacetanilides as a novel class of potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg & Med Chem Lett. 2008;18:5368–5371. doi: 10.1016/j.bmcl.2008.09.055. [DOI] [PubMed] [Google Scholar]
- 16.Zhan P, Liu X, Li Z, Fang Z, Li Z, Wang D, et al. Novel 1,2,3-thiadiazole derivatives as HIV-1 NNRTIs with improved potency: Synthesis and preliminary SAR studies. Bioorg & Med Chem Lett. 2009;17:5920–5927. doi: 10.1016/j.bmc.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Zhan P, Liu X, Fang Z, Li Z, Pannecouque C, Clercq ED. Synthesis and anti-HIV activity evaluation of 2-(4-(naphthalen-2-yl)-1,2,3- thiadiazol-5-ylthio)-N-acetamides as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Eur J Med Chem. 2009;44:4648–4653. doi: 10.1016/j.ejmech.2009.06.037. [DOI] [PubMed] [Google Scholar]
- 18.Ogretir C, Mihci B, Bereket G. Quantum chemical studies of some pyridine derivatives as corrosion inhibitors. J Mol Struct: Theochem. 1999;488:223–231. [Google Scholar]
- 19.Bereket G, Ogretir C, Ozsahin C. Quantum chemical studies on the inhibition efficiencies of some piperazine derivatives for the corrosion of steel in acidic medium. J Mol Struct: Theochem. 2003;663:39–46. [Google Scholar]
- 20.Obot IB, Obi-Egbedi NO. HSAB descriptors of thiadiazole derivatives calculated by DFT: possible relationship as mild steel corrosion inhibitors. Der Pharma Chemica. 2009;1:106–123. [Google Scholar]
- 21.Lee C, Yang W, Parr RG. Local Softness and chemical reactivity in the molecules CO, SCN- and H2CO. J Mol Struct : Theochem. 1998;163:305–313. [Google Scholar]
- 22.Yang W, Parr RG. Hardness, softness, and the Fukui function in the electronic theory of metals and catalysis. Proc Nati Acad Sci USA. 1985;82:6723–6726. doi: 10.1073/pnas.82.20.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parr RG, Yang W. Density Functional Theory of Atoms and Molecules. Oxford: Oxford University Press; 1989. [Google Scholar]
- 24.Cavalli A, Carloni P, Recanatini M. Target-related applications of first principles quantum chemical methods in drug design. Chem Rev. 2006;106:3498–3519. doi: 10.1021/cr050579p. [DOI] [PubMed] [Google Scholar]
- 25.Faver J, Merz KM. Utility of the hard/soft acid-base principle via the Fukui function in biological systems. J Chem Theory Comput. 2010;6:548–559. doi: 10.1021/ct9005085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melin J, Aparicio F, Subramanian, Galvan M, Chattaraj PK. Is the Fukui function a right descriptor of hard-hard interactions. J Phys Chem A. 2004;108:2487–2491. [Google Scholar]
- 27.Contreras R, Fuentealba P, Galvan M, Perez P. A direct evaluation of regional Fukui functions in molecules. Chem Phys Lett. 1999;304:405–413. [Google Scholar]
- 28.Cardenas C, Rabi N, Ayers PW, Morell C, Jaramillo P, Fuentealba P. Chemical reactivity descriptors for ambiphilic reagents: Dual descriptor, local hypersoftness, and electrostatic potential. J Phys Chem A. 2009;113:8660–8667. doi: 10.1021/jp902792n. [DOI] [PubMed] [Google Scholar]
- 29.Morell C, Grand A, Toro-Labbe A. New dual descriptor for chemical reactivity. J Phys Chem A. 2005;109:205–212. doi: 10.1021/jp046577a. [DOI] [PubMed] [Google Scholar]
- 30.Boys SF, Bernardi F. The calculation of small molecular interactions by the differences of separate total energies: Some procedures with reduced errors. Mol Phys. 1970;19:553–556. [Google Scholar]
- 31.Duijneveldt FBV, Rijdt JGCMvD-vd, Lenthe JHv. State of the art in counterpoise theory. Frans Chem Rev. 1994;94:1873–1885. [Google Scholar]
- 32.Senent ML, Wilson S. Intramolecular basis set superposition errors. Int J Quant Chem. 2001;82:282–292. [Google Scholar]
- 33.Horton HR, Moran LA, Ochs RS, Rawn JD, Scrimgeour KG. Principles of Biochemistry. 2nd. New Jersey: Prentice Hall; 1996. [Google Scholar]
- 34.Reed AE, Curtiss LA, Weinhold F. Intermolecular interactions from a natural bond orbital, donor-acceptor, viewpoint. Chem Rev. 1988;88:899–926. [Google Scholar]
- 35.Gutowski M, Lenthe JHV, Duijneveldt FBV. Accuracy of the Boys and Bernardi function counterpoise method. J Chem Phys. 1993;98:4728–4738. [Google Scholar]
- 36.Yang C, Wang H. Ab initio and DFT theory studies of interaction of thymine with formaldehyde. Struct Chem. 2008;19:843–847. [Google Scholar]
- 37.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, et al. In: GAUSSIAN03. B.05 R, editor. Pittsburgh, PA: Pople, Gaussian, Inc.; 2003. [Google Scholar]
- 38.Mendoza-Wilson AM, Glossman-Mitnik D. CHIH-DFT determination of the molecular structure, infrared and ultraviolet spectra of the flavonoid quercetin. J Mol Struct: Theochem. 2004;681:71–76. [Google Scholar]
- 39.Manne R, Aerg T. Koopmans, theorem for inner-shell ionization. Chem Phys Lett. 1970;7:282–284. [Google Scholar]
- 40.Heinrich N, Koch W, Frenking G. On the use of Koopmans, theorem to estimate negative electron affinity. Chem Phys Lett. 1986;124:20–25. [Google Scholar]
- 41.Cruz J, Martýnez-Aguilera LMR, Salcedo R, Castro M. Reactivity Properties of derivatives of 2-imidazoline: An ab initio DFT study. Int J Quantum Chem. 2001;85:546–556. [Google Scholar]