

Abstract
Food intake is generally accepted to be regulated by the melanocortin system, however recent data suggests that mesolimbic dopaminergic neurons also influence food intake. Whether dopamine signaling is crucial for the acute effect of leptin on feeding is unknown. Using pharmacological and genetic strategies, we tested the hypothesis that the acute inhibitory effect of leptin on food intake is partially mediated by dopamine. Dopamine D2 but not D1 receptor blockade attenuated the acute hypophagic effect of leptin in fasted mice. Additionally, mice lacking the D2R (D2R KO) exhibited an attenuated response to leptin. Conversely, dopamine receptor blockade had no effect on the acute hypophagic effect of melanocortin stimulation or the hyperphagic effect of ghrelin. These findings suggest that dopaminergic pathways do not constitute a normal part of melanocortin-dependent feeding regulation and that the dopaminergic neurocircuitry typically associated with regulation of hedonic feeding likely contributes to feeding regulation by leptin.
Keywords: Dopamine, Leptin, Melanocortin system, Hypothalamus, Mesolimbic dopamine system, Food intake
1. Introduction
Leptin directly affects melanocortin neurons in the arcuate nucleus of the hypothalamus (ARH) [1–5]; therefore the anorectic effects of leptin are generally attributed to signal transduction at the level of the basomedial hypothalamus. Recent studies demonstrate that leptin can also influence feeding and locomotor behavior via direct actions on mesolimbic dopaminergic neurons [6,7]. Whether dopamine signaling is critical for the effect of leptin on energy balance remains to be determined.
Leptin is an adiposity signal that circulates in proportion with fat mass [8]. Leptin administration can decrease food intake, increase energy expenditure and cause weight loss, whereas deficiencies in leptin are associated with obesity [9,10]. Although the anorexic effects of leptin are attributed to leptin signaling in the ARH [1–3], recent evidence suggests direct leptin action on mesolimbic dopaminergic systems can influence aspects of energy balance that include feeding behavior and locomotor activity. In the rodent midbrain, leptin receptors colocalize with tyrosine hydroxylase [6,7,11] and direct leptin administration to the ventral tegmental area (VTA) decreases food intake [7]. In humans, leptin deficiency is associated with increased striatal activity in response to food images that is normalized by leptin administration [12]. Furthermore, leptin-deficient ob/ob mice appear to have impaired mesolimbic dopamine signaling and a decreased locomotor response to amphetamine [6]. By demonstrating an extrahypothalamic site for leptin-mediated regulation of feeding and locomotor activity, these studies challenge the autonomy of the melanocortin system as the primary site of leptin action.
Through the use of pharmacological and genetic manipulations, we addressed whether the acute anorectic effect of leptin requires dopaminergic signaling. Because dopamine signaling is mediated by 2 different classes of dopamine receptors, D1-like and D2-like [13], we investigated whether the acute anorexic effect of leptin would be attenuated by a D1R or a D2R antagonist. We also assessed if the acute hypophagic effect of a melanocortin agonist also requires dopamine signaling. Finally, we tested whether dopamine receptors are important for the acute hyperphagic effects of the orexigenic hormone, ghrelin.
2. Experimental procedures
2.1. Animal care and housing
All animal procedures were approved by the Oregon National Primate Research Center Institutional Animal Care and Use Committee or Monash University Animal Ethics Committee. At 24 weeks of age, male C57Bl/6J mice (Jackson Labs, Bar Harbor, ME or Monash University Animal Services) were individually housed under a 12 h light/dark cycle and constant temperature (22±3 °C). Food (Purina Lab Chow, #5001 or Specialty Feeds, Glen Forrest, WA, Australia) and water were available ad libitum, unless specified otherwise. Adult male mice lacking the D2R and wild type mice, backcrossed to C57BL/6J for 20 generations, were generously donated by Dr. David K. Grandy. The average body weight for D2R KO mice was 23 g and the average body weight for wild type mice was 26 g.
2.2. Behavioral tests: drugs
All drugs and stock solutions were prepared fresh on day of use. Drug doses were based on published data and pilot studies to determine the highest effective dose that did not significantly alter 1h food intake from vehicle following an overnight (16 h) fast. SKF83566 (SKF; Tocris), melanotan II (MTII; Bachem) and recombinant murine leptin (Peprotech) were dissolved in sterile nonpyrogenic 0.9% NaCl. Stock solutions in H2O were made fresh for L741,626 (L741; Tocris), and Pergolide (Tocris) which were further diluted with saline to the final injection concentration. All drugs and vehicle were administered by intraperitoneal (i.p.) injection in a volume of 0.1±0.02 mL (according to body weight).
2.3. Behavioral test paradigm
Mice were habituated to behavioral testing by daily i.p. injection of 0.1mL sterile saline and 16h fasting every third day for 2 weeks; mice that received ghrelin were not habituated to overnight fasting. All mice were assigned to treatment groups balanced for body weight. On the morning of testing, 16 h fasted mice received i.p. injection of freshly prepared drug or vehicle, were returned to their cage and given 6 pre-weighed food pellets; mice that received ghrelin were not fasted. Food was weighed at 1, 2, 4 h, 8 h and 24 h following drug administration. For the antagonist studies, mice were habituated to overnight fasting as described above, but received 2 i.p. injections of 0.1 mL sterile saline 15 min apart on either side of the abdomen. On the morning of testing, 16 h fasted mice received an i.p. injection of freshly prepared drug or vehicle, followed by drug or vehicle 15 min later and food was re-introduced to the mice. Food weight was measured at 1 h, 2 h and 4 h, 8 h and 24 h. Body weight was measured 24 h post injection.
2.4. Statistical analysis
Data sets were analyzed using Prism Software (Graph-Pad Software, San Diego, CA). Two-way ANOVA, with Bonferroni post hoc test was used to determine significant interactions between D1R or D2R antagonists and leptin, MTII or ghrelin. A P value of P<0.05 was considered significant. Data are expressed as mean ± SEM.
3. Results
3.1. Blockade of D2R limits leptin-induced hypophagia in lean mice
16h-fasted lean mice were pretreated with a dopamine receptor antagonist, followed by leptin or vehicle 15 min later. Leptin reduced food intake in lean mice at 1 h and 2 h but not at 4 h post injection (Fig. 1A–C; also no significant effect at 8 h or 24 h post injection, data not shown). Pretreatment with the selective D2R antagonist L741 had no effect on food intake but blocked the hypophagic action of leptin (Fig. 1A–C). This suggests that leptin acts through the D2R to produce anorexic actions. Leptin also reduced acute body weight regain at 24 h (Fig. 1D). Mice treated with the D2R antagonist had less weight regain at 24 h post injection (Fig. 1D).
Fig. 1.

Blockade of D2R but not D1R reduces the acute hypophagic effect of leptin in lean mice. Acute pretreatment with the D2R antagonist (L741; 1.5 mg/kg) decreased the hypophagic efficacy of leptin (5 mg/kg) at (A) 1h post injection, (B) 2 h post injection, (C) 4 h post injection. Acute pretreatment with vehicle or the D1R antagonist (SKF; 0.25 mg/kg) failed to reduce the hypopagic effects of leptin (A–C). The D2R antagonist (L741; 1.5 mg/kg) attenuated the reduction in weight gain cause by leptin while the D1R antagonist had no effect. (Two way ANOVA, Bonferroni post hoc test, *P<0.05, **P<0.01, ***P<0.001 for treatment vs. vehicle; n=18–21 per group).
3.2. Blockade of D1R does not alter leptin induced hypophagia
In contrast to D2R antagonism, acute pretreatment with a D1R antagonist (SKF; 0.25 mg/kg) did not attenuate the acute hypophagic effect of systemic leptin (5 mg/kg) treatment at 1 h, 2 h, 4 h, 8 h, or 24 h (Fig. 1A–C; 8 h and 24 h data not shown) in mice. Acute pretreatment with a D1R antagonist alone did not significantly alter food intake from control. Similarly, pretreatment with a D1R antagonist did not influence the reduction in weight gain with leptin at 24 h post injection (Fig. 1D).
3.3. D2R deficient mice have an attenuated hypophagic response to leptin
To verify that the D2R is important for the acute hypophagic effect of leptin, leptin was administered to mice with genetic deletion of the D2R [14]. Male D2R KO mice and their wild type littermates were treated with either saline or leptin (5 mg/kg). Because D2R KO mice are significantly smaller [15] (23 g vs. 26 g), food intake was normalized to body weight. Systemic leptin treatment significantly reduced food intake in wild type mice by 20% at 1 h but only caused a 10% reduction in food intake in D2R KO mice that was not significantly different from vehicle-treated animals (Fig. 2A). Leptin also reduced food intake significantly in wild type mice at 2 h and 4 h post injection (Figs. 2B and C) but not 8 h or 24 h (data not shown). Leptin did not significantly affect food intake at any time point in D2R KO mice.
Fig. 2.
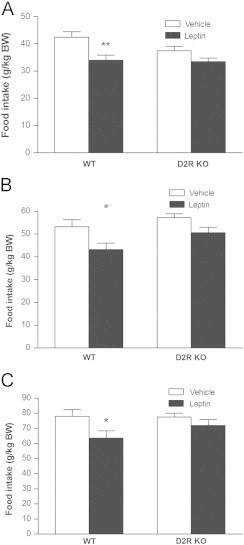
Hypophagic effect of leptin is attenuated in mice lacking the D2R. Leptin significantly decreased food intake in wild type but not D2R KO mice at (A) 1h post injection, (B) 2 h post injection and (C) 4 h post injection. (Two way ANOVA, Bonferroni post hoc test, *P<0.05, **P<0.01 for leptin vs. vehicle; n=11–13 per group). Food intake was normalized to body weight because the D2 RKO mice are smaller than WT mice.
3.4. The hypophagic effect of MTII does not require dopamine signaling
These acute feeding studies demonstrate, via pharmacological and genetic manipulations, that the D2R partially mediates the acute hypophagic effect of leptin. Because leptin directly stimulates POMC neurons [1], which project to mesolimbic dopaminergic structures like the VTA, [16,17], it is possible that leptin influences midbrain dopamine signaling indirectly via the melanocortin system. To determine if dopamine is required for the acute reduction in feeding caused by stimulation of melanocortin signaling, the effect of D2R or D1R blockade on the acute hypophagic effect of a melanocortin agonist was studied. Overnight fasted mice were pretreated with either the D2R antagonist L741 (1.5 mg/kg) or the D1R-like antagonist SKF (0.25 mg/kg), or vehicle, followed 15 min later by the melanocortin agonist, melanotan-II (MTII; 1 mg/kg). MTII significantly reduced food intake at 1 h post injection in all treatment groups (Fig. 3A) and also at 2 h (Fig. 3B, D; P<0.001) and 4 h (Fig. 3C,F; P<0.01) post injection. Neither D1R nor D2R receptor blockade attenuated the acute anorectic effect of MTII at any later time points (data not shown).
Fig. 3.

Neither D2R nor D1R blockade affects the acute hypophagic effect of MTII. Pretreatment with a D2R antagonist (L741; 1.5 mg/kg) or D1R antagonist (SKF; 0.25 mg/kg) did not affect the reduction in food intake caused by MTII (1 mg/kg) at (A) 1 h post injection, (B) 2 h post injection, (C) 4h post injection. (Two way ANOVA, Bonferroni post hoc test, **P<0.01, ***P<0.001 for treatment vs. vehicle; n=9–11 per group).
3.5. The D2R mediates the hypophagic effect of a dopamine agonist
The D2R appears necessary for dopamine's hypophagic effect, because D2R antagonism with L741 also blocked the hypophagic effect of the broad spectrum (D1R and D2R) dopamine agonist, pergolide, at 1 h (Fig. 4A); pergolide did not significantly reduce food intake at 2 h (Fig. 4C). Blockade of D1R did not attenuate the hypophagic effect of pergolide at 1 h (Fig. 4B) and potentiated the anorectic effect of pergolide as evidenced by a reduction in food intake at 2 h post injection (Fig. 4D). There was no effect of any drug on food intake at 4 h post injection Fig. 4E, F).
Fig. 4.
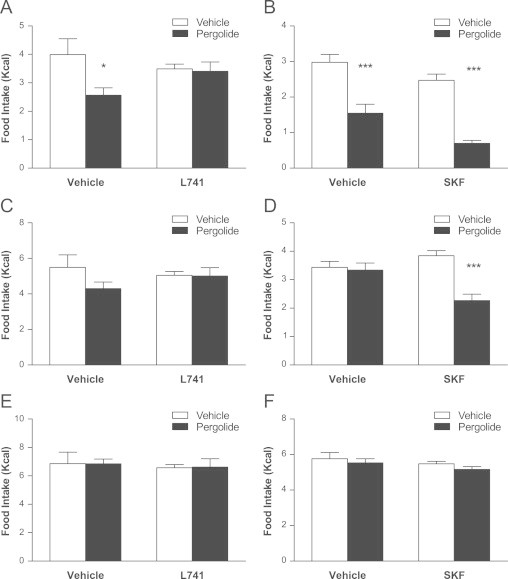
D2R reduces the hypophagic effect of the dopamine agonist, pergolide. Pretreatment with the D2R antagonist (L741; 1.5 mg/kg) blocked the reduction in food intake caused by pergolide (0.1 mg/kg) at (A) 1 h post injection. Pergolide did not reduce food intake at (C) 2 h post injection or (E) 4 h post injection. Pretreatment with the D1R antagonist (SKF; 0.25 mg/kg) was associated with significant reductions in food intake only in the presence of pergolide at (A) 1 h post injection and (B) 2 h post injection but not at (C) 4 h post injection. (Two way ANOVA, Bonferroni post hoc test, ***P<0.001 for treatment vs. vehicle; L741 n=6 per group. SKF n=12 per group).
3.6. Ghrelin does not require the D1R or D2R to increase food intake
After determining that leptin requires the D2R to modulate some of its anorexigenic actions, we tested whether dopamine signaling is required for the hyperphagic effect of the hormone, ghrelin. In wild type mice, either the D1R (SKF; 0.25 mg/kg) or D2R (L741;1.5 mg/kg) antagonist was administered followed by ghrelin (0.36 mg/kg) 15 min later. Ghrelin significantly increased food intake at 1 h post injection (Fig. 5A), and this effect was not affected by pretreatment with either D1R or D2R antagonists (Fig. 5A). The hyperphagic effect of ghrelin was evident in SKF treated mice at up to 2 h post injection (Fig. 5B) but was gone by 4 h post injection (Fig. 5C; 8 h and 24 h data not shown). These results suggest that unlike leptin, ghrelin does not require functional D1R or D2Rs.
Fig. 5.
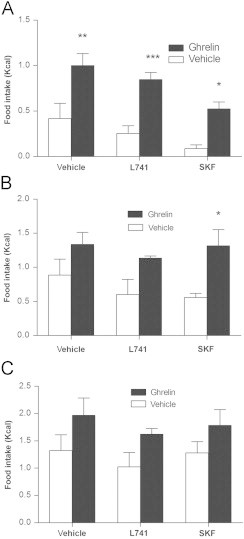
Neither D2R nor D1R blockade affects the acute hyperphagic effect of ghrelin. Pretreatment with either the D2R antagonist (L741; 1.5 mg/kg) or D1R antagonist (SKF; 0.25 mg/kg) did not affect the hyperphagic efficacy of ghrelin (0.36 mg/kg) at (A) 1 h post injection, (B) 2 h post injection, (C) 4 h post injection. (Two way ANOVA, Bonferroni post hoc test, *P<0.05, **P<0.01, ***P<0.001 for treatment vs. vehicle; n=4–5 per group).
To further test this effect, D2R KO mice and their WT littermates where treated with systemic ghrelin (0.36 mg/kg). Ghrelin significantly elevated food intake in both D2R KO and WT mice at 1 h, 2 h and 4 h post injection (Fig. 6A–C) but not at 8 h and 24 h (data not shown).
Fig. 6.
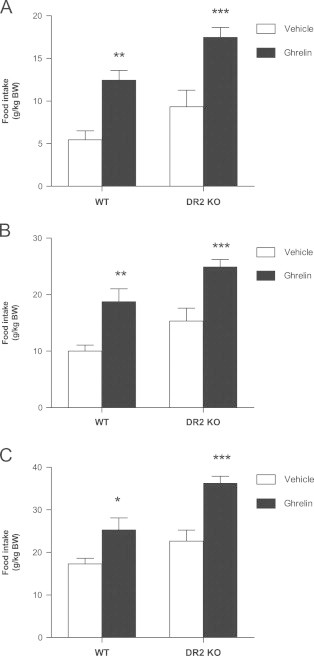
Hyperphagic effect of ghrelin is maintained in mice lacking the D2R. Ghrelin (0.36 mg/kg) significantly increased food intake in both wild type and D2R KO mice at (A) 1 h post injection, (B) 2 h post injection, (C) 4h post injection. Two way ANOVA, Bonferroni post hoc test, *P<0.05, **P<0.01, ***P<0.001 for treatment vs. vehicle; n=6–9 per group).
Together these results suggest that D2Rs are critical mediators of leptin's anorexigenic actions and that neither the D1R nor the D2R are required for ghrelin to induce hyperphagia.
4. Discussion
By demonstrating that dopaminergic signaling is necessary for a significant part of the acute inhibitory effect of leptin on deprivation-induced intake of normal diet, this study provides evidence for an extra-hypothalamic site of leptin action that regulates homeostatic feeding. We rule out the possibility that dopaminergic signaling mediates the acute anorectic effect of melanocortin stimulation, showing that the dopamine-dependent reduction in feeding caused by leptin involves systems other than the melanocortin system. Furthermore, we show that the increase in food intake caused by the hyperphagic hormone, ghrelin, does not appear to require dopamine signaling. This work strengthens growing evidence that mesolimbic dopaminergic signaling contributes to homeostatic food intake and body weight regulation [6,7,18].
Although traditionally believed to primarily regulate locomotion and reward, mesolimbic dopamine systems are increasingly demonstrated to be involved in regulation of energy balance. For example, leptin, ghrelin, and insulin are all known to influence energy balance via hypothalamic actions; however dopamine neurons in the VTA express receptors for leptin, insulin, and ghrelin [6,7,11,19–22], and recent evidence also demonstrates that mesolimbic dopamine neurons respond to caloric availability [18]. Furthermore, leptin and ghrelin signaling in the VTA can influence short-term feeding in mice, and midbrain leptin signaling is important for locomotor sensitization to amphetamine [6,7,19]. In humans, leptin and ghrelin can influence striatal activity in response to food images [12,23,24]. Recent work by Davis and colleagues indicate that hypothalamic leptin actions reduce overconsumption of a palatable diet while midbrain leptin regulates responding for food reward [25]. One interpretation of this data is that mesolimbic dopamine signaling regulates hedonic feeding while homeostatic feeding is regulated by the melanocortin system [26]. This idea stems from the role of mesolimbic signaling in drug addiction and reward [17] and the role of the basomedial hypothalamus in energy homeostasis [27]. According to this theory, mesolimbic systems are separate and ancillary to the primary role of the mediobasal hypothalamus in regulation of energy intake and expenditure.
Recent discoveries dispute even this dual-purpose hypothesis for feeding regulation, and contest the autonomy of the melanocortin system in homeostatic feeding regulation. One such study demonstrated that young mice lacking the MC4 receptor have a normal hypophagic response to peripheral leptin [28]. This finding is consistent with a recent study in which van de Wall and colleagues demonstrated that adult mice with genetic deletion of the leptin receptor in POMC and AgRP/NPY neurons maintain a partial response to the hypophagic effect of leptin [29]. Additionally, in obese leptin receptor deficient mice, targeted re-expression of the leptin receptor in POMC cells only produces modest weight-loss [30]. These findings agree with the conclusion that leptin can regulate feeding via extrahypothalamic pathways, one of which may be the mesolimbic dopamine system. Furthermore, mesolimbic dopamine neurons may confer information about energy availability in the absence of taste feedback, which is generally assumed to be important for reward-based feeding. de Araujo and colleagues demonstrated that mice unable to taste sucrose still preferred to consume sucrose-containing solution and that ingestion of the sucrose solution was associated with increased dopamine release in the midbrain [18]. Taken together, these studies challenge the idea that mesolimbic dopamine signaling only mediates behavior motivated by acute sensory reward, and suggests a role for this system in homeostatic regulation of food intake.
In addition to identifying the critical role of dopamine in acute regulation of feeding by leptin, the current study implicates the D2R specifically. Systemic administration of dopamine receptor antagonists prior to leptin treatment demonstrated that the D2R but not the D1R is important for part of the acute effect of leptin on feeding. Doses of the D1R and D2R antagonists were selected that did not affect food intake, indicating that the effect on leptin induced hypophagia was probably not due to an effect of the antagonist itself. This finding is supported by the finding that D2R KO mice had a diminished acute response to leptin. The results from these studies are in agreement with other data demonstrating that stimulation of the D2R has an overall anorectic effect, while antagonism of the D2R is associated with hyperphagia and weight gain [31]. The finding that leptin administration can modulate striatal D2R binding in mice [32] suggests that the present findings may be due to a striatal effect of leptin.
Seemingly in contrast to our results, another group concluded that D2R KO mice exhibited increased sensitivity to centrally administered leptin, and that leptin action is opposed by the central administration of the D2R antagonist haloperidol [33]. These divergent findings may be accounted for by differences in methodology. Kim et al administered leptin and the D2R antagonist centrally (in order to target the hypothalamus). In the present study, leptin and dopamine receptor antagonists were administered systemically. Thus, while we cannot rule out the exact location of the effects, we may be reporting results that depend on the location of action. We also normalized food intake to body weight because the D2R KO mice are significantly smaller than WT mice; this is a transformation that some investigators have questioned [34]. Further studies may elucidate the differences between the datasets.
Systemic administration of a D1R antagonist had no effect on the acute anorectic effect of leptin and had a tendency to slightly decrease food intake; consistent with other data demonstrating that systemic D1R blockade can produce small reductions in feeding [35,36]. We believe that the dose of SKF was sufficient to achieve D1R blockade, as the dose was determined based on dose-response studies conducted to determine the highest drug dose that did not significantly affect behavior or food intake. Furthermore, D1R blockade appeared to potentiate the reduction in food intake caused by both leptin and pergolide.
Although many of ghrelin's effects oppose that of leptin, there is an important divergence in the actions of these hormones. Ghrelin does not merely produce opposite effects from leptin in the same neural pathways. Consistent with this, pharmacological and genetic D2R blockade failed to reduce the increased food intake caused by ghrelin. It is known that ghrelin requires intact hypothalamic pathways to increase food intake [37], the present data shows that ghrelin does not require dopamine signaling.
Antipsychotics, which are D2R antagonists, are associated with increased appetite, weight gain, and increased risk for insulin resistance, hyperglycemia, dyslipidemia and type II diabetes [38–40]. Conversely, administration of D2R agonists to obese humans decreases circulating leptin levels, elevates metabolism, reduces blood glucose and insulin, and decreases blood pressure [41,42]. We have shown that D2R antagonism can acutely reduce the normal response to leptin. It is possible that chronic D2R antagonism may chronically reduce leptin sensitivity, which may influence body weight control during antipsychotic treatment.
Leptin-dopamine interactions that influence various aspects of feeding behavior have been documented in other regions of the brain. The hypothalamus contains both dopamine and leptin receptors and increased dopaminergic activity in the ARH and lateral hypothalamic area (LHA) is associated with decreased food intake [36,43]. In leptin-deficient mice, leptin activation of LHA, but not VTA, neurons was shown to decrease feeding by restoring mesolimbic dopamine [44]. This finding suggests that the site of leptin action in regulating homeostatic feeding may be the LHA, however the effect of chronic leptin deficiency on this result is not known. Although we interpret the current data to describe mesolimbic dopamine actions, it remains possible that some of these effects of dopamine signaling may occur in the hypothalamus or other brain regions. Further studies involving localized dopamine receptor blockade in the hypothalamus or other brain regions may shed light on the location of this leptin-dopamine interaction.
Because dopamine is involved in food reward and motivation, many studies focus on the role of dopamine in mediating reward-based feeding, including acquisition of flavor preference [45],food addiction and overconsumption of calorically dense food [46] and salience [47]. Our studies address a different aspect of feeding behavior, the role of dopamine receptor signaling on leptin's effects on intake of standard diet following a fast. Certainly, the role of dopamine in regulation of food intake could be influenced by many factors such as food palatability (normal vs. high-fat or high-sugar chow), fasting, obesity, and species. The present studies do not address these aspects of feeding. Additionally, brain reward circuitry and dopamine receptor expression can be modified by chronic food restriction [48], thus overnight fasting may have influenced outcomes of this study, as there have been reports of increased D2R expression in food restricted states [49]. Chronic obesity is associated with altered D2R availability [50,51], so it is possible that the response to D1R or D2R antagonism would be different in obese mice, although in our preliminary studies, both lean and obese mice exhibited the same response to leptin in the presence of dopamine receptor blockade. Of course, it is well documented that leptin and ghrelin have reduced efficacy in DIO animals [52–54], so the design of such further studies will be challenging.
This study presents novel evidence that the dopamine system is a potential mediator of leptin effects on energy balance, and that dopamine signaling is not critical to melanocortin regulation of food intake. This highlights an important divergence between the pathways utilized by leptin or ghrelin, which appears to depend on ARH circuits [37]. Although dopaminergic systems are known to influence reward-based feeding [55], the current findings strengthen the idea that mesolimbic dopamine systems are important for homeostatic regulation of feeding. They also encourage further research on the contribution of mesolimbic neurocircuitry to physiologic feeding regulation and how they may contribute to obesity.
Acknowledgments
The authors thank Dr. David K. Grandy for the generous donation of D2R KO mice. This work was funded by the US National Institute of Health grants RR01163 and DK62202, by the National Health and Medical Research Foundation of Australia, National Heart Foundation of Australia and Pfizer Australia.
References
- 1.Cowley M.A. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411(6836):480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz M.W. Identification of targets of leptin action in rat hypothalamus. The Journal of Clinical Investigation. 1996;98(5):1101–1106. doi: 10.1172/JCI118891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartz M.W. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46(12):2119–2123. doi: 10.2337/diab.46.12.2119. [DOI] [PubMed] [Google Scholar]
- 4.Elias C.F. Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron. 1998;21(6):1375–1385. doi: 10.1016/s0896-6273(00)80656-x. [DOI] [PubMed] [Google Scholar]
- 5.Stephens T.W. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature. 1995;377(6549):530–532. doi: 10.1038/377530a0. [DOI] [PubMed] [Google Scholar]
- 6.Fulton S. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron. 2006;51(6):811–822. doi: 10.1016/j.neuron.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Hommel J.D. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51(6):801–810. doi: 10.1016/j.neuron.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 8.Maffei M. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nature Medicine. 1995;1(11):1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 10.Halaas J.L. Weight-reducing effects of the plasma protein encoded by the obese gene. Science (New York, NY) 1995;269(5223):543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 11.Figlewicz D.P. Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat. Brain Research. 2003;964(1):107–115. doi: 10.1016/s0006-8993(02)04087-8. [DOI] [PubMed] [Google Scholar]
- 12.Farooqi I.S. Leptin regulates striatal regions and human eating behavior. Science (New York, NY) 2007;317(5843):1355. doi: 10.1126/science.1144599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Missale C. Dopamine receptors: from structure to function. Physiological Reviews. 1998;78(1):189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 14.Kelly M.A. Pituitary lactotroph hyperplasia and chronic hyperprolactinemia in dopamine D2 receptor-deficient mice. Neuron. 1997;19(1):103–113. doi: 10.1016/s0896-6273(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Tornadu I. New insights into the endocrine and metabolic roles of dopamine D2 receptors gained from the Drd2 mouse. Neuroendocrinology. 2010;92(4):207–214. doi: 10.1159/000321395. [DOI] [PubMed] [Google Scholar]
- 16.Hsu R. Blockade of melanocortin transmission inhibits cocaine reward. The European Journal of Neuroscience. 2005;21(8):2233–2242. doi: 10.1111/j.1460-9568.2005.04038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelley A.E., Berridge K.C. The neuroscience of natural rewards: relevance to addictive drugs. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience. 2002;22(9):3306–3311. doi: 10.1523/JNEUROSCI.22-09-03306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Araujo I.E. Food reward in the absence of taste receptor signaling. Neuron. 2008;57(6):930–941. doi: 10.1016/j.neuron.2008.01.032. [DOI] [PubMed] [Google Scholar]
- 19.Abizaid A. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. The Journal of Clinical Investigation. 2006;116(12):3229–3239. doi: 10.1172/JCI29867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pardini A.W. Distribution of insulin receptor substrate-2 in brain areas involved in energy homeostasis. Brain Research. 2006;1112(1):169–178. doi: 10.1016/j.brainres.2006.06.109. [DOI] [PubMed] [Google Scholar]
- 21.Scott M.M. Leptin targets in the mouse brain. The Journal of Comparative Neurology. 2009;514(5):518–532. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan X.M. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Research Molecular Brain Research. 1997;48(1):23–29. doi: 10.1016/s0169-328x(97)00071-5. [DOI] [PubMed] [Google Scholar]
- 23.Malik S. Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metabolism. 2008;7(5):400–409. doi: 10.1016/j.cmet.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Baicy K. Leptin replacement alters brain response to food cues in genetically leptin-deficient adults. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(46):18276–18279. doi: 10.1073/pnas.0706481104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis J.F. Leptin regulates energy balance and motivation through action at distinct neural circuits. Biological Psychiatry. 2011;69(7):668–674. doi: 10.1016/j.biopsych.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saper C.B., Chou T.C., Elmquist J.K. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002;36(2):199–211. doi: 10.1016/s0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 27.Cone R.D. Anatomy and regulation of the central melanocortin system. Nature Neuroscience. 2005;8(5):571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 28.Marsh D.J. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nature Genetics. 1999;21(1):119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- 29.van de Wall E. Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology. 2008;149(4):1773–1785. doi: 10.1210/en.2007-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berglund E.D. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. The Journal of Clinical Investigation. 2012;122(3):1000–1009. doi: 10.1172/JCI59816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pijl H. Reduced dopaminergic tone in hypothalamic neural circuits: expression of a thrifty genotype underlying the metabolic syndrome? European Journal of Pharmacology. 2003;480(1–3):125–131. doi: 10.1016/j.ejphar.2003.08.100. [DOI] [PubMed] [Google Scholar]
- 32.Pfaffly J. Leptin increases striatal dopamine D2 receptor binding in leptin-deficient obese (ob/ob) mice. Synapse (New York, NY) 2010;64(7):503–510. doi: 10.1002/syn.20755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim K.S. Enhanced hypothalamic leptin signaling in mice lacking dopamine D2 receptors. The Journal of Biological Chemistry. 2010;285(12):8905–8917. doi: 10.1074/jbc.M109.079590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butler A.A., Kozak L.P. A recurring problem with the analysis of energy expenditure in genetic models expressing lean and obese phenotypes. Diabetes. 2010;59(2):323–329. doi: 10.2337/db09-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clifton P.G., Rusk I.N., Cooper S.J. Effects of dopamine D1 and dopamine D2 antagonists on the free feeding and drinking patterns of rats. Behavioral Neuroscience. 1991;105(2):272–281. doi: 10.1037//0735-7044.105.2.272. [DOI] [PubMed] [Google Scholar]
- 36.Rusk I.N., Cooper S.J. Parametric studies of selective D1 or D2 antagonists: effects on appetitive and feeding behaviour. Behavioural Pharmacology. 1994;5(6):615–622. doi: 10.1097/00008877-199410000-00007. [DOI] [PubMed] [Google Scholar]
- 37.Chen H.Y. Orexigenic action of peripheral ghrelin is mediated by neuropeptide Y and agouti-related protein. Endocrinology. 2004;145(6):2607–2612. doi: 10.1210/en.2003-1596. [DOI] [PubMed] [Google Scholar]
- 38.Newcomer J.W. Metabolic considerations in the use of antipsychotic medications: a review of recent evidence. The Journal of Clinical Psychiatry. 2007;68(Suppl 1):20–27. [PubMed] [Google Scholar]
- 39.Correll C.U., Malhotra A.K. Pharmacogenetics of antipsychotic-induced weight gain. Psychopharmacology (Berl) 2004;174(4):477–489. doi: 10.1007/s00213-004-1949-9. [DOI] [PubMed] [Google Scholar]
- 40.Birt J. Management of weight gain associated with antipsychotics. Annals of Clinical Psychiatry: Official Journal of the American Academy of Clinical Psychiatrists. 2003;15(1):49–58. doi: 10.1023/a:1023280610379. [DOI] [PubMed] [Google Scholar]
- 41.Kok P. Activation of dopamine D2 receptors simultaneously ameliorates various metabolic features of obese women. American Journal of Physiology, Endocrinology and metabolism. 2006;291(5):E1038–E1043. doi: 10.1152/ajpendo.00567.2005. [DOI] [PubMed] [Google Scholar]
- 42.Kok P. Activation of dopamine D2 receptors lowers circadian leptin concentrations in obese women. The Journal of Clinical Endocrinology and Metabolism. 2006;91(8):3236–3240. doi: 10.1210/jc.2005-2529. [DOI] [PubMed] [Google Scholar]
- 43.Yang Z.J. Bilateral hypothalamic dopamine infusion in male Zucker rat suppresses feeding due to reduced meal size. Pharmacology, Biochemistry and Behavior. 1997;58(3):631–635. doi: 10.1016/s0091-3057(97)00022-1. [DOI] [PubMed] [Google Scholar]
- 44.Leinninger G.M. Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metabolism. 2009;10(2):89–98. doi: 10.1016/j.cmet.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Touzani K., Bodnar R.J., Sclafani A. Neuropharmacology of learned flavor preferences. Pharmacology, Biochemistry, and Behavior. 2010;97(1):55–62. doi: 10.1016/j.pbb.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Avena N.M., Rada P., Hoebel B.G. Sugar and fat bingeing have notable differences in addictive-like behavior. The Journal of Nutrition. 2009;139(3):623–628. doi: 10.3945/jn.108.097584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berridge K.C., Robinson T.E. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Research Brain Research Reviews. 1998;28(3):309–369. doi: 10.1016/s0165-0173(98)00019-8. [DOI] [PubMed] [Google Scholar]
- 48.Figlewicz D.P., Benoit S.C. Insulin, leptin, and food reward: update 2008. American Journal of Physiology Regulatory, Integrative and Comparative Physiology. 2009;296(1):R9–R19. doi: 10.1152/ajpregu.90725.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thanos P.K. Food restriction markedly increases dopamine D2 receptor (D2R) in a rat model of obesity as assessed with in-vivo muPET imaging ([11C] raclopride) and in-vitro ([3H] spiperone) autoradiography. Synapse (New York, NY) 2008;62(1):50–61. doi: 10.1002/syn.20468. [DOI] [PubMed] [Google Scholar]
- 50.Wang G.J. Brain dopamine and obesity. Lancet. 2001;357(9253):354–357. doi: 10.1016/s0140-6736(00)03643-6. [DOI] [PubMed] [Google Scholar]
- 51.Volkow N.D. Low dopamine striatal D2 receptors are associated with prefrontal metabolism in obese subjects: possible contributing factors. Neuroimage. 2008;42(4):1537–1543. doi: 10.1016/j.neuroimage.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Briggs D.I. Diet-induced obesity causes ghrelin resistance in arcuate NPY/AgRP neurons. Endocrinology. 2010;151(10):4745–4755. doi: 10.1210/en.2010-0556. [DOI] [PubMed] [Google Scholar]
- 53.Enriori P.J. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metabolism. 2007;5(3):181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 54.Enriori P.J. Leptin action in the dorsomedial hypothalamus increases sympathetic tone to brown adipose tissue in spite of systemic leptin resistance. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2011;31(34):12189–12197. doi: 10.1523/JNEUROSCI.2336-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Berthoud H.R. Interactions between the cognitive and metabolic brain in the control of food intake. Physiology and Behavior. 2007;91(5):486–498. doi: 10.1016/j.physbeh.2006.12.016. [DOI] [PubMed] [Google Scholar]